

Abstract
2-(4′-Methylaminophenyl)benzothiazole (BTA-1) is an uncharged derivative of thioflavin-T that has high affinity for Aβ fibrils and shows very good brain entry and clearance. In this study, we asked whether BTA-1, at concentrations typical of those achieved during positron emission tomography (PET) studies, could specifically bind to amyloid deposits in the complex milieu of human brain or whether amyloid binding was overshadowed by nonspecific binding, found even in brains that did not contain amyloid deposits. We quantitatively assessed [3H]BTA-1 binding to crude homogenates of postmortem brain obtained from nine Alzheimer's disease (AD) subjects, eight controls, and six subjects with non-AD dementia. BTA-1 binding was >10-fold higher in AD brain, and the majority (94%) of the binding was specific (displaceable). High-affinity [3H]BTA-1 was observed only in AD brain gray matter and was not present in control brain gray matter, AD brain white matter, or cerebellum. The Kd of [3H]BTA-1 for binding to AD brain (5.8 ± 0.90 nm) was very similar to theKd for binding to synthetic Aβ fibrils. In addition, the Ki of various BTA analogs for inhibition of [3H]BTA-1 binding to AD brain homogenates was very similar to their Ki for inhibition of [3H]BTA-1 binding to synthetic Aβ fibrils. Nanomolar concentrations of [3H]BTA-1 did not appear to bind to neurofibrillary tangles. Finally, BTA-1 did not appear to bind significantly to common neuroreceptors or transporter sites. These data suggest that the binding of BTA-1 to AD brain is dominated by a specific interaction with Aβ amyloid deposits.
Keywords: Alzheimer's disease, neuroimaging, plaques, neurofibrillary tangles, positron emission tomography, PET, thioflavin-T, benzothiazole, postmortem, binding
Introduction
Alzheimer's disease (AD) is defined pathologically by the characteristic deposits found in the brain (Mirra et al., 1991), including amyloid plaques and neurofibrillary tangles (NFTs). Both the amyloid-β (Aβ) protein found in plaques and the tau protein found in NFTs have a predominantly β-sheet structure (Kirschner et al., 1986). Current evidence suggests that both Aβ and tau deposits play important roles in the pathophysiological cascade of AD (Goate et al., 1991; Mullan et al., 1992; Hardy et al., 1998;Goedert and Spillantini, 2000; Golde et al., 2000).
Aβ deposition has become an important therapeutic target in AD research (Schenk et al., 1999; Bard et al., 2000; DeMattos et al., 2001; Olson et al., 2001). Although promising anti-amyloid therapies are being intensely pursued, there is currently no accepted tool to directly assess their success in delaying or reversing amyloid deposition in humans. It will be critical to show that a clinical treatment effectively reduces plaque load, before positive or negative effects on clinical cognitive measures can be accurately interpreted. In addition, because the clinical effectiveness of these anti-amyloid therapies will serve as a test of the “amyloid hypothesis” of AD (Hardy and Allsop, 1991; Hardy, 1992), it also will be critical to show that the treatments actually decrease amyloid load before we can make inferences about the validity of the amyloid hypothesis.
Our laboratory has been interested in the development of a noninvasive method for direct and quantitative assessment of amyloid deposition in living subjects (Klunk et al., 1994; Klunk, 1998). We have reported the potential of lipophilic thioflavin-T analogs as amyloid imaging agents (Klunk et al., 2001) and have described a particularly promising carbon-11-labeled derivative of thioflavin-T, namely 2-(4′-methylaminophenyl)benzothiazole (BTA-1) (see Fig. 1) (Mathis et al., 2002). BTA-1 shows very good brain entry and clearance and has been used for in vivo imaging of amyloid deposits in living presenilin-1/amyloid precursor protein transgenic mice using multiphoton microscopy (Mathis et al., 2002). These properties justified further pursuit of BTA-1 as an in vivo human amyloid-imaging agent for positron emission tomography (PET).
Fig. 1.

Chemical structures of thioflavin-T and BTA-1. The uncharged compound, BTA-1, differs from thioflavin-T by the lack of three methyl groups, including the methyl group imparting the positive charge to the quaternary heterocyclic nitrogen of thioflavin-T (arrow).
A critical issue in the development of a PET imaging agent is whether binding to brain tissue is dominated by the specific target (in this case, amyloid deposits) or by nonspecific, background binding. Here, we further assessed the utility of BTA-1 for in vivo PET imaging studies by quantitatively assessing its binding to homogenates of postmortem brain from nine AD subjects, eight controls, and six subjects with non-AD dementia (NAD). We found that BTA-1 binding was much higher in amyloid-containing areas of AD brain tissue, and most of the binding appeared to be specific. In addition, theKi of various BTA analogs for inhibition of [3H]BTA-1 binding to AD brain homogenates was very similar to theirKi for inhibition of [3H]BTA-1 binding to synthetic Aβ fibrils. These data suggest that the binding of BTA-1 to AD brain is dominated by specific interaction with amyloid deposits and strengthen the case for use of BTA-1 as an in vivo imaging agent for AD diagnosis and treatment assessment.
