

Abstract
Huntington's disease (HD) mouse models that express N-terminal huntingtin fragments show rapid disease progression and have been used for developing therapeutics. However, light microscopy reveals no significant neurodegeneration in these mice. It remains unclear how mutant huntingtin induces neurodegeneration. Using caspase staining, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, and electron microscopy, we observed that N171-82Q mice, which express the first 171 aa of mutant huntingtin, displayed more degenerated neurons than did other HD mouse models. The neurodegeneration was also evidenced by increased immunostaining for glial fibrillary acidic protein and ultrastructural features of apoptosis. R6/2 mice, which express exon 1 of mutant huntingtin, showed dark, nonapoptotic neurons and degenerated mitochondria associated with mutant huntingtin. In HD repeat knock-in mice (HdhCAG150), which express full-length mutant huntingtin, degenerated cytoplasmic organelles were found in both axons and neuronal cell bodies in association with mutant huntingtin that was not labeled by an antibody to huntingtin amino acids 342–456. Transfection of cultured cells with mutant huntingtin revealed that an N-terminal huntingtin fragment (amino acids 1–208 plus a 120 glutamine repeat) caused a greater increase in caspase activity than did exon 1 huntingtin and longer huntingtin fragments. These results suggest that context-dependent neurodegeneration in HD may be mediated by different N-terminal huntingtin fragments. In addition, this study has identified neurodegenerative markers for the evaluation of therapeutic treatments in HD mouse models.
Keywords: Huntington, polyglutamine, neurodegeneration, apoptosis, ultrastructure, transgenic
Introduction
Huntington's disease (HD) is characterized by progressive neurodegeneration that occurs primarily in the striatum and extends to other brain regions as the disease progresses (Vonsattel et al., 1985). These other regions include the cerebral cortex, hypothalamus, and brainstem (Cudkowicz and Kowall, 1990; Kremer et al., 1990; Sotrel et al., 1993; Jackson et al., 1995). As a result, patients with HD show progressive movement disorder, memory loss, and psychiatric abnormalities until death (Harper, 1991). However, the lack of well preserved postmortem human brain tissue has been an obstacle to detailed characterization of neurodegeneration at the ultrastructural level and during the early stages of HD.
Studies of several HD mouse models have confirmed that polyglutamine (Q) expansion in huntingtin causes neurological phenotypes. HD transgenic mice expressing smaller N-terminal mutant huntingtin (Davies et al., 1997; Schilling et al., 1999) develop more progressive phenotypes than those expressing longer N-terminal huntingtin (Laforet et al., 2001) or full-length mutant huntingtin (Reddy et al., 1998; Hodgson et al., 1999). Of these HD transgenic mice, R6/2 and N171-82Q mice have been extensively studied and used for drug screens because of their rapid disease progression (Andreassen et al., 2001; Ferrante et al., 2002, Hockly et al., 2002; Keene et al., 2002). R6/2 mice express the HD exon 1 protein containing 115-150Q, and N171-82Q mice express the first 171 aa with 82Q. Although these HD mice display severe phenotypes and early death, usually at 3–6 months after birth, no prominent neurodegeneration has been found in their brains. In contrast, HD transgenic mice expressing full-length mutant huntingtin with a shorter repeat than 150Q or at a lower expression level than that of endogenous huntingtin develop neurodegeneration despite their slow disease progression (Reddy et al., 1998; Hodgson et al., 1999). Because N-terminal mutant huntingtin fragments are more toxic than full-length mutant huntingtin (Hackam et al., 1998) and may accumulate in different subcellular sites to mediate cellular toxicity (Lunkes et al., 2002), it would be necessary to examine different HD mice using more sensitive assays. Examination at the ultrastructural level should reveal whether neurodegeneration in vivo is caused by full-length huntingtin or its N-terminal fragments. Identification of specific neuropathological changes in various HD mouse models will also help in evaluating the therapeutic effects of drugs on these models.
In this study, we examined the neuropathology in various HD mouse models including N171-82Q mice and HdhCAG150 mice, which express 150Q in the endogenous mouse huntingtin (Lin et al., 2001). With the antibody EM48, which sensitively detects mutant huntingtin in the brain, we performed electron microscopy and found that N171-82Q mice had significantly more apoptotic neurons than other HD mouse models examined. The results suggest that N-terminal huntingtin sequences may specify different types of neuronal degeneration in HD and also provide neurodegenerative markers for evaluating therapeutic effects on HD mouse models.
Materials and Methods
HD mice. R6/1 and R6/2 mice [B6CBA-TgN (HDexon1)61 and 62], which express exon 1 of the human mutant HD gene containing 115–150 CAGs (Mangiarini et al., 1996), were obtained from The Jackson Laboratory (Bar Harbor, ME). N171-82Q mice [B6C3F1/−TgN(HD82Gln)81Dbo], which express the first 171 aa with 82 glutamines (Schilling et al., 1999), were also obtained from The Jackson Laboratory. HD repeat knock-in mice (HdhCAG150), which have a repeat length of 150 CAGs in the endogenous mouse HD gene, were generated as described previously (Lin et al., 2001). Breeding pairs of HdhCAG150 mice were provided by Dr. Peter Detloff (University of Alabama, Birmingham, AL). All mice were bred and maintained in the animal facility at Emory University. The genotyping of transgenic mice was performed using methods described previously (Mangiarini et al., 1996; Schilling et al., 1999; Lin et al., 2001).
Antibodies. EM48, a rabbit polyclonal antibody against the N-terminal region (amino acids 1–256) of human huntingtin, was generated during our previous studies (Gutekunst et al., 1999). EM121 was generated using glutathione-S-transferase fusion proteins containing amino acids 342–456 of human huntingtin. Purified fusion proteins were used as antigens for Covance Inc. (Denver, PA) to generate rabbit antisera. Other antibodies used included a mouse monoclonal antibody against glial fibrillary acidic protein (GFAP; Chemicon, Temecula, CA) and a rabbit polyclonal antibody against the activated form of caspase-3 (Cell Signaling Technology, Beverly, MA).
Light microscopy and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Mice were anesthetized and then perfused intracardially with PBS, pH 7.2, for 30 sec, followed by 4% paraformaldehyde in 0.1m phosphate buffer (PB, pH 7.2). The brains were removed, cryoprotected in 30% sucrose at 4°C, and sectioned at 40 μm using a freezing microtome. Free-floating sections were preblocked in 4% normal goat serum (NGS) in PBS, 0.1% Triton X-100, and avidin (10 μg/ml) and then incubated with huntingtin antibody at room temperature for 24 hr. For light microscopy, the immunoreactive product was visualized with the avidin–biotin complex kit (ABC Elite; Vector Laboratories, Burlingame, CA). Controls included brain sections from age-matched wild-type mice.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed on brain sections using an in situ cell death detection kit from Roche Molecular Biochemicals (Indianapolis, IN) according to the manufacturer's instructions. After perfusion with 4% paraformaldehyde, the mouse brain was cut into sections using a microtome. The tissue sections were fixed with 4% paraformaldehyde in PBS and then treated with 0.3% Triton X-100 in PBS for 30 min. After washes with PBS, the sections were incubated with fluorescein-12-dUTP and terminal transferase in the buffer provided with the kit. Counterstaining with Nissl (cresyl violet) was also performed to reveal the total number of neurons examined.
