

Abstract
Neuronal apoptosis plays a critical role in the normal development of the mammalian brain and is thought to contribute to the pathogenesis of several neurologic disorders. However, the intracellular mechanisms underlying apoptosis of neurons remain incompletely understood. In the present study, we characterized a cell-cycle-based mechanism by which neuronal activity deprivation induces apoptosis of postmitotic neurons. Activity deprivation, but not growth factor withdrawal, was found to induce Cdc2 expression and consequent Cdc2-mediated apoptosis in granule neurons of the developing rat cerebellum. We found that activity deprivation induces cdc2 transcription in neurons via an E2F-binding element (EBE) within the cdc2 promoter. The transcription factor E2F1 that is expressed in granule neurons was found in DNA binding assays to bind to the EBE of the cdc2gene. In chromatin immunoprecipitation analysis, endogenous E2F1 forms a complex with the promoter of the endogenous cdc2 gene in granule neurons, indicating that endogenous E2F1 is poised to activate transcription of the endogenous cdc2 gene in neurons. Consistent with this conclusion, a dominant interfering form of E2F, when expressed in granule neurons, blocked activity deprivation-inducedcdc2 transcription. In other experiments, we found that the expression of E2F1 in granule neurons induces Cdc2 expression and promotes neuronal apoptosis via the activation of Cdc2. Remarkably, in contrast to inducing the E2F-mediated expression and activation of Cdc2 in granule neurons, activity deprivation fails to stimulate the expression of E2F-target genes that trigger DNA synthesis and replication. Together, our findings define a novel apoptotic mechanism whereby E2F selectively couples an activity deprivation-induced signal to cdc2 transcription in the absence of stimulating DNA synthesis and thus culminating in Cdc2-mediated apoptosis of postmitotic neurons.
Keywords: Cdc2, E2F, apoptosis, neuron, activity, transcription, cell cycle, promoter
Introduction
During the development of the mammalian brain, the generation of neurons from their precursor cells coincides with their exit from the cell cycle (Caviness et al., 1995;McConnell, 1995). Although postmitotic neurons do not undergo cell division, they continue to express components of the cell cycle for some time after their terminal differentiation (Freeman et al., 1994;Vincent et al., 1997; Busser et al., 1998; Padmanabhan et al., 1999). Accumulating evidence suggests that these cell-cycle proteins play a key role in programmed cell death or apoptosis of postmitotic neurons that is critical to the normal development of the mammalian brain (for review, see Liu and Greene, 2001a). However, the intracellular mechanisms that trigger reactivation of components of the cell cycle in postmitotic neurons remain to be elucidated. In addition, the mechanisms by which cell-cycle proteins induce apoptosis of neurons remain incompletely understood.
Within the developing brain, the cerebellar granule neuron population has been widely used to characterize the cellular and molecular mechanisms of neuronal apoptosis. Proliferating granule neuron precursors differentiate into postmitotic granule neurons within the external granule cell layer (EGL) of the developing cerebellum (Hatten and Heintz, 1995). Newly generated granule neurons migrate from the EGL into the underlying internal granule cell layer (IGL) where they take on the properties of mature granule neurons (Altman and Bayer, 1997). Granule neuron apoptosis is observed in both the EGL and IGL, peaking in the first to second week of the postnatal rat cerebellum (Wood et al., 1993).
Granule neurons cultured from the rat cerebellum undergo apoptosis that is suppressed by neuronal activity and growth factors (D'Mello et al., 1993, 1997; Galli et al., 1995; Miller and Johnson, 1996). Growth factors are typically provided by serum or by a high concentration of insulin that activates the insulin-like growth factor 1 (IGF1) receptor (Cohick and Clemmons, 1993; Alessi et al., 1996). Neuronal activity is mimicked by a high concentration of extracellular KCl that triggers membrane depolarization leading to the entry of calcium through voltage-sensitive calcium channels (VSCCs) (Catterall, 2000). Granule neurons undergo apoptosis when they are deprived of membrane-depolarizing concentrations of KCl or when they are deprived of growth factors. The mechanisms by which activity deprivation or growth factor withdrawal induces apoptosis are beginning to be characterized (Watson et al., 1998; Levkovitz and Baraban, 2001;Yamagishi et al., 2001; Putcha et al., 2002). An important question that remains to be addressed is whether there is specificity in the mechanisms that mediate activity deprivation-induced or growth factor withdrawal-induced neuronal apoptosis.
Recently, we reported that in newly differentiated cerebellar granule neurons, neuronal activity deprivation triggers the activation of the protein kinase Cdc2, a cyclin-dependent kinase with an established function in driving proliferating cells through mitosis (Konishi et al., 2002). In granule neurons, activated Cdc2 mediates activity deprivation-induced apoptosis by directly phosphorylating the BH3-only protein Bcl-2-associated death promoter (BAD) at a distinct site, serine 128, and thereby activating BAD-mediated apoptosis (Konishi et al., 2002). Thus, reactivation of the cell-cycle component Cdc2 by neuronal activity deprivation directly activates the cell death machinery. A major question that was raised by these findings is: how is Cdc2 activity regulated in postmitotic neurons? In particular, how does neuronal activity deprivation induce the activity of Cdc2 in postmitotic neurons, thereby triggering apoptosis?
In this study, we characterized a specific mechanism by which activity deprivation induces neuronal apoptosis. We found that activity deprivation, but not growth factor withdrawal, induces the expression of Cdc2 in granule neurons, and that Cdc2 activity specifically mediates activity deprivation-induced neuronal apoptosis. Activity deprivation was found to induce cdc2 transcription in granule neurons via a regulatory site within the cdc2promoter that binds the transcription factor E2F1. In chromatin immunoprecipitation analyses, we found that endogenous E2F1 in cerebellar granule neurons forms a complex with the promoter of the endogenous cdc2 gene, suggesting that the cdc2gene is a target of endogenous E2F1 in cerebellar granule neurons. Consistent with this interpretation, the expression of a dominant interfering form of E2F was found to inhibit activity deprivation-induced cdc2 transcription in granule neurons. In other experiments, we found that the expression of E2F1 in granule neurons induces the expression of endogenous Cdc2 and triggers apoptosis. The E2F1-induced apoptosis was found to be reduced significantly by coexpression of a dominant interfering form of Cdc2. Together, our findings define the E2F–Cdc2 cell-cycle pathway as a novel and specific mechanism by which activity deprivation induces apoptosis of postmitotic neurons. In proliferating cells, E2F is believed to regulate the expression of a large set of genes that drive these cells through the cell cycle (DeGregori et al., 1995; Dyson, 1998; Harbour and Dean, 2000; Ishida et al., 2001; Ren et al., 2002). In contrast, activity deprivation of granule neurons fails to induce the set of E2F-regulated genes serving a role in DNA synthesis and replication. We conclude that the cdc2 gene represents a select target of E2F in activity-deprived postmitotic neurons, and that the activation of Cdc2 in the absence of DNA synthesis in postmitotic neurons might signal apoptosis in a similar manner to unscheduled Cdc2 activation in proliferating cells (Shi et al., 1994).
Materials and Methods
Antibodies. Mouse monoclonal antibodies to Cdc2 and actin and rabbit polyclonal antibody to BAD, E2F1, proliferating cell nuclear antigen (PCNA), Cdk2, and hemagglutinin (HA) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The mouse monoclonal antibody to β-galactosidase (Promega, Madison, WI) and cleaved caspase-3 (Cell Signaling, Beverly, MA) were also purchased. The phospho128-BAD antibody was described previously (Konishi et al., 2002).