Materials and Methods
Chemicals. Chemicals were obtained from Sigma (St. Louis, MO) unless noted otherwise. Tritium-labeled and unlabeled BTA-1, and BTA-1 derivatives, were synthesized and characterized as described previously (Klunk et al., 2001; Mathis et al., 2002).
Postmortem tissue. Autopsy tissue was obtained from the University of Pittsburgh Alzheimer Disease Research Center Brain Bank. Clinical and postmortem diagnoses were made by standardized procedures as described previously in detail (Lopez et al., 2000). Clinical characteristics are shown in Table 1.
Table 1.
Demographic and neuropathological characteristics
| Brain no. | Primary neuropath Dx | Other neuropath Dx | Braak stage | Cortical NP1-a | Age | PMI (hr) |
|---|---|---|---|---|---|---|
| AD 01 | AD1-a | AA (severe) | VI | Frequent | 83 | 3 |
| AD 02 | AD1-a | AA (severe) | VI | Frequent | 57 | 4 |
| AD 03 | AD1-a | AA (mild) | VI | Frequent | 78 | 3 |
| AD 04 | AD1-a | AA (mild), DLB (1/10)1-b | VI | Frequent | 80 | 4 |
| AD 05 | AD1-a | AA (moderate) | VI | Frequent | 78 | 9 |
| AD 06 | AD1-a | Chronic subdural hematoma | IV | Frequent | 85 | 3 |
| AD 07 | AD1-a | AA (severe), old infarct right putamen | VI | Frequent | 79 | 4 |
| AD 08 | AD1-a | AA (mild), old infarct left basis pontis | IV | Frequent | 83 | 4 |
| AD 09 | AD1-a | Infarcts, right putamen (3 old);
Left thalamus (1 subacute) |
IV | Frequent | 84 | 4 |
| Average: | 78.6 | 4.2 | ||||
| SD | 8.5 | 1.9 | ||||
| Cntl 01 | Normal brain1-a | Moderate atherosclerosis, circle of Willis | 0 | None | 72 | 18 |
| Cntl 02 | Normal brain1-a | Persistent posterior fetal circulation, bilaterally
Incidental choroid plexus cyst |
I | None | 82 | 4 |
| Cntl 03 | Normal brain1-a | Moderate congestion of leptomeninges | 0 | None | 75 | 8 |
| Cntl 04 | Normal brain1-a | Incidental focal hypoplasia of corpus callosum | II | None | 82 | 3 |
| Cntl 05 | Normal brain1-a | None | I | None | 74 | 5 |
| Cntl 06 | Normal brain1-a | Atherosclerosis, circle of Willis
Old infarcts cerebellar cortex, inferior olivary nucleus |
II | Rare | 72 | 4 |
| Cntl 07 | Normal brain1-a | Subacute infarct right brainstem | I | None | 74 | 6 |
| Cntl 08 | Normal brain1-a | None | 0 | None | 53 | 5 |
| Average: | 73.0 | 6.6 | ||||
| SD | 9.0 | 4.8 | ||||
| p = (vs AD) | 0.21 | 0.22 | ||||
| NAD 01 | Pure DLB(7/10)1-b | None | I | None | 69 | 10 |
| NAD 02 | Huntington's | Status post evacuation subdural hematoma,
Atherosclerosis, circle of Willis |
I | None | 64 | 7 |
| NAD 03 | Pick's | Atherosclerosis, circle of Willis | 0 | None | 66 | 4 |
| NAD 04 | MND-inclusion
Dementia |
Severe cortical atrophy, atherosclerosis, circle of Willis | 0 | None | 75 | 8 |
| NAD 05 | DLDHF | Mesial temporal sclerosis | I | None | 56 | 4 |
| NAD 06 | DLB(6/10)1-b | Parkinson's disease | I | None | 87 | 8 |
| Average: | 69.5 | 6.8 | ||||
| SD | 10.6 | 2.4 | ||||
| p= (vs AD) | 0.11 | 0.05 |
NP, Neuritic plaques; PMI, postmortem interval; AD, Alzheimer's disease; AA, amyloid angiopathy; DLB, dementia with Lewy Bodies; SD, standard deviation; p = (vs AD), p values compared with corresponding values for AD cases; Cntl, control; NAD, demented, non-Alzheimer's disease; MND, motor neuron disease-inclusion dementia (Jackson et al., 1996); DLDHF, dementia lacking distinctive histologic features (Mann, 1998); neuropath; Dx, neuropathological diagnosis.
CERAD criteria (Mirra et al., 1991).
Scored according to consensus criteria (McKeith et al., 1996).
