

Abstract
Proneuropeptides are packaged into dense-core vesicles in which they are processed into active peptides by copackaged enzymes. Proprotein convertases (PCs) cleave precursors after dibasic residues, and carboxypeptidases remove basic residues from the C terminals. We show here that the Caenorhabditis elegans egl-21 gene encodes a protein that is very similar to carboxypeptidase E (CPE) and is broadly expressed in the nervous system. Mutants lacking either egl-21 CPE oregl-3, which encodes the C. elegansortholog of PC type 2 (PC2), were defective for processing endogenously expressed FMRFamide (Phe-Met-Arg-Phe-NH2)-related peptides (FaRPs). Mutants lacking the unc-104 kinesin motor protein were defective for anterograde movement of dense-core vesicle components, including egl-3 PC2, egl-21 CPE, and FaRPs. We provide evidence that egl-3 PC2 andegl-21 CPE mutants have diminished acetylcholine release at neuromuscular junctions (NMJs). Taken together, these results suggest that egl-21 CPE and egl-3 PC2 process endogenous neuropeptides that facilitate acetylcholine release at C. elegans NMJs.
Keywords: carboxypeptidase E, CPE, proprotein convertase, PC2, egl-21, egl-3, neuromuscular junction, neuropeptide, dense-core vesicle, DCV, synapse, C. elegans
Introduction
Neuropeptides represent an extensive and diverse set of neuronal and endocrine chemical transmitters. Although neuropeptides and classical neurotransmitters are secreted by a similar calcium-dependent mechanism, the mechanisms by which neuropeptides are synthesized and packaged into vesicles are quite distinct. Classical transmitters are packaged in small, clear, synaptic vesicles that are clustered near release sites, whereas large dense-core vesicles filled with neuropeptides are seen throughout the presynaptic compartment. Neuropeptides are initially synthesized as large preproteins that are packaged into dense-core vesicle precursors in the trans-Golgi network. Once packaged, proneuropeptides are subsequently processed into active forms by copackaged enzymes. Two critical processing steps are proteolytic cleavage after dibasic residues by proprotein convertases (PCs) and removal of the dibasic residues from the C terminals of the cleaved peptides by carboxypeptidases. The Caenorhabditis elegans unc-104 kinesin motor protein, and its mouse ortholog (KIF1A), are required for anterograde transport of small synaptic vesicle precursors (Hall and Hedgecock, 1991; Yonekawa et al., 1998), whereas the anterograde motor for dense-core vesicle precursors has not been identified.
Secretion of neuromodulatory peptides has often been proposed as a mechanism for regulating synaptic efficacy and producing adaptive changes in behavior; however, genetic studies of neuropeptide function have focused primarily on endocrine functions of these peptides. Thefat/fat mutant mouse lacks carboxypeptidase type E (CPE) activity, develops late onset obesity, is sterile, and accumulates C-terminally extended neuroendocrine peptides (Naggert et al., 1995;Fricker et al., 1996; Rovere et al., 1996; Cain et al., 1997; Lacourse et al., 1997, 1998; Udupi et al., 1997; Friis-Hansen et al., 2001). Loss of PC type 2 (PC2) function in mice produces a similar accumulation of proinsulin, proglucagon, and prosomatostatin (Furuta et al., 1997; Westphal et al., 1999). Deletions of theDrosophila PC2 (amontillado) or neuropeptide amidating enzyme (PHM) result in embryonic lethality, which are caused by defects in hatching behavior foramontillado and molting defects for PHM (Siekhaus and Fuller, 1999; Jiang et al., 2000). Mutations in theDrosophila silver gene, orthologous to carboxypeptidase D, cause cuticular defects (Settle et al., 1995). In Drosophilaand C. elegans, FMRF (Phe-Met-Arg-Phe)-related peptides (FaRPs) have been implicated in regulating several behaviors (Hewes et al., 1998; Nelson et al., 1998). In Drosophila, thepdf neuropeptide is required for producing behavioral circadian rhythms (Renn et al., 1999), whereas amnesiac has been implicated in learning and alcohol intoxication (Feany and Quinn, 1995; Moore et al., 1998). Given the diversity of neuropeptides, much remains to be learned about how these peptides regulate behavioral circuits.
