

Abstract
We characterized synaptosomal glutamate transport activity in a recently developed transgenic rat model of amyotrophic lateral sclerosis (ALS) overexpressing the G93A Cu2+/Zn2+ superoxide dismutase (SOD1) mutation. Using spinal cord synaptosomes, a significant reduction (43%) in the maximal velocity for high-affinity, Na+-dependent glutamate uptake was observed at disease end stage in G93A rats compared with age-matched controls. Similarly, a 27% reduction in maximum velocity (Vmax) was measured at disease onset, but no difference in spinal cord Vmax values were observed with presymptomatic animals compared with controls. In comparison, we observed no differences in theVmax for glutamate clearance at disease end stage with synaptosomes from cortex, hippocampus, striatum, cerebellum, and brainstem, indicating a specific deficit in the spinal cord. The pharmacological sensitivity of spinal cord uptake to dihydrokainate suggests that the GLT-1 (glutamate transporter-1) subtype primarily mediates the transport activity. Expression analysis revealed a loss of GLT-1 as well as qualitative changes in GLAST (glutamate/aspartate transporter) but no measurable changes in EAAC1 (excitatory amino acid carrier 1) in spinal cord of end-stage G93A rats, indicating that deficits in glutamate transporters in this rat model may be glial specific. Riluzole, a neuroprotective agent used clinically to slow the progression of ALS, produced an enhancement of spinal cord synaptosomal glutamate uptake in control animals and early-stage disease G93A rats, but this effect was lost in end-stage animals. Altered expression of astroglial glutamate transporters accompanied by reduced capacity for spinal cord clearance of extracellular glutamate in the G93A SOD1 transgenic rat may account for a dampened effect of riluzole to enhance glutamate uptake at end-stage disease.
Keywords: Cu2+/Zn2+superoxide dismutase, ALS, GLT-1, GLAST, glutamate, riluzole
Introduction
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disease involving progressive motor neuron degeneration, paralysis, and death (Brown, 2001; Cleveland and Rothstein, 2001; Cluskey and Ramsden, 2001). A glutamatergic hypothesis has been proposed as one of the mechanisms contributing to the degeneration of motor neurons in ALS (for review, see Plaitakis et al., 1996). Several independent lines of evidence have provided support for an impaired capacity for the clearance of extracellular glutamate as a contributing factor in ALS. Elevated extracellular glutamate levels have been documented in plasma and CSF from ALS patients (Rothstein et al., 1990, 1991) and in cortical microdialysates from a transgenic mouse model of ALS (Alexander et al., 2000). Impaired glutamate uptake capacity has been described using spinal cord and motor cortex synaptosomes from human postmortem ALS tissue (Rothstein et al., 1992). Recently, it was shown that 41% of ALS patients have elevated CSF glutamate levels, which correlated to a spinal onset of disease and severity of symptoms (Spreux-Varoquaux et al., 2002). At the molecular level, a selective loss of the predominant CNS glutamate transporter EAAT2 (GLT-1) in spinal cord as well as motor cortex from ALS patients (Rothstein et al., 1995) suggests a potential mechanism for the elevated extracellular levels of glutamate and impaired capacity for uptake.
Transgenic mice harboring a human Cu2+/Zn2+superoxide dismutase 1 (SOD1) transgene containing the G93A mutation (Gurney et al., 1994) exhibit impaired capacity for spinal cord high-affinity glutamate uptake (Canton et al., 1998). No data however were presented regarding glutamate uptake in tissue other than spinal cord in the G93A mouse. With respect to expression of the glutamate transporter GLT-1, conflicting results have been reported in G93A mice. Consistent with the reported decrease in spinal cord glutamate uptake, a decrease in GLT-1 immunoreactivity in spinal cord has been reported with no change in the levels of the other astroglial transporter GLAST (Bendotti et al., 2001). However, another group recently reported no change in the net amount of GLT-1 protein in the spinal cord, cortex, and brainstem of G93A mice but rather observed a qualitative molecular weight change in the GLT-1 monomer as well as increases in dimeric GLT-1 (Deitch et al., 2002).
Recently, transgenic rats expressing human SOD1 G93A have been generated (Howland et al., 2002) that develop an ALS-like phenotype, including motor neuron degeneration in the spinal cord. A pronounced loss of GLT-1 occurs in the ventral horn of G93A rats at end stage (Howland et al., 2002). In the current study, we extend these observations by evaluating functional glutamate uptake capacity in specific regions of the nervous system in transgenic G93A rats. Our findings reveal a correlation between altered expression of glial glutamate transporters to loss in maximal velocity (Vmax) of glutamate uptake. Finally, we report an attenuated effect of the ability of riluzole to enhance spinal cord synaptosomal glutamate uptake as a function of disease progression in the G93A rat and implicate loss of GLT-1 as the cause.
Materials and Methods
Materials. Transgenic G93A rats (Howland et al., 2002) were maintained according to Institutional Animal Care and Use Committee approved protocols, and age-matched control Sprague Dawley rats were either bred (littermate controls) or purchased from Taconic (Germantown, NY).l-[3H]Glutamate (specific activity, 40–60 Ci/mmol) was obtained from Amersham Biosciences (Buckinghamshire, UK);l-trans-pyrrolidine-2,4-dicarboxylate (trans-PDC), dihydrokainate (DHK), and riluzole were from Tocris (Ballwin, MO), and l-glutamate was from Sigma (St. Louis, MO). Primary antibodies for immunoblots were obtained from the following sources: GLT-1, Chemicon(Temecula CA); GLAST, Abcam (Cambridge, UK); EAAC1 (excitatory amino acid carrier 1), Zymed (South San Francisco, CA) and J. Rothstein (Johns Hopkins School of Medicine, Baltimore, MD); synaptophysin, Santa Cruz Biotechnology (Santa Cruz, CA); actin, Sigma; and glial fibrillary acidic protein (GFAP), Research Diagnostics (Flanders NJ). Secondary antibodies, linked to horseradish peroxidase (HRP), were obtained from Bio-Rad (Hercules, CA) (anti-mouse, anti-rabbit, and anti-goat) or Research Diagnostics (anti-guinea pig).
Preparation of synaptosomes. Specific regions of the nervous system, including spinal cord, cortex, hippocampus, striatum, cerebellum, and brainstem, were dissected, collected in ice-cold isolation medium (310 mm sucrose and 10 mm HEPES, pH 7.4), and homogenized with a Teflon–glass homogenizer, followed by centrifugation at 1000 × g for 5 min. The resulting supernatant was collected and centrifuged at 20,000 × g for 20 min to obtain the crude synaptosomal P2 pellet which was used at a protein concentration of 0.75 mg/ml in HEPES-buffered saline (HBS) (in mm: 10 HEPES, 5 Tris base, 140 NaCl, 2.5 KCl, 1.2 CaCl2, 1.2 MgCl2, 1.2 K2HPO4, and 10 glucose, pH 7.4). Protein concentration was determined by a modified Bradford method using the commercially available Bio-Rad protein assay kit. Synaptosomal pellets were kept on ice under isolation medium and were resuspended in HBS at room temperature 15–30 min before the assay ofl-[3H]glutamate uptake.