All nine AD brains represented fairly advanced stages of pathology as is typical of postmortem tissue. The eight control and six NAD brains were selected for having no significant evidence of neuritic plaques or cerebrovascular amyloid. In addition, there were no NFTs in the frontal cortex of any control or NAD case. Samples of medial-frontal cortex gray matter, medial-frontal cortex white matter, entorhinal cortex (EC), or cerebellum were dissected at autopsy from 1-cm-thick coronal slices and frozen at −80°C. The tissue was homogenized with a Polytron tissue homogenizer (PT 10/35; Brinkman Instruments, Westbury, NY) at room temperature for 30 sec at setting 6 in PBS (137 mm NaCl, 3 mm KCl, 10 mm sodium phosphate, pH 7.0) at a concentration of 10 mg of brain per milliliter. The homogenates were aliquoted and frozen at −80°C until used for binding assays within 2 months. No significant changes in binding characteristics were observed in samples assayed repeatedly over this time period.
Neuroreceptor screening. Binding assays for receptors and transporters were performed as described previously using the resources of the National Institute of Mental Health Psychoactive Drug Screening Program (Roth et al., 2000, 2001).
Binding studies. Aβ(1–40) fibrils were prepared, andKi determinations were performed as described previously (Klunk et al., 2001) using [N-methyl-3H]BTA-1 ([3H]BTA-1; 61 Ci/mmol) as the radioligand (Mathis et al., 2002). Binding to brain homogenates for determination of Ki andKd was performed with slight modifications. Briefly, 10 mg/ml frozen brain aliquots were thawed and diluted 10-fold in PBS to 1 mg/ml. Unlabeled test compounds were dissolved in DMSO at 400 μm (to yield <1% DMSO in the final assay). The appropriate concentrations of unlabeled test compounds were combined with [3H]BTA-1 (∼1.2 nm) in a volume of 900 μl of PBS, pH 7.0. The assay was begun by addition of 100 μl of the 1 mg/ml brain homogenate to achieve a final concentration of 100 μg tissue per milliliter. After incubation for 60 min at room temperature, the binding mixture was filtered through a Whatman GF/B glass filter via a Brandel M-24R cell harvester (Gaithersburg, MD) and rapidly washed five times with 3 ml PBS. The filters were counted in Cytoscint-ES after thorough vortexing and sitting overnight. Complete (100%) inhibition of specific binding was defined as the number of counts displaced by 3 μm unlabeled BTA-1. All assays were performed at least in triplicate. For determination ofKi values, inhibition curves were fit (using the RS/1 statistical package, version 6.1; Brooks Automation, Chelmsford, MA) to the following equation, which is derived from the Hill equation (Bennett and Yamamura, 1985): (F(x) =M(Ki)H/{[L]H+ (Ki)H}, where M = maximal percentage [3H]BTA-1 bound (typical fit gave 100–104% for M), H is the Hill coefficient (typically 0.85–1.0), and [L] is the concentration of inhibitor compound. TheKd value for BTA-1 binding to Aβ(1–40) fibrils and brain homogenates was determined similarly using increasing concentrations of [3H]BTA-1 between 0.2 and 1.2 nm, and, for BTA-1 concentrations from 1.2 nm to 1000 nm, a constant concentration of [3H]BTA-1 (1.2 nm) plus additional unlabeled BTA-1. For comparison of [3H]BTA-1 binding to brain homogenates among the three groups (AD, control, and NAD), 1.2 nm [3H]BTA-1 was incubated with 100 μg tissue in 1 ml PBS with and without 3 μm unlabeled BTA-1 as described above. Results were corrected for nonspecific, nondisplaceable binding in the presence of 3 μm BTA-1 and expressed as picomoles of [3H]BTA-1 bound per milligram of wet tissue weight in the homogenate. Linearity of [3H]BTA-1 binding was determined using 1.2 nm [3H]BTA-1 and 20–500 μg/ml of the AD brain homogenate with the highest level of [3H]BTA-1 binding. Likewise, linearity with respect to [3H]BTA-1 was determined using 100 μg of tissue per milliliter and 1.2–6 nm[3H]BTA-1. Binding results between AD and control and AD and NAD were compared using an unpaired sample, unequal variance t test.
Tissue staining. Postmortem tissue from three of the autopsy-confirmed AD cases used for the binding study was obtained through the University of Pittsburgh Alzheimer Disease Research Center. Samples of medial-frontal cortex, entorhinal cortex, or cerebellum were dissected from 1-cm-thick coronal slices and placed into 10% buffered formalin (7 d). Paraffin blocks were prepared by sequential dehydration in graded alcohol and vacuum infiltrated in paraffin before embedding and serial sectioning at a thickness of 8 μm. Tissue was processed for staining, and quenching of autofluorescence was accomplished according to previously described methods (Styren et al., 2000). Quenched tissue sections were taken from PBS into a solution of 100 nm BTA-1 in PBS, pH 7.0, for 45 min. The sections were then dipped into water for ∼5 sec before coverslipping with Fluoromount-G (Electron Microscopy Sciences, Fort Washington, PA). Fluorescent sections were viewed using an Olympus Vanox AH-RFL-LB fluorescence microscope using a UV filter set: U-filter (excites 360–370 nm, dichroic mirror DM400, 420 nm long-pass filter). X-34 staining was performed according to previously described methods (Styren et al., 2000). X-34-stained tissue sections were optimally viewed with a violet filter set: V-filter (excites <460 nm, DM455 mirror, 455 nm long-pass filter).