To further study the role of neuropeptides in modulating synaptic transmission and behavior, we have analyzed mutations in two C. elegans neuropeptide processing enzymes. We showed previously that the egl-3 gene encodes the C. elegans ortholog of PC2 and that egl-3 PC2 regulates mechanosensory behaviors (Kass et al., 2001). Here we show that the egl-21 gene encodes a protein that is very similar to CPE, and we describe the effects of egl-21 mutations on processing of endogenous neuropeptides and on locomotion behavior.
Materials and Methods
Strains
Strain maintenance and genetic manipulation were performed as described (Brenner, 1974). Animals were cultivated at 20°C, unless noted otherwise. The animals described as wild type were C. elegans, variety Bristol, strain N2. The following strains were used in this work: egl-21(n476), egl-21(n576), egl-3(nr2090), egl-3(nu349), unc-104(e1265), and nuIs93 [a transgenic strain expressing green fluorescent protein (GFP)-tagged synaptobrevin (SNB) in ventral cord motor neurons].
Positional cloning of egl-21
Mapping data. The alleles n476 andn576 were isolated in a genetic screen for egg laying-defective mutants (Trent et al., 1983). We mappedegl-21 to a small region on the right arm of chromosome IV. A cosmid clone (F01D4) from this region corrected the defecation defect of egl-21 mutants in transgenic animals (data not shown).
Sequencing of egl-21 alleles. Sequence changes in mutant alleles were determined by amplifying exons and exon–intron boundaries from mutant strains and direct sequencing of the amplified products by cycle sequencing. Mutations found in each allele are indicated in Figure 1.
Fig. 1.

Cloning of egl-21 CPE. A, Genomic structure of egl-21 CPE, including 5′ and 3′ regions used in the genomic rescuing construct. The numbered black boxes show exon positions confirmed by sequenced cDNA from RT-PCR. Sequence changes in egl-21 alleles are indicated by an arrow for the point mutation and abar under the deletion. n576 is a splice donor mutation from G to A at nucleotide 2467 in intron 3. Then476 deletion results in a frame shift mutation in codon 121, leading to a predicted protein that is truncated at residue 132. The site of the GFP fusion is shown. B, Alignment of the amino acid sequences of EGL-21 (as predicted from cDNA sequence) with human (GenBank accession no. AAH33866.1) and mouse CPE (accession no. AAH10197) orthologs. Shaded regionsindicate identity; boxed regions show similarity. The 14 amino acid proregion for mouse and human CPE isbracketed and labeled, as well as the C-terminal membrane association (memb assoc) domain. Ser202 and Glu300 are conserved residues known to be necessary for catalytic activity.
RT-PCR of cDNA. The EGL-21 CPE exons and introns depicted in Figure 1 were determined by sequencing of cDNA generated by RT-PCR with RNA isolated from wild-type animals.
Transgenes and germline transformation
Plasmids were constructed by standard techniques, and sequences were verified when appropriate; full details are available on request. Transgenic animals were constructed by coinjecting each transgene withttx-3:: gfp (at concentrations of 50–100 μg/ml) as a marker (O. Hobert, Columbia Presbyterian, New York, NY). For each array, at least three transgenic lines were obtained, and data from a representative line are shown. Plasmids and transgenic strains were constructed as follows.
egl-21 constructs
Three plasmid subclones were shown to rescue all behavioral defects caused by egl-21(n476): KP#701, KP#702, and KP#867. KP#701 is an egl-21 genomic construct that contains nucleotides 24612–31161 of the cosmid F01D4 and spans theegl-21 coding region, 1.3 kb of promoter region and 2.2 kb of the 3′-untranslated region. KP#702 is the egl-21 genomic construct with GFP inserted in-frame between codons 30 and 31 of KP#701. Different promoters were used to drive expression of GFP-tagged EGL-21 in the following classes of neurons: all neurons (snb-1 promoter, KP#867); in type A and B ventral cord motor neurons (acr-2 promoter, KP#680); in A and C ventral cord motor neurons (unc-4, KP#681); and in ventral cord interneurons (glr-1 promoter, KP#703). Together the A, B, and C class motor neurons account for 45 of the 56 cholinergic motor neurons in the ventral cord.
