

Abstract
Analysis of cellular morphology is the most general approach to neuronal classification. With the increased use of genetically engineered mice, there is a growing need for methods that can selectively visualize the morphologies of specified subsets of neurons. This capability is needed both to define cell morphologic phenotypes and to mark cells in a noninvasive manner for lineage studies. To this end, we describe a bipartite genetic system based on a Cre-estrogen receptor (ER) fusion protein that irreversibly activates a plasma membrane-bound alkaline phosphatase reporter gene by site-specific recombination. Because the efficiency and timing of gene rearrangement is controlled pharmacologically, a sparse subset of labeled cells can be generated from the set of CreER-expressing cells at any time during development. Histochemical visualization of alkaline phosphatase activity reveals neuronal morphology with strong and uniform labeling of all processes.
Keywords: neuronal morphology, tamoxifen, Cre recombinase, lineage tracing, brain development, cell labeling
Introduction
There is now a wealth of data regarding the development and function of the nervous system. However, the roles of many individual neuronal types are still poorly understood, in large part because of difficulties in correlating their physiological, anatomical, and molecular properties. This problem is compounded by the absence of highly selective molecular markers for classifying many neuronal cell types.
Currently, the most general approach for classifying neurons is based on morphology, and a wide variety of methods are now available for this purpose. Classical methods for revealing neuronal morphology include Golgi staining, diI tracing, and single-cell injection with neurobiotin or HRP. Recent improvements to cell injection methodology include the use of fluorescent stains or retrograde tracers to visualize subsets of neurons before injection (Tauchi and Masland, 1985; Rodieck and Watanabe, 1993) and sparse delivery of lipophilic dyes by particle bombardment (Gan et al., 2000). A second general class of methods uses genetically encoded histochemical or fluorescent reporters, principally β-galactosidase, placental alkaline phosphatase (AP), or green fluorescent protein (GFP) and its derivatives. Expression in a sparse subset of cells is effected by low efficiency infection with a replication incompetent virus (Slack and Miller, 1996) or by DNA delivery using particle bombardment or electroporation (Lo et al., 1994; Haas et al., 2001). These methods are especially well suited to slice or explant cultures because they require access to the tissue for viral infection or DNA delivery. An important recent addition to this genetically based class of methods, and one that does not require tissue access for cell marking, has emerged from the observation that transgenic lines carrying GFP or its derivatives under the control of the Thy-1 promoter often exhibit sparse expression in diverse subsets of neurons (Feng et al., 2000).
Marking individual cells is also the central methodology needed for lineage tracing, an application that has been of longstanding interest in developmental neurobiology. For lineage tracing, the marker should ideally be delivered to or activated within a single cell and then passed with little or no modification or dilution to all of the progeny of that cell. Microinjection of fluorescent or other inert markers has been used with great success in early embryonic lineage studies inXenopus and other nonplacental animals (Lane and Sheets, 2002). However, this approach can only be used with difficulty at later stages of development when cells are small and inaccessible, and it is impractical in placental animals. As an alternative, engraftment of genetically marked cells, as in the classic chick–quail chimera system (Burns and Le Douarin, 2001), has been used with great success for lineage analysis in avian embryos, but this method is difficult in placental animals (Wichterle et al., 2001). For experiments in both mammals and birds, tagging of mitotically active progenitors by integration of a replication-incompetent retrovirus is the current method of choice for CNS lineage analysis (Cepko et al., 1998; Noctor et al., 2001; Reid and Walsh, 2002). However, for use in the mammalian brain, viral injection requires surgical access to the ventricular zone, a technically challenging problem, especially at early times in gestation (McCarthy et al., 2001).
At present, three noninvasive methods exist for lineage tracing in mammals. Two of these involve the analysis of highly chimeric animals, produced either by embryonic stem cell injection at the blastocyst stage (Kuan et al., 1997) or by scoring differentially marked patches of X-chromosome inactivation in heterozygous females (Tan et al., 1995). Both methods suffer from a lack of flexibility with respect to the ratio of marked to unmarked cells and the timing of the marking event. A third method uses transgenic mice that express a β-galactosidase (lacZ) gene with an internal duplication within the coding region (“laacZ”) driven by the neuron-specific enolase promoter (Nicolas et al., 1996). Rare intragenic recombination events eliminate the duplication, restore the wild-type lacZ coding sequence, and allow histochemical visualization of clonally related cells at later times. This system has been used to analyze both neural and non-neural lineages (Eloy-Trinquet and Nicolas, 2002) but is limited by the low frequency of recombination [approximately one event per 20 mice when analyzed at embryonic day (E) 11–12], the inability of the experimenter to control the timing of recombination events, and the generally poor definition of neuronal morphology afforded by β-galactosidase.
In this paper, we describe a combined genetic and pharmacologic method that (1) does not require surgical or mechanical access to the target cells, (2) results in high quality definition of neuronal morphology with good visualization of distant processes, (3) produces a durable stain using a simple, reliable, and rapid protocol, (4) targets predetermined subsets of cells for labeling based on specificity of a gene-targeted or transgenic promoter, and (5) allows pharmacologic control of the timing and efficiency of the genetic event that marks cell lineage. When cells are marked late in development or in adulthood, the morphologies of hundreds of labeled cells can be analyzed in a single animal, suggesting that this method could be used for routine phenotyping of genetically altered mice.
Materials and Methods
Cre-estrogen receptor knock-in mice. The R26 Cre-estrogen receptor (ER) knock-in mouse line was generated by standard procedures and maintained in the homozygous state. The lacZ/placental alkaline phosphatase (ZAP) reporter mouse was a kind gift of Corine Lobe and Andras Nagy (University of Toronto). This line must be maintained in the heterozygous state because ZAP homozygotes are nonviable. Crosses and genotyping were performed by standard procedures. 4-Hydroxytamoxifen (4HT) (Sigma, St. Louis, MO) was dissolved in ethanol at a concentration of 10 mg/ml and stored in aliquots at −80°C. Aliquots were emulsified in 5 vol of sunflower seed oil by vortexing for several minutes, after which the ethanol was evaporated in a speed-vac (Savant, Holbrook, NY). Injections were intraperitoneal, with the doses noted in the figure legends.