Cerebellar granule neuron cultures and transfections.Cerebellar granule neurons were prepared as described previously (Bonni et al., 1999). One day after preparation, cytosine arabinofuranoside (AraC) (10 μm) was added to inhibit proliferation of non-neuronal cells. Transfection of granule neuron cultures was done using a modified calcium phosphate method as described previously (Konishi et al., 2002). In the experiments in Figure 2A, 0.1 μg of cdc2-firefly luciferase reporter plasmid and 0.02 μg of elongation factor (EF)-renilla luciferase together with 0.5 μg of carrier plasmid were transfected. In the experiments in Figure 3A, 0.1 μg ofcdc2-firefly luciferase plasmid, 0.02 μg of EF-renilla luciferase plasmid, and 1 μg of E2F1 expression plasmid were transfected. In the experiments in Figure 3, B andC, 1 μg of green fluorescent protein (GFP) plasmid, 0.25 μg of β-galactosidase expression plasmid, and 1 μg of E2F1 expression plasmid were transfected. In the experiments in Figure4A–G, 2 μg of E2F1 expression plasmid was transfected. In the experiments in Figure 4I, 0.1 μg of cdc2-firefly luciferase plasmid, 0.02 μg of EF-renilla luciferase, and 0.1 μg of E2F-Rb expression plasmid together with 0.5 μg of carrier plasmid were transfected. In the experiments in Figure 4J, 1 μg of E2F-Rb or the vector plasmid together with 0.25 μg of β-galactosidase expression plasmid were transfected. In the experiments in Figure5A–E, 0.5 μg of E2F1 expression plasmid and 0.25 μg of β-galactosidase expression plasmid were transfected. In the experiments in Figure 5F, 0.5 μg of E2F1 expression plasmid, 0.5 μg of a dominant interfering form of Cdc2 (Cdc2-DN) expression plasmid, and 0.25 μg of β-galactosidase expression plasmid were transfected.
Fig. 2.

Activity deprivation induces cdc2transcription via an EBE within the cdc2 promoter.A, Schematic representation of cdc2promoter–luciferase reporter constructs and their activity in cerebellar granule neurons. Cerebellar granule neurons (P6 plus 2 DIV) were transfected with the indicated cdc2 firefly-luciferase reporter plasmid and reporter gene that is controlled by the elongation factor promoter (EF-renilla) and incubated for 4–5 hr in conditioned full medium (30 mm KCl plus serum). Transfected neurons were then placed in full medium (30 mm KCl plus serum;black bars) or deprived of neuronal activity (5 mm KCl plus serum; gray bars) for 36 hr and then subjected to dual-luciferase assay (Promega). The normalized firefly-luciferase activity of each cdc2 luciferase reporter gene is shown relative to the activity of −94/cdc2luciferase gene in membrane-depolarized granule neuron cultures. Values shown are mean ± SEM (n = 4). Activity deprivation significantly increased the expression of the −3200/cdc2 and −245/cdc2 reporter genes (ANOVA;p < 0.05) but not of the −245mEBE/cdc2or the −94/cdc2 reporter genes. B, Nuclear extracts prepared from cerebellar granule neuron cultures (P6 plus 2 DIV) maintained in full survival medium (30 mm KCl plus serum) or deprived of neuronal activity (5 mm KCl plus serum) for 24 hr were subjected to an EMSA. The radiolabeled probe contains the EBE within the cdc2 promoter. For competition analysis, excess amount of wild-type cold probe (lanes 4and 5) or a probe that contains a point mutation within the EBE (lanes 6 and 7) was added to the reaction. C, A rabbit antibody to E2F1 (2 μg,lane 3; 5 μg, lane 4) or a control antibody (anti-HA antibody, 2 μg, lane 5; 5 μg, lane 6) was added to the EMSA reaction. Three protein–DNA complexes that are shown as specific EBE–protein complexes in B (arrowheads) were blocked or super-shifted (bracket) by the addition of E2F1 antibody but not by control antibody.
Fig. 3.
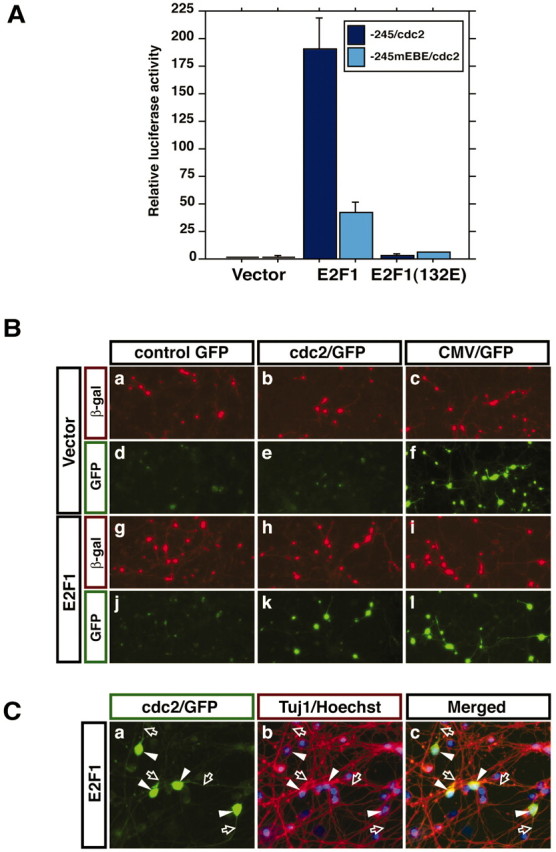
E2F1 activates cdc2 promoter-mediated transcription in postmitotic cerebellar granule neurons.A, Cerebellar neuron cultures (P6 plus 2 DIV) were transfected with the −245/cdc2 or −245/mEBE/cdc2 luciferase reporter plasmids together with the EF-renilla reporter gene and an expression plasmid encoding E2F1, an E2F1 mutant in which the DNA-binding domain is mutated [E2F1(132E)], or their control vector. One day after transfection, transfected cells were harvested and subjected to a dual-luciferase assay. Normalized reporter activities are shown relative to the reporter activity of the −245/cdc2 luciferase gene that was cotransfected with the vector control plasmid. Values shown are mean ± SEM (n = 4). The activity of the −245/cdc2 reporter gene was significantly increased by E2F1 (ANOVA; p < 0.001) but not by E2F1(132E). Mutation within the EBE (−245/mEBE/cdc2) significantly decreased E2F1 induction of cdc2 transcription (ANOVA;p < 0.001). B, Cerebellar granule neurons were transfected with the control GFP, cdc2/GFP reporter gene, or CMV/GFP reporter gene together with an expression plasmid encoding E2F1 or its control vector and an expression plasmid encoding β-galactosidase (β-gal). Two days after transfection, cells were subjected to indirect immunofluorescence with an antibody to β-galactosidase, and GFP reporter gene activity was monitored by visualizing GFP in transfected cells. E2F1 induced thecdc2/GFP reporter. C, Cerebellar granule neuron cultures were transfected with the E2F1-expression vector together with the cdc2/GFP reporter gene. Transfected cultures were subjected to indirect immunofluorescence with an antibody to β-tubulin type III (Tuj1) that is expressed in postmitotic neurons, and GFP visualization was used to monitor thecdc2/GFP reporter activity. Cell bodies (arrowheads) and neurites (open arrows) of all GFP-positive cells also stained with the Tuj1 antibody.
Fig. 4.