egl-3 constructs
KP#871 is a 10.5 kb genomic construct containing 4.2 kb of 5′ and 2.3 kb of 3′-untranslated region, spanning nucleotides 10871–21418 of cosmid C26B6. Vectors driving the expression of the egl-3PC2 genomic construct are as follows: KP#677 contains theacr-2 promoter; KP#509 contains the glr-1promoter (Kass et al., 2001); KP#678 contains the unc-4promoter. KP#454 contains a rescuing GFP-tagged egl-3construct, in which GFP was fused in-frame at the C terminus of the 10.5 kb egl-3 genomic construct (Kass et al., 2001).
snb-1 synaptobrevin constructs
KP#704 encodes a GFP-tagged SNB-1, in which GFP was inserted at the N terminus (J. Dittman and J. Kaplan, unpublished observations), expressed by the acr-2 promoter. The nuIs93strain carries an integrated version of the KP#704 transgene.
Analysis of behaviors and drug sensitivities
Acute sensitivities to aldicarb (1 mm; Chem Services) and levamisole (400 μm; Sigma, St. Louis, MO) were determined as described previously (Nurrish et al., 1999). In brief, we assayed the time course of paralysis after exposure of a population of animals to these drugs. In each experiment, 20–25 worms per genotype were placed on drug plates, and paralysis was assessed by prodding animals with a platinum wire every 10 min over a 2 hr period. Worms that did not respond were classified as paralyzed. In all cases, assays were performed by an experimenter unaware of the genotypes of the animals. Each experiment was repeated at least three times.
Antibodies, immunostaining, and GFP reporters
Anti-FMRFamide related peptide (FaRP) antibodies were provided by Chris Li (Boston University, Boston, MA). For FaRP immunofluorescence, animals were fixed and stained as described (Li and Chalfie, 1990). Anti-GFP antibodies were prepared as described (Burbea et al., 2002). Anti-EGL-3 PC2 antibodies were prepared as described (Kass et al., 2001). Whole-mount immunofluorescence of fixed worms was done using Bouin's fixative, as described (Nonet et al., 1997).
Microscopy
GFP-expressing animals were mounted on agarose pads and viewed on a Zeiss Axiovert microscope, using a ZeissPlanapo 63× (numerical aperture 1.4) objective, as in Burbea et al. (2002). Antibody-stained animals were placed directly on slides. Images were captured with a Hamamatsu ORCA digital camera. Digital images were processed to remove out of focus light and to give maximum intensity projections of a z series, using Metamorph 4.5 image processing software (Universal Imaging).
Results
The egl-21 gene encodes a CPE
Mutations in egl-21 were isolated previously in a screen for egg laying-defective mutants (Trent et al., 1983). Animals carrying egl-21 mutations were also defective for defecation and had uncoordinated locomotion. The spectrum of behavioral defects observed in egl-21 mutants was similar to that found inegl-3 PC2 mutants (Trent et al., 1983; Kass et al., 2001). Therefore, we scanned the genome sequence in the egl-21region for genes that play a role in neuropeptide processing or secretion. We found a gene (F01D4.4) that is predicted to encode a protein that is very similar to vertebrate CPE. We did several experiments to determine whether F01D4.4 and egl-21correspond to the same gene. Transgenes containing the cosmid F01D4, or a 6.5 kb subclone spanning F01D4.4 (with or without a GFP tag), were able to rescue several of the phenotypic defects observed inegl-21 mutants (as detailed below; see Figs.4D, 5A) (Table1). Next, we showed that bothegl-21 alleles, n576 and n476,corresponded to mutations in F01D4.4 (Fig.1A). Then576 allele altered a splice donor consensus in intron 3. The n476 allele corresponded to a 123 base pair deletion (comprising nucleotides 28029–28151 in cosmid F01D4) that shifts the reading frame and is predicted to encode a truncated mutant protein lacking most of the catalytic domain. Therefore, the n476allele is likely to produce a severe or complete loss of CPE activity. These results showed that the F01D4.4 gene corresponds to theegl-21 genetic locus. Hereafter, we refer to this gene asegl-21 CPE.
Fig. 4.