Tissue processing. Tissues were collected either fresh or after cardiac perfusion. For retinal histology, adult mice were anesthetized with ether and their eyes were enucleated. All incubations were performed at room temperature, except where noted. Whole eyeballs were prefixed for 15 min in PBS containing 2% paraformaldehyde, 0.5% glutaraldehyde, 2 mm MgCl2. The eyes were opened by removing the cornea and lens and flattened with a series of four lateral cuts, and the retinas were dissected and placed between two small circular plastic meshes. The retinas were fixed for 1 hr at room temperature in the same fixative, washed twice in PBS with 2 mm MgCl2, transferred to PBS without MgCl2, and heated in a water bath for 1 hr at 65°C to inactivate endogenous AP activity. AP staining was performed in 0.1 m Tris, 0.1m NaCl, 50 mmMgCl2, pH 9.5, 0.34 μg/ml nitroblue tetrazolium (NBT), and 0.175 μg/ml 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) (Boehringer Mannheim, Indianapolis, IN), for 1 hr to overnight at room temperature with gentle agitation. After staining, tissues were washed three times for 20 min in PBS, 0.1% Tween 20, and postfixed in PBS with 4% paraformaldehyde overnight. Before imaging, samples were dehydrated through an ethanol series and then cleared with 2:1 benzyl benzoate (BB)/benzyl alcohol (BA). For postnatal brain histology, mice were anesthetized with ketamine–xylazine and perfused with the fixative described above, and the brain was dissected, embedded in 3% low melting point agarose in PBS with 2 mmMgCl2, and sectioned on a vibratome at 250–300 μm thickness. Thereafter sections were processed as described for retinal flat mounts. Whole embryos, embryonic heads, or dissected embryonic brains were fixed and heat treated intact, and then vibratome sectioned for staining and analysis. Staining of whole embryos up to E13 was performed similarly, except embryos were incubated in PBS, 2 mm MgCl2, 0.5% Tween 20 overnight, washed several times with PBS, 2 mmMgCl2, and then washed twice in AP staining buffer without substrates before incubation in the complete staining solution.
Tissues can be stored for months after fixation and heat inactivation and before staining, or after staining and postfixation, with no apparent loss in quality. However, after tissue clearing in BB/BA, the NBT/BCIP precipitate dissolves over several days.
Microscopy and image analysis. Cleared tissues were imaged using a Zeiss Axiophot microscope, with a black and white Axiocam CCD, with or without a CRI color filter set. For serial sections through the tissue, a Ludl stage Z-drive was adapted to the microscope. Image acquisition and Z-focus control were controlled by Openlab software (Improvision). Tracing and three-dimensional reconstruction were performed with Objectimage (an enhanced version of NIHimage), with the Treetrace program written By D. K. Hartline (Bekesy Laboratory of Neurobiology, University of Hawaii).
Results
Principle of the cell marking method
The marking system consists of two genetic components (Fig. 1A). The first is a Cre recombinase fused to a mutated estrogen receptor ligand binding domain (CreER) that selectively binds the estrogen analog 4-hydroxytamoxifen (4HT) and is inactive in the absence of 4HT (Feil et al., 1996). We targeted the CreER coding region to the Rosa26 locus (Friedrich and Soriano, 1991) to obtain ubiquitous or nearly ubiquitous expression of the CreER recombinase (Fig.1B). We have derived two lines of mice that differ with respect to the efficiency with which the CreER message is translated. In one line the initiator methionine codon of the CreER coding region is in an optimal translation initiation context (GCCACCATGT), and in the second line it is in a context that is far from optimal (CCCTTTATGT). The experiments reported here used the first of these lines, referred to hereafter as R26CreER.
Fig. 1.
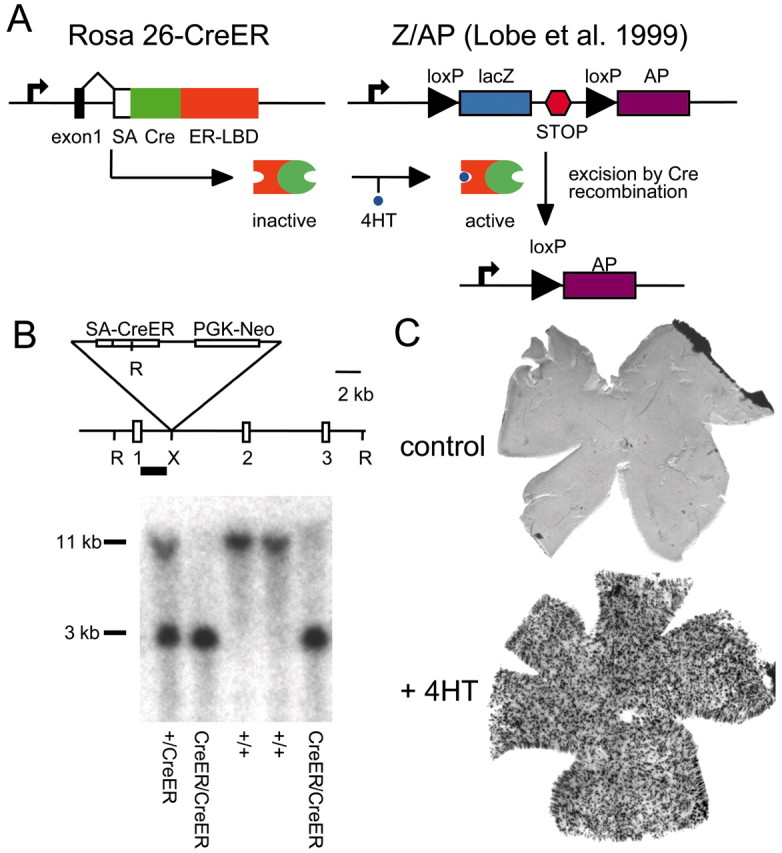
General strategy and generation of knock-in mice.A, Outline of the R26CreER;ZAP labeling strategy. The CreER fusion protein is expressed ubiquitously from the Rosa26 locus. After systemic exposure to 4HT, activated CreER catalyzes recombination between loxP sites at the ZAP reporter locus, allowing transcription of the downstream AP coding region. The efficiency of recombination is dependent on the amount of 4HT delivered. ER-LBD, Estrogen receptor ligand-binding domain mutated to recognize 4HT; SA, splice acceptor;STOP, three-tandem polyA addition sites.B, Generation of the R26CreER knock-in mouse.Top, Map of the Rosa26 locus showing theXbaI site (X) used for insertion of the CreER-PGK-neo cassette, the EcoRV restriction sites (R) used for genotyping, and exons 1–3 of the Rosa26 gene. The Southern blot probe is shown as ablack bar. The expected sizes of the hybridizing bands from knock-in and wild-type alleles are 3 and 11 kb, respectively.Bottom, Southern blots of DNA from mice of the indicated genotypes. C, Flat mounts of adult retinas from R26CreER/+;ZAP/+ mice stained with NBT/BCIP. Top, Uninjected control; bottom, after three intraperitoneal injections of 250 μg of 4HT at P7, P8, and P9.
The second genetic component, referred to as ZAP, is a ubiquitously expressed Cre-sensitive reporter transgene developed by Lobe et al. (1999) (Fig. 1A). The ZAP reporter contains a cytomegalovirus enhancer/β-actin promoter driving a lacZ coding region followed by three transcription termination sites. The lacZ coding region and termination signals are flanked by loxP sites. An AP coding region is located immediately distal to the loxP flanked sequences but is not expressed unless the upstream cassette is removed by Cre recombinase. As determined by 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (Xgal) staining, both the Rosa26 locus and the ZAP transgene in the mouse line developed by Lobe et el. (1999) are expressed ubiquitously or nearly ubiquitously within the CNS throughout development, although expression levels vary to some extent among different regions (Zambrowicz et al., 1997; Lobe et al., 1999) (data not shown).