Results
Paraffin sections (8 μm) from the frontal and entorhinal cortex of AD brain were stained with 100 nm BTA-1 (Figs. 1 , 2). Like the parent compound, thioflavin-T (Klunk et al., 2001), BTA-1 stained plaques, cerebrovascular amyloid, and NFTs at these relatively high concentrations. BTA-1 appeared to stain plaques and cerebrovascular amyloid more intensely than it stained NFTs. Very little background staining was observed. Most of the background fluorescence seen in Figure 2 was caused by residual lipofuscin autofluorescence (distinguished by the fact that it is visible in all filter sets). Equivalent results were observed in frozen sections. Essentially no staining was observed in control brain, which had been shown to contain no plaques or NFTs by the standard CERAD neuropathology protocol (Mirra et al., 1991) (data not shown).
Fig. 2.

Paraffin sections (8 μm) from AD brain stained with 100 nm BTA-1. Left panel shows plaques and cerebrovascular amyloid in the frontal cortex. Right panel shows plaques and neurofibrillary tangles in the entorhinal cortex. Scale bar, 100 μm. Most smaller bright spots are residual lipofuscin autofluorescence.
The tissue staining results supported the specificity of BTA-1 for β-sheet deposits in a qualitative manner, but the 100 nmBTA-1 concentration used for staining was not representative of the low or subnanomolar concentrations achieved by typical PET ligands in vivo. In addition, it is possible that the fluorescence properties of BTA-1 change after binding to amyloid in a way that enhances the fluorescence similar to that observed for thioflavin-T (LeVine, 1993). This could make the staining appear more specific than it actually was. Therefore, quantitative binding studies were performed in tissue homogenates using 1.2 nm[3H]BTA-1.
[3H]BTA-1 binding was linear with tissue concentration (r = 0.993) and [3H]BTA-1 concentration (r = 0.999) throughout the range used in this study. In homogenates from frontal gray, significantly more [3H]BTA-1 was bound to amyloid-containing AD brain than to the control or NAD brains, which lacked observable amyloid deposits (p < 0.0002) (Fig. 3). Using 1.2 nm [3H]BTA-1, the average amount of [3H]BTA-1 bound to AD frontal gray (0.73 ± 0.21 pmol/mg wet weight) was >10-fold higher than that bound to frontal gray obtained from either control brain (0.061 ± 0.023 pmol/mg wet weight) or NAD brain (0.072 ± 0.021 pmol/mg wet weight).
Fig. 3.

A, Comparison of [3H]BTA-1 binding to homogenates from control (open bars, circles), AD (filled bars, squares), and NAD brain (hatched bars, triangles) frontal gray (Fr) or cerebellum (Cb).B, Ratio of [3H]BTA-1 binding to the Fr and Cb for each individual brain. Bars represent the mean, and error bars represent the SD. Also shown are the individual data points.
As expected, and confirming the specificity of BTA-1, a very different finding was observed in the cerebellum where fibrillar (i.e., Congo red or thioflavin-S positive) amyloid deposits are very rarely observed (Joachim et al., 1989; Yamaguchi et al., 1989; Mann et al., 1996) (Fig.3A). Binding was low in the cerebellar homogenates of all three groups, and there was complete overlap between binding to AD cerebellum and binding to cerebellum from control and NAD brains.
The lack of difference in [3H]BTA-1 binding in cerebellum is important because it identifies this brain area as a potential reference tissue that could be used to simplify the quantitation of future PET studies. To more precisely examine this issue, ratios of [3H]BTA-1 binding to frontal gray and cerebellum were determined for each individual brain studied, and the data were expressed as the frontal/cerebellar ratio for each brain (Fig. 3B). When expressed in this manner, the clear differentiation between amyloid-containing AD brain and the nonamyloid-containing control groups remained. The ratio in AD brain (7.8 ± 4.0) was >10-fold higher than the ratio calculated in either control brain (0.59 ± 0.40; p < 0.0006) or NAD brain (0.78 ± 0.26; p < 0.0007).
Approximately 94% of [3H]BTA-1 binding to homogenates of AD frontal gray was displaceable by 3 μm unlabeled BTA-1 (i.e., is specific). Specific binding represented only 36–52% of binding in all samples except AD frontal gray. The absolute value of nonspecific binding was not significantly different in the frontal gray or cerebellum of any of the groups studied, although it was lowest in AD frontal gray.