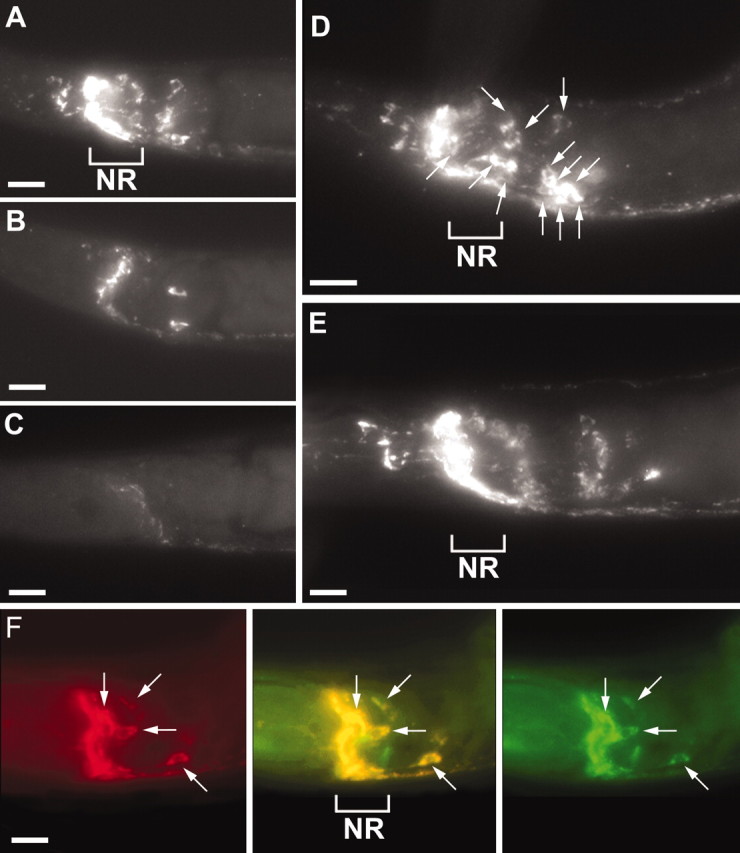
FaRPs are substrates for processing byegl-3 PC2 and egl-21 CPE.A, Anti-FaRP immunostaining in wild-type animals, with the nerve ring labeled (NR). B, Anti-FaRP immunostaining is decreased in an egl-3(nr2090) mutant (B) compared with wild type. egl-3PC2 mutants have lost FaRP staining in several head neurons (B). The same FaRP expression pattern was also seen in egl-3(nu349) (data not shown). C, FaRP staining was nearly abolished in egl-21(n476) CPE mutants. The egl-21(n576) allele showed a similar absence of FaRP staining (data not shown). D, Thesnb-1 promotedgfp:: egl-21 transgene restores wild-type FaRP expression to an egl-21(n476) mutant animal. FaRP staining is restored in a large number of neurons; several cell bodies are indicated by the small arrows. E, Theegl-3 PC2 genomic construct similarly restores wild-type FaRP expression pattern to egl-3(nr2090) mutants.F, A glr-1 promotedgfp:: egl-21 transgene restores FaRP expression to a subset of head neurons in egl-21(n476) mutants. Theleft panel shows FaRP staining, which coincides with the anti-GFP labeling in the right panel. Themiddle merged image shows that the FaRP staining colocalizes with gfp:: egl-21 staining. The nerve ring (NR) is labeled, andarrowheads indicate the double-labeled neuronal cell bodies. Scale bars, 10 μm.
Fig. 5.