Systemic exposure of R26CreER;ZAP mice to 4HT relieves the cytoplasmic sequestration of the CreER fusion protein, allowing it to translocate to the nucleus and irreversibly activate AP expression in a fraction of the cells. Previous in vivo studies with CreER that were aimed at developing highly efficient temporal control of gene rearrangement reported disappointingly inefficient recombination with systemic 4HT exposure (Feil et al., 1996; Brocard et al., 1997;Danielian et al., 1998). In the present application, low-efficiency recombination is desirable and readily achievable with nontoxic levels of 4HT. Figure 1C illustrates the effect of intraperitoneal 4HT exposure during early postnatal life: ∼1 month later, several hundred AP+ cells are seen uniformly distributed across the retina, whereas in a control retina that was not exposed to 4HT there are no AP+ cells.
Cell labeling in the adult and embryonic retina and brain
For the present application we have chosen AP as the reporter because of its plasma membrane localization (via a glycosylphosphatidylinositol anchor) and its robust histochemical stain. Figure 2 illustrates the range of cell types and the morphologic definition seen in retinal flat mounts from R26CreER/+;ZAP/+ mice exposed postnatally to 4HT. Conveniently, the high stability of the NBT/BCIP precipitate allows one to view the same cells first in flat mount and then in cross section after the retina is cut into strips. Figure2B–H shows perpendicular sections containing AP+ photoreceptors, bipolar cells, and an amacrine cell. Figure 2I–Q shows successive serial sections from a flat mount of a starburst amacrine cell and a ganglion cell, together with digitized reconstructions of their arbors. A preliminary catalog of morphologic classes within the mouse retina based on this method suggests that cells are labeled in rough proportion to their abundance and that all major classes are represented among the labeled cells as judged by a comparison with work on other mammalian retinas (Masland, 2001) (T. Badea and J. Nathans, unpublished observations). This observation, together with that described below for the brain, indicates that the combination of R26CreER and ZAP labels a wide variety of neuronal cell types and appears to have no deleterious effect on cell viability.
Fig. 2.
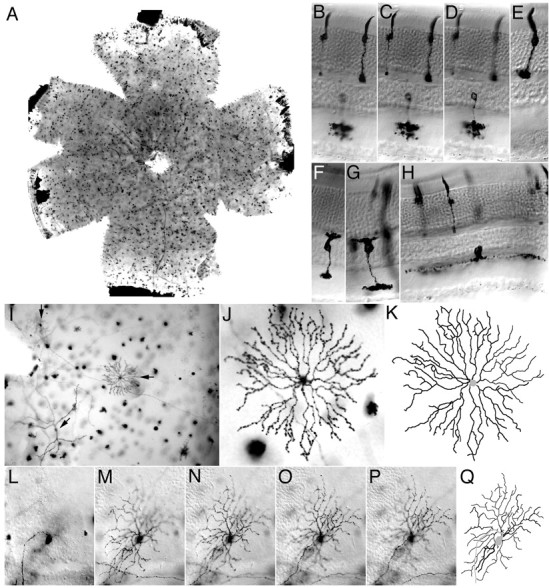
Morphology of retinal cells. Retinas from adult CreER/+;ZAP/+ mice that had received 3 mg of 4HT ∼1 week before staining (A–H) or 0.8 mg of 4HT at P20 (I–Q). These doses of 4HT produce a relatively low density of AP+ cells, appropriate for characterizing individual cell morphologies. Retinal whole mounts were stained with NBT/BCIP, and images were collected with Nomarsky optics to reveal the retinal cell layers. A, Retina flat mount. B–H,Perpendicular sections at a thickness of ∼500 μm. The photoreceptor layer is at the top. B–D, Three focal planes centered on a cone (right photoreceptor with large pedicle), a rod (left photoreceptor with small spherule), and a narrow-field amacrine cell (bottom).E, Cone; F, G, cone bipolar cells;H, starburst amacrine cell. I–Q, Flat-mount images and computerized image reconstructions.I, Image (10×) of a retina flat mount revealing a ganglion cell (vertical arrow), a starburst amacrine cell (horizontal arrow), and a wide field amacrine cell (arrow at 45°). Mueller glia, bipolar, and narrow field amacrine cells are also seen in this plane of focus. In flat mounts, most of the abundant and highly compact cells either traverse the full thickness of the retina (Mueller glia) or are confined to the photoreceptor layer (rods and cones). J, Image (40×) of the starburst amacrine cell shown in I and its reconstructed image (K). The neurites of this cell were distributed in a narrow plane of ∼4 μm thickness.L–P, Successive focal planes through the ganglion cell in I and its reconstructed image (Q).
An evenly distributed sampling of diverse cell types is also obtained in the adult R26CreER/+;ZAP/+ brain after systemic 4HT exposure (Fig.3). The larger, intensely stained, and compact cells distributed throughout the brain appear to be glia; smaller finely branched neurons are readily apparent among them (Fig.3B). We typically view the adult brain in 300 μm vibratome sections as shown in Figure 3. These sections are sufficiently thin that they provide good optical clarity (when equilibrated in benzyl benzoate/benzyl alcohol), but they are of sufficient thickness that for many neurons most or all of the dendritic arbors are located within a single section, thereby facilitating three-dimensional reconstruction of cell morphology from optical sections. AP labeling of cell processes appears to be highly efficient; for example, individual pyramidal cell axons can be followed for several millimeters, ending only at the plane of section. Examples of a CA1 neuron, a cortical pyramidal cell, and three cerebellar granule cells are shown in Figure 3,C–H, I–N, andO–Q, respectively.
Fig. 3.
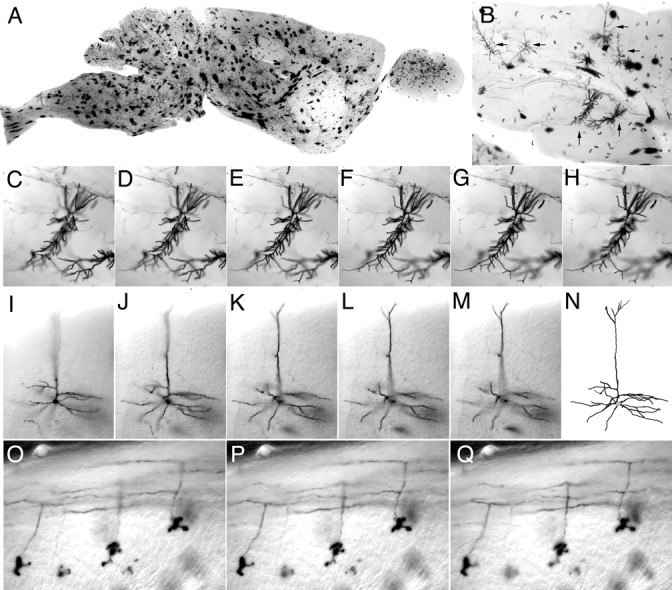
Morphology of adult CNS neurons. Brain from an adult R26CreER/+;ZAP/+ mouse that received a single intraperitoneal injection of 0.8 mg of 4HT at P21. A, Parasagittal section (300 μm) through the brain. Most of the intensely labeled cells are glia. B, Section (300 μm) through cortex (top) and hippocampus (center) reveals labeled pyramidal neurons (horizontal arrows) and CA1 neurons (vertical arrows). C–H, Successive focal planes through one of the CA1 neurons shown inB. I–M, Successive focal planes through a pyramidal neuron and its reconstructed image (N). O–Q, Three cerebellar granule cells. The cell bodies and compact dendritic trees are seen in the bottom half of each panel; the characteristic T-shaped branching of the axon that gives rise to parallel fibers is seen in the top half of each panel. In C–Q, successive focal planes are at intervals of 5 μm.