Glutamate uptake assays. Uptake studies in synaptosomes were performed with synaptosomal P2 fractions isolated from cervical spinal cord or the specified brain regions.l-[3H]Glutamate uptake was assayed in a final volume of 250 μl of HBS containing 75 μg of synaptosomal protein, 1 μml-glutamate, and 0.25 μCi/assayl-[3H]glutamate in the absence and presence of inhibitors for the determination of IC50 values. Kinetic experiments were undertaken in the presence of 1–30 μml-glutamate in the presence of 0.25 μCi/assayl-[3H]glutamate as tracer. The effect of riluzole was evaluated by preincubating synaptosomes with the indicated concentrations for 10 min, followed by measurement ofl-[3H]glutamate uptake, as described above for the inhibitors, in the continued presence of riluzole. Reactions were incubated for 4 min at room temperature and then terminated by filtration using a Packard Filtermate 96-well harvester, followed by rapid washing with ice-cold Na+-free HBS (prepared by equimolar replacement with choline). Radioactivity retained on the filters was determined by scintillation counting. In all uptake experiments, the radioactivity retained after incubation in Na+-free HBS was used to correct all data to represent Na+-dependent uptake.
Histochemical analysis. Animals were anesthetized using approved animal welfare protocols and perfused by cardiac puncture with 4% paraformaldehyde–PBS. Brain and spinal cord were removed, followed by regional dissection. Tissue blocks were embedded in paraffin for sectioning (7 μm). Hematoxylin and eosin stains of brain and spinal cord were performed on paraffin sections. Immunostaining was performed with antibody to glial fibrillary acidic protein, GFAP (1:50; Dako, Copenhagen, Denmark).
Immunoblot analysis of glutamate transporters. The expression of GLT-1, GLAST, and EAAC1 in whole spinal cord and brainstem homogenates was examined by immunoblot. Whole cervical spinal cord or brainstem (∼0.2 gm) was homogenized in 2 ml of 50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, and 1% SDS, pH 7.4, with protease inhibitors (Complete; Roche, Indianapolis, IN) and centrifuged at 14,000 × g. Five to 25 μg of total extract protein was electrophoresed on 7.5% SDS–polyacrylamide gels. Immunoblots were probed with anti-GLT-1 (1:5000), anti-GLAST (1:2000), anti-EAAC1 (1:500), anti-GFAP (1:5000), anti-actin (C4; 1:5000), or anti-synaptophysin (1:5000) antibodies. Secondary antibodies conjugated to HRP used at 1:2000 were followed by signal detection using ECL (Amersham Biosciences, Piscataway NJ).
Data analysis. All data for uptake assays were corrected to represent Na+-dependent uptake by subtraction of the uptake observed in the absence of extracellular Na+. Kinetic data were determined by nonlinear regression analysis of the saturation curves using the following Michaelis-Menten equation: velocity =Vmax × [S]/Km + [S], where [S] is substrate concentration, using Origin 6.0 software (Microcal Software, Northampton, MA). In the pharmacological experiments, control uptake was calculated as the total Na+-dependent uptake measured over 4 min, and drug effects were expressed as a percentage of the control response. Log concentration–response curves were constructed for the determination of IC50 values using the following four-parameter logistic function: y = (Top − Bottom/1 + (x/IC50)p)) + Bottom, where p represents the Hill coefficient.
Quantitation of immunoblots was done using Scion Image software. Statistical analyses were performed using between-groups ANOVA, followed by Dunnett's t test.
Results
Spinal cord synaptosomal uptake of glutamate
High-affinity Na+-dependent glutamate uptake was examined in spinal cord synaptosomes prepared from presymptomatic (8–10 weeks), disease onset (13–15 weeks), and end-stage (15–17 weeks) G93A rats and compared with the activity measured in age-matched control (nontransgenic) animals. Onset of the disease was typified by the appearance of hindlimb abnormal gait, followed by a rapid progression (<2 weeks) to complete hindlimb paralysis, with end-stage animals characterized with a loss of righting reflex concomitant with paralysis extending to at least one forepaw (Table 1). No difference in theVmax of glutamate transport in spinal cord synaptosomes was observed with presymptomatic animals compared with controls; however, a reduction in theVmax was observed coincident with disease onset and was more dramatic at end stage (Fig.1). Vmaxvalues were 161 ± 16 and 92 ± 17 pmol · min−1 · mg−1for control and disease end-stage G93A rats, respectively, representing a 43% decrease in total transport capacity. At disease onset, the reduction in Vmax in the G93A animals was 27% relative to controls, and no changes in the affinity for glutamate were observed over developmental time between transgenic and control rats (Km values were in the range of 3–8 μm).
Table 1.
Clinical classification of G93A SOD1 transgene-induced disease progression
| Group classification | Clinical signs |
|---|---|
| Presymptomatic | Full mobility and no observable difference in behavior from age-matched controls |
| Onset | Observation of abnormal gait or hindlimb weakness |
| End stage | Overall muscle wasting and complete hindlimb paralysis
indexed as an inability to right themselves in a 30 sec time window; typically accompanied by paralysis of at least one front limb |
Fig. 1.
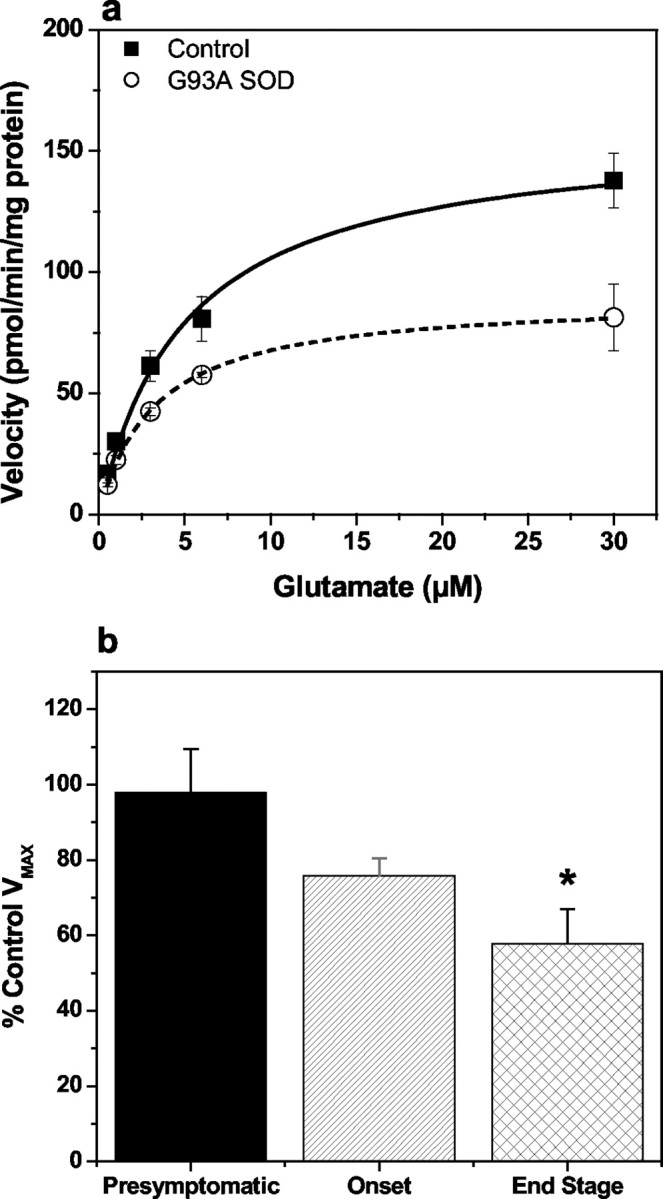
Saturation isotherm for the high-affinity Na+-dependent transport of glutamate in spinal cord synaptosomes prepared from disease end-stage G93A transgenic rats and age-matched controls (a) and effect onVmax relative to control as a function of disease progression (b). Glutamate uptake into P2 synaptosomal fractions prepared from presymptomatic, disease onset, and end-stage G93A rats, together with age-matched controls, was measured as described in Materials and Methods for the estimation of the kinetic parameters Km andVmax. Solid line ina depicts data from control animals, and thedashed line represents the G93A data. Initial rates of Na+-dependent glutamate uptake were expressed as picomoles per minute per milligram of synaptosomal protein after subtraction of the uptake observed in the absence of extracellular Na+ (equimolar replacement of NaCl with choline chloride). Kinetic constants Km andVmax were estimated from the saturation isotherm using the Michaelis-Menten equation, and data inb represent the percentage controlVmax values for the G93A animals at each stage of the disease evaluated. Data represent mean ± SEM values from three to four independent experiments. * indicates statistically significant difference from presymptomatic, by ANOVA with Dunnett's test for multiple comparisons at the 5% significance level.