Detailed Scatchard analysis of [3H]BTA-1 binding to homogenates of frontal gray from three AD and three control brains showed high-affinity binding to be present only in AD frontal gray. [3H]BTA-1 appeared to bind to a single, high-affinity binding site in AD frontal gray with a meanKd of 5.8 ± 0.90 nm and a meanBmax of 5.4 ± 1.1 pmol/mg wet weight. (Fig. 4). This affinity was very similar to the high-affinity binding of [3H]BTA-1 to synthetic Aβ(1–40) or Aβ(1–42) fibrils (Kd = 2.8 ± 0.35 nm; data not shown).
Fig. 4.

Scatchard plots showing the binding of [3H]BTA-1 to homogenates from AD frontal gray matter (▪) and underlying frontal white matter from the same AD brain (▵). In this AD frontal gray matter homogenate, theKd was 4.4 nm and theBmax was 6.9 pmol/mg wet weight. In the frontal white matter immediately underlying this gray matter sample, the Kd was 111 nm and theBmax was 27 pmol/mg wet weight. Also shown is a Scatchard plot showing the binding of [3H]BTA-1 to a homogenate from control brain frontal gray (○) in which the Kd was 180 nm and the Bmax was 5.9 pmol/mg wet weight.
Control brains showed only low-affinity binding, having a meanKd of 132 ± 59 nm and a meanBmax of 11.3 ± 5.2 pmol/mg wet weight (Fig. 4). The frontal gray matter samples used in this study were dissected to remove the underlying white matter as much as possible. Comparing the frontal gray matter from an AD brain with the white matter immediately underlying it showed a marked difference in high-affinity [3H]BTA-1 binding. Figure4 includes a Scatchard analysis of the binding of [3H]BTA-1 to the white matter dissected from directly beneath the gray matter of the AD brain also shown in Figure 4. In contrast to the frontal gray matter, the frontal white matter showed only a low-affinity binding component, similar to that seen in control brain frontal gray. This low-affinity binding component in control frontal gray and AD frontal white matter would not play a significant role in in vivo imaging studies using high specific-activity radiotracers present at low nanomolar concentrations.
To address the relative contributions of [3H]BTA-1 binding to Aβ and tau deposits in the frontal gray of AD brain we compared [3H]BTA-1 binding in homogenates from entorhinal cortex, frontal gray, and cerebellum from a typical Braak stage VI AD brain (Table 1, AD 01) and a Braak stage II control brain (Table 1, Cntl 04), matched for age and postmortem interval. This control brain had frequent NFTs in the entorhinal cortex (Fig. 5A), similar in numbers to those found in many AD cases (Fig.5B); however, it had no neuritic or diffuse plaques in any area of the brain (Fig.5A,C,E). [3H]BTA-1 binding in the plaque-free and NFT-rich EC region of this Cntl 04 brain was no greater than [3H]BTA-1 binding in the plaque- and NFT-free cerebellum and frontal gray from this brain (Table2). A similar survey of these same brain areas in a Braak VI AD brain (Table 2) showed low binding in NFT-free cerebellum and NFT-rich EC and >10-fold higher levels in frontal gray, where there are frequent numbers of neuritic plaques (Fig.5D). The extensive NFT pathology in the EC of the Cntl 04 and AD 01 brains, coupled with the low [3H]BTA-1 binding in the EC, suggests that at low nanomolar concentrations the amount of [3H]BTA-1 binding to NFT deposits is very small in comparison with the amount of [3H]BTA-1 bound to Aβ in the plaques and cerebrovascular amyloid of AD frontal gray. The AD 01 brain showed diffuse amyloid plaque deposits in the EC (Fig. 5B), which also did not appear to produce significant [3H]BTA-1 binding. The frontal cortex had extensive amyloid plaques that were both compact and diffuse and associated with high levels of [3H]BTA-1 binding (Fig. 5D, Table 2). The qualitative BTA-1 staining of NFTs, seen at 100 nm concentrations (Fig. 2), is apparently not reflected by significant amounts of [3H]BTA-1 binding at a concentration of 1.2 nm.
Fig. 5.