E2F1 mediates activity deprivation-inducedcdc2 transcription and apoptosis in cerebellar granule neurons. A–F, Cerebellar granule neurons (P6 plus 2 DIV) were transfected with an expression plasmid encoding HA-tagged E2F1 or HA-tagged E2F1(132E). Two days after transfection, cultures were subjected to indirect immunofluorescence using a rabbit anti-HA antibody and the mouse monoclonal antibody to Cdc2 together with the DNA dye bisbenzimide (Hoechst 33258) to reveal cell nuclei.Arrowheads indicate HA–E2F1- or HA–E2F1(132E)-expressing neurons. G, Quantification of experiments in A–F showing the percentages of E2F1- or E2F1(132E)-expressing neurons that exhibit high endogenous Cdc2 immunoreactivity. The percentage of transfected neurons with high Cdc2 immunoreactivity was significantly higher in E2F1-expressing neurons than in E2F1(132E)-expressing neurons (mean ± SEM;n = 3; p < 0.001;t test). H, Chromatin immunoprecipitation from cerebellar granule neuron cultures using no antibody (lane 1), a rabbit antibody to HA to serve as a control (lane 2), and an antibody against E2F1 (lane 3). Precipitated chromatin was subjected to PCR analysis using a set of primers encompassing the EBE within the cdc2 promoter (as shown in the figure). Endogenous cdc2 promoter was specifically coprecipitated with immunoprecipitated E2F1 (lane 3). I, Cerebellar granule neurons (P6 plus 2 DIV) were transfected with the −3200/cdc2–luciferase reporter plasmid together with a control vector or an expression plasmid encoding the dominant interfering form of E2F (E2F-Rb), in which the E2F1 DNA-binding region (1–368) was fused to Rb (379–972). Transfected cultures were processed as in Figure2A. Activity deprivation induced cdc2promoter-mediated transcription in vector-transfected (mean ± SEM; n = 4; ANOVA; p < 0.0001) but not in E2F-Rb-expressing granule neurons. J, Cerebellar granule neurons (P6 plus 2 DIV) were transfected with the E2F-Rb expression plasmid or the control vector together with an expression plasmid encoding β-galactosidase. One day after transfection, granule neurons were placed in full medium (30 mm KCl plus serum) or deprived of neuronal activity (5 mm KCl plus serum) for 2 d. Transfected neurons were subjected to indirect immunofluorescence using the monoclonal antibody to β-galactosidase and the DNA dye bisbenzimide (Hoechst 33258). Cell survival and death were measured in β-galactosidase-expressing neurons based on the integrity of neurites and nucleus. KCl withdrawal significantly reduced the survival of vector-transfected granule neurons (n = 3; ANOVA; p < 0.05) but not of E2F-Rb-expressing granule neurons.
Fig. 5.
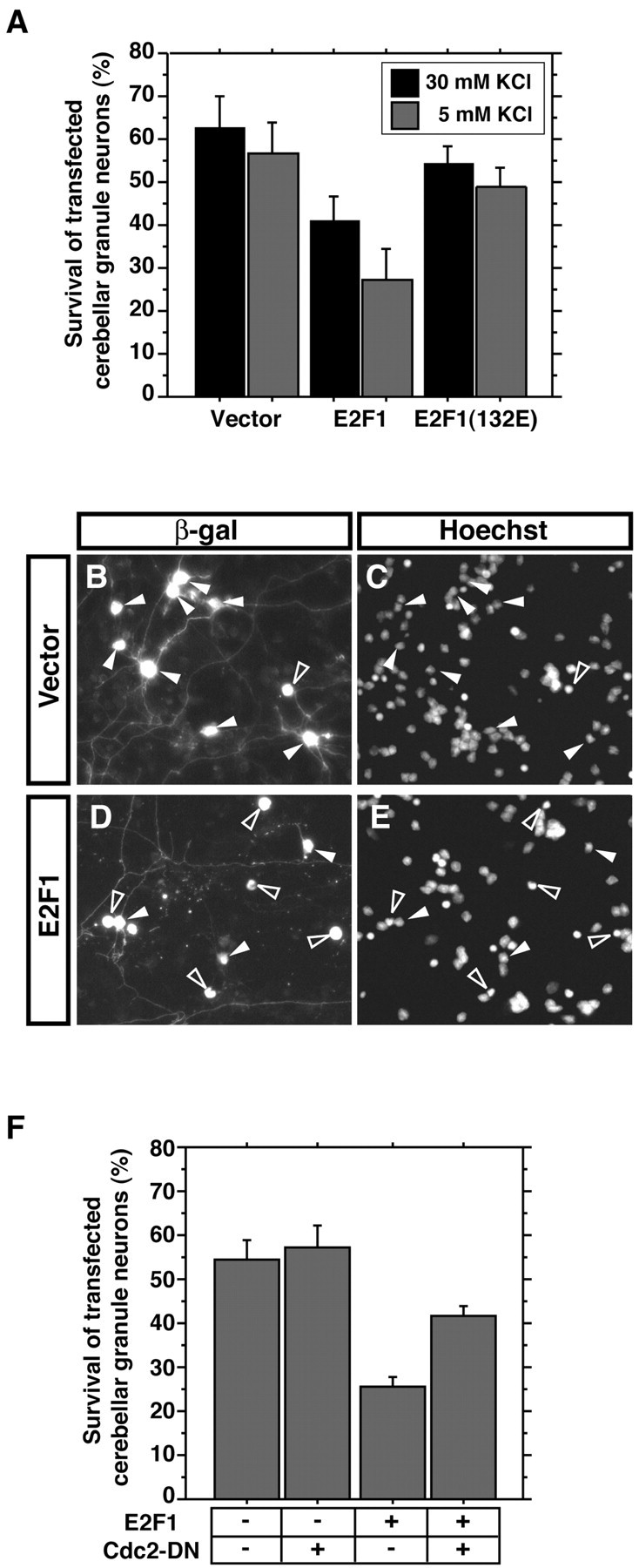
Cdc2 mediates E2F1-induced neuronal apoptosis.A, Cerebellar granule neurons (P6 plus 2 DIV) were transfected with the expression plasmid encoding E2F1, E2F1(132E), or their control vector together with an expression plasmid encoding β-galactosidase. One day after transfection, granule neurons were placed in full medium (30 mm KCl plus serum) or deprived of neuronal activity (5 mm KCl plus serum) for 24 hr. Transfected neurons were subjected to indirect immunofluorescence and cell survival assays as in Figure 4J. The expression of E2F1 significantly reduced the survival of cerebellar granule neurons [mean ± SEM; n = 3; ANOVA;p < 0.05 (30 mm KCl),p < 0.005 (5 mm KCl)]. However, E2F1(132E) had little effect on the survival of cerebellar granule neurons.B–E, Representative images of cerebellar granule neurons transfected with the control vector (B,C) or the E2F1 expression plasmid (D,E). In the vector control, several healthy neurons appear (closed arrowheads), whereas several E2F1-transfected neurons are undergoing apoptosis (open arrowheads). F, Cerebellar granule neurons were transfected with the E2F1 expression plasmid together with Cdc2-DN or its vector control together with the β-galactosidase expression plasmid. Transfected neurons were deprived of neuronal activity (5 mm KCl plus serum) for 24 hr and subjected to indirect immunofluorescence and cell survival assay as in Figure4J. Cdc2-DN significantly reduced E2F1-induced apoptosis (mean ± SEM; n = 4; ANOVA;p < 0.01).
Western blot analysis. For Western blot analysis, cerebellar granule neurons were harvested with a lysis buffer containing 20 mm Tris-HCl, pH 7.5, 150 mmNaCl, 1% Triton X-100, 5 mm EGTA, 25 mm β-glycerophosphate, 1 mm DTT, 1 mm PMSF, 1 mm NaVO3, and protease inhibitors. Protein amounts were measured by a Bradford assay. Equal amounts of proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was performed as described previously (Bonni et al., 1999).
Immunocytochemistry. In the experiments in Figures 3,B and C, and 4A–G, cerebellar granule neurons were fixed in 4% paraformaldehyde for 20 min at room temperature. Neurons were washed twice with PBS and permeabilized with PBS containing 0.4% Triton X-100 for 20 min, followed by blocking with TBS containing 0.02% Tween 20 and 10% milk for 1 hr at room temperature. Primary antibody incubations were performed at 4°C overnight in TBS containing 0.02% Tween 20 and 3% bovine serum albumin. In experiments in which bromodeoxyuridine (BrdU) incorporation was measured in Figure 6A–D, cells were fixed with a mixture of ethanol and glycine solution at −20°C for 20 min. Immunocytochemistry for BrdU was performed as described in the Labeling and Detection Kit I (Roche Diagnostics, Indianapolis, IN).
Fig. 6.