Quantification of GFAP- and TUNEL-positive cells. Light microscopic graphs were taken using a Zeiss (Oberkochen, Germany) Axioskop 2 microscope connected to a Spot-RT digital camera (Diagnostic Instruments, Sterling Heights, MI). All of the images were captured using the same parameters and saved in a computer for analysis. Each group of HD mouse models consisted of three to four mice. Every sixth brain section (240 μm distance) of these mice was selected through the striatum or cortex, providing six to eight sections for examining each mouse brain. We adopted a grid stereologic method as reported previously (Manoonkitiwongsa et al., 2001) to measure GFAP staining of glial cell bodies and processes. Briefly, all micrographs were taken at a magnification of 630×, providing high-resolution images (53.62 × 38.1 cm or 1520 × 1080 pixels). Using Adobe Photoshop 5.0 software (Adobe Systems, San Jose, CA), the images were adjusted at a threshold level of 128 so that GFAP signals could be unambiguously identified. The adjusted images were analyzed in a 5040 point grid in which points on the GFAP-labeled areas were counted. The percentage of these points of the 5040 point grid in each micrograph was used to represent the relative density of GFAP labeling in glial cells and also used for comparison.
To count TUNEL-positive neurons and cells labeled by anti-caspase-3, each micrograph (53.62 × 38.1 cm) at a magnification of 200× was examined. The percentage of TUNEL-positive neurons of cresyl violet-labeled cells was used for comparison. For each brain, six to eight randomly selected sections were counted, and the mean ± SD was obtained from three mice for each group.
Electron microscopic immunocytochemistry. For electron microscopy, mice were fixed by perfusion with PBS containing 4% paraformaldehyde and 0.2% glutaraldehyde. After perfusion, the brain was removed, postfixed with 4% paraformaldehyde in PB for 6–8 hr, and then sectioned using a vibratome. For immunogold labeling, brain sections were incubated with EM48 in PBS containing 4% NGS for 24–48 hr at 4°C. After washes with PBS, Fab fragments of goat anti-rabbit secondary antibodies (1:50) conjugated to 1.4 nm gold particles (Nanoprobes Inc., Stony Brook, NY) in PBS with 4% NGS were added to the section and incubated overnight at 4°C. After rinsing in PBS, sections were fixed again in 2% glutaraldehyde in PB for 1 hr, silver-intensified using the IntenSEM kit (Amersham Biosciences, Buckinghamshire, UK), and osmicated in 1% OsO4 in PB.
All sections used for electron microscopy were dehydrated in ascending concentrations of ethanol and propylene oxide/Eponate 12 (1:1) and embedded in Eponate 12 (Ted Pella Inc., Redding, CA). Ultrathin sections (60 nm) were cut using a Leica Ultracut S ultramicrotome. Thin sections were counterstained with 5% aqueous uranyl acetate for 5 min followed by Reynolds lead citrate for 5 min and examined using a Hitachi (Tokyo, Japan) H-7500 electron microscope.
Huntingtin transfection and Western blotting. PRK expression vectors encoding the HD exon 1 protein containing 150Q (HD-exon 1), the first 208 aa of human huntingtin containing 23 (23Q-208) or 120 (120Q-208) glutamine repeats, or truncated huntingtin [amino acids 1–508 (120Q-508) and 1–945 (120Q-945)] with a 120 glutamine repeat were obtained from our previous study and transfected into human embryonic kidney (HEK) 293 cells (Gutekunst et al., 1999; H.Li et al., 2000). Transfected cells were resuspended in PBS with a protease inhibitor mixture (1×, P8340; Sigma, St. Louis, MO), PMSF (100 μg/ml), and 1% Triton X-100. The cells were homogenized for 10 sec and centrifuged at 800 × g for 5 min at 4°C. The supernatant was used for Western blotting with an ECL kit (Amersham Biosciences). Approximately 50 μg of protein was loaded on each lane of the SDS gel.
Caspase activity and cell viability assays. Fluorometric assays of caspase-3 and caspase-9 activity were performed using kits obtained from Bio-Rad (Hercules, CA), as reported previously (Zhou et al., 2001). Cultured HEK293 cells were transiently transfected with mutant huntingtin or the PRK vector for 48 hr in six well plates. After washing with PBS, the cells were lysed in lysis buffer containing (in mm): 10 Tris-HCl, 10 NaH2PO4/NaHPO4, pH 7.5, 130 NaCl, and 10 sodium pyrophosphate, as well as 1% Triton X-100. To measure caspase activity, 200 μl of 2× assay buffer containing (in mm): 40 PIPES, pH 7.2, 200 NaCl, 20 dithiothreitol, and 2 EDTA, as well as 0.2% w/v CHAPS and 20% sucrose were added to a tube, with a final concentration of 10 ng/μl of the peptide substrate (Ac-DEVD-AFC for caspase-3 and LEHD-7-amino-4-trifluoromethyl cumarin for caspase-9). Cell lysates (40 μg of protein) were added to the tube to start the reaction. When the caspase inhibitor (DEVD or LEHD-fmk) was used to measure the specificity of the assay, it was added to cell lysates at a concentration of 10 or 20 μm for 30 min before adding the specific caspase substrate. The background was obtained with the same assay buffer without cell lysate or substrate. The reaction was incubated at 37°C for 1 hr, followed by measuring the cleaved caspase products with a fluorescence plate reader (Fluostar Galaxy; BMG Labtechnologies, Durham, NC) set at 460 nm excitation and 520 nm emission.
HEK293 cells were plated in six well dishes and transfected with 1 μg per well of huntingtin constructs as described above. HEK293 cells transfected with the PRK vector alone served as a control. After transfection for 72 hr using Lipofectamine, the viability of the transfected cells was then determined by a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTS) assay using a microplate reader (SPECTRAmax Plus; Molecular Devices, Sunnyvale, CA) (S. H. Li et al., 2000).
Statistical analysis. Statistical significance was assessed by using Student's t test and Sigma Plot 4.11 software, with p < 0.05 indicating statistical significance.
Results
GFAP staining of HD mouse brain
Immunocytochemical staining of GFAP has been used to identify reactive gliosis, an early marker of CNS damage in HD (Hedreen and Folstein, 1995). We compared GFAP staining among different HD mouse models, with a focus on the striatum because this region is a primary site of injury in HD. The densities of GFAP labeling in glial cell bodies and processes in these HD mice were measured. Compared with age-matched wild-type mouse striatum, N171-82Q mouse striatum showed a remarkable increase in GFAP immunoreactivity, indicated by intense labeling throughout astroglial cell bodies and their fibrous processes. The increased GFAP staining was found in the brains of N171-82Q mice at 3 months of age and became prominent at 4–5 months of age (Fig.2A). The striatum of R6/2 mice at 12 weeks of age displayed some glial cells with increased GFAP staining. However, the overall number of GFAP-labeled glial cells was not increased in R6/2 mice compared with that of their littermates or other HD mice. We also examined R6/1 mice, which express the same exon 1 huntingtin but live longer than R6/2 mice. Even at 9 months of age, there was still little GFAP staining in the striatum in R6/1 mice (data not shown). HdhCAG150 mice at 5–6 months of age did not show intense GFAP immunoreactivity. A significant increase in GFAP immunoreactivity was observed in the striatum of HdhCAG150 mice at 14 months of age, as reported previously (Lin et al., 2001). However, this increase was not as great as that in N171-82Q mice. Of the HD mice examined, N171-82Q mice showed the strongest induction of GFAP in the striatum (Fig. 1B). The increased GFAP staining was also observed in other brain regions in N171-82Q mice, including the cortex, hippocampus, and hypothalamus (data not shown), suggesting that N171-82Q mice have the most severe gliosis.
Fig. 2.