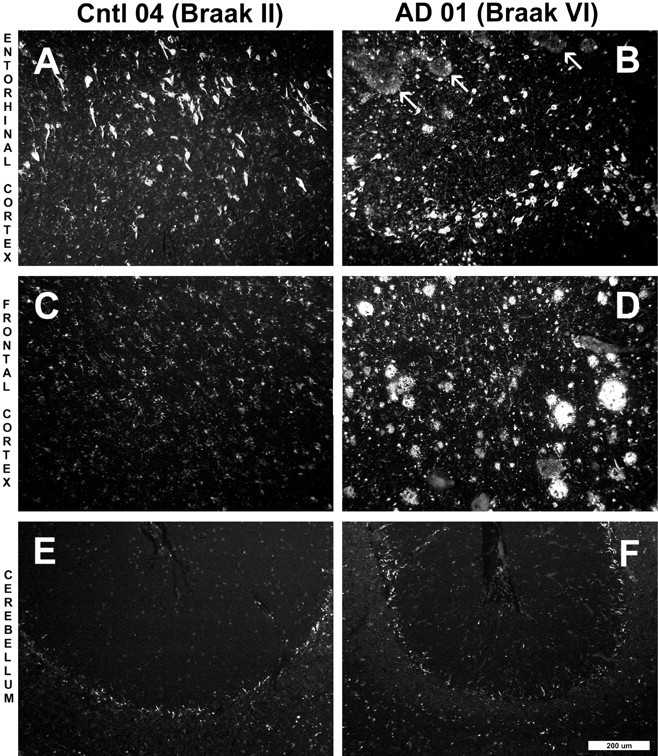
Paraffin sections (8 μm) from the entorhinal cortex (A, B), frontal cortex (C, D), and cerebellum (E,F) of a Braak stage II control brain (A, C, E, Cntl 04) and a Braak stage VI AD brain (B,D, F, AD 01) stained with X-34. X-34 is a highly sensitive, fluorescent Congo red derivative that intensely stains plaques, NFTs, and amyloid deposits in general (Styren et al., 2000). Marked atrophy of AD tissue was notable in all regions. In the entorhinal cortex of the control subject (A), frequent numbers of NFTs were seen, whereas there were no amyloid deposits. In the AD case (B), there also were frequent NFTs along with diffuse amyloid deposits (arrows). X-34-positive NFTs and amyloid plaques were absent from the frontal cortex of the control case (C), whereas they were abundant in AD (D). There was no detectable X-34 staining of plaques or NFT in control (E) and AD (F) cerebellum. Control frontal cortex (C) and cerebellum from both control and AD (E, F) show small stellate cells that stain with X-34. Scale bar, 200 μm.
Table 2.
[3H]BTA-1 binding to specified areas of a Braak stage II control brain (Cntl 04) and a Braak stage VI AD brain (AD 01)
| Brain area | pmol BTA-1/mg wet weight (mean ± SD) | p value2-a | |
|---|---|---|---|
| Cntl 04 | AD 01 | ||
| EC | 0.078 ± 0.006 | 0.082 ± 0.008 | 0.489 |
| Fr | 0.093 ± 0.004 | 0.887 ± 0.011 | 0.00001 |
| Cb | 0.078 ± 0.0001 | 0.043 ± 0.004 | 0.005 |
See Table 1 for details. EC, Entorhinal cortex; Fr, frontal cortex; Cb, cerebellum.
Student's t test comparison of control and AD values.
As further evidence that [3H]BTA-1 binding to brain homogenates was dominated by the Aβ component, we compared the binding of 10 structurally related BTA derivatives to synthetic Aβ(1–40) fibrils and to AD brain frontal gray. To get a broad range of affinities for the correlation, we chose compounds withKi values for inhibition of [3H]BTA-1 binding to Aβ(1–40) fibrils ranging from 1.8 to 3030 nm. A good correlation was found between the Ki values determined for [3H]BTA-1 binding to synthetic Aβ(1–40) fibrils and to the AD brain homogenate (Fig.6). In fact, the correlation betweenKi values determined in AD brain and synthetic Aβ binding assays (r = 0.88) was no worse than the correlation of Ki values determined with two separately aggregated lots of synthetic Aβ(1–40) (r = 0.89) (Fig. 6). This finding suggested that the [3H]BTA-1 binding site in AD brain homogenates had the same structure–affinity relationship as the binding site in suspensions of pure, synthetic Aβ(1–40) fibrils and was consistent with the hypothesis that [3H]BTA-1 binding to AD brain homogenates was dominated by binding to fibrillar Aβ deposits.
Fig. 6.

Comparison of the Kivalues of 10 BTA-1 derivatives for inhibition of [3H]BTA-1 binding to Aβ(1–40) fibrils and homogenates of AD frontal gray (filled diamonds,solid line). Also shown is a comparison of the same data for Aβ(1–40) fibrils compared with Kivalues previously determined in a separate, older lot of Aβ(1–40) fibrils (open squares, dashed line).
Additional experiments were performed using the resources of the NIMH Psychoactive Drug Screening Program to assess whether BTA-1 interacts with any of a representative array of neurotransmitter receptors and transporters (Rothman et al., 2000; Roth et al., 2001). The array has been published previously (Roth et al., 2001) and includes adrenergic, serotonergic, muscarininc, dopaminergic, opiate, vasopressin, oxytocin, GABA, glutamate, and imidazoline receptor sites. Using 10 μm BTA-1, no inhibition was observed at any receptor site except at the norepinephrine transporter, where theKi of BTA-1 was 4504 ± 31 nm, a value 1000-fold higher than theKd for Aβ(1–40) or AD brain.