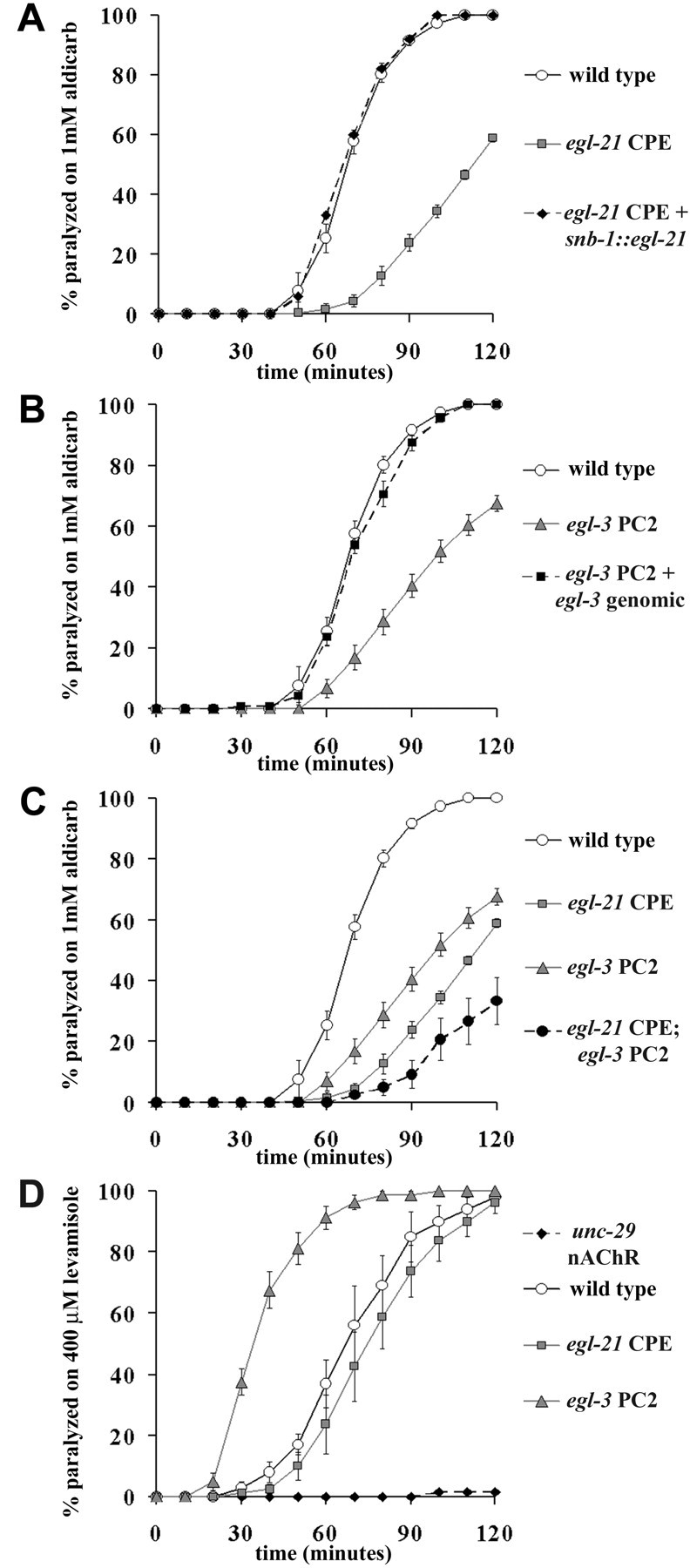
The egl-21 CPE andegl-3 PC2 mutants have decreased steady-state acetylcholine release at ventral cord NMJs. Steady-state release of acetylcholine from ventral cord NMJs was assayed by measuring the time course of paralysis induced by aldicarb, as described in Materials and Methods. A, The egl-21 CPE mutant was resistant to aldicarb (indicated by the delayed paralysis). The aldicarb resistance was rescued with a transgene driving the expression of GFP-tagged EGL-21 in all neurons (using the snb-1promoter). B, The egl-3 PC2 mutant was resistant to aldicarb, and the aldicarb resistance was rescued with theegl-3 PC2 genomic construct. C, Theegl-21 CPE;egl-3 PC2 double mutant was more aldicarb resistant than either single mutant. Aldicarb resistance could reflect a decrease in release of acetylcholine by motor neurons or a decrease in muscle responsiveness. Muscle sensitivity to acetylcholine was determined by measuring the time course of paralysis induced by levamisole, an acetylcholine receptor agonist.D, egl-3 PC2 is hypersensitive to levamisole, whereas egl-21 CPE has normal levamisole sensitivity. Control animals lacking the unc-29nicotinic acetylcholine receptor were resistant to levamisole paralysis. The levamisole sensitivity of egl-21 CPE andegl-3 PC2 mutants indicates that the aldicarb resistance