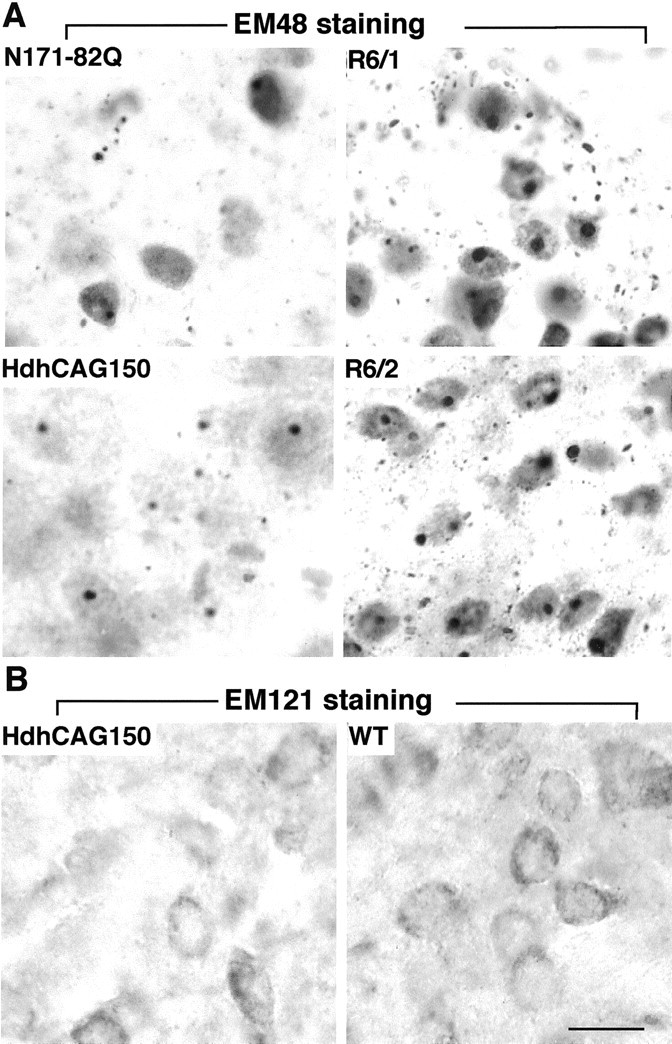
Immunostaining of HD mouse striatum.A, The striatum of N171-82Q (4 months old), HdhCAG150 knock-in (14 months old), R6/1 (8 months old), and R6/2 (3 months old) mice were stained with EM48, which specifically reacts with mutant huntingtin and its aggregates. Note that both nuclear inclusions and neuropil aggregates are present in all of the HD mouse brains examined, although they are more abundant in R6/2 and R6/1 mice.B, The striatum of HdhCAG150 was also labeled by the antibody EM121, which reacts with huntingtin amino acids 359–429. Only diffuse cytoplasmic staining was seen in HD mice, with a pattern similar to that of the wild-type (WT) mice. Scale bar, 10.8 μm.
Fig. 1.
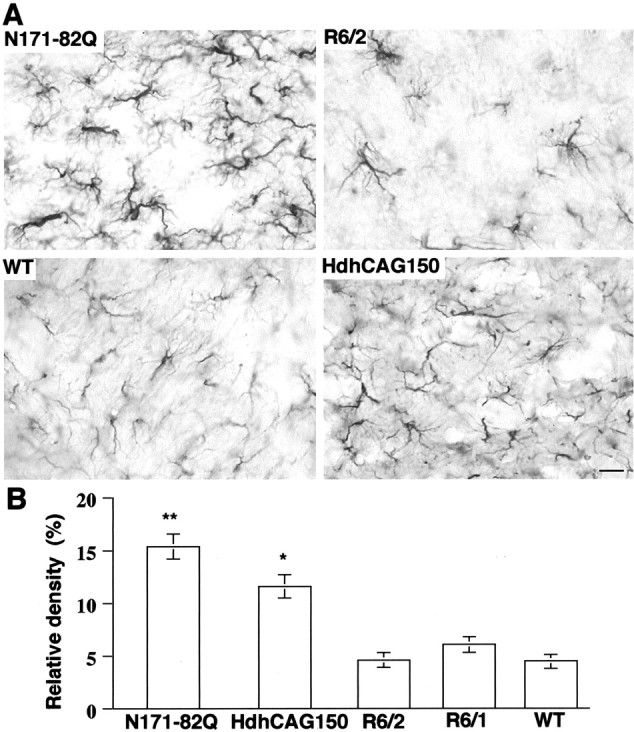
GFAP staining of HD mouse striatum.A, Brain striatal sections from N171-82Q (4 months old), age-matched control [wild type (WT), 4 months old], R6/2 (3 months old), and HdhCAG150 knock-in (14 months old) mice were immunostained with an antibody against GFAP. Increased GFAP immunoreactivity of astroglial cell bodies and their fibrous processes is present in N171-82Q and HdhCAG150 mouse striatum. Scale bar, 84.6 μm. B, Density of GFAP staining in the striatum of different HD mouse models. The density was determined by counting grid points containing GFAP immunoreactive signal in micrographs at 630× magnification. Data are presented as the percentage (means ± SD) of a 5040 point grid in each micrograph and were obtained from three to four mice for each group. WT, Wild-type littermates (4 months old) of the same strain as N171-82Q mice. *p< 0.05 and **p < 0.01 compared with WT.
Expression of mutant huntingtin in HD mouse brain
To examine whether the increased GFAP immunoreactivity observed is indeed associated with different types of mutant huntingtin rather than the different expression levels of these transgenic proteins, we immunostained HD mouse brains with EM48, an antibody that was generated with the first 256 aa of human huntingtin (Gutekunst et al., 1999). It is apparent that R6/2 and R6/1 mouse brains showed more intense EM48 immunoreactivity than the brains of N171-82Q mice and HdhCAG150 mice, because more nuclear EM48 labeling and a greater density of nuclear and neuropil aggregates were seen in R6/2 and R6/1 brains. Hdh150CAG mice at 14 months of age showed the weakest nuclear staining and the fewest nuclear or neuropil inclusions in the brain (Fig.2A). Despite the lower EM48 immunoreactivity in their brains, both N171-82Q and HdhCAG150 mice had a greater number of GFAP-positive cells than R6/2 and R6/1 mice (Fig. 1), suggesting that the increased GFAP staining is more related to the nature, not the quantity, of expressed mutant huntingtin.
Because only small N-terminal huntingtin fragments are able to form huntingtin inclusions in the nucleus and neuropil (DiFiglia et al., 1997; Gutekunst et al., 1999; H. Li et al., 2000), we wanted to examine whether aggregates seen in HdhCAG150 mice were formed by small N-terminal huntingtin fragments. To do so, we developed a rabbit antibody (EM121) against huntingtin amino acids 342–456. This antibody reacted with diffuse huntingtin in the cytoplasm of neurons. No aggregates or inclusions were observed with EM121 in HdhCAG150 mice, suggesting that huntingtin aggregates may be formed of N-terminal mutant huntingtin smaller than the first 342 aa (Fig.2B).