Activity deprivation does not stimulate DNA synthesis in cerebellar granule neurons. A–D, Cerebellar granule neuron cultures (P6 plus 2 DIV) were labeled with BrdU for 24 hr in cultures that were in full medium (30 mmKCl plus serum) or that were deprived of activity (5 mm KCl plus serum) in the presence or absence of the antimitotic agent AraC. Incorporated BrdU was detected by immunofluorescence using a mouse monoclonal antibody that recognizes BrdU. Postmitotic granule neurons in culture were identified using a rabbit antibody that recognizes microtubule-associated protein 2 (MAP2). MAP2-negative non-neuronal cells incorporated BrdU in the absence of AraC, whereas postmitotic granule neurons did not incorporate BrdU in the presence or absence of AraC. E, Lysates of cerebellar granule neurons (P6 plus 2 DIV) that were placed in full medium or deprived of activity for 24 or 48 hr were subjected to immunoblotting using an antibody to PCNA, Cdk2, Cdc2, or actin. Activity deprivation induced the level of Cdc2 protein but failed to induce the expression of PCNA or Cdk2.
Neuronal survival and apoptosis assays. In the experiments in Figures 4J and 5, transfected cerebellar granule neurons were fixed in 4% paraformaldehyde. Indirect immunofluorescence was performed using an antibody to β-galactosidase to visualize transfected neurons. Neuronal survival and apoptosis were assessed in β-galactosidase-expressing neurons based on the integrity of neurites and integrity of the nucleus as determined by the staining with DNA dye bisbenzimide (Hoechst 33258) as described previously (Konishi et al., 2002). Cell counts were performed in a blinded manner.
Electrophoretic mobility shift assays. Electrophoretic mobility shift assays (EMSAs) were performed as described previously (Campanero et al., 1999). Nuclear extracts from cerebellar granule neurons were prepared as described previously (Schreiber et al., 1989). The DNA probe was prepared by annealing the following oligonucleotides: 5′-TTCCTCTTTCTTTCG CGCTCTAGCCACC-3′ and 5′-GGGTGGGCTAGAGCGCGAAAGAAAGAGGA-3′. Nuclear extracts of cerebellar granule neurons (5 μg) were incubated in 10 μl of a preincubation mixture containing 20 mm HEPES/KOH, pH 7.9, 20% glycerol, 0.1 m KCl, 0.2 mm EDTA, 0.5 mm DTT, and 0.2 μg of sheared herring sperm DNA for 30 min on ice. After preincubation, 0.1 ng of the radiolabeled probe was added to the reaction mixture and incubated for 30 min on ice. In competition assays, cold wild-type E2F-binding element (EBE) probe or a mutant EBE probe in which CG (underlined in the sequence above) was replaced by AT was added to the preincubation mixture. For reactions including antibodies, rabbit antibody that recognizes E2F1 specifically or a control rabbit anti-HA antibody was added to the preincubation mixture. DNA–protein complexes were analyzed on a 4% polyacrylamide–0.1% bisacrylamide nondenaturing gel in 0.5% Tris–borate–EDTA at 4°C. The gel was dried and analyzed by autoradiography.
Chromatin immunoprecipitation assays. Chromatin immunoprecipitation assays were performed as described previously (Takahashi et al., 2000; Rayman et al., 2002). Approximately 4.5 × 107 cerebellar granule neurons were used in each reaction. Chromatin prepared from granule neurons was precipitated using 4 μg of rabbit antibody that recognizes E2F1 specifically or control anti-HA antibody together with protein A-Sepharose at 4°C overnight. Precipitated chromatin was treated with proteinase K and RNase A at 55°C for 3 hr followed by overnight incubation at 65°C to reverse cross-linking. Primers used for PCR that anneal to the rat cdc2 promoter in a region encompassing the EBE were designed as follows: 5′-CTGAGCTCAAGAGTCAGTTGGCG-3′ and 5′-CGCCAATCCGATTGCACGTAGAC-3′. PCRs were performed using 1/20th of chromatin precipitates. PCR products were labeled with [α32-P]dCTP and detected by autoradiography. Experiments were performed three times using chromatin that was prepared independently.
Results
Cdc2 specifically mediates activity deprivation-induced granule neuron apoptosis
We recently characterized the Cdc2–BAD signaling pathway as a novel apoptotic mechanism that is activated in newly generated cerebellar granule neurons with the suppression of neuronal activity (Konishi et al., 2002). Because common signaling mechanisms typically mediate apoptosis of neurons during trophic factor withdrawal, we tested whether the Cdc2–BAD apoptotic pathway is also activated in neurons during growth factor withdrawal.
Cerebellar granule neurons were prepared from postnatal day 6 (P6) rat pups and cultured in the presence of serum and high concentrations of KCl (30 mm) that induce membrane depolarization and consequent activation of VSCCs. We first characterized the activation of the Cdc2–BAD pathway in activity-deprived granule neurons. After 2 d in vitro (DIV), granule neuron cultures (P6 plus 2 DIV) were placed in survival medium [Basal Medium Eagle (BME) plus 30 mm KCl plus serum] or were deprived of membrane-depolarizing concentrations of KCl (BME plus 5 mm KCl plus serum). After 24 hr, cultures were subjected to Western blot analysis using an antibody to Cdc2, an antibody that recognizes the serine 128 phosphorylated form of BAD specifically (phospho128-BAD antibody), or an antibody that recognizes BAD regardless of its phosphorylation state (N-20 BAD antibody). Activity deprivation induced the expression of endogenous Cdc2 protein and induced the phosphorylation of endogenous BAD at serine 128 in granule neurons (Fig.1A, lanes 1and 2) (Konishi et al., 2002).
Fig. 1.

Cdc2 specifically mediates activity deprivation-induced apoptosis of cerebellar granule neurons.A, Lysates of granule neuron cultures (P6 plus 2 DIV) that were in survival medium [30 mm KCl plus serum,lane 1; 30 mm KCl plus insulin (Ins), lane 3)], that were deprived of KCl (5 mm KCl plus serum, lane 2), or that were deprived of growth factors (30 mm KCl, lane 4) for 24 hr were immunoblotted with a mouse monoclonal antibody that recognizes Cdc2 (top), the rabbit phospho128 (P128)-BAD antibody (middle), or an antibody that recognizes BAD regardless of its phosphorylation (bottom). Activity withdrawal significantly induced the expression of Cdc2 (1.4 ± 0.1 fold; n = 3;p < 0.001; ANOVA) and the phosphorylation of BAD at serine 128 (1.9 ± 0.1 fold; n = 3; ANOVA;p < 0.001), but insulin withdrawal failed to induce Cdc2 expression (0.8 ± 0.1 fold; n = 3) or to induce the BAD serine 128 phosphorylation (1.1 ± 0.2 fold; n = 3). B, Cerebellar granule neurons (P6 plus 2 DIV) were kept in survival medium (30 mmKCl plus serum, left; 30 mm KCl plus insulin, right) or were deprived for 48 hr of KCl (5 mm KCl plus serum, left) or insulin (30 mm KCl, right) in the presence of the Cdc2 inhibitor roscovitine (Rosc; 10 μm) or its vehicle [dimethylsulfoxide (DMSO)]. Roscovitine inhibited neuronal activity deprivation-induced apoptosis (mean ± SEM;n = 3; ANOVA; p < 0.01) but not growth factor withdrawal-induced apoptosis. C, Lysates of cerebellar granule neurons (P6 plus 2 DIV) that were treated as in B in the presence of DMSO (D) or roscovitine (R) were immunoblotted using an antibody to the cleaved form of caspase-3 (top) or an antibody that recognizes actin (bottom) to serve as control for equal protein loading.D, Lysates of cerebellar granule neurons that were placed in full medium (30 mm KCl plus serum) or deprived of KCl (5 mm KCl plus serum) for the indicated times were immunoblotted with the antibody to Cdc2 (top) or the antibody to actin (bottom). E, Northern blot analysis of total RNA from cerebellar granule neuron cultures that had either been maintained in full medium (30 mm KCl plus serum, lanes 1 and 2) or deprived of activity (5 mm KCl plus serum, lanes 3 and4) for 24 hr. RNA was subjected to Northern blot analysis using a cdc2 probe (top) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH;bottom).