With each preparation of synaptosomes, we also examined the sensitivity of the transport process to the nonselective glutamate uptake blockertrans-PDC and the selective GLT-1/EAAT2 inhibitor DHK. Inhibitor sensitivity, in combination with Na+ dependency of uptake, was evaluated as an index of the integrity of the synaptosomal preparations, and the analysis was also performed to determine any changes in pharmacological sensitivity. Glutamate uptake activity in spinal cord synaptosomes from presymptomatic G93A and age-matched control rats was blocked in a concentration-dependent manner by both trans-PDC and DHK, with equivalent IC50 values between G93A animals and controls (Fig. 2a, Table2). No changes in the inhibitor sensitivity were observed with spinal cord synaptosomes prepared from disease onset or end-stage G93A rats compared with either their corresponding age-matched controls (Fig.2b,c) or the results obtained for presymptomatic animals. A summary of the pharmacological comparison for spinal cord synaptosomes is provided in Table 2.
Fig. 2.
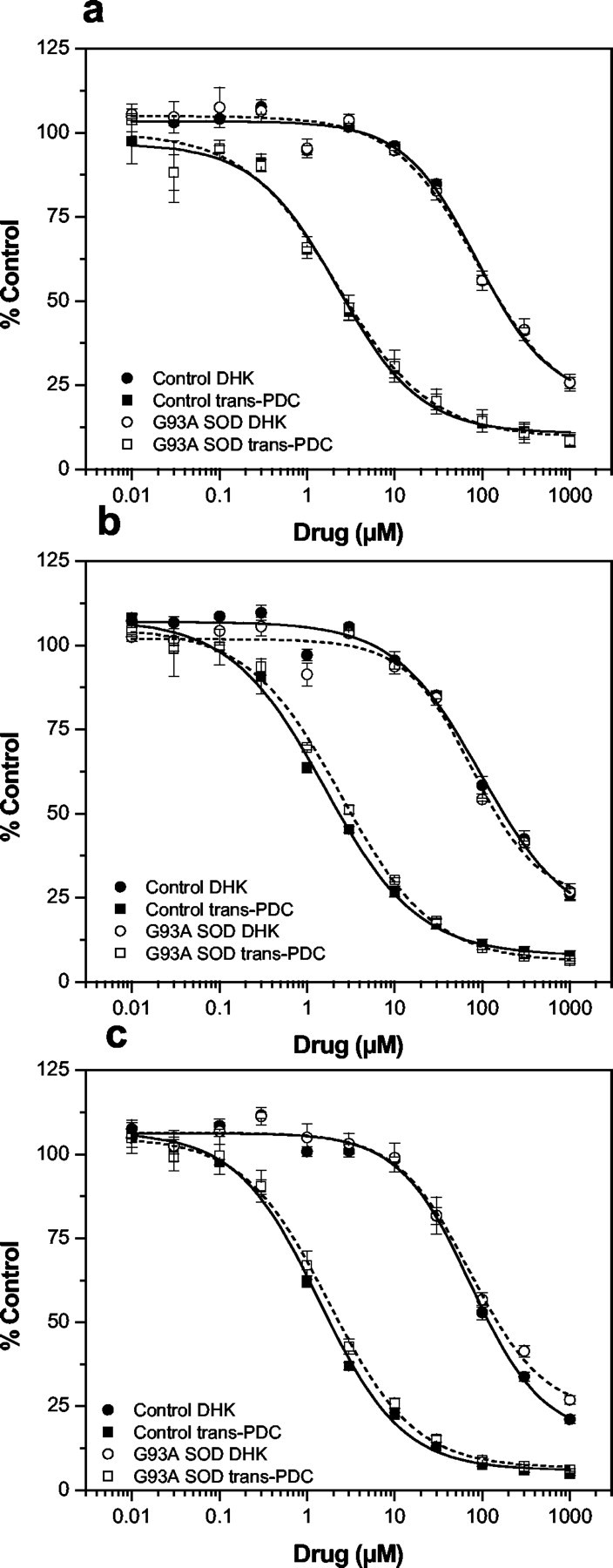
Log concentration–response curves for the inhibition of glutamate uptake into spinal cord synaptosomes prepared from G93A transgenic rats and age-matched controls. Glutamate uptake into P2 synaptosomal fractions prepared from presymptomatic (a), disease onset (b), and end-stage (c) G93A rats, together with age-matched controls, was measured in the absence and presence of the nonselective transport inhibitor trans-PDC or the selective GLT-1/EAAT2 inhibitor DHK. Solid lines depict data from control animals, and dashed lines rep resent the G93A data. The net Na+-dependent glutamate uptake values were expressed as a percentage of the activity observed in the absence of drug, and IC50 values were determined from the log concentration–response curves by nonlinear regression analysis using the four-parameter logistic function. Averaging the percentage control values obtained from three independent experiments generated each curve shown, and the IC50 values are summarized in Table 2.
Table 2.
Pharmacological characterization of glutamate uptake into spinal cord synaptosomes prepared from transgenic G93A rats and age-matched controls
| PDC (μm) | DHK (μm) | |
|---|---|---|
| End stage | ||
| CON | 1.4 ± 0.1 | 74.2 ± 6.0 |
| G93A | 1.8 ± 0.1 | 75.3 ± 0.9 |
| Onset | ||
| CON | 1.6 ± 0.3 | 98.2 ± 5.3 |
| G93A | 2.4 ± 0.1 | 79.9 ± 8.1 |
| Presymptomatic | ||
| CON | 2.4 ± 0.5 | 87.9 ± 3.2 |
| G93A | 2.3 ± 0.3 | 89.8 ± 13.1 |
Pharmacological sensitivity of glutamate uptake in synaptosome fractions prepared from spinal cord of G93A and age-matched control (CON) rats was evaluated at various stages of disease progression. IC50 values were generated by nonlinear regression analysis of log concentration–response curves using the four-parameter logistic model. Data are mean ± SEM values from three independent experiments.