Apoptotic neurons in N171-82Q mouse brain
Previous studies using hematoxylin and eosin and silver stains or counting cell numbers did not reveal obvious neuronal loss in N171-82Q or HdhCAG150 mice (Schilling et al., 1999; Lin et al., 2001). We decided to examine HD mice with a more sensitive assay that detects activated caspase-3, an executive molecule whose cleavage and activation are involved in various apoptotic pathways. We could not find any significant labeling of R6/2 mouse brains by the antibody against activated caspase-3. However, we observed a number of intensely labeled neurons in the striatum and cortex of N171-82Q mouse brains at 4.5 months of age, but not in the brains of wild-type mice even at 14 months of age (Fig. 3A–C). Similar positive neurons were also seen in the hypothalamus and preoptic area, consistent with the widespread expression of transgenic mutant huntingtin in the brains of N171-82Q mice (data not shown). However, very few neurons showed immunoreactivity for activated caspase-3 in HdhCAG150 mice at 4 to 14 months of age (data not shown). The increased immunostaining of neurons for activated caspase-3 appeared to be specific to N171-82Q mice.
Fig. 3.
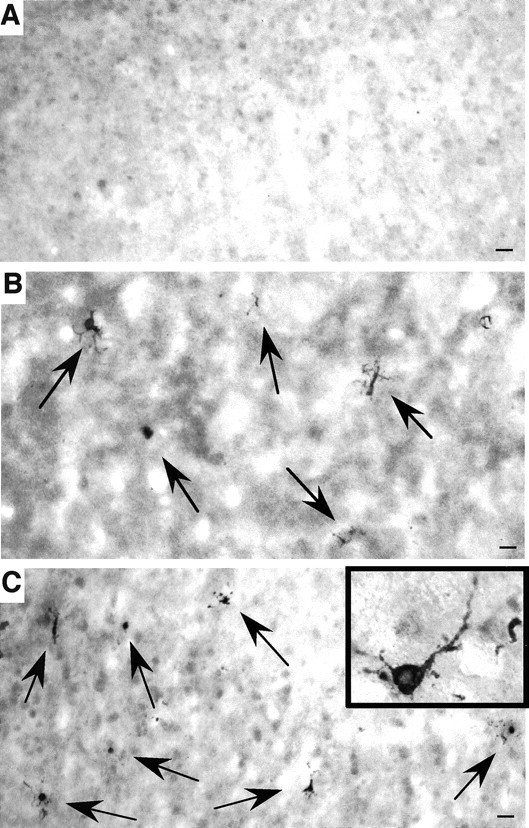
Caspase-3-activated neurons in N171-82Q mouse brain. The cortex of a wild-type control mouse at 14 months of age (A) and the striatum (B) and cortex (C) of N171-82Q mice at 4.5 months of age were stained with an antibody against the activated form of caspase-3. Positively stained neurons (arrows) are present in the N171-82Q mouse brain regions. Inset, High-magnification micrograph of a neuron containing activated caspase-3. Scale bars, 32 μm.
We also performed TUNEL to examine the relative percentage of apoptotic neurons in different HD mouse brains. The result showed that N171-82Q mice had the greatest number of TUNEL-positive neurons compared with wild-type littermates, HdhCAG150, and R6/2 mice (Fig.4). TUNEL-labeled product was found in the nucleus, reflecting a nuclear DNA strand break or an apoptotic event (Fig. 4A). Furthermore, more TUNEL-positive neurons in N171-82Q mice appeared as the animals became older (Fig.4B). This result is consistent with the progression of HD phenotypes in N171-82Q mice. The cortex appeared to have more TUNEL-positive neurons than the striatum, perhaps because of the higher expression level of the transgene in the cortex (Schilling et al., 1999).
Fig. 4.
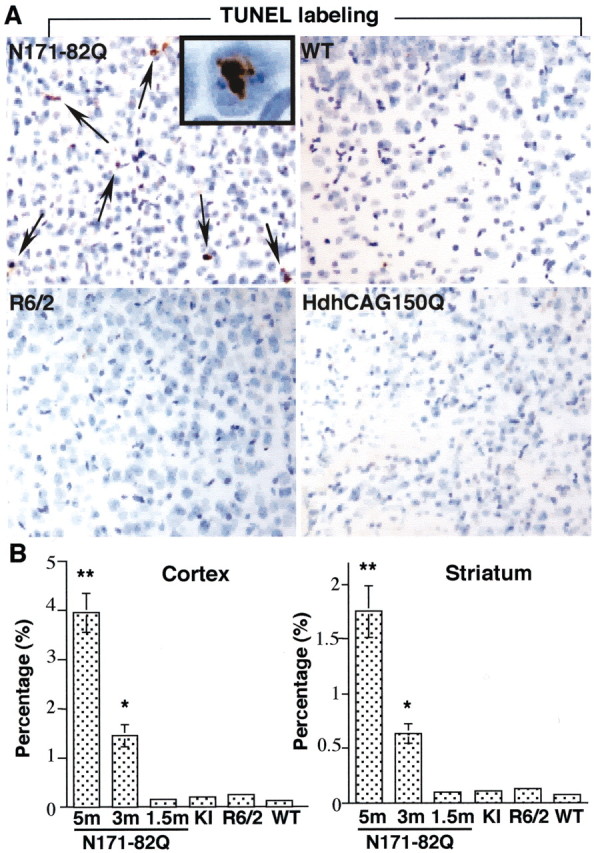
TUNEL-positive neurons in N171-82Q mouse brain.A, TUNEL of the cortex of N171-82Q (5 months old), age-matched control (5 months old), R6/2 (12 weeks old), and Hdh150CAG (14 months old) mice. The brain sections were also counterstained with cresyl violet (blue). Arrows indicate TUNEL-positive neurons. The inset shows a high-magnification (630×) image in which a neuronal nucleus contains TUNEL-positive product. B, Quantitative measurement of the percentage of TUNEL-positive neurons in the cortex and striatum of HD mouse brains. The total number of neurons was assessed by cresyl violet staining in each micrograph (200×). N171-82Q mice at the age of 1.5, 3, and 5 months were examined. The ages of other HD mice are the same as those in A. KI, HdhCAG150 mice;WT, wild-type mice. *p < 0.05 and **p < 0.01 compared with wild-type control mice (n = 3–4).
We then performed electron microscopy to provide ultrastructural evidence for degenerating neurons in N171-82Q mice. Electron microscopy of the cortex frequently revealed degenerated neurons with a number of apoptotic features. These neurons showed an abnormal nuclear shape with obvious chromatin margination and condensation, which are features of apoptosis (Fig.5A). The cytoplasm was mildly condensed, but most cytoplasmic organellar structures remained intact (Fig. 5B). At a more advanced stage of apoptosis, more condensed chromatin or fragmented chromatin was seen in the nucleus (Fig. 5C). Degenerated and dark cytoplasmic bodies were also seen in some neurons, and these bodies were associated with the appearance of chromatin condensation (Fig. 5D). These dark cytoplasmic bodies may be degenerated lysosomal structures. We also performed EM48 immunogold labeling. Mutant huntingtin and its inclusions were not found frequently in neurons that appeared to have reached a late stage of apoptosis. However, in the neurons that began to show nuclear chromatin margination and condensation or were undergoing early apoptosis, mutant huntingtin and its inclusions could be observed in their nuclei (Fig. 5E). Similar ultrastructural signs of apoptosis were also evident in the striatum and hypothalamus of the mutant mice, but not in the brains of age-matched control mice (data not shown), suggesting that apoptotic neurons are widely distributed in the brains of N171-82Q mice.
Fig. 5.