We next tested whether growth factor withdrawal also induces Cdc2 expression and BAD serine 128 phosphorylation in neurons. Activation of the IGF1 receptor by a high concentration of insulin (10 μg/ml) in granule neurons provides a prototypical growth factor-induced survival signal in granule neurons (D'Mello et al., 1993; Dudek et al., 1997). P6 plus 2 DIV granule neurons were placed in survival medium (BME plus 30 mm KCl plus insulin) or were deprived of growth factor signaling (BME plus 30 mm KCl). Surprisingly, we found that growth factor withdrawal failed to induce the expression of Cdc2 and failed to induce the phosphorylation of BAD at serine 128 (Fig.1A, lanes 3 and 4).
In assays of cell survival, inhibition of Cdc2 activity by roscovitine protected cerebellar granule neurons from activity deprivation-induced apoptosis (Fig. 1B) (Konishi et al., 2002) but failed to protect granule neurons against growth factor withdrawal-induced apoptosis (Fig. 1B), suggesting that Cdc2 is not required for growth factor withdrawal-induced neuronal apoptosis. In other experiments, both activity deprivation and growth factor withdrawal triggered the accumulation of the cleaved product of the protease caspase-3 that reflects caspase-3 activation (Fig.1C) (Di Cunto et al., 2000). However, consistent with results of the cell-survival assays, although the inhibition of Cdc2 by roscovitine inhibited activity deprivation-induced caspase-3 cleavage, roscovitine failed to inhibit growth factor withdrawal-induced caspase-3 cleavage (Fig. 1C). We also found that expression of Cdc2-DN that blocks activity deprivation-induced neuronal apoptosis (van den Heuvel and Harlow, 1993; Konishi et al., 2002) failed to inhibit granule neuron apoptosis after insulin withdrawal (data not shown). Together, these results indicate that activity deprivation selectively induces the Cdc2–BAD apoptotic pathway and that Cdc2 promotes apoptosis specifically after neuronal activity deprivation but does not mediate growth factor withdrawal-induced apoptosis of cerebellar granule neurons.
Activity deprivation induces cdc2 transcription in granule neurons via an E2F-binding element within the cdc2 promoter
Having identified the Cdc2–BAD pathway as a specific mediator of activity deprivation-induced neuronal apoptosis, we focused our attention on the characterization of the mechanism by which activity deprivation induces the expression and consequent activation of Cdc2 in granule neurons. We performed a kinetic analysis of activity deprivation-induced Cdc2 protein expression in granule neurons, and found that the induction of Cdc2 protein was not apparent in the first 12 hr but became evident 24 hr after KCl withdrawal (Fig.1D). The elevated levels of Cdc2 were maintained thereafter at 48 and 72 hr after the KCl withdrawal (see Fig. 6) (data not shown). The kinetics of activity deprivation-induced Cdc2 expression is characteristic of a late response gene. To distinguish between the two major possibilities that activity deprivation induction of Cdc2 occurs via a mechanism that is predominantly at the level of protein turnover or translation or at the level of transcription or mRNA processing, we characterized the effect of activity deprivation on the amount of cdc2 mRNA in granule neurons. We found that the expression of cdc2 mRNA was robustly induced in granule neurons 24 hr after KCl withdrawal (Fig. 1E). Together, these results suggest that activity deprivation induces the expression of Cdc2 via an alteration in cdc2 transcription or mRNA turnover.
We next determined whether activity deprivation regulatescdc2 expression at the transcriptional level. In transient transfection experiments in granule neurons, we tested the effect of activity deprivation on the activity of a luciferase reporter gene that contains 3.2 kb of the 5′ regulatory sequence of the cdc2gene (−3200/cdc2) (Sugarman et al., 1995). We found that KCl withdrawal robustly stimulated the expression of the −3200/cdc2–luciferase reporter gene in granule neurons (Fig. 2A). These results suggest that activity deprivation stimulates Cdc2 expression by inducing cdc2 transcription.
Deletion analysis of the cdc2 promoter in granule neurons revealed that the region of the promoter between nucleotide −245 and nucleotide −94 relative to the transcriptional start site of thecdc2 gene plays a critical role in activity withdrawal-induced cdc2 transcription. The basal level ofcdc2 promoter-mediated transcription in membrane-depolarized granule neurons was not affected by the 5′ deletion mutants (Fig.2A). These results argue against the interpretation that activity deprivation in granule neurons triggers the derepression of the cdc2 promoter. Rather, these results suggest that activity deprivation induces the activity of a transcription factor that stimulates cdc2 transcription in granule neurons.
We next focused our effort on identifying the site within the −245 to −94 region of the cdc2 promoter that mediates activity deprivation-induced cdc2 transcription. This region of thecdc2 promoter contains an EBE at −128 that is conserved in the rat and human cdc2 genes. The EBE plays an important role in mediating cdc2 transcription in proliferating cells (Dalton, 1992; Shimizu et al., 1995). We therefore asked whether the EBE might mediate cdc2 transcription in the postmitotic granule neurons after KCl withdrawal. We found that mutation of the EBE within the context of the −245/cdc2 reporter gene reduced significantly the ability of KCl withdrawal to induce cdc2promoter-mediated transcription (Fig. 2A). These results suggest that the EBE within the cdc2 promoter plays a critical role in mediating activity deprivation-inducedcdc2 transcription.
Members of the E2F family of transcription factors bind the EBE in proliferating cells (Tommasi and Pfeifer, 1995; Takahashi et al., 2000;Rayman et al., 2002). We therefore tested whether the EBE within thecdc2 promoter binds to an E2F family member in granule neurons. In EMSAs, the EBE formed protein–DNA complexes when incubated with nuclear extracts of cerebellar granule neurons (Fig.2B). The incubation of excess cold wild-type EBE in the DNA binding reaction, but not of mutant EBE known to disrupt its binding to E2Fs, reduced partly or completely the formation of three protein–DNA complexes (Fig. 2B,arrowheads). These results suggest that the EBE within thecdc2 promoter binds E2F transcription factors that are present in nuclear extracts of granule neurons. The three EBE–protein complexes were disrupted or supershifted with the addition of an antibody that recognizes E2F1 specifically (Fig. 2C,bracket), but not by a control antibody. There was an increased amount of EBE-binding activity in nuclear extracts of activity-deprived granule neurons (Fig. 2B), suggesting that activity deprivation stimulates the activity of endogenous E2F1 in neurons. These data suggest that granule neurons express E2F1 that binds to the EBE within the cdc2 promoter and raised the possibility that E2F1 might mediate activity deprivation-induced cdc2 transcription in cerebellar granule neurons.
E2F mediates activity deprivation-induced cdc2transcription in postmitotic granule neurons
The finding that the EBE of the cdc2 promoter binds to E2F1 and mediates activity deprivation-induced cdc2transcription led us next to address the question of whether E2F mediates activity deprivation-induced cdc2 transcription in cerebellar granule neurons. First, we asked whether E2F1 has the capacity to induce cdc2 transcription in granule neurons, which are in a postmitotic state. We imagined that E2F1 that is expressed in membrane-depolarized granule neurons is maintained in a repressed state to prevent the induction of cdc2 and of genes that might trigger reentry of neurons into the cell cycle. Therefore, to investigate the ability of E2F1 to induce transcription of the cdc2 gene in granule neurons, we overexpressed E2F1 in granule neurons together with the cdc2–luciferase (−245/cdc2) reporter plasmid. We found that overexpressed E2F1 strongly enhanced cdc2 promoter-mediated transcription (Fig. 3A). In contrast to wild-type E2F1, a mutant E2F1 that is unable to bind to the EBE because of an amino acid substitution within the DNA binding domain of E2F1 [E2F1(132E)] (Johnson et al., 1993) failed to induce thecdc2 promoter effectively (Fig. 3A). In addition, mutation of the EBE within the cdc2 promoter (−245mEBE/cdc2) significantly inhibited E2F1-dependent transcriptional activation of the cdc2promoter, arguing against the interpretation that E2F1-inducedcdc2 transcription occurs via the squelching of transcriptional corepressors or through a mechanism that is independent of E2F binding to the cdc2 promoter. Together, these results demonstrate the ability of E2F1 to induce cdc2 transcription in granule neuron cultures.