We next asked whether this method could be applied to embryonic neurons, and in particular whether a period of several days between 4HT exposure and time the animals were killed would be sufficient to permit high level accumulation of the AP protein over the entire surface of dendritic and axonal processes. Figure4 shows an E18 brain from a R26CreER/+;ZAP/+ mouse that was exposed at E12 to a maternal injection of 600 μg of 4HT. Intense AP staining of cell bodies, dendrites, and long fiber tracts is apparent. Importantly, the relatively sparse distribution of AP+ cells permits clear visualization of overlapping or interdigitating fiber tracts (Fig. 4A–D), thereby providing a convenient survey of the major tracts in a single brain. By contrast, it is often difficult to clearly distinguish overlapping tracts by immunostaining for general axonal markers such as neurofilament. In the cortex, immature pyramidal cells show few if any basal dendrites and minimal arborization of the apical dendrite (Fig.4E), in contrast to the mature pyramidal cell morphology seen in Figure 3I–N. Within the E18 retina, labeled cells are arrayed in radial clusters with little tangential migration (Fig. 4F,G), consistent with previous retroviral tagging and X-inactivation studies (Turner et al., 1990; Reese et al., 1995). Complete AP labeling of neuronal processes is also seen with R26CreER/+;ZAP/+ embryos at E12 that were exposed to a single maternal injection of 25 μg of 4HT at E8 (data not shown). For example, in these embryos, individual AP+ spinal and cranial sensory axons are strongly labeled along their entire lengths.
Fig. 4.
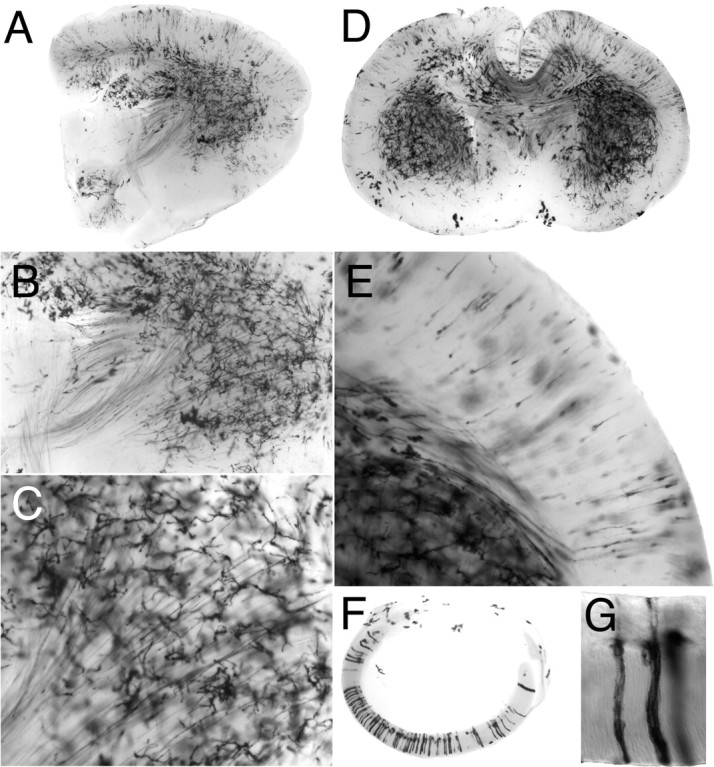
Morphology of brain and retinal neurons at E18. An R26CreER/R26CreER homozygous female was mated with a ZAP/+ male and injected at day 12 of gestation with 600 μg of 4HT. Embryos were harvested at E18. Parasagittal (A–C) and coronal (D, E) sections from two R26CreER/+;ZAP/+ littermates. B, C, The striatum at successively higher magnification showing fiber tracts (B) and individual axons originating from striatal neurons (C). E, Pyramidal neurons exhibit simple morphology, with unbranched apical dendrites and scarce basal dendrites; axons are seen descending to subcortical regions. F, G, Columnar arrangement of labeled cells within the retina. Cells within each vertical cluster of cells are likely to be clonally related.
Production of marked clones
For lineage tracing, the marked progenitors should be sufficiently sparse so that later in development all of the cells in each AP+ cluster have a high probability of being clonally related. To determine the optimal concentration of 4HT for lineage analysis, we injected pregnant females with 0.5, 1, or 4 μg of 4HT per gram of body weight (equivalent to 12.5, 25, and 100 μg of 4HT for a 25 gm mouse) at day 8 of gestation and examined the pattern of labeled cells in the brains and retinas of their adult progeny.
Figure 5A shows three adult retinas from R26CreER/+;ZAP/+ mice exposed in utero to 4 μg/gm 4HT (retinas a–c) and one retina from a mouse exposed to 1 μg/gm 4HT (retina d). Several large clusters of AP+ cells are present in retinas a–c, and a single small cluster of AP+ cells is present in retina d. Overall, six of six retinas from littermates exposed to 4 μg/gm 4HT had multiple AP+ cell clusters per retina; three of eight retinas from littermates exposed to 1 μg/gm 4HT had only one AP+ cell cluster per retina, and the remaining five of eight retinas had no AP+ cells; and zero of eight retinas from littermates exposed to 0.5 μg/gm 4HT had labeled AP+ cells. In the E18 retina, a similar dose dependence of 4HT exposure at mid-gestation governs the number of AP+ radial cell clusters (data not shown). The steep 4HT dose–response relation most likely reflects the requirement for four Cre monomers to catalyze the site-specific DNA cleavage and exchange reaction (Guo et al., 1997). Thus, if the nuclear concentration of Cre monomer varies linearly with 4HT concentration, then recombination efficiency should be proportional to the fourth power of the 4HT concentration. This nonlinear dose–response relation should significantly sharpen the time course of recombinase action after a single 4HT injection.
Fig. 5.