Loss of glutamate uptake enhancement by riluzole in end-stage spinal cord from G93A rats
Riluzole, a neuroprotective agent used clinically to slow the progression of ALS, has been reported to increase the glutamate transport capacity in spinal cord synaptosomes (Azbill et al., 2000). Coincubation with riluzole (10–300 μm) produced a 25–30% increase in the glutamate uptake measured with spinal cord synaptosomes prepared from G93A rats at disease onset, and the enhancing effect was similar in the corresponding control animals (Fig.3a). In contrast, the stimulatory effect of riluzole was blunted with spinal cord synaptosomes prepared from end-stage G93A rats in which a significantly reduced (<10%) increase in the glutamate transport activity was observed compared with a 30% increase observed with the age-matched controls (Fig. 3b).
Fig. 3.
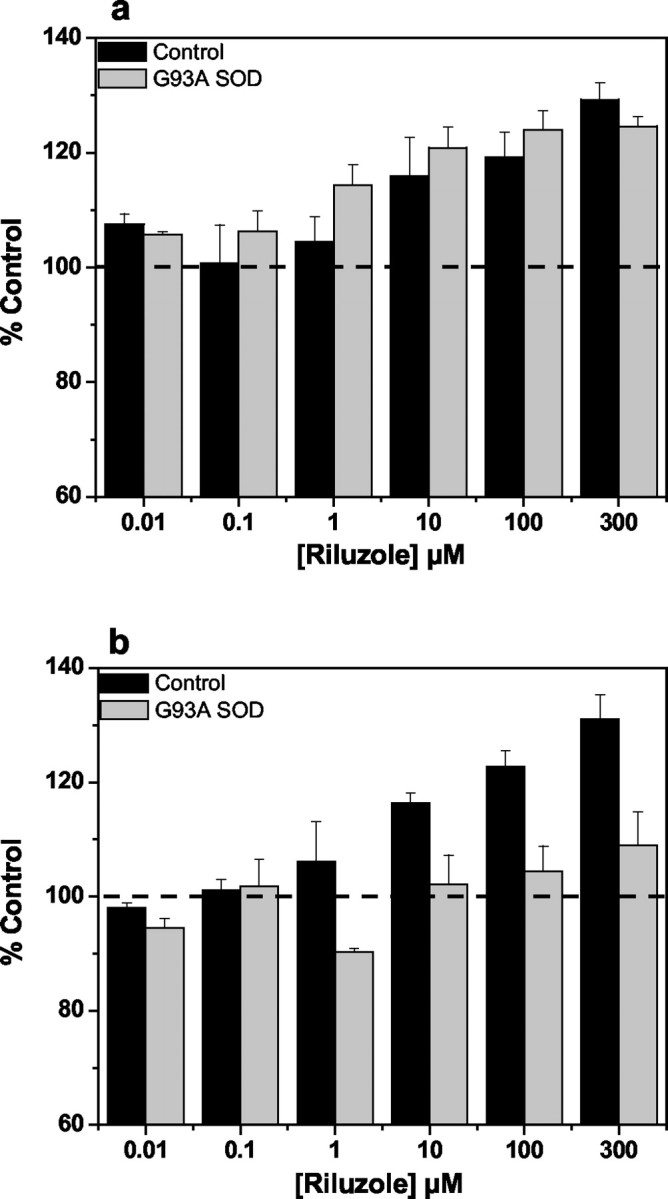
Effect of riluzole on the high-affinity Na+-dependent uptake of glutamate in spinal cord synaptosomes prepared from G93A transgenics and age-matched controls. Glutamate uptake into P2 synaptosomal fractions prepared from disease onset (a) and end-stage (b) G93A rats, together with age-matched controls, was measured in the absence or presence of riluzole (0.1–300 μm) after a 10 min pretreatment with the drug. The net Na+-dependent glutamate uptake data were expressed as a percentage of the activity observed in the absence of drug. A significant enhancement of glutamate uptake by riluzole was measured for control tissue (a, b) and G93A transgenic tissue at disease onset (a) (ANOVA;p < 0.05) but not for G93A transgenic tissue at end-stage disease (b).
Disease changes and synaptosomal glutamate uptake in various brain regions
Spinal cord from end-stage G93A transgenic rats shows a complete loss of ventral large motor neuron cell bodies (Fig.4A) as well as dramatic increases in gliosis (Fig. 4A,B) but only few vacuoles. Vacuoles appear in higher abundance in earlier stages of disease in these rats (Howland et al., 2002) but disappear by end stage, possibly reflecting the severity of damage. Brainstem of G93A rats show a profound vacuolization (Fig. 4C), as well as an increased gliosis at end-stage disease; however, neuronal cell loss is not as profound as that observed in the end-stage diseased spinal cord (Fig. 4) (Howland et al., 2002). Other brain regions surveyed (Fig. 4D,E, neocortex and hippocampus, respectively) show no degenerative signs of pathology.
Fig. 4.
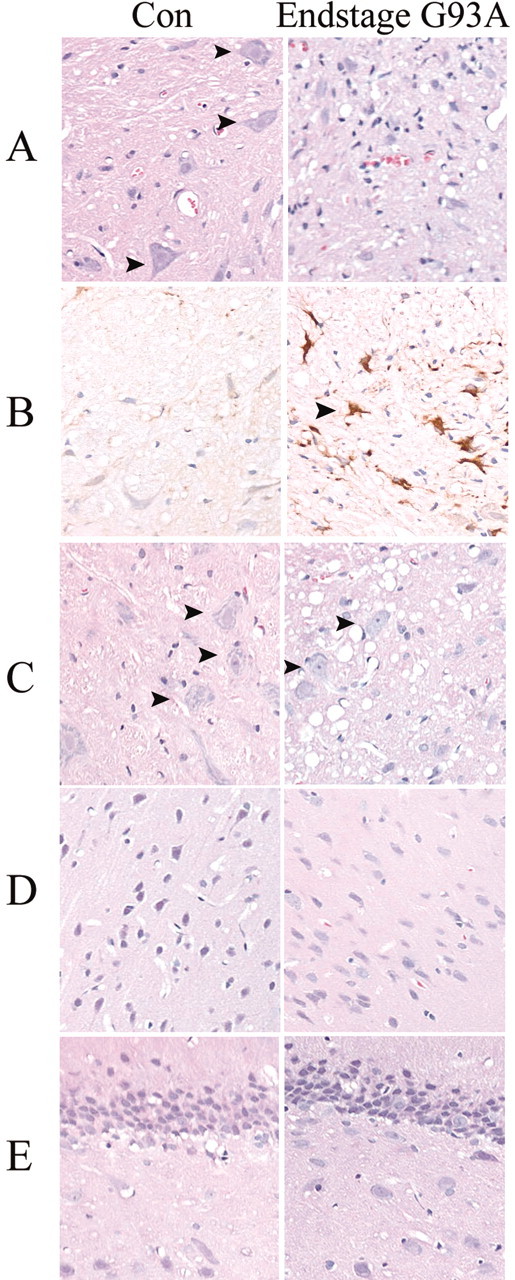
Histochemical analysis of pathology in end-stage G93A transgenic rat spinal cord and brain regions. Cervical spinal cord (A) or brain areas including brainstem (C), neocortex (D), and hippocampus (E) were stained with hematoxylin and eosin to show extent of neuronal loss, degenerative structures (vacuoles), as well as gliosis in end-stage G93A rats compared with age-matched control rats (Con). Arrows inA and C denote the presence of large motor neuron cell bodies, rarely found in end-stage diseased spinal cord. Representative spinal cord sections from control and end-stage G93A rats are also shown stained with anti-GFAP antibody (B). Arrows in Bhighlight a hypertrophic astrocyte, frequently found in end-stage diseased spinal cord.