Granule neuron cultures contain >95% granule neurons, and the introduction of plasmids into these cultures in our transfections targets granule neurons selectively, because 99% of transfected cells are granule neurons (Konishi et al., 2002). Nevertheless, because E2F-mediated transcription might be much more effective in non-neuronal cells, we investigated the possibility that E2F1 induction ofcdc2 transcription in our experiments might arise predominantly from cdc2 promoter activity in the non-neuronal cells. We therefore generated a construct in which the GFP reporter gene is driven by the cdc2 promoter (cdc2/GFP) (Fig. 3B). We transfected cerebellar granule neuron cultures with a GFP, cdc2/GFP, or cytomegalovirus (CMV)/GFP reporter gene together with an expression plasmid encoding β-galactosidase. Cultures were fixed 2 d after transfection and subjected to indirect immunofluorescence using a mouse monoclonal antibody to β-galactosidase. The activity of the cdc2 promoter was monitored by visualizing GFP fluorescence. In the absence of E2F1, the activity of GFP was very weak and near background fluorescence levels. In contrast, the expression of E2F1 strongly induced the cdc2/GFP reporter gene but had little effect on the control GFP reporter or CMV/GFP reporter gene (Fig. 3B). In other experiments, we found that the cells that expressed the E2F1-induced cdc2 promoter-mediated GFP also expressed the neuron specific β-tubulin type III (Fig.3C). Together, these results establish that E2F1 possesses the capacity to activate cdc2 promoter-mediated transcription in postmitotic cerebellar granule neurons.
We next examined whether E2F1 has the ability to induce transcription of the endogenous cdc2 gene in cerebellar granule neurons. We transfected cerebellar granule neurons with an expression plasmid encoding HA-tagged E2F1 or HA-tagged E2F1(132E). Two days after transfection, granule neurons were subjected to indirect immunofluorescence using a rabbit antibody to HA and a mouse monoclonal antibody to Cdc2. Robust endogenous Cdc2 immunoreactivity was detected in granule neurons in which wild-type E2F1 was expressed (Fig.4A–C). In contrast, we detected weak endogenous Cdc2 immunoreactivity in E2F1(132E)-expressing granule neurons (Fig. 4D–F). The percentage of strong Cdc2-positive granule neurons was quantified and revealed that strong endogenous Cdc2 was detected in a large fraction (82%) of E2F1-expressing granule neurons but only in 10% of E2F1(132E)-expressing granule neurons (Fig. 4G). These results suggest that the expression of E2F1 stimulates endogenous Cdc2 expression in cerebellar granule neurons.
We next characterized the role of endogenous E2F in mediatingcdc2 expression in activity-deprived cerebellar granule neurons. To examine whether endogenous E2F might mediate transcription of the endogenous cdc2 gene, we performed chromatin immunoprecipitation analysis. Chromatin was prepared from cerebellar granule neurons and was immunoprecipitated using no antibody, a control antibody, or an anti-E2F1 antibody together with protein A-Sepharose beads. Immunoprecipitated chromatin was subjected to PCR using primers designed to anneal to the cdc2 promoter in a region encompassing the EBE (Fig. 4H). We found that the endogenous cdc2 promoter was coprecipitated with endogenous E2F1 (Fig. 4H). The results of control experiments using no antibody or control antibody supported the interpretation that the endogenous cdc2 promoter and endogenous E2F1 coprecipitated specifically (Fig. 4H). These results suggest that the endogenous cdc2 gene is a target of endogenous E2F1 in postmitotic neurons. Next, we tested the effect of a dominant interfering form of E2F, in which the DNA-binding domain of E2F1 was fused to the repressor Rb (E2F-Rb) (Sellers et al., 1995,1998), on the ability of KCl withdrawal to induce the cdc2promoter in cerebellar granule neurons. E2F-Rb is expected to inhibit endogenous E2F proteins by blocking their ability to induce transcription (Sellers et al., 1995, 1998). We found that E2F-Rb, when coexpressed with the −3200/cdc2–luciferase reporter gene, abolished KCl withdrawal induction of the cdc2 promoter (Fig. 4I). Together with the finding of chromatin immunoprecipitation experiments, these results suggest that endogenous E2F mediates neuronal activity deprivation-dependent cdc2transcription.
The E2F–cdc2 cell-cycle pathway promotes granule neuron apoptosis
Our results suggesting that E2F1 mediates activity deprivation-induced cdc2 transcription raised the question of whether E2F1 and Cdc2 lie on a biochemical pathway that mediates activity deprivation-induced apoptosis of neurons. E2F1 has been demonstrated to promote apoptosis in postmitotic neurons including cerebellar granule neurons (O'Hare et al., 2000; Gendron et al., 2001;Liu and Greene, 2001b). Consistent with these observations, we found that the dominant interfering form of E2F (E2F-Rb), when expressed in granule neurons, blocked the ability of KCl withdrawal to induce apoptosis (Fig. 4J), suggesting that endogenous E2F proteins mediate activity deprivation-induced apoptosis of neurons.
The molecular mechanisms by which E2F1 promotes apoptosis remain to be elucidated. Because activity deprivation induces E2F-mediatedcdc2 transcription (this study) and because the activity of Cdc2 promotes granule neuron apoptosis (Konishi et al., 2002), we asked whether the proapoptotic function of E2F1 in cerebellar granule neurons is mediated via the induction and activation of Cdc2. We first established that the expression of E2F1 induces granule neuron apoptosis in our cultures. Granule neurons were transfected with an E2F1 expression plasmid or the control vector and were then maintained in full medium or deprived of membrane-depolarizing concentrations of KCl for just 24 hr to obtain a negligible amount of activity withdrawal-induced apoptosis in vector-transfected cultures to allow the detection of apoptosis on E2F1 overexpression (Fig.5A). Although the expression of E2F1 induced apoptosis in membrane-depolarized cultures, E2F1-mediated apoptosis was more marked in cultures that were deprived of depolarizing concentrations of KCl (Fig. 5A–E), suggesting that activity deprivation further activates the ability of E2F1 to induce apoptosis or that additional activity deprivation-induced mechanisms cooperate with E2F1 to induce apoptosis. The expression of the mutant E2F1 protein [E2F1(132E)], that poorly binds to the EBE, failed to effectively induce neuronal apoptosis, suggesting that the binding of E2F1 to the promoter of responsive genes is necessary for E2F1 to stimulate transcription of apoptotic genes and thereby trigger apoptosis. We next asked whether Cdc2 might constitute an apoptotic gene target of E2F1 in granule neurons. We determined whether the inhibition of Cdc2 activity rescues granule neurons from E2F1-induced apoptosis. We found that a dominant interfering form of Cdc2 in which an amino acid is mutated in the catalytic region (Cdc2-DN), when coexpressed with E2F1 in granule neurons, significantly reduced E2F1-induced apoptosis (Fig. 5F). These results indicate that Cdc2 is functionally downstream of E2F1, and that the E2F–Cdc2 cell-cycle pathway thus provides an apoptotic signal in postmitotic neurons.