Electron microscopy showing degenerated neurons in the cortex of N171-82Q mouse brain. A, B, An apoptotic neuron with condensed cytoplasm and abnormal nuclear shape showing margination and condensation of chromatin. Note that most cytoplasmic organelles remain intact. The rectangular area inA is shown in B at higher magnification, indicating that the mitochondria (arrowhead) appear normal, whereas the nuclear membrane has chromatin margination (arrow). C, The nucleus of an advanced stage of apoptosis showing chromatin condensation and fragmentation.D, A degenerating neuron with condensed nuclear chromatin (arrow) and an increased number of dark cytoplasmic bodies resembling lysosomes (arrowheads).E, EM48 immunogold labeling showing that a neuron contains mutant huntingtin and its inclusion (arrow) in the nucleus. Note that this neuron is undergoing early apoptosis, characterized by the disintegration of the nuclear membrane and mild chromatin margination (arrowheads). Scale bars:A, 1.54 μm; C, 1.77 μm;D, 0.92 μm: E, 1.45 μm.
Dark neurons and degenerated mitochondria in R6/2 mice
We could not identify typical apoptotic neurons in R6/2 mice despite our extensive electron microscopic examination. However, quite a few dark neurons were found in the brains of these mice, as reported previously (Turmaine et al., 2000). The degeneration profile of these neurons was apparently different from that in N171-82Q mice. Dying neurons in R6/2 mice exhibited a condensed cytoplasm and nucleus (Fig.6A). However, these darkened structures never showed membrane blebbing, chromatin margination and fragmentation, apoptotic bodies, or other apoptotic features. Dark neurons with similar ultrastructural appearance were also found in the striatum of R6/1 mice (Fig.6B).
Fig. 6.
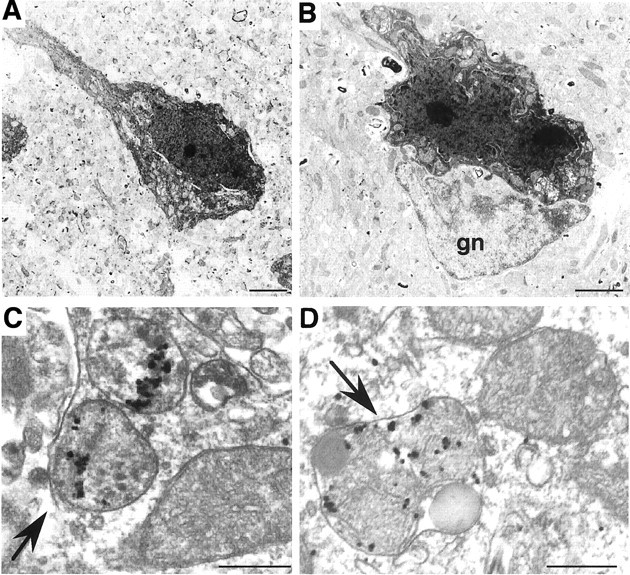
Dark neurons and degenerated mitochondria in R6/2 mice. A, B, Dark degenerating neurons, which do not have typical chromatin margination and nuclear fragmentation, are present in an R6/2 mouse at 3 months of age (A) and in an R6/1 mouse at 8 months of age (B). A glial cell (gn) is also indicated. C, D, Degenerated mitochondria were observed in association with EM48 immunogold particles in the brain cortex at 8–10 weeks of age. Note that the cytoplasmic swelling, vacuolization, enlargement, and condensation of mitochondria (arrows) are associated with huntingtin immunogold particles. Degenerated mitochondria are also enclosed in a lysosome-like structure (D). Scale bars: A, 4 μm; B, 1.6 μm, C, D, 0.3 μm.
In addition, degenerated mitochondria were observed in neurons that showed normal nuclear and cytoplasmic appearance in the brains of R6/2 mice. Mitochondrial degeneration was evident by their swollen appearance, disintegration of double membrane and internal cristas, and darkened internal structures (Fig.6C,D). Although similar degenerated mitochondria were also found in N171-82Q and HdhCAG150 mice, degenerated mitochondria in R6/2 mouse brains were more frequently associated with EM48 immunogold particles. These immunogold particles were either clustered within the degenerating mitochondria or associated with the surface of the mitochondrial membrane. A recent study shows that mutant huntingtin is associated with the surface of mitochondrial membrane in HD mouse brain (Panov et al., 2002). We observed that mutant huntingtin could also be trapped inside degenerated mitochondria, supporting the role of mutant huntingtin in mitochondrial dysfunction (Beal, 2000; Panov et al., 2002). Degenerating mitochondria were also often surrounded by lysosomal structures (Fig. 6D), suggesting an advanced stage of degeneration. Such degenerated mitochondria were found more frequently in R6/2 mice that were >8 weeks of age. The identification of degenerated mitochondria provides a pathological basis for the protective effects of drugs against mitochondrial toxicity in HD mice (Ferrante et al., 2002).
Degenerated neurons and axons in Hdh150CAG mice
In HdhCAG150 mice, very few degenerated neurons with typical apoptotic features were seen under electron microscopy. However, neurons containing degenerated structures in the cytoplasm were frequently identified. These cytoplasmic structures, which often surrounded vacuoles, might be derived from lysosomes. However, their presence was not associated with obvious nuclear condensation or blebbing of the cytoplasm. Neurons containing the degenerated cytoplasmic organelles showed huntingtin inclusions in their nucleus (Fig. 7A). High-magnification micrographs showed that clusters of electron-dense bodies surrounded a large number of small vacuoles. Some of these dark bodies had a double membrane and visible internal cristas (Fig. 7B,arrows), indicating a mitochondrial origin. Other, darker structures might represent secondary lysosomes or autolysosomes (Fig.7B). Despite their unknown nature, these degenerated structures were observed only in the HD brain and not in the wild-type control mice.
Fig. 7.
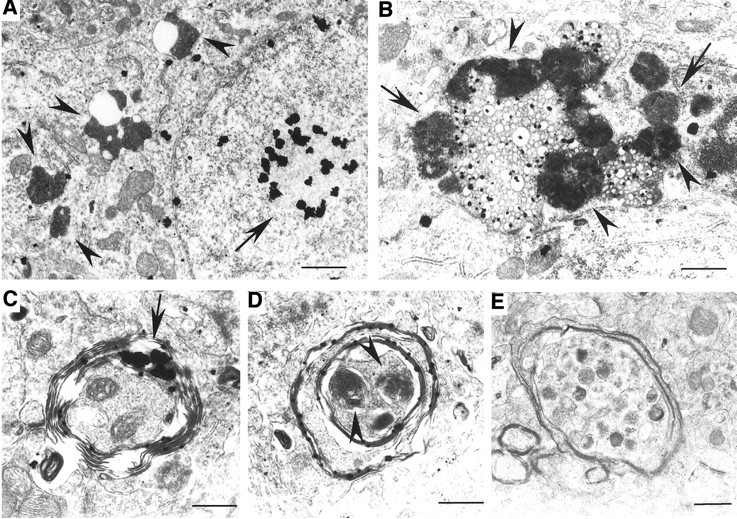
Degenerated neurons and axons in Hdh150CAG knock-in mice. A, B, Cytoplasmic organellar degeneration in HdhCAG150 mouse striatum. A, Several dark cytoplasmic organelles surround vacuoles (arrowheads). The nucleus that contains an intranuclear inclusion (arrow) appears intact, without nuclear fragmentation or condensation.B, A high-magnification micrograph shows clusters of electron-dense bodies and dark lysosomal structures that surround many cytoplasmic vacuoles. Some of these dense bodies show a mitochondrial origin with internal cristas (arrows). Others may represent secondary lysosomal bodies (arrowheads).C–E, Various types of degenerating axons are present in the striatum of HdhCAG150 mice. Axons are surrounded by disrupted myelin in which huntingtin aggregates (arrow) are trapped (C). Degenerated axons contain dark and swollen organelles (D, E), probably derived from mitochondria (arrowheads in D). Scale bars: A, 1 μm; B, 0.63 μm;C, 0.42 μm; D, 0.41 μm;E, 0.71 μm.