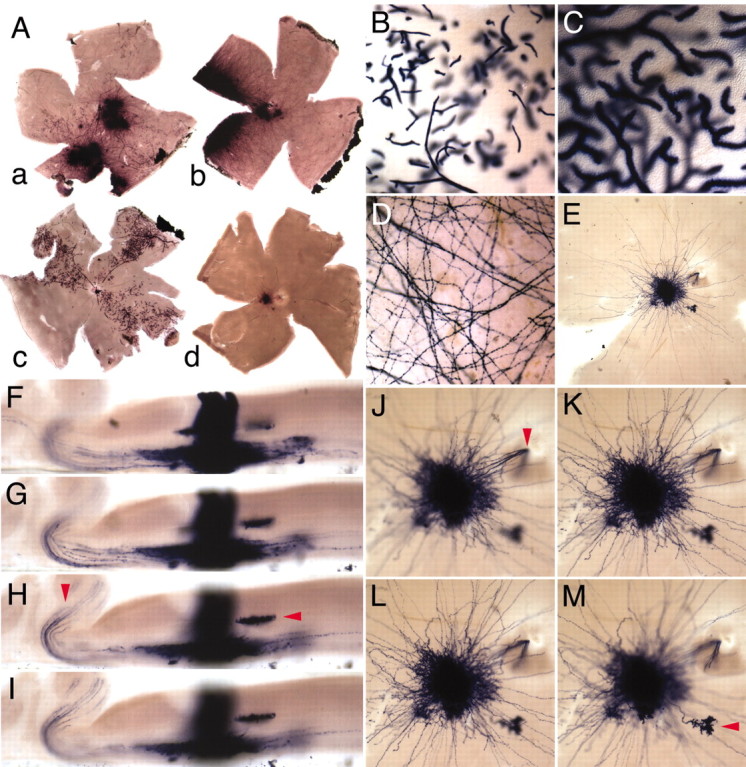
Genetically marked clones in the retina. R26CreER/R26CreER females were mated with ZAP/+ heterozygous males and injected with 1 μg/gm 4HT or 4 μg/gm 4HT at day 8 of gestation. Pregnancies were carried to term, and retinas from the progeny were analyzed in adulthood and viewed as flat mounts (A–E, J–M) or vertically sectioned at a thickness of ∼500 μm (F–I). A, Retinasa–c are from 4 μg/gm 4HT injections; retinad is from a 1 μg/gm 4HT injection. Retinaa has both neuronal/glial and vascular clones, retinasb and d have only neuronal/glial clones, and retina c has only vascular clones. B, C, Progressively higher magnification views of blood vessels from retina c. D, Higher magnification view of retina b on the side of the retina distal to the labeled cell bodies; long dendritic processes are well labeled and extend across the entire retina. E, Higher magnification view of the single clone in retina d. The fibers with the greatest lateral extent are likely to originate from wide-field amacrine cells. Vertical sections (F–I) and flat mounts (J–M) of the clone in retina d at successive focal planes are shown. Two different horizontal cell arbors are indicated by horizontal arrowheads in H and M, and ganglion cell axons entering the optic nerve are indicated byvertical arrowheads in H andJ. F–I also show that within this clone the labeled cell bodies are tightly clustered and occupy a cross-sectional area far smaller that that occupied by the cell processes seen in the flat mount.
Given their paucity, each AP+ cell cluster in the 1 μg/gm 4HT group very likely represents a single clone. Interestingly, all of the AP+ cells in retina c are endothelial cells within the intraretinal vasculature (Fig. 5A–C). Both AP+ endothelial cells and neurons are seen in retina a. The AP+ endothelial cells are confined to several large wedge-shaped zones of the intraretinal vascular tree, consistent with a clonal origin followed by restricted migration of marked cells. The alternating pattern of AP+ and AP− cells along the vascular tree suggests the existence of a small local population of endothelial progenitors, the progeny of which intermingle extensively during intraretinal angiogenesis. In retina b, strong AP labeling is observed in the dendritic processes of wide-field amacrine cells, and these spread efficiently over the entire surface of the retina (Fig.5D).
The single cluster of AP+ cells in retina d, seen both in flat mount (Fig.5E,J–M) and in cross section (Fig. 5F–I), has a tight radial cluster of cell bodies spanning the full thickness of the retina, with several horizontal cell processes in the outer plexiform layer (Fig. 5H,M, horizontal arrows), both narrow and wide field amacrine cell dendrites (Figs.5K,L), and 10–20 ganglion cell axons projecting into the optic nerve (Fig.5H,J, vertical arrows). As seen here, the close apposition of adjacent cells within a cluster hinders the identification of individual cell morphologies, a limitation that might potentially be overcome by serial reconstruction from semithin or ultrathin sections.
Similar analyses of the adult brain reveal rare clusters of cells in a mouse exposed to 1 μg/gm 4HT at E8 (Fig.6); higher densities of labeled cells are observed with 4 μg/gm 4HT (data not shown). In consecutive parasagittal sections, a single cluster of cortical cells is seen to occupy an extended mediolateral arc (Fig. 6A), with both subcortical and intracortical fibers emanating from it (Fig.6B). No other cortical cells are labeled in this hemisphere. A single morphologic class of interneurons is present at low density throughout the striatum (Fig.6A,D). The possibility that such widely dispersed striatal interneurons derive from one or a few precursors is consistent with recent retroviral lineage studies (Reid and Walsh, 2002). Interestingly, the dorsal cochlear nucleus is heavily labeled, whereas adjacent brainstem and cerebellar structures are not (Fig. 6A,E,F), suggesting a distinct progenitor for this structure.
Fig. 6.
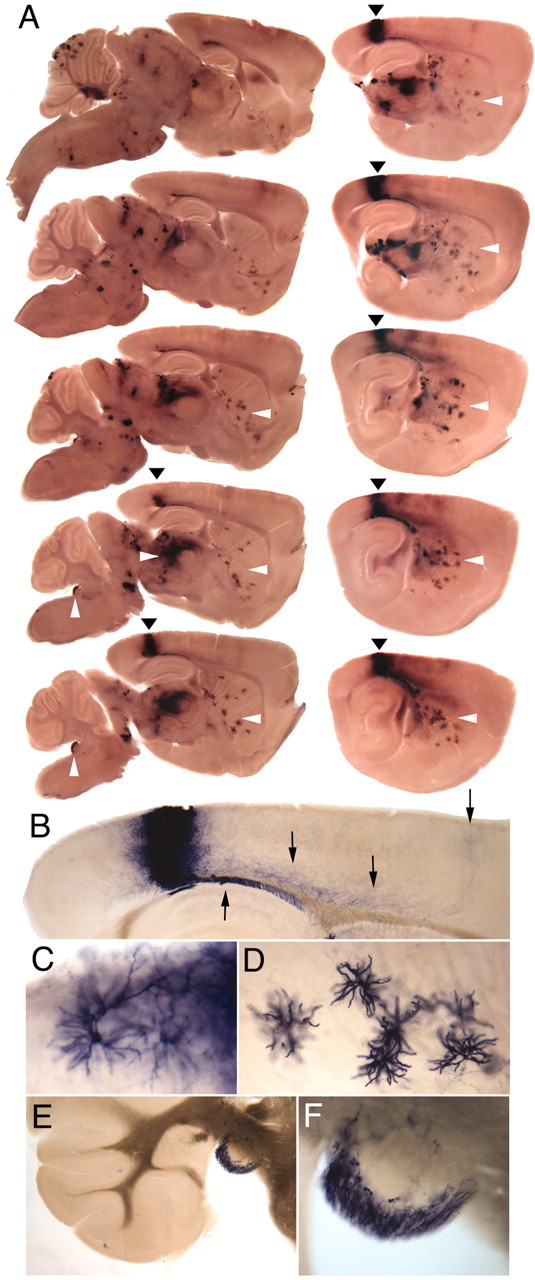
Embryonic clones visualized in the adult brain. Adult brain from a R26CreER/+;ZAP/+ mouse that had been exposed at E8 to a maternal injection of 1 μg/gm 4HT, as described in the legend of Figure 5. A, Consecutive 250 μm parasagittal sections, arrayed from medial (top left) to lateral (bottom right). In the cortex, a single contiguous cluster of stained cells is indicated by arrowheads and shown at higher magnification in B. One group of fibers from this clone descends to the external capsule (B, upward arrow); a second group of fibers targets adjacent regions of cortex (B, downward arrows). At the edge of the clone, individual interneurons can be resolved from the mass of labeled cells (C). The striatum (A, leftward arrowheads) has a single morphologic class of labeled interneurons, shown at higher magnification in D. Examination of the striatum at high magnification shows that each labeled cell has, in addition to numerous thick dendritic processes, a single thin axon that either passes out of the plane of section or terminates tens to hundreds of micrometers away in a halo of fine arbors. This morphology identifies these cells as neurons rather than glia. The dense labeling of the thalamus (A,rightward arrowhead) is derived primarily from neurites; few labeled cell bodies are seen. Densely stained fibers and cell bodies are seen in the dorsal cochlear nucleus (A,vertical arrowheads), shown at progressively higher magnifications in E and F.