The similarity in the ability of E2F1 to induce cdc2 in both postmitotic and proliferating cells raised the question of the extent of similarity in the regulation of E2F1 function in two cell types. In proliferating cells, E2F1 regulates the expression of a large set of genes that promote DNA synthesis and replication (DeGregori et al., 1995; Dyson, 1998; Harbour and Dean, 2000; Ishida et al., 2001; Ren et al., 2002). We therefore asked whether the activation of the E2F–Cdc2 cell-cycle pathway in activity-deprived granule neurons also leads to the induction of genes that serve a role in DNA synthesis. We determined whether activity deprivation stimulates DNA synthesis in granule neurons. Cerebellar granule neuron cultures were incubated with bromodeoxyuridine for 24 hr. As expected, non-neuronal cells incorporated BrdU. However, we failed to detect BrdU-positive neurons in these cultures. These results indicate that despite the activation of the E2F–Cdc2 cell-cycle pathway in neurons on activity deprivation, the DNA synthesis machinery is not activated in granule neurons (Fig.6A–D). Consistent with this conclusion, we found that although activity deprivation induced Cdc2 expression, activity deprivation failed to induce the expression of the DNA-synthesis related factors PCNA and Cdk2 in granule neurons (Fig. 6E). Together, our results suggest that activity deprivation induces the E2F-mediated expression of Cdc2 in neurons but fails to induce E2F-regulated genes that turn on the DNA synthesis machinery. The activation of the mitotic kinase Cdc2 in postmitotic neurons in the absence of DNA synthesis may thus comprise the trigger for apoptosis.
Discussion
In this study, we have characterized a cell-cycle mechanism that specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. We have found that the mitotic kinase Cdc2 mediates apoptosis of cerebellar granule neurons with the deprivation of neuronal activity but not with growth factor withdrawal. Activity deprivation induces the expression of Cdc2, leading to Cdc2-mediated neuronal apoptosis, by stimulating transcription of the cdc2gene in granule neurons. An EBE within the cdc2 promoter was found to bind to E2F1 and mediate activity deprivation-inducedcdc2 transcription in granule neurons. We found that endogenous E2F1 in granule neurons occupies the promoter of the endogenous cdc2 gene in chromatin-immunoprecipitation analysis. Consistent with these results, we also found that inhibition of E2F protein by a dominant interfering form of E2F blocks activity deprivation-induced cdc2 transcription. In other experiments, we found that the expression of E2F1 in granule neurons induces the expression of Cdc2, which in turn mediates E2F1-induced apoptosis. Finally, although activity deprivation induces the E2F-mediated expression of Cdc2 in granule neurons, activity deprivation in neurons fails to induce the expression of genes that drive DNA synthesis and replication. Together, our findings define E2F–Cdc2 as a novel cell-cycle pathway in postmitotic neurons that selectively propagates an apoptotic signal during neuronal activity deprivation.
A role for reactivation of the cell-cycle machinery in apoptosis of postmitotic neurons was suggested nearly a decade ago (for review, seeLiu and Greene, 2001a). However, the regulation of the cell-cycle machinery in postmitotic neurons and the mechanisms by which cell-cycle activation mediates neuronal apoptosis are just beginning to be elucidated. One of the early observations in this field was the finding that the withdrawal of nerve growth factor (NGF) from sympathetic neurons induces the expression of cyclin D1 expression in these neurons (Freeman et al., 1994). This observation was extended to a number of paradigms of neuronal apoptosis, including activity deprivation of cerebellar granule neurons and the exposure of neurons to DNA-damaging agents and other pathogenic stimuli (Park et al., 1997, 1998;Padmanabhan et al., 1999). In these studies, the induction of cyclin D1 was found to lead to the activation of D1-associated cdk4/6, and inhibition of these kinases protected neurons against apoptosis (Park et al., 1997, 1998; Padmanabhan et al., 1999).
As in proliferating cells, an important target of CDK4/6 in neurons is the protein Rb. The CDK4/6-induced phosphorylation of Rb is thought to control the G1–S transition in proliferating cells (Weinberg, 1995). During the early G1 phase of the cell cycle, hypophosphorylated Rb binds to the transcription factor E2F and thereby represses E2F-mediated transcription (Chellappan et al., 1991; Dyson, 1998). However, the CDK-induced phosphorylation of Rb induces the derepression and transactivation of E2F-regulated genes leading to cell-cycle progression (Harbour et al., 1999). In postmitotic neurons, the CDK4/6-induced phosphorylation of Rb and consequent disinhibition of E2F have been linked to neuronal apoptosis in a number of settings including activity withdrawal-induced apoptosis of granule neurons (Padmanabhan et al., 1999; O'Hare et al., 2000).
The mechanisms by which E2F mediates apoptosis of postmitotic neurons are beginning to be characterized. In a recent study by Liu and Greene (2001b), the repression of E2F-responsive genes was demonstrated to be required for neuronal survival. In this study, NGF withdrawal in sympathetic neurons and chemotoxin stimulation of cortical neurons was demonstrated to trigger the derepression of E2F-regulated genes leading to apoptosis (Liu and Greene, 2001b). It was also suggested in this study that transactivation of E2F-responsive genes is not required for neuronal apoptosis (Liu and Greene, 2001b).
In our study, we show that the transactivation function of E2F leads to the induction of Cdc2 expression and apoptosis in cerebellar granule neurons with activity deprivation. Our findings provide a novel link in neurons between the activity deprivation-induced G1 CDK-Rb-E2F1 pathway and the mitotic kinase Cdc2 culminating in the direct activation of the cell-death machinery via phosphorylation of the BH3-only protein BAD. We have found that activity deprivation but not growth factor withdrawal induces the expression of Cdc2. Consistent with this result, we found that Cdc2 mediates activity deprivation-induced but not growth factor withdrawal-induced neuronal apoptosis. We also found that E2F1 triggers apoptosis of cerebellar granule neurons, and that E2F1-induced Cdc2 mediates the ability of E2F1 to promote apoptosis. Together, these findings suggest that the E2F–Cdc2 cell-cycle pathway specifically mediates the activity deprivation-induced neuronal apoptotic response.
In future studies, it will be important to address the question of whether in other neurons Cdc2 might be activated with apoptotic stimuli in addition to activity withdrawal. If Cdc2 but not E2F1 is the specificity determinant of activity deprivation-induced apoptosis, thecdc2 promoter may provide a valuable biochemical endpoint in future studies for the identification and characterization of transcription factors that cooperate with E2F1 to specifically induce Cdc2 in activity-deprived postmitotic neurons. However, distinct E2F1-mediated mechanisms may mediate activity deprivation-induced apoptosis of granule neurons (this study) and apoptosis of sympathetic neurons with growth factor withdrawal or chemotoxin-induced apoptosis of cortical neurons (Liu and Greene, 2001b). The characterization of the distinct mechanisms regulating E2F function in postmitotic neurons in the different paradigms of neuronal apoptosis in these two studies may reveal additional specific signals that mediate activity deprivation-induced neuronal apoptosis.
The observation that neuronal activity deprivation induces E2F1-mediated cdc2 transcription points to the similarity in the role of E2F1 in inducing Cdc2 in postmitotic neurons and proliferating cells. However, we found that much of the function of E2Fs, and E2F1 in particular, to stimulate S phase genes in proliferating cells is not recapitulated in postmitotic neurons. These observations carry a number of implications. First, they suggest that Cdc2 represents a select target of E2F in postmitotic neurons and point to specificity mechanisms that exist in postmitotic CNS neurons that confer E2F with the selective ability to induce cdc2transcription or that silence the majority of E2F-regulated genes. Investigation of these mechanisms might shed light on the postmitotic neuronal state. Second, our observation of Cdc2 triggering apoptosis of postmitotic neurons in the absence of DNA synthesis points to a similarity of Cdc2-mediated neuronal apoptosis and the role of premature Cdc2 activation in cycling cells culminating in apoptosis (Shi et al., 1994). It will be important to determine whether lessons from one situation might be applied to the other. Finally, it is interesting to compare the role of the cell-cycle machinery in apoptosis of neurons in the CNS and peripheral nervous system (PNS). A recent study revealed distinct responses of CNS and PNS neurons to expression of the transcription factor N-myc that promotes cell-cycle progression in dividing cells (Wartiovaara et al., 2002). Whereas N-myc induced successful re-entry of sympathetic neurons into the cell cycle, N-myc induced apoptosis of cortical neurons (Wartiovaara et al., 2002). It will be interesting to compare the cell-cycle protein responses of PNS and CNS neurons to activity deprivation.