Discussion
Although a previous study showed good pharmacokinetic properties for BTA-1 in normal mice (Mathis et al., 2002), it remained to be shown that BTA-1 could specifically bind to its amyloid target in human brain and to more precisely characterize that target in the complex milieu of human AD brain. The ability of BTA-1 to fluorescently stain amyloid deposits in postmortem tissue sections gives some indication that this compound would specifically bind amyloid deposits. However, this staining is a qualitative technique and certainly does not prove sufficient specificity for use as an in vivo PET amyloid-imaging agent in humans. In an effort to address this question, we quantitatively examined the binding of [3H]BTA-1 to homogenates of postmortem human brain at concentrations typical of in vivo PET radiotracer studies. We found that 12-fold more [3H]BTA-1 bound to homogenates from AD brain frontal gray than to similar homogenates from control and NAD brain. There was no overlap between the AD group and the other groups. Binding of [3H]BTA-1 to cerebellar homogenates was identical in all three groups, making cerebellum a good candidate for a reference tissue for PET studies. More than 90% of [3H]BTA-1 binding to AD frontal gray was specific. The affinity of [3H]BTA-1 binding to AD frontal gray was relatively high (∼6 nm) and very similar to the affinity of [3H]BTA-1 for synthetic Aβ fibrils (∼3 nm). Control brain frontal gray and frontal white matter from AD showed essentially no high-affinity binding. NFTs alone did not appear to increase [3H]BTA-1 binding above control levels, suggesting that most of the [3H]BTA-1 binding represents an interaction with Aβ deposits. Consistent with this, for a series of 10 BTA-1 derivatives having a 1000-fold range of affinity for binding amyloid, the Kivalues for the inhibition of [3H]BTA-1 binding to Aβ(1–40) fibrils correlated very well with theirKi values for inhibition of [3H]BTA-1 binding to AD brain homogenates. BTA-1 did not bind significantly to a wide array of neurotransmitter receptors or transporters. All of these data are consistent with the notion that [3H]BTA-1 binding in AD brain is mainly accounted for by binding to fibrillar Aβ.
Although [3H]BTA-1 binding clearly distinguished AD brain homogenates from the control and NAD groups, it is important to note that the goal of this study was not to test whether we could use [3H]BTA-1 to establish a diagnosis. The validation of BTA-1, or any other compound, as an accurate tool to quantify amyloid is a complex process composed of many steps such as the one described in this study. In addition, it should be recognized that the frontal cortex does not represent the area of maximal pathologic change for several of the diseases included in the NAD group, and one would need to examine other brain areas to adequately address diagnostic issues in these cases.
The purpose of this study was solely to determine whether the binding of [3H]BTA-1 to areas of human brain with abundant amyloid deposits (e.g., AD frontal gray) was dominated by a specific interaction with the amyloid deposits. It was possible that [3H]BTA-1 could have nonspecific interactions with binding sites on non-amyloid components in brain that would be of sufficient magnitude to mask the specific, amyloid binding observed in AD brain. Therefore, both control groups were chosen for the absence of visible Aβ deposits to assess the contribution of the non-amyloid components in isolation. The NAD group was included to control for the possibility that [3H]BTA-1 binding was affected by general neurodegenerative processes in a manner unrelated to amyloid deposition. Likewise, it is possible that highly lipophilic brain areas such as white matter from both AD and control brains could have large amounts of nonspecific, lipophilic interactions with [3H]BTA-1 that would overshadow the specific, amyloid binding in the overlying cortex. Our results indicated that high-affinity [3H]BTA-1 binding was much greater in AD frontal gray than either control frontal gray or AD frontal white matter and suggested that the binding of [3H]BTA-1 to AD brain mainly represented a specific interaction with Aβ deposits.
Taken together with the previously reported pharmacokinetic properties of [11C]BTA-1 in mice and successfulin vivo imaging of plaques in transgenic mice using BTA-1 and multiphoton microscopy (Mathis et al., 2002), the findings of the present study suggest that BTA-1 is a good candidate as an in vivo PET amyloid-imaging agent. Further, detailed, quantitative validation studies in transgenic mice will need to be performed to confirm the potential of BTA-1. In addition, pharmacokinetic studies are underway in primate brain to verify the mouse studies. Promising results from all of these studies and an acceptable toxicological profile will justify preliminary studies in human subjects with and without AD.
Footnotes
This work was supported in part by the Alzheimer's Association (Grants IIRG-95-076 and TLL-01-3381 to W.E.K. and NIRG-00-2335 to Y.W.), the National Institutes of Health (Grants AG01039 and AG20226 to W.E.K., AG18402 to C.A.M., and AG05133 to S.T.D.), and Institute for the Study of Aging/American Federation of Aging Research (Y.W.). We thank Dr. Bryan Roth (Case Western Reserve University) and Dr. Linda Brady [National Institute of Mental Health (NIMH)] for allowing us access to the NIMH Psychoactive Drug Screening Program. We thank Dr. Robert Sweet for his help in procuring the appropriate tissue samples and the families that made brain tissue available to the University of Pittsburgh Alzheimer Disease Research Center Brain Bank.