To systematically determine the effect of 4HT injection time on the size of AP+ cell clusters and on the types of cells that are labeled, and to assess the degree of animal-to-animal variability within an experiment, CreER/+;ZAP/+ embryos were exposed to a single maternal injection of 200 μg of 4HT at days 7, 8, 9, 10, 11, or 13, and the embryos were analyzed at either E14 or E18. Figure7 shows that (1) injection with 4HT at progressively later times in gestation leads to a corresponding decrease in the number of AP+ cells per cluster and (2) a constant dose of 4HT (200 μg) produces a larger number of AP+ clusters per brain when delivered later in gestation, presumably because the larger embryo presents a correspondingly greater number of target cells for Cre-mediated recombination. We note that for the present purpose of defining the effect of 4HT exposure time on cell cluster size and composition, large numbers of labeled clusters are useful; for lineage tracing applications in which rare and widely separated clusters are required, a lower 4HT dose is desirable.
Fig. 7.
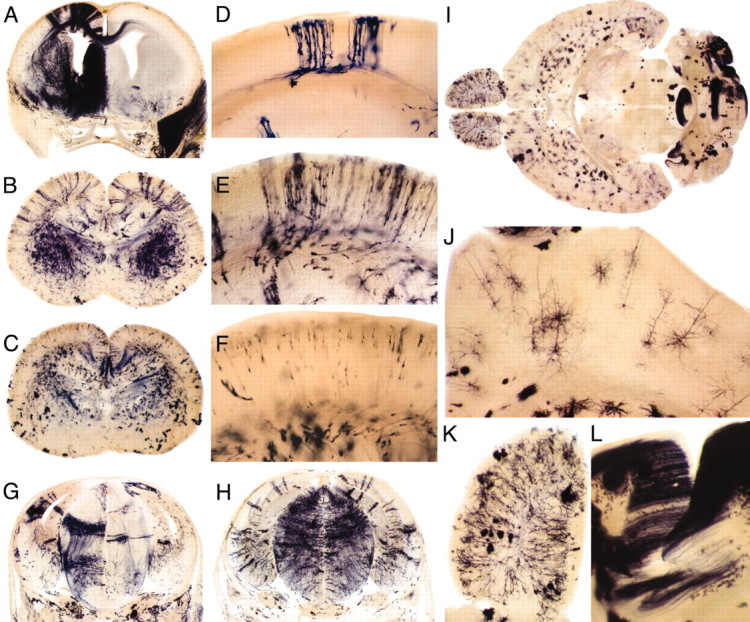
Effect of 4HT injection time on the composition and size of labeled clones. Matings were performed as for Figure 5, except some of the progeny are additionally heterozygous for knock-out alleles either of frizzled3 or of frizzled4, neither of which appears to affect development in the heterozygous state (Wang et al., 2001,2002). A–C, Coronal sections of E18 embryos exposed to a maternal injection of 200 μg of 4HT on days 7 (A), 11 (B), or 13 (C) of gestation. In A, interhemispheric fibers from the anterior commissure (bottom) and corpus callosum (top) can be seen in the right hemisphere. A large cluster of labeled muscle fibers is apparent on the right side of the head.D–F, Clusters of AP+ cortical cells from E18 embryos exposed to a maternal injection of 200 μg of 4HT on days 10 (D), 11 (E), or 13 (F) of gestation. Compare this with Figure 4, which shows an E18 embryo that was exposed to a maternal injection of 400 μg of 4HT on day 12 of gestation. G, H, Coronal sections of E14 embryos exposed to a maternal injection of 200 μg of 4HT on day 8 (G) or 9 (H). I–L, Horizontal section of an adult brain exposed to a maternal injection of 200 μg of 4HT on day 13 of gestation; higher magnification view of cortex (J), olfactory bulb (K), and cerebellum (L). L, Large numbers of closely apposed parallel fibers from AP+ cerebellar granule cells in one of the regions that is heavily labeled. Compare this with Figure 3, which shows the adult brain from a mouse that was injected with 800 μg of 4HT on P21.
Assuming that Cre-mediated recombination is a stochastic event that occurs independently in each cell, the absence of labeled cells in one hemisphere in Figure 7A indicates that the labeled cells in the other hemisphere arose from Cre-mediated recombination in one progenitor cell, or at most, several progenitor cells. Figure7A also shows AP+ interhemispheric fibers from the anterior commissure and corpus callosum, indicating effective labeling of long-range axons. As seen in Figures 7A–H, the average number of cells within an individual AP+ cluster decreases with progressively later 4HT injection times. This can be seen by comparing whole-brain coronal sections at E18 after 4HT injection at E7 (Fig.7A), E11 (Fig. 7B), and E13 (Fig. 7C); the cerebral cortex at E18 after 4HT injection at E10 (Fig.7D), E11 (Fig. 7E), and E13 (Fig.7F); or whole-brain coronal sections at E14 after 4HT injection at E8 (Fig. 7G) and E9 (Fig.7H). Within the cortex, the locations of cells within a cluster conform to the classic pattern of radial migration of cortical precursors (Fig. 7B,D) (Rakic, 1972).
A comparison among littermate embryos reveals nearly identical overall patterns and densities of AP+ cells after exposure to 200 μg of 4HT after day 10 of gestation. These data indicate that delivery of 4HT via maternal injection results in uniform or nearly uniform drug exposure among littermate embryos. At earlier times, the patterns of AP+ cells shows greater animal-to-animal variability, presumably attributable to statistical fluctuations that reflect the small number of labeled precursors generated by 200 μg of 4HT exposure in the early embryo.