High-affinity Na+-dependent glutamate uptake was examined in synaptosomes prepared from cortex, hippocampus, striatum, cerebellum, and brainstem using end-stage G93A rats and compared with the activity measured in the same tissues from age-matched control animals. In contrast to what we observed in G93A rat spinal cord, there was no difference in either theVmax orKm values for glutamate uptake in cortical, hippocampal, striatal (Fig.5), cerebellar, or brainstem (Fig. 6) synaptosomes between end-stage disease G93A and age-matched control animals. Similarly, we observed no difference in the pharmacological sensitivity to either trans-PDC or DHK using synaptosomes prepared from these same brain regions when comparing G93A rats with control animals (Table 3). Bothtrans-PDC and DHK were effective blockers of glutamate uptake in all regions examined, with the exception of DHK in cerebellar synaptosomes in which 50% inhibition of uptake was not achieved, even at 1 mm.
Fig. 5.
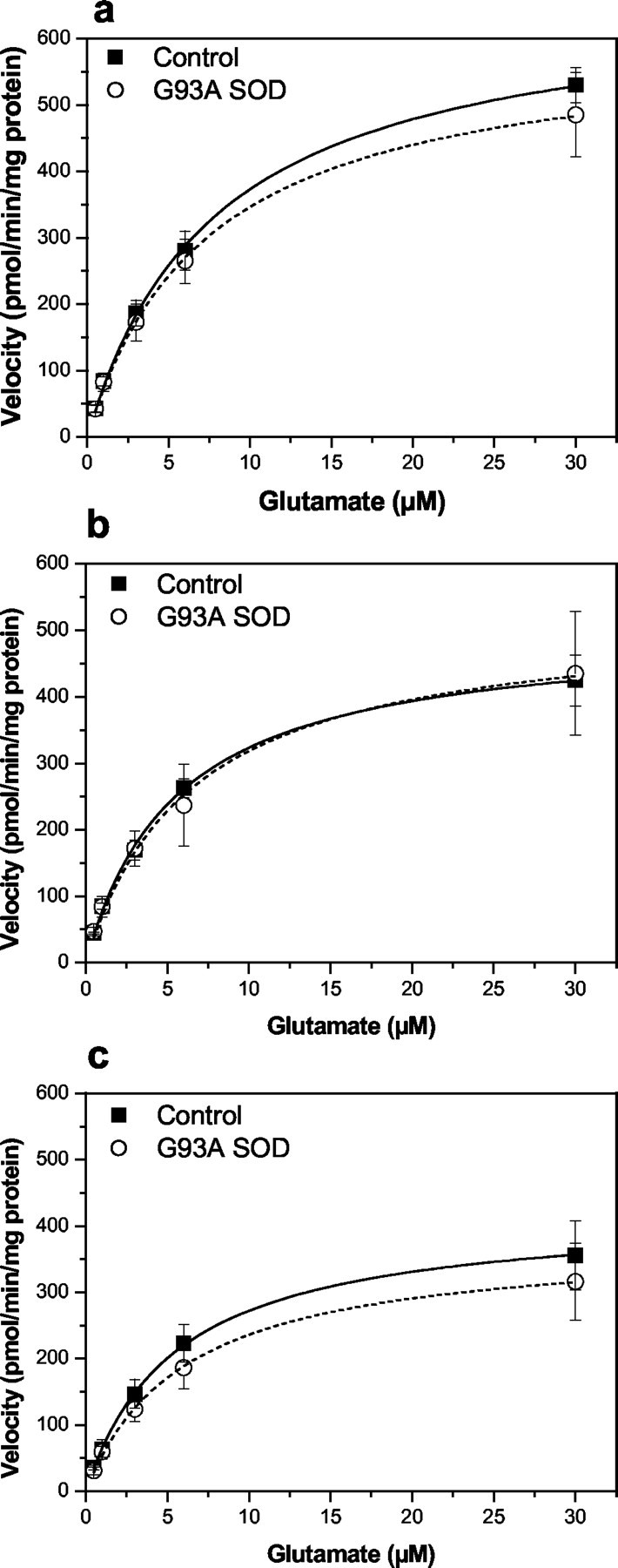
Saturation isotherms for the high-affinity Na+-dependent transport of glutamate in cortical (a), hippocampal (b), and striatal (c) synaptosomes prepared from end-stage G93A transgenic rats and age-matched controls. Glutamate uptake into P2 synaptosomal fractions was measured as described in Materials and Methods for the estimation of the kinetic parametersKm and Vmax. Initial rates of Na+-dependent glutamate uptake were expressed as picomoles per minute per milligram of synaptosomal protein after subtraction of the uptake observed in the absence of extracellular Na+ (equimolar replacement of NaCl with choline chloride). Kinetic constants Kmand Vmax were estimated from the saturation isotherm using the Michaelis-Menten equation and are presented in Table3. Data represent mean ± SEM values from three independent experiments.
Fig. 6.
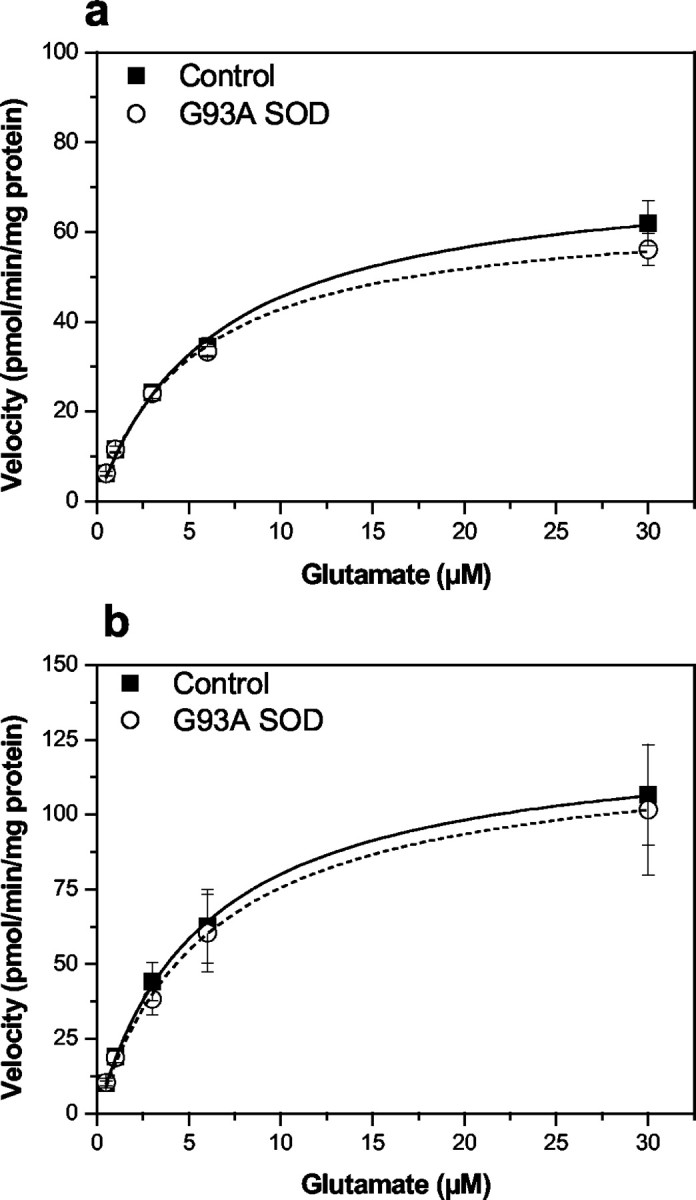
Saturation isotherms for the high-affinity Na+-dependent transport of glutamate in cerebellar (a) and brainstem (b) synaptosomes prepared from end-stage G93A transgenic rats and age-matched controls. Note that the scale of the y-axis in this figure is lower than tissue analyzed in Figure 5. Brainstem and cerebellum typically exhibited lower overall glutamate uptake capacity. Glutamate uptake measurements and calculations ofVmax and Km were identical to that as described in Figure 5. Kinetic constantsKm and Vmax were estimated from the saturation isotherm using the Michaelis-Menten equation and are presented in Table 3. Data represent mean ± SEM values from three independent experiments.