Similar cytoplasmic degenerative structures were also found in the brains of R6/2 mice. Some old HdhCAG150 mice (>14 months of age) showed very few dark degenerating neurons with a profile similar to that in R6/2 mice (data not shown). Thus, HdhCAG150 mice shared some pathological changes that were found in N-terminal huntingtin transgenic mice. The more specific pathological change in HdhCAG150 mice might be of axonal degeneration. Our previous studies revealed axonal degeneration in 17- to 22-month-old HD knock-in mice that express 80 CAG repeats (H. Li et al., 2000). We observed similar degenerating axons in HdhCAG150 mice at an earlier age (14 months). These degenerated axons were often surrounded by a disrupted myelin structure. Although cross sections of axons might not always reveal mutant huntingtin within axons, we did find some cases in which mutant huntingtin was associated with degenerated axons (Fig.7C–E). Mutant huntingtin was trapped or formed aggregates in the degenerating myelin. Organelles within the degenerated axons appeared dark and swollen, with no clear membrane-bound structures. However, their size and shape suggested that they might be derived from mitochondria. EM121 immunogold labeling was unable to reveal any mutant huntingtin specifically associated with degenerated myelin and axons in HdhCAG150 mouse brains (data not shown). Electron microscopic examination of R6/2 and N171-82Q mouse brains did not reveal such obvious axonal degeneration. Thus, degenerated axons appear to be associated with N-terminal mutant huntingtin smaller than the first 342 aa and might occur only in aged HD mice.
N-terminal mutant huntingtin activates caspases differently in transfected cells
The prominent apoptotic neurons in N171-82Q mice suggest that different N-terminal huntingtin fragments may activate caspases to different extents. To test this hypothesis, we examined caspase activities in huntingtin-transfected cells that expressed various N-terminal mutant huntingtin fragments containing 120–150Q [amino acids 1–67 (150Q-exon 1), 1–208 (120Q-208), 1–508 (120Q-508), and 1–945 (120Q-945)]. Western blots showed that these transfected huntingtin proteins were expressed at similar levels (Fig.8A). Only small N-terminal mutant huntingtin fragments (150Q-exon 1 and 120Q-208) formed aggregates that remained in the stacking gel. Consistent with other studies (Hackam et al., 1998; S. H. Li et al., 2000), transient transfection of small N-terminal huntingtin into non-neuronal cells resulted in cytoplasmic localization of the majority of mutant huntingtin and its aggregates (Fig. 8B), allowing for comparison of the cytoplasmic effects of transfected huntingtins containing different protein contexts. To examine whether these transfected proteins could activate caspase cascades, we first used fluorometric assays to measure caspase activity quantitatively. The mutant N-terminal huntingtin (120Q-208) produced the greatest activity of caspase-3. HD exon 1 huntingtin (150Q-exon 1) also increased caspase-3 activity, but the degree of this increase was less than that of the larger N-terminal huntingtin (120Q-208). As the length of N-terminal huntingtin was elongated further, caspase-3 activity decreased and became similar to that of vector transfection (Fig.8C).
Fig. 8.
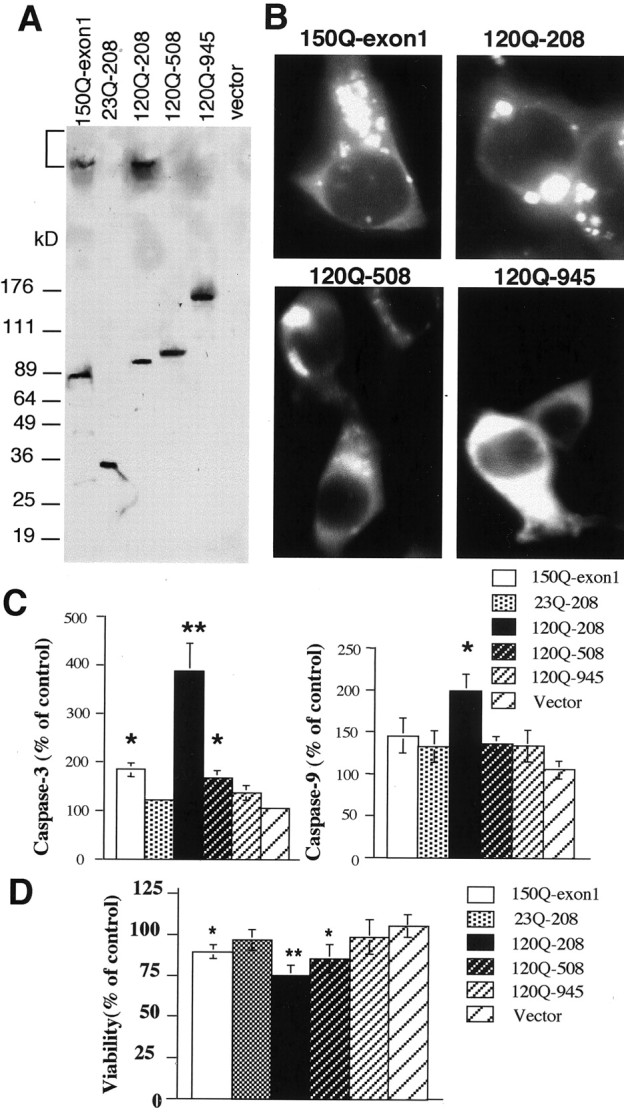
The effect of huntingtin context on the activation of caspases in transfected HEK293 cells. A, Western blot showing the expression of transfected huntingtin containing different protein sizes. The bracket indicates the stacking gel in which aggregated huntingtin is present.B, Immunofluorescence labeling of transfected HEK293 cells expressing 150Q-exon 1, 120Q-208, 120Q-508, and 120Q-945.C, Caspase-3 and caspase-9 activities of transfected cells that expressed different huntingtin fragments as indicated. The data were obtained from the 48 hr transfection of cells of three independent transfections and were presented as means ± SEM of control. The control is the activity of nontransfected cells.D, MTS assays of the viability of huntingtin-transfected cells. The control is the viability of nontransfected cells. The data (means ± SD) were obtained from three to four transfections for 72 hr. *p < 0.05 and **p < 0.01 compared with the activity of the vector-transfected cells.
We also measured the activity of caspase-9, an upstream caspase that is primarily activated by cytochrome c and in turn activates caspase-3. We saw that 120Q-208 could also increase the activity of caspase-9 significantly (Fig. 8C), suggesting that this increase may contribute to the higher activity of caspase-3 in these cells. Because caspase-3 is also activated by caspase-8 and other apoptotic pathways, transfected mutant huntingtin that did not significantly increase caspase-9 activity could trigger other apoptotic pathways to increase caspase-3 activity. Previous studies showed that the decreasing protein length of mutant huntingtin could increase the cellular toxicity of transfected cells in response to apoptotic stimulation (Hackam et al., 1999). This idea was also supported by our cell viability assays that, by measuring mitochondrial dysfunction caused by transfected huntingtin, showed the most reduced viability in 120Q-208-transfected cells (Fig.8D). 150Q-exon 1 or 120Q-508 transfection also reduced cell viability, although its transfection had less of an effect than 120Q-208 transfection on caspase activity and cell viability.