A comparison of labeled cell types in the adult brain as a function of 4HT injection time can be seen by comparing Figures 3 and7I–L. As noted above, 4HT injection at postnatal day (P) 21 (Fig. 3) results in extensive labeling of both glia and neurons with essentially no clustering of AP+ cells, as expected for a predominantly postmitotic population. By contrast, 4HT injection at E13, near the height of cortical and striatal neurogenesis (Bayer and Altman, 1995), labels considerably more neurons than glia throughout the forebrain and midbrain, and these labeled cells are evenly dispersed (Fig. 7J). In the cerebellum, 4HT exposure at E13 gives rise to large clusters of labeled cells, most of which are granule cells as judged by their distinctive pattern of axonal arborization parallel to the plane of the cerebellar surface (Fig. 7L). The absence of labeling in some folia and the heavy labeling in others (Fig. 7I) argue that the labeled cells in each cluster likely arose from Cre-mediated recombination in one progenitor cell, or at most, several progenitor cells. The labeling pattern after 4HT exposure at E13 is consistent with the proliferation of cerebellar precursors in late embryonic and early postnatal life (Bayer and Altman, 1995).
Analyses similar to those described above have been conducted in the adult retina after 4HT injection at early postnatal ages. A single injection of 80 μg of 4HT at E10, P4, P6, P7, P9, P10, P11, P12, P13, P14, P15, or P16 shows a progressive shift of labeled cell types from ganglion cells, amacrine cells, and cone photoreceptors with earlier injection times, to rods, bipolar cells, and Mueller glia with later injection times (data not shown), a pattern that parallels the known birth dates for these different cell types (Polley et al., 1989). These observations, together with the analyses described above in the adult brain, suggest that by a judicious choice of injection times, it should be possible to selectively enrich for labeling of particular cell types. At present there is no evidence that the efficiency of Cre-mediated recombination differs in mitotic versus postmitotic cells. In this respect, the CreER/ZAP marking method differs from retroviral tagging, in which case actively dividing precursors are selectively labeled both by virtue of the mechanism of retroviral integration and by their selective exposure to virus particles after injection of virus into the ventricular or subretinal space.
Discussion
In this paper we describe a combined genetic–pharmacologic method for obtaining durable Golgi-like staining of both neuronal and non-neuronal cells throughout development and adulthood. Analogous methods exist for Drosophila and have been used to great effect in studying neural development and function (Marin et al., 2002). We note that the use of a ubiquitously expressed CreER as a general tool for widespread and roughly synchronous genetic modification of somatic cells in the mouse has been described independently by Guo et al. (2002), Hayashi and McMahon (2002), andZirlinger et al. (2002). In the present study, we focus on developing the CreER technology for high-resolution morphologic analysis. This system should prove especially useful for identifying cells in CNS lineage tracing experiments and for phenotypic characterization of genetic or environmental perturbations. In particular, the extremely low frequency of AP+ cells observed in the absence of 4HT should facilitate lineage analysis in animals with small numbers of marking events, because clusters of labeled cells can be confidently assumed to derive from a single marked progenitor.
A principal experimental virtue of the CreER;ZAP marking method is that AP labeling can be targeted to specific cell types by selective expression of the CreER reporter. For example, selective visualization of cholinergic or dopaminergic neurons could be effected by expression of CreER under the control of the choline acetyltransferase or tyrosine hydroxylase promoters, respectively. Similarly, selective visualization of neurons but not glia could be effected by expression of CreER under the control of a neuron-specific promoter. In the experiments presented here we have chosen to demonstrate the method by labeling the widest possible variety of cell types with R26CreER.
One simple extension of this method would be to use cell labels other than AP, for example GFP or its derivatives (Novak et al., 2000). In a more significant extension, one could derive mouse lines analogous to ZAP in which the AP (or GFP) reporter is followed by an internal ribosome entry site–cDNA that codes for a protein capable of altering the development or function of the expressing cell. By generating a sparse set of AP+ or GFP+ neurons that express the particular protein of interest at a defined time during development, one could reveal, in a single experiment, the cell-autonomous morphologic effect of its expression in diverse developmental contexts throughout the CNS.
As a histochemical marker, AP has a number of favorable attributes for large scale morphologic analyses. The NBT/BCIP reaction is simple and rapid, effectively stains relatively thick tissue sections, and produces a precipitate that remains unaltered over many months. The high signal-to-noise ratio and efficient labeling of small and distant processes permits the use of thick tissue sections, which greatly facilitates tracing of fiber tracts and three-dimensional reconstruction of large and complex dendritic trees. The use of a photostable histochemical reaction allows data sets to be collected by conventional microscopy, circumventing problems related to photobleaching of fluorescent labels that arise with confocal microscopy. The AP histochemistry is compatible with electron microscopic visualization (Gustincich et al., 1997) and with immunostaining for cell type-specific markers visualized with horseradish peroxidase. Finally, AP+ cells can also be visualized by conventional anti-AP immunostaining (data not shown). A limitation of AP histochemistry is that its spatial resolution does not equal that of intracellular filling methods (GFP, Lucifer yellow, or Golgi staining) or of plasma membrane labeling by diI, most likely because of extracellular deposition of the AP reaction product.
As presently conceived, the CreER;ZAP labeling method has several limitations. First, the time course of CreER action after a single exposure to 4HT is likely to extend over 1–2 d, limiting the temporal resolution of the marking event (Brocard et al., 1997; Guo et al., 2002; Hayashi and McMahon, 2002), although, as noted above, the nonlinear 4HT dose–response curve should sharpen the time window of Cre activity. Second, CreER-mediated recombination may be more effective in some cell types than in others, thereby biasing the population of labeled cells. Third, as with viral or particle delivery of genetic reporters, there is an obligatory several day delay between the time at which gene activation begins and the accumulation and transport of sufficient AP to visualize distal processes. Fourth, the current genetic system uses two independently segregating loci. For marking cells in the context of other genetic alterations, it would be convenient to have a single locus carrying both the CreER and ZAP cassettes. Fifth, in targeting selected cell types, the method is limited by the range of promoter specificities that are currently available. In this regard, the diversity of neuronal expression patterns exhibited by Thy-1-GFP transgenic mice (Feng et al., 2000) suggests that empiric sampling of diverse promoters or combinations of promoter elements may resolve this limitation. We therefore anticipate that over the next several years the neuroscience community will have access to an increasingly diverse and specific set of well characterized CreER-expressing mouse lines for cell-selective marking and modification.
Footnotes
This work was supported by the Howard Hughes Medical Institute. We thank Corrinne Lobe and Andras Nagy for ZAP mice; Phil Soriano for Rosa26 plasmids; Elaine Fuchs for the CreER construct; Mitra Cowan and Chip Hawkins for blastocyst injections; Haiqing Zhao and Randy Reed for advice; and David Ginty, Alex Kolodkin, Amir Rattner, Randy Reed, and David Valle for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Jeremy Nathans, 805 Preclinical Teaching Building, 725 North Wolfe Street, Johns Hopkins University School of Medicine, Baltimore, MD 21205. E-mail:jnathans@jhmi.edu.