Table 3.
Kinetic parameters and inhibitor IC50 values for glutamate uptake in synaptosomes prepared from various brain regions of transgenic G93A rats and age-matched controls
| Kinetics | Pharmacology | |||
|---|---|---|---|---|
| Vmax (pmol · min−1 · mg−1) | Km(μm) | PDC (μm) | DHK (μm) | |
| Cortex | ||||
| CON | 669 ± 16 | 8 ± 1 | 2.1 ± 0.3 | 38.2 ± 0.5 |
| G93A | 603 ± 74 | 7 ± 0 | 2.0 ± 0.1 | 31.9 ± 7.2 |
| Hippocampus | ||||
| CON | 503 ± 39 | 5 ± 0 | 1.8 ± 0.1 | 35.7 ± 10.9 |
| G93A | 523 ± 96 | 6 ± 0 | 1.7 ± 0.2 | 35.1 ± 6.2 |
| Striatum | ||||
| CON | 422 ± 60 | 5 ± 0 | 1.7 ± 0.1 | 32.0 ± 6.1 |
| G93A | 378 ± 72 | 6 ± 0 | 1.8 ± 0.4 | 31.5 ± 5.0 |
| Cerebellum | ||||
| CON | 74 ± 8 | 6 ± 0 | 3.6 ± 0.3 | >1000 |
| G93A | 65 ± 5 | 5 ± 0 | 3.3 ± 0.3 | >1000 |
| Brainstem | ||||
| CON | 127 ± 20 | 6 ± 0 | 2.0 ± 0.2 | 87.6 ± 22.4 |
| G93A | 123 ± 29 | 6 ± 1 | 2.0 ± 0.1 | 79.3 ± 19.5 |
Glutamate uptake in synaptosome fractions prepared from the indicated brain regions of G93A and age-matched control (CON) rats was evaluated at end stage of disease progression. Kinetic parameters and IC50 values are mean ± SEM values from three independent experiments.
Expression analysis of GLT-1, GLAST, and EAAC1 in spinal cord and brainstem of G93A rats
Whole cervical spinal cord from end-stage G93A rats and nontransgenic age-matched controls (16–20 weeks) were analyzed for levels of glial glutamate transporters GLT-1 and GLAST, as well as the neuronal transporter EAAC1, by immunoblot analysis (Fig.7A). In addition, immunoblots were probed with antibodies to GFAP to index the extent of gliosis, synaptophysin to index the degree of neuronal cell loss, and actin to control for protein loading. A significant decrease of GLT-1 of 45% was observed in end-stage cervical spinal cord of the G93A rats compared with controls (Fig. 7A). No consistent changes in gel migration of the 72 kDa GLT-1 monomeric species were observed between control and end-stage disease tissue, in contrast to that reported by Deitch et al. (2002) for the G93A transgenic mice. Qualitative changes in GLAST immunoreactivity, however, were consistently observed in end-stage spinal cord of the G93A rats, but total immunoreactivity was not significantly different from controls. GLAST in control spinal cord migrated as a doublet, with the major band migrating at 65 kDa (Fig. 7A). However, a more complex pattern was observed in end-stage G93A rat spinal cord. A new band of intermediate molecular weight appeared in end-stage G93A spinal cords. The intensity of this species was more pronounced in animals that had most severe clinical symptoms (Fig. 7A). No significant changes in EAAC1 immunoreactivity in end-stage disease G93A spinal cord versus controls was evident using two different antisera (Zymed) (Rothstein et al., 1995). End-stage G93A rats exhibit extensive gliosis as well as neuron cell loss in the spinal cord (Fig. 4A,B) (Howland et al., 2002). We probed spinal cord immunoblots with anti-GFAP as well as anti-synaptophysin antibodies to attempt to correlate the degree of gliosis and cell loss in G93A spinal cord to changes in glutamate transporter immunoreactivity. End-stage G93A rats exhibit significantly increased (2.5-fold) GFAP immunoreactivity as well as decreased (53%) synaptophysin immunoreactivity in cervical spinal cord (Figs.7A, 8). The extent of these changes was most marked in most severely affected animals.
Fig. 7.

Immunoblot analysis of GLT-1, GLAST, and EAAC1 protein expression in whole cervical spinal cord (A) and brainstem (B) homogenates. Expression of glutamate transporters (GLT-1, GLAST, and EAAC1), as well as GFAP, actin, and synaptophysin is shown in nontransgenic animals (Con) and end-stage SOD G93A rats between 16 and 20 weeks of age and depict typical results obtained.Asterisks denote G93A rats that were scored as most severely affected in this group. Five micrograms of extract protein were immunoblotted for GLT-1, GFAP, synaptophysin (SYPH), and actin (ACT), and 25 μg of protein were immunoblotted for EAAC1 and GLAST.
Fig. 8.

Immunoblot analysis of GLT-1 and GLAST, as well as GFAP, synaptophysin, and actin expression in whole cervical spinal cord through disease progression [nontransgenic (Con), presymptomatic G93A (8 weeks), onset G93A (16 weeks), and end-stage G93A (18 weeks) rats]. Five micrograms of extract protein were immunoblotted for GLT-1, GFAP, synaptophysin (SYPH), and actin, and 25 μg of protein were immunoblotted for GLAST.
In contrast to GLT-1 changes in spinal cord, we observed no consistent changes in GLT-1 immunoreactivity in brainstem of end-stage G93A rats (Fig. 7B). A slightly decreased mobility for the 65 kDa form of GLAST was evident in brainstem but was not as dramatic as that seen in spinal cord from end-stage diseased rats. No significant changes in EAAC1 immunoreactivity were noted in the brainstem of the G93A rats. As expected, GFAP immunoreactivity was significantly elevated in brainstem (twofold) of the G93A rats; however, no significant changes in synaptophysin immunoreactivity were noted, consistent with a lower degree of neuron cell loss in brainstem compared with spinal cord. Cortex of the G93A end-stage rats as well as controls were also analyzed in a similar manner as for spinal cord and brainstem; however, no changes in any of the transporters was noted in this brain region (data not shown).