Discussion
Our studies demonstrate that transgenic huntingtin containing the first 171 aa with an expanded glutamine repeat (82Q) causes apoptosis in the brain. However, expression of the shorter N-terminal huntingtin (the HD exon 1) and the full-length mutant huntingtin did not result in typical apoptotic neurons. In vitro experiments using similar N-terminal huntingtin fragments also showed increased caspase activity in transfected cells. In addition, axonal degeneration was frequently found in HD repeat knock-in mice. Thus, mutant huntingtin may cause different types of neurodegeneration in a context-dependent manner.
The different types of neurodegeneration seen in our studies appear not to be dependent on the expression levels of transgenic huntingtin or on having a very long polyglutamine tract. First, it has been reported that in N171-82Q mice, the expression level of the transgene is approximately one-third the level of endogenous mouse huntingtin (Schilling et al., 1999). Immunostaining of brain using EM48 also showed less huntingtin staining in N171-82Q mice than in R6/2 mice. Second, the expression of mutant huntingtin with a long repeat (150Q) in R6/2 and HD repeat knock-in mice did not result in significant neurodegeneration. Furthermore, transfection of various N-terminal huntingtin fragments into cultured cells revealed that an N-terminal huntingtin fragment (1–208), which is similar to the N171-82Q transgenic protein, produced more caspase activity than the smaller or longer huntingtin fragments. All of these results suggest that N-terminal huntingtin sequences are important for various types of neurodegeneration.
The study of HdhCAG150 mice also suggests that N-terminal mutant huntingtin causes neurodegeneration. It is known that in HD repeat knock-in models, huntingtin aggregates are formed by N-terminal mutant huntingtin and become prominent and abundant as the mouse's age increases (H. Li et al., 2000; Wheeler et al., 2000), perhaps because the gradual accumulation of N-terminal mutant huntingtin is required for the formation of huntingtin aggregates. Consistent with that idea, degenerating neurons and axons were found only in old HdhCAG150 mice. More importantly, mutant huntingtin proteins associated with the degeneration were not labeled by the antibody against huntingtin amino acids 342–456, suggesting that they were small N-terminal mutant huntingtin fragments. A recent study shows that N-terminal huntingtin fragments generated from the cleavage at amino acids 104–114 and 146–214 are localized differently in cells (Lunkes et al., 2002). It remains to be determined whether the same N-terminal huntingtin fragments are present in HdhCAG150 mice.
In addition to protein context, the toxicity of mutant huntingtin fragments is also determined by a number of variables. First, polyglutamine length is certainly a primary determinant for cellular toxicity, because expanded polyglutamine repeats can induce neurological phenotypes in transgenic mice (Ordway et al., 1997) and apoptosis in cultured cells (Sanchez et al., 1999). Second, cellular factors that remove huntingtin fragments could inhibit huntingtin toxicity. Of these factors, chaperones and proteasomes prevent protein misfolding and degrade misfolded polyglutamine proteins, respectively (Orr, 2001). Third, aging may affect the function of these cellular factors and contribute to the late-onset pathology in Hdh150CAG mice.
However, the protein context of huntingtin is likely to determine the specific types of cellular toxicity by its influence on protein interactions or modulations. In support of this idea, a number of proteins, including huntingtin-associated protein 1 and huntingtin-interacting protein 1, bind to N-terminal mutant huntingtin, and their binding is altered by polyglutamine expansion (Li et al., 1995; Kalchman et al., 1997). Interactions of huntingtin with other proteins are important not only for the regulation of the function of a protein but also for the subcellular localization of huntingtin. N171-82Q may carry N-terminal huntingtin sequences that confer pathological events more specific to HD. In contrast, the exon 1 protein, which is shorter than other transgenic huntingtins, is more likely to be translocated into the nucleus. Its initial effects may primarily affect gene expression in the nucleus, because a number of studies have shown that this fragment produces a severe defect at the transcriptional level in R6/2 mice (Cha et al., 1998; Luthi-Carter et al., 2002) and stably transfected cells (Li et al., 1999). The smaller size of the N-terminal region with a longer repeat confers a more misfolded and aggregated state that can cause cellular toxicity via multiple pathological pathways, such as recruiting caspases, inhibiting proteasome function, increasing cellular stress, and affecting mitochondrial function (Ona et al., 1999; Sanchez et al., 1999; S. H. Li et al., 2000; Bence et al., 2001; Kouroku et al., 2002; Nishitoh et al., 2002; Panov et al., 2002). A global transcriptional dysregulation and multiple cytosolic effects mediated by exon 1 huntingtin in the brain may give rise to a severe neurotoxic phenotype without obvious apoptotic events before the animals die.
Although several previous studies were also unable to show obvious apoptotic neurons in R6/2 mice (Iannicola et al., 2000; Turmaine et al., 2000), others using different reagents and assays reported that R6/2 mouse brain had an increase in the number of TUNEL-positive cells (Keene et al., 2002) or caspase activity on Western blots (Chen et al., 2000). It seems that the identification of apoptotic events in HD mouse brains is also dependent on the experimental conditions and reagents used. However, under the same conditions, we found that N171-82Q mice had a greater number of apoptotic neurons than other HD mice, supporting the role of protein context in mediating neurodegeneration in HD. The context-dependent cytotoxicity was also observed in huntingtin-transfected cells in response to apoptotic stimulation (Hackam et al., 1998, 1999). We have presented new data showing context-dependent activation of caspases in huntingtin-transfected cells, which is consistent with the role of huntingtin context in different degrees of apoptosis in various HD mouse models.
In HD repeat knock-in mice, a large repeat (150Q) appears to facilitate disease progression, giving rise to more detectable neurological phenotypes (Lin et al., 2001) and earlier occurrence of neurodegeneration than those in HD repeat 80Q knock-in mice (H. Li et al., 2000). In addition to protein context, aging may contribute to certain types of neurodegeneration such as axonal degeneration. The short life span of the mouse might prevent obvious neurodegeneration from occurring, perhaps because the concentration of accumulated N-terminal huntingtin fragments never becomes high enough to induce neurodegeneration. In contrast, the expression of N171-80Q huntingtin results in more apoptotic neurons than do other transgenic huntingtins. As the length of huntingtin is reduced to exon 1 containing a large glutamine repeat, polyglutamine-mediated and non-context-dependent toxicity arises and becomes dominant. As N-terminal huntingtin fragments become larger, they are less misfolded and less toxic. Alternatively, larger huntingtin fragments may contain the sequences that protect against huntingtin toxicity. For example, a recent study has shown that phosphorylation of serine 421 of huntingtin is required for protecting against huntingtin toxicity by IGF-1 (Humbert et al., 2002).
Full-length or other forms of mutant huntingtin may also be pathogenic, because HdhCAG150 mice show neurological symptoms before the appearance of neurodegeneration and the formation of huntingtin aggregates (Lin et al., 2001; Menalled et al., 2002). Neuronal dysfunction and toxicity can occur in the absence of neurodegeneration. Because neurodegeneration in the brain provides an objective hallmark for HD, the study of its relationship with mutant huntingtin in HD mouse models will be especially helpful in uncovering the mechanisms of the neuropathology of HD. Identification of the neurodegenerative markers also helps with the evaluation of therapeutic effects of drugs on HD mouse models.