References
- 1.Bayer SA, Altman J. Neurogenesis and neuronal migration. In: Paxinos G, editor. The rat nervous system. Academic; San Diego: 1995. pp. 1041–1078. [Google Scholar]
- 2.Brocard J, Warot X, Wendling O, Messaddeq N, Vonesch JL, Chambon P, Metzger D. Spatio-temporally controlled site-specific somatic mutagenesis in the mouse. Proc Natl Acad Sci USA. 1997;94:14559–14563. doi: 10.1073/pnas.94.26.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burns AJ, Le Douarin NM. Enteric nervous system development: analysis of the selective developmental potentialities of vagal and sacral neural crest cells using quail-chick chimeras. Anat Rec. 2001;262:16–28. doi: 10.1002/1097-0185(20010101)262:1<16::AID-AR1007>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 4.Cepko CL, Fields-Berry S, Ryder E, Austin C, Golden J. Lineage analysis using retroviral vectors. Curr Top Dev Biol. 1998;36:51–74. doi: 10.1016/s0070-2153(08)60495-0. [DOI] [PubMed] [Google Scholar]
- 5.Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 6.Eloy-Trinquet S, Nicolas JF. Clonal separation and regionalisation during formation of the medial and lateral myotomes in the mouse embryo. Development. 2002;129:111–122. doi: 10.1242/dev.129.1.111. [DOI] [PubMed] [Google Scholar]
- 7.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci USA. 1996;93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 9.Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 10.Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- 11.Guo C, Yang W, Lobe CG. A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis. 2002;32:8–18. doi: 10.1002/gene.10021. [DOI] [PubMed] [Google Scholar]
- 12.Guo F, Gopaul DN, van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 13.Gustincich S, Feigenspan A, Wu DK, Koopman LJ, Raviola E. Control of dopamine release in the retina: a transgenic approach to neural networks. Neuron. 1997;18:723–736. doi: 10.1016/s0896-6273(00)80313-x. [DOI] [PubMed] [Google Scholar]
- 14.Haas K, Sin WC, Javaherian A, Li Z, Cline HT. Single-cell electroporation for gene transfer in vivo. Neuron. 2001;29:583–591. doi: 10.1016/s0896-6273(01)00235-5. [DOI] [PubMed] [Google Scholar]
- 15.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 16.Kuan CY, Elliott EA, Flavell RA, Rakic P. Restrictive clonal allocation in the chimeric mouse brain. Proc Natl Acad Sci USA. 1997;94:3374–3379. doi: 10.1073/pnas.94.7.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lane MC, Sheets MD. Primitive and definitive blood share a common origin in Xenopus: a comparison of lineage techniques used to construct fate maps. Dev Biol. 2002;248:52–67. doi: 10.1006/dbio.2002.0717. [DOI] [PubMed] [Google Scholar]
- 18.Lo DC, McAllister AK, Katz LC. Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron. 1994;13:1263–1268. doi: 10.1016/0896-6273(94)90412-x. [DOI] [PubMed] [Google Scholar]
- 19.Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A. Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- 20.Marin EC, Jefferis GS, Komiyama T, Zhu H, Luo L. Representation of the glomerular olfactory map in the Drosophila brain. Cell. 2002;109:243–255. doi: 10.1016/s0092-8674(02)00700-6. [DOI] [PubMed] [Google Scholar]
- 21.Masland RH. Neuronal diversity in the retina. Curr Opin Neurobiol. 2001;11:431–436. doi: 10.1016/s0959-4388(00)00230-0. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy M, Turnbull DH, Walsh CA, Fishell G. Telencephalic neural progenitors appear to be restricted to regional and glial fates before the onset of neurogenesis. J Neurosci. 2001;21:6772–6781. doi: 10.1523/JNEUROSCI.21-17-06772.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicolas JF, Mathis L, Bonnerot C, Saurin W. Evidence in the mouse for self-renewing stem cells in the formation of a segmented longitudinal structure, the myotome. Development. 1996;122:2933–2946. doi: 10.1242/dev.122.9.2933. [DOI] [PubMed] [Google Scholar]
- 24.Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- 25.Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- 26.Polley EH, Zimmerman RP, Fortney RL. Neurogenesis and maturation of cell morphology in the development of the mammalian retina. In: Finlay BL, Sengelaub DR, editors. Development of the vertebrate retina. Plenum; New York: 1989. pp. 3–29. [Google Scholar]
- 27.Rakic P. Mode of cell migration to the superficial layers of the fetal monkey neocortex. J Comp Neurol. 1972;145:61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 28.Reese BE, Harvey AR, Tan SS. Radial and tangential dispersion patterns in the mouse retina are cell-class specific. Proc Natl Acad Sci USA. 1995;28:2494–2498. doi: 10.1073/pnas.92.7.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid CB, Walsh CA. Evidence of common progenitors and patterns of dispersion in rat striatum and cerebral cortex. J Neurosci. 2002;22:4002–4014. doi: 10.1523/JNEUROSCI.22-10-04002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodieck RW, Watanabe M. Survey of the morphology of macaque retinal ganglion cells that project to the pretectum, superior colliculus, and parvicellular laminae of the lateral geniculate nucleus. J Comp Neurol. 1993;338:289–303. doi: 10.1002/cne.903380211. [DOI] [PubMed] [Google Scholar]
- 31.Slack RS, Miller FD. Viral vectors for modulating gene expression in neurons. Curr Opin Neurobiol. 1996;6:576–583. doi: 10.1016/s0959-4388(96)80088-2. [DOI] [PubMed] [Google Scholar]
- 32.Tan SS, Faulkner-Jones B, Breen SJ, Walsh M, Bertram JF, Reese BE. Cell dispersion patterns in different cortical regions studied with an X-inactivated transgenic marker. Development. 1995;121:1029–1039. doi: 10.1242/dev.121.4.1029. [DOI] [PubMed] [Google Scholar]
- 33.Tauchi M, Masland RH. Local order among the dendrites of an amacrine cell population. J Neurosci. 1985;5:2494–2501. doi: 10.1523/JNEUROSCI.05-09-02494.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner DL, Snyder EY, Cepko CL. Lineage-independent determination of cell type in the embryonic mouse retina. Neuron. 1990;4:833–845. doi: 10.1016/0896-6273(90)90136-4. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Huso D, Cahill H, Ryugo D, Nathans J. Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J Neurosci. 2001;21:4761–4771. doi: 10.1523/JNEUROSCI.21-13-04761.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Thekdi N, Smallwood PM, Macke JP, Nathans J. Frizzled-3 is required for the development of major fiber tracts in the rostral CNS. J Neurosci. 2002;22:8563–8573. doi: 10.1523/JNEUROSCI.22-19-08563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- 38.Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci USA. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zirlinger M, Lo L, McMahon J, McMahon AP, Anderson DJ. Transient expression of the bHLH factor neurogenin-2 marks a subpopulation of neural crest cells biased for a sensory but not a neuronal fate. Proc Natl Acad Sci USA. 2002;99:8084–8089. doi: 10.1073/pnas.122231199. [DOI] [PMC free article] [PubMed] [Google Scholar]