Glutamate transporter levels were also assessed in the G93A rat spinal cord at different stages of disease (nontransgenic, presymptomatic, onset, and end stage) (Fig. 8). Reduced levels of GLT-1 were evident as well as GLAST, showing altered gel migration in all G93A rats at end stage. GLT-1 changes, however, were not typically evident in G93A rats at disease onset or in presymptomatic rats when analyzing whole spinal cord tissue. However, we showed previously that, when rat spinal cord is microdissected into ventral and dorsal horns, decreased levels of GLT-1 are evident as early as 100 d old (late presymptomatic) (Howland et al., 2002). Sampling whole spinal cord most likely dilutes the magnitude of the measurable decrease in GLT-1 immunoreactivity, which appears to be ventral horn specific, and results in a shift to later time points when quantitative differences can be measured. A similar phenomena was reported by Bendotti et al. (2001) in transgenic SOD1 mice. GFAP immunoreactivity increased with disease progression in G93A spinal cord, reaching its peak in end-stage rats. Decreased synaptophysin was not evident until end-stage disease, with the exception of one animal at disease onset (Fig. 8).
Discussion
We describe a kinetic and pharmacological characterization of synaptosomal glutamate transport capacity in multiple regions of the nervous system using a recently developed transgenic rat model of ALS overexpressing the human G93A mutant Cu2+/Zn2+superoxide dismutase 1 gene (Howland et al., 2002). Our results indicate a selective deficit in glutamate uptake activity in synaptosomes prepared from spinal cord, with no changes detected in various brain regions, including cortex, hippocampus, striatum, cerebellum, and brainstem. The reduced glutamate transport capacity measured in spinal cord synaptosomes was restricted to animals exhibiting signs of disease onset (13–15 weeks) and at end stage of the disease (15–17 weeks). At end stage, the loss of glutamate transport capacity was most dramatic, with a 43% reduction in theVmax detected compared with a 27% loss observed with animals at disease onset. Although a reduced maximal capacity for glutamate uptake was demonstrated in both disease onset and end-stage G93A rats, there was no change in the affinity (Km) for the substrate at any developmental time point of the disease, a result consistent with no change in the pharmacological specificity of glutamate uptake as disease progresses.
We examined glutamate uptake in tissue known to contain ALS-like lesions in the G93A transgenic rats (spinal cord and brainstem) as well as regions devoid of pathology (cortex, hippocampus, striatum, and cerebellum). Functional glutamate transport is only deficient in spinal cord of the G93A rats, is evident as early as disease onset, and becomes more severe by end stage. Our results in end-stage diseased spinal cord are similar to those described previously for transgenic mice harboring the same G93A mutant form of SOD1 (Canton et al., 1998). That study, however, did not report on the effect of the G93A mutation on glutamate transport in the brainstem as well as other brain regions. Here, we showed that no measurable change in glutamate transport occurs in the brainstem, despite the presence of pathological changes. Although vacuolization is quite extensive in end-stage G93A brainstem, the extent of neuron cell loss is not as severe as that observed in the spinal cord. Sparing of glutamate transport function, despite some pathological changes, may be related to lesser degree of neuron cell loss in the brainstem of the G93A rat.
Studies with synaptosomal preparations prepared from postmortem human ALS tissue have described a marked reduction in the maximal velocity of glutamate transport in spinal cord (−59%), motor cortex (−70%), and somatosensory cortex (−39%) synaptosomes, with no accompanying change in Km (Rothstein et al., 1992). The data reported here on the G93A rat are consistent with a deficit in glutamate uptake in spinal cord of ALS patients and lack of a deficit in brain regions known to be spared of ALS-like lesions. However, the transgenic rat differs in that glutamate uptake capacity is spared in brainstem, a region that shows some pathological changes in the G93A rat as well as in ALS.
A molecular mechanism for the deficits in glutamate clearance capacity was initially suggested in the human study with the demonstration that expression levels of the glial-specific glutamate transporter EAAT2 (GLT-1) were dramatically reduced in both spinal cord and motor cortex (Rothstein et al., 1995). Because GLT-1 is considered to account for a large proportion of the glutamate transport capacity in the nervous system, its loss is consistent with both a reduction in glutamate uptake capacity and elevated extracellular glutamate, as demonstrated in both CSF and plasma in ALS patients (Rothstein et al., 1992;Spreux-Varoquaux et al., 2002). Consistent with the loss of GLT-1 protein described in human ALS patients (Rothstein et al., 1995), a 50% loss of spinal cord homogenate GLT-1 protein has also been detected in transgenic mice expressing another mutant form of SOD1 incorporating a G85R mutation, at end stage (Bruijn et al., 1997).
Immunohistochemical methods were also used to demonstrate reductions of GLT-1 in G93A transgenic mouse spinal cords at 14–18 weeks in age, a time when motor impairments is evident (Bendotti et al., 2001). However, the loss of total GLT-1 protein in the G93A transgenic mouse has been questioned recently given the observations of Deitch et al. (2002), who were unable to detect a net loss of GLT-1 protein but rather reported a mobility shift of GLT-1 monomer to a higher molecular weight, thought to result from an unidentified posttranslational modification. This latter observation is not inconsistent with the loss of functional uptake in these animals because aberrant processing of the protein could result in altered function. We demonstrate a loss of GLT-1 immunoreactivity in end-stage diseased G93A rat spinal cords. We did not consistently observe gel migration differences in GLT-1 in the G93A rat spinal cord or brainstem, even under extraction and gel conditions identical to that reported by Deitch et al. (2002).
Our results with the G93A transgenic rats support a model of dysfunction of astroglial glutamate transporter expression represented by loss of total GLT-1 protein, accompanied by the appearance of GLAST at an intermediate molecular weight, possibly reflecting a posttranslational modification. These changes in glutamate transporters in end-stage diseased spinal cord from the G93A rats were accompanied by decreased synaptophysin immunoreactivity and markedly reduced numbers of large motor neurons in spinal cord (Howland et al., 2002). In contrast however, we were unable to consistently demonstrate a change in GLT-1 levels in brainstem, a region that, despite the presence of vacuoles, does not exhibit a significant loss in neurons. Together, these data underscore a potential causal relationship of GLT-1 loss to neuronal survival in this rat model.
Pharmacological sensitivity of spinal cord glutamate transport to both the nonselective glutamate uptake blocker trans-PDC and the selective GLT-1/EAAT2 inhibitor DHK was observed in our study at all developmental time points evaluated, with no changes in inhibitor IC50 observed as the disease progressed. Sensitivity to DHK in the spinal cord preparation suggests that the GLT-1 subtype of glutamate transporter predominantly mediates the measured transport activity. This conclusion is consistent with the reported high expression of GLT-1 in spinal cord homogenates by immunoblot analysis and the relatively low expression of GLAST or EAAC1 immunoreactivity. The loss of glutamate transport capacity observed in the spinal cord of G93A rats together with the pharmacological identification of GLT-1 as the predominant subtype provides indirect evidence for the loss of GLT-1 in this model as a contributing factor to the deficit. More directly, the 45% loss of GLT-1 protein correlates well with the 43% reduction in net glutamate uptake capacity.