The present study provides in vivo evidence that mutant huntingtin induces different forms of neurodegeneration that are dependent on protein context. Consistent with this, protein context-dependent dysregulation of gene expression was reported recently in HD transgenic mice (Chan et al., 2002). Considering that a number of N-terminal huntingtin fragments have been found in vitro and in vivo (DiFiglia et al., 1997; Wellington et al., 2000; Kim et al., 2001; Mende-Mueller et al., 2001; Gafni and Ellerby 2002), it is likely that proteolysis produces a wide range of N-terminal huntingtin fragments, each of which may mediate specific forms of neuropathology. This may explain the various types of neurodegeneration found in the brains of patients with HD and HD animal models, including apoptotic neurons (Dragunow et al., 1995;Portera-Cailliau et al., 1995; Thomas et al., 1995) and nonapoptotic neurons (Turmaine et al., 2000). Investigation of the mechanisms by which these N-terminal sequences confer specific forms of neurodegeneration would help us to understand the pathogenesis of HD and to develop effective therapeutic strategies.
Footnotes
This work was supported by National Institutes of Health Grants AG19206 and NS41669. We thank Dr. Peter Deltloff for providing HD repeat knock-in mice and Huu-Phuc Nguyen for technique assistance.
Correspondence should be addressed to Dr. Xiao-Jiang Li, Department of Human Genetics, Emory University School of Medicine, 615 Michael Street, Atlanta, GA 30322. E-mail: xiaoli@genetics.emory.edu.
References
- 1.Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF. Creatine increases survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis. 2001;8:479–491. doi: 10.1006/nbdi.2001.0406. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 3.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 4.Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan EY, Luthi-Carter R, Strand A, Solano SM, Hanson SA, DeJohn MM, Kooperberg C, Chase KO, DiFiglia M, Young AB, Leavitt BR, Cha JH, Aronin N, Hayden MR, Olson JM. Increased huntingtin protein length reduces the number of polyglutamine-induced gene expression changes in mouse models of Huntington's disease. Hum Mol Genet. 2002;11:1939–1951. doi: 10.1093/hmg/11.17.1939. [DOI] [PubMed] [Google Scholar]
- 6.Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- 7.Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990;27:200–204. doi: 10.1002/ana.410270217. [DOI] [PubMed] [Google Scholar]
- 8.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 9.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 10.Dragunow M, Faull RL, Lawlor P, Beilharz EJ, Singleton K, Walker EB, Mee E. In situ evidence for DNA fragmentation in Huntington's disease striatum and Alzheimer's disease temporal lobes. NeuroReport. 1995;6:1053–1057. doi: 10.1097/00001756-199505090-00026. [DOI] [PubMed] [Google Scholar]
- 11.Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hackam AS, Singaraja R, Zhang T, Gan L, Hayden MR. In vitro evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington's disease. Hum Mol Genet. 1999;8:25–33. doi: 10.1093/hmg/8.1.25. [DOI] [PubMed] [Google Scholar]
- 16.Harper PS. Huntington's disease. Saunders; London: 1991. [Google Scholar]
- 17.Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington's disease. J Neuropathol Exp Neurol. 1995;54:105–120. doi: 10.1097/00005072-199501000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Hockly E, Cordery PM, Woodman B, Mahal A, van Dellen A, Blakemore C, Lewis CM, Hannan AJ, Bates GP. Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol. 2002;51:235–242. doi: 10.1002/ana.10094. [DOI] [PubMed] [Google Scholar]
- 19.Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 20.Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 21.Iannicola C, Moreno S, Oliverio S, Nardacci R, Ciofi-Luzzatto A, Piacentini M. Early alterations in gene expression and cell morphology in a mouse model of Huntington's disease. J Neurochem. 2000;75:830–839. doi: 10.1046/j.1471-4159.2000.0750830.x. [DOI] [PubMed] [Google Scholar]
- 22.Jackson M, Gentleman S, Lennox G, Ward L, Gray T, Randall K, Morrell K, Lowe J. The cortical neuritic pathology of Huntington's disease. Neuropathol Appl Neurobiol. 1995;21:18–26. doi: 10.1111/j.1365-2990.1995.tb01024.x. [DOI] [PubMed] [Google Scholar]
- 23.Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- 24.Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc Natl Acad Sci USA. 2002;99:10671–10676. doi: 10.1073/pnas.162362299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, Qin ZH, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kouroku Y, Fujita E, Jimbo A, Kikuchi T, Yamagata T, Momoi MY, Kominami E, Kuida K, Sakamaki K, Yonehara S, Momoi T. Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Hum Mol Genet. 2002;11:1505–1515. doi: 10.1093/hmg/11.13.1505. [DOI] [PubMed] [Google Scholar]
- 27.Kremer HP, Roos RA, Dingjan G, Marani E, Bots GT. Atrophy of the hypothalamic lateral tuberal nucleus in Huntington's disease. J Neuropathol Exp Neurol. 1990;49:371–382. doi: 10.1097/00005072-199007000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21:9112–9123. doi: 10.1523/JNEUROSCI.21-23-09112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- 30.Li SH, Cheng AL, Li H, Li XJ. Cellular defects and altered gene expression in PC12 cells stably expressing mutant huntingtin. J Neurosci. 1999;19:5159–5172. doi: 10.1523/JNEUROSCI.19-13-05159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li SH, Lam S, Cheng AL, Li XJ. Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis. Hum Mol Genet. 2000;9:2859–2867. doi: 10.1093/hmg/9.19.2859. [DOI] [PubMed] [Google Scholar]
- 32.Li XJ, Li SH, Sharp AH, Nucifora FC, Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- 33.Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 34.Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 35.Luthi-Carter R, Strand AD, Hanson SA, Kooperberg C, Schilling G, La Spada AR, Merry DE, Young AB, Ross CA, Borchelt DR, Olson JM. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum Mol Genet. 2002;11:1927–1937. doi: 10.1093/hmg/11.17.1927. [DOI] [PubMed] [Google Scholar]
- 36.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 37.Manoonkitiwongsa PS, McMillan PJ, Schultz RL, Jackson-Friedman C, Lyden PD. A simple stereologic method for analysis of cerebral cortical microvessels using image analysis. Brain Res Brain Res Protoc. 2001;8:45–57. doi: 10.1016/s1385-299x(01)00087-3. [DOI] [PubMed] [Google Scholar]
- 38.Menalled LB, Sison JD, Wu Y, Olivieri M, Li XJ, Li H, Zeitlin S, Chesselet MF. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mende-Mueller LM, Toneff T, Hwang SR, Chesselet MF, Hook VY. Tissue-specific proteolysis of Huntingtin (htt) in human brain: evidence of enhanced levels of N- and C-terminal htt fragments in Huntington's disease striatum. J Neurosci. 2001;21:1830–1837. doi: 10.1523/JNEUROSCI.21-06-01830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 42.Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, Wiener HW, Dure LS, IV, Lindsey R, Hersch SM, Jope RS, Albin RL, Detloff PJ. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91:753–763. doi: 10.1016/s0092-8674(00)80464-x. [DOI] [PubMed] [Google Scholar]
- 43.Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15:925–932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- 44.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 45.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 48.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 49.Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington's disease: a quantitative Golgi study. Neurology. 1993;43:2088–2096. doi: 10.1212/wnl.43.10.2088. [DOI] [PubMed] [Google Scholar]
- 50.Thomas LB, Gates DJ, Richfield EK, O'Brien TF, Schweitzer JB, Steindler DA. DNA end labeling (TUNEL) in Huntington's disease and other neuropathological conditions. Exp Neurol. 1995;133:265–272. doi: 10.1006/exnr.1995.1029. [DOI] [PubMed] [Google Scholar]
- 51.Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 53.Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 54.Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- 55.Zhou H, Li SH, Li XJ. Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J Biol Chem. 2001;276:48417–48424. doi: 10.1074/jbc.M104140200. [DOI] [PubMed] [Google Scholar]