Correspondence should be addressed to Dr. William E. Klunk, University of Pittsburgh, 705 Parran Hall– GSPH, 130 DeSoto Street, Pittsburgh, PA 15213. E-mail: klunkwe@msx.upmc.edu.
References
- 1.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Schenk D, Yednock T. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 2.Bennett JP, Yamamura HI. Neurotransmitter, hormone, or drug receptor binding methods. In: Yamamura HI, Enna SJ, Kuhar MJ, editors. Neurotransmitter receptor binding. Raven; New York: 1985. pp. 61–89. [Google Scholar]
- 3.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 5.Goedert M, Spillantini MG. Tau mutations in frontotemporal dementia FTDP-17 and their relevance for Alzheimer's disease. Biochim Biophys Acta. 2000;1502:110–121. doi: 10.1016/s0925-4439(00)00037-5. [DOI] [PubMed] [Google Scholar]
- 6.Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta. 2000;1502:172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 7.Hardy J. An “anatomical cascade hypothesis” for Alzheimer's disease. Trends Neurosci. 1992;15:200–201. doi: 10.1016/0166-2236(92)90033-5. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 9.Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- 10.Jackson M, Lennox G, Lowe J. Motor neurone disease-inclusion dementia. Neurodegeneration. 1996;5:339–350. doi: 10.1006/neur.1996.0046. [DOI] [PubMed] [Google Scholar]
- 11.Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer's disease. Am J Pathol. 1989;135:309–319. [PMC free article] [PubMed] [Google Scholar]
- 12.Kirschner DA, Abraham C, Selkoe DJ. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation. Proc Natl Acad Sci USA. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klunk WE. Biological markers of Alzheimer's disease. Neurobiol Aging. 1998;19:145–147. doi: 10.1016/s0197-4580(98)00013-x. [DOI] [PubMed] [Google Scholar]
- 14.Klunk WE, Debnath ML, Pettegrew JW. Development of small molecule probes for the beta-amyloid protein of Alzheimer's disease. Neurobiol Aging. 1994;15:691–698. doi: 10.1016/0197-4580(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 15.Klunk WE, Wang Y, Huang G-F, Debnath ML, Holt DP, Mathis CA. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 2001;69:1471–1484. doi: 10.1016/s0024-3205(01)01232-2. [DOI] [PubMed] [Google Scholar]
- 16.LeVine H. Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez OL, Becker JT, Klunk WE, Saxton J, Hamilton RL, Kaufer DI, Sweet RA, Cidis-Meltzer C, Wisniewski S, Kamboh MI, DeKosky ST (2000) Research evaluation and diagnosis of probable Alzheimer's disease over the last two decades. I. Neurology 55:1854–1862. [DOI] [PubMed]
- 18.Mann DM. Dementia of frontal type and dementias with subcortical gliosis. Brain Pathol. 1998;8:325–338. doi: 10.1111/j.1750-3639.1998.tb00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann DM, Iwatsubo T, Snowden JS. Atypical amyloid (A beta) deposition in the cerebellum in Alzheimer's disease: an immunohistochemical study using end-specific A beta monoclonal antibodies. Acta Neuropathol. 1996;91:647–653. doi: 10.1007/s004010050479. [DOI] [PubMed] [Google Scholar]
- 20.Mathis CA, Bacskai BJ, Kajdasz ST, Mclellan ME, Frosch MP, Hyman BT, Holt DP, Wang Y, Huang G-F, Debnath ML, Klunk WE. A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett. 2002;12:295–298. doi: 10.1016/s0960-894x(01)00734-x. [DOI] [PubMed] [Google Scholar]
- 21.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 22.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 23.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 24.Olson RE, Copeland RA, Seiffert D. Progress towards testing the amyloid hypothesis: inhibitors of APP processing. Curr Opin Drug Discov Devel. 2001;4:390–401. [PubMed] [Google Scholar]
- 25.Roth BL, Ernsberger P, Steinberg SA, Rao S, Rauser L, Savage J, Hufeisen S, Berridge MS, Muzic RF. The in vitro pharmacology of the beta-adrenergic receptor pet ligand (s)-fluorocarazolol reveals high affinity for cloned beta-adrenergic receptors and moderate affinity for the human 5-HT1A receptor. Psychopharmacology. 2001;157:111–114. doi: 10.1007/s002130100844. [DOI] [PubMed] [Google Scholar]
- 26.Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation. 2000;102:2836–2841. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]
- 27.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 28.Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer's disease pathology. J Histochem Cytochem. 2000;48:1223–1232. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Nakazato Y. Diffuse type of senile plaques in the cerebellum of Alzheimer-type dementia demonstrated by beta protein immunostain. Acta Neuropathol. 1989;77:314–319. doi: 10.1007/BF00687584. [DOI] [PubMed] [Google Scholar]