Additional characterization of the pharmacological properties of spinal cord glutamate transport was explored in this study by examining the facilitatory effect of riluzole on the uptake mechanism. Riluzole is used clinically in delaying the progression of ALS (Desai and Swash, 1997), and multiple mechanisms of action might account for its neuroprotective properties, including both presynaptic and postsynaptic effects on a variety of target proteins, including glutamate receptors, Ca2+, Na+, and K+ channels (Benavides et al., 1985;Mizoule et al., 1985; Doble, 1996; Huang et al., 1997; Zona et al., 1998; Duprat et al., 2000; Xu et al., 2001). Generally speaking, the compound acts as an antagonist of glutamatergic neurotransmission. Riluzole treatment, in the drinking water, of SOD1 G93A transgenic mice has been shown to increase survival without affecting age of disease onset (Gurney et al., 1996). More recently, riluzole has been reported to produce both an increase (31%) inVmax and a decrease (21%) in the apparent Km for glutamate transport in spinal cord synaptosomes (Azbill et al., 2000). That study also demonstrated significant increases in sodium-dependent glutamate uptakein vivo. Synaptosomes prepared from rats after two separate injections of riluzole showed a 49% increase in glutamate uptake. We also observed an enhancement of glutamate uptake activity in spinal cord synaptosomes treated with riluzole in vitro, although we were unable to demonstrate an effect with 0.1 μm riluzole, in contrast to the study of Azbill and coworkers, with concentrations in the range of 10–300 μm being effective in the present study at producing a 25–30% increase in the uptake of 1 μm glutamate. More interestingly, we observed a consistent reduction in the enhancing effect of riluzole in spinal cord synaptosomes prepared from end-stage G93A rats, although similar studies with tissue derived from disease onset animals revealed an equivalent enhancement by riluzole in transgenic compared with control animals. Because the neuroprotective activity associated with riluzole might be attributed at least in part to this facilitated extracellular glutamate clearance, the attenuation of the enhancing activity demonstrated at end-stage disease in the current study raises the intriguing possibility that the efficacy of this drug might be reduced with disease progression in ALS patients.
In summary, we demonstrated marked deficits in glutamate uptake in spinal cord, but not brainstem and other brain areas not affected by disease, in the G93A transgenic rat. Reductions in spinal cord glutamate transport capacity are accompanied by altered expression of astroglial glutamate transporters GLT-1 and GLAST. Significant deficits in glutamate uptake mediated by glial cells is likely to be the cause for loss of efficacy of riluzole to potentiate glutamate uptake in end-stage G93A spinal cord compared with rats at earlier stages of disease. On the basis of these data, it is possible that the effectiveness of riluzole in ALS patients in latter stages of disease may be dampened compared with patients at earlier stages of disease on this drug therapy.
Footnotes
We are grateful to John Kulik, George Psaltis, and Lisa DeVito for assistance with the transgenic rat colony. We thank Drs. Seung Kwak and Erika Holzbaur for technical assistance and advice.
Correspondence should be addressed to Dr. David Howland, Wyeth Research, Neuroscience, CN8000, Princeton, NJ 08543. E-mail:howland@wyeth.com.
References
- 1.Alexander GM, Deitch JS, Seeburger JL, Del Valle L, Heiman-Patterson TD. Elevated cortical extracellular fluid glutamate in transgenic mice expressing human mutant (G93A) Cu/Zn superoxide dismutase. J Neurochem. 2000;74:1666–1673. doi: 10.1046/j.1471-4159.2000.0741666.x. [DOI] [PubMed] [Google Scholar]
- 2.Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 3.Benavides J, Camelin JC, Mitrani N, Flamand F, Uzan A, Legrand JJ, Gueremy C, Le Fur G. 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission-II. Biochemical properties. Neuropharmacology. 1985;24:1085–1092. doi: 10.1016/0028-3908(85)90196-0. [DOI] [PubMed] [Google Scholar]
- 4.Bendotti C, Tortarolo M, Suchak SK, Calvaresi N, Carvelli L, Bastone A, Rizzi M, Rattray M, Mennini T. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J Neurochem. 2001;79:737–746. doi: 10.1046/j.1471-4159.2001.00572.x. [DOI] [PubMed] [Google Scholar]
- 5.Brown RH. Amyotrophic lateral sclerosis: lessons from mouse and human genetics. Clin Neurosci Res. 2001;1:84–90. [Google Scholar]
- 6.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 7.Canton T, Pratt J, Stutzmann J-M, Imperato A, Boireau A. Glutamate uptake is decreased tardively in the spinal cord of FALS mice. NeuroReport. 1998;9:775–778. doi: 10.1097/00001756-199803300-00001. [DOI] [PubMed] [Google Scholar]
- 8.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 9.Cluskey S, Ramsden DB. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol Pathol. 2001;54:386–392. [PMC free article] [PubMed] [Google Scholar]
- 10.Deitch JS, Alexander GM, Del Valle L, Heiman-Patterson TD. GLT-1 glutamate transporter levels are unchanged in mice expressing G93A human mutant SOD1. J Neurol Sci. 2002;193:117–126. doi: 10.1016/s0022-510x(01)00656-6. [DOI] [PubMed] [Google Scholar]
- 11.Desai J, Swash M. Clinical studies of riluzole in amyotrophic lateral sclerosis. Drugs Today. 1997;33:595–600. [Google Scholar]
- 12.Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47 [Suppl 4]:S233–S241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- 13.Duprat F, Lesage F, Patel AJ, Fink M, Romey G, Lazdunski M. The neuroprotective agent riluzole activates the two P domain K+ channels TREK-1 and TRAAK. Mol Pharmacol. 2000;57:906–912. [PubMed] [Google Scholar]
- 14.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 15.Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin on a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- 16.Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1997;282:1280–1290. [PubMed] [Google Scholar]
- 18.Mizoule J, Meldrum B, Mazadier M, Croucher M, Ollat C, Uzan A, Legrand JJ, Gueremy C. 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission-I. Anticonvulsant properties. Neuropharmacology. 1985;24:767–773. doi: 10.1016/0028-3908(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 19.Plaitakis A, Fesdjian CO, Shashidharan P. Glutamate antagonists in amyotrophic lateral sclerosis—a review of their therapeutic potential. CNS Drugs. 1996;5:437–456. [Google Scholar]
- 20.Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornbath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 21.Rothstein JD, Kuncl R, Chaudhry V, Clawson L, Cornblath DR, Coyle JT, Drachman DB. Excitatory amino acids in amyotrophic lateral sclerosis: an update. Ann Neurol. 1991;30:224–225. doi: 10.1002/ana.410300223. [DOI] [PubMed] [Google Scholar]
- 22.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 23.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 24.Spreux-Varoquaux O, Bensimon G, Lacomblez L, Salachas F, Pradat PF, Le Forestier N, Marouan A, Dib M, Meininger V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci. 2002;193:73–78. doi: 10.1016/s0022-510x(01)00661-x. [DOI] [PubMed] [Google Scholar]
- 25.Xu L, Enyeart JA, Enyeart JJ. Neuroprotective agent riluzole dramatically slows inactivation of Kv1.4 potassium channels by a voltage-dependent oxidative mechanism. J Pharmacol Exp Ther. 2001;299:227–237. [PubMed] [Google Scholar]
- 26.Zona C, Siniscalchi A, Mercuri NB, Bernardi G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience. 1998;85:931–938. doi: 10.1016/s0306-4522(97)00604-0. [DOI] [PubMed] [Google Scholar]