In conclusion, our findings suggest that the E2F–Cdc2 cell-cycle pathway plays a critical role in neuronal apoptosis. Our study raises several questions. It will be important in future studies to determine the role of the E2F–Cdc2 cell-cycle pathway in the normal development of the mammalian brain. In addition, it will be important to determine how the E2F–Cdc2 cell-cycle pathway interacts with other signals that mediate activity deprivation-induced neuronal apoptosis. In this regard, the Jun-N-terminal kinase (JNK) signaling pathway is thought to play a critical role in apoptosis of granule neurons with the suppression of neuronal activity (Watson et al., 1998; Le-Niculescu et al., 1999). Interestingly, we found recently that JNK induces the phosphorylation of BAD at serine 128 and thereby activates BAD-mediated apoptosis (Donovan et al., 2002), raising the possibility that Cdc2 and JNK may converge on BAD to effectively induce the cell-death machinery in neurons, or that these kinases target BAD in a temporally specific manner in granule neurons (Donovan et al., 2002). Whether the regulation of the Cdc2 and JNK signaling pathways in activity-deprived neurons is coordinated remains to be determined. Finally, because neuronal death is thought to be a major pathophysiologic feature of several common neurologic disorders, including neurodegenerative diseases and stroke (Cotman, 1998; Pettmann and Henderson, 1998; Snider et al., 1999), it will also be interesting to determine whether the E2F–Cdc2 cell-cycle pathway contributes to neuronal apoptosis in these neurological diseases.
Footnotes
This work was supported by a Burroughs Wellcome Career Development Award (A.B.) and by National Institutes of Health Grant R01-NS41021-01 (A.B.). A.B. is the recipient of an Alfred P. Sloan Foundation Fellowship, a Robert H. Ebert Clinical Scholar Award from the Esther A. and Joseph Klingenstein Fund, an EJLB Foundation Award, and a Sidney Kimmel Foundation Award. We thank B. Gaudilliere for critical reading of this manuscript, P. Hinds for helpful discussions, C. K. Glass for providing thecdc2–luciferase plasmids, W. Sellers for providing the E2F1 expression plasmids, and S. Vasquez for technical assistance.
Correspondence should be addressed to Azad Bonni, Department of Pathology, Harvard Medical School, 200 Longwood Avenue, Boston, MA 02115. E-mail: azad_bonni@hms.harvard.edu.
References
- 1.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 2.Altman J, Bayer S. Development of the cerebellar system: in relation to its evolution, structure, and functions. CRC; New York: 1997. [Google Scholar]
- 3.Bonni A, Brunet A, West A, Datta SR, Takasu M, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 4.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campanero MR, Armstrong M, Flemington E. Distinct cellular factors regulate the c-myb promoter through its E2F element. Mol Cell Biol. 1999;19:8442–8450. doi: 10.1128/mcb.19.12.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 7.Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- 8.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 9.Cohick WS, Clemmons DR. The insulin-like growth factors. Annu Rev Physiol. 1993;55:131–153. doi: 10.1146/annurev.ph.55.030193.001023. [DOI] [PubMed] [Google Scholar]
- 10.Cotman CW. Apoptosis decision cascades and neuronal degeneration in Alzheimer's disease. Neurobiol Aging. 1998;19:S29–32. doi: 10.1016/s0197-4580(98)00042-6. [DOI] [PubMed] [Google Scholar]
- 11.Dalton S. Cell cycle regulation of the human Cdc2 gene. EMBO J. 1992;11:1797–1804. doi: 10.1002/j.1460-2075.1992.tb05231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, Atzori C, Turco E, Triolo R, Dotto GP, Silengo L, Altruda F. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron. 2000;28:115–127. doi: 10.1016/s0896-6273(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 14.D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;10:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donovan N, Becker EBE, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–40949. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- 17.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 18.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 19.Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of Cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 20.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gendron TF, Mealing GA, Paris J, Lou A, Edwards A, Hou ST, MacManus JP, Hakim AM, Morley P. Attenuation of neurotoxicity in cortical cultures and hippocampal slices from E2F1 knockout mice. J Neurochem. 2001;78:316–324. doi: 10.1046/j.1471-4159.2001.00423.x. [DOI] [PubMed] [Google Scholar]
- 22.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 23.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 24.Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- 25.Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21:4684–4699. doi: 10.1128/MCB.21.14.4684-4699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- 27.Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- 28.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levkovitz Y, Baraban JM. A dominant negative inhibitor of the Egr family of transcription regulatory factors suppresses cerebellar granule cell apoptosis by blocking c-Jun activation. J Neurosci. 2001;21:5893–5901. doi: 10.1523/JNEUROSCI.21-16-05893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu DX, Greene LA. Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell Tissue Res. 2001a;305:217–228. doi: 10.1007/s004410100396. [DOI] [PubMed] [Google Scholar]
- 31.Liu DX, Greene LA. Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron. 2001b;32:425–438. doi: 10.1016/s0896-6273(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 32.McConnell SK. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761–768. doi: 10.1016/0896-6273(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 33.Miller TM, Johnson EM. Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS, Park DS. Induction and modulation of cerebellar granule neuron death by E2F-1. J Biol Chem. 2000;275:25358–25364. doi: 10.1074/jbc.M001725200. [DOI] [PubMed] [Google Scholar]
- 35.Padmanabhan J, Park D, Greene LA, Shelanski ML. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19:8747–8756. doi: 10.1523/JNEUROSCI.19-20-08747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park DS, Levine B, Ferrari G, Greene LA. Cyclin-dependent kinase inhibitors and dominant negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 39.Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson EM., Jr Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol. 2002;157:441–543. doi: 10.1083/jcb.200110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, te Riele H, Dynlacht BD. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts,” prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sellers WR, Rodgers JW, Kaelin WG., Jr A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proc Natl Acad Sci USA. 1995;92:11544–11548. doi: 10.1073/pnas.92.25.11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG., Jr Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 1998;12:95–106. doi: 10.1101/gad.12.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi L, Nishioka WK, Th'ng J, Bradbury EM, Lithfield DW, Greenberg AH. Premature p34cdc2 activation required for apoptosis. Science. 1994;263:1143–1145. doi: 10.1126/science.8108732. [DOI] [PubMed] [Google Scholar]
- 46.Shimizu M, Ichikawa E, Inoue U, Nakamura T, Nakajima T, Nojima H, Okayama H, Oda K. The G1/S boundary-specific enhancer of the rat Cdc2 promoter. Mol Cell Biol. 1995;15:2882–2892. doi: 10.1128/mcb.15.5.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snider BJ, Gottron FJ, Choi DW. Apoptosis and necrosis in cerebrovascular disease. Ann NY Acad Sci. 1999;893:243–253. doi: 10.1111/j.1749-6632.1999.tb07829.x. [DOI] [PubMed] [Google Scholar]
- 48.Sugarman JL, Schonthal AH, Glass CK. Identification of a cell-type-specific and E2F-independent mechanism for repression of Cdc2 transcription. Mol Cell Biol. 1995;15:3282–3290. doi: 10.1128/mcb.15.6.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- 50.Tommasi S, Pfeifer GP. In vivo structure of the human Cdc2 promoter: release of a p130–E2F-4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of Cdc2 expression. Mol Cell Biol. 1995;15:6901–6913. doi: 10.1128/mcb.15.12.6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 52.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic Cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wartiovaara K, Barnabe-Heider F, Miller FD, Kaplan DR. N-myc promotes survival and induces S-phase entry of postmitotic sympathetic neurons. J Neurosci. 2002;22:815–824. doi: 10.1523/JNEUROSCI.22-03-00815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 56.Wood KA, Dipasquale B, Youle RJ. In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- 57.Yamagishi S, Yamada M, Ishikawa Y, Matsumoto T, Ikeuchi T, Hatanaka H. p38 Mitogen-activated protein kinase regulates low potassium-induced c-Jun phosphorylation and apoptosis in cultured cerebellar granule neurons. J Biol Chem. 2001;276:5129–5133. doi: 10.1074/jbc.M007258200. [DOI] [PubMed] [Google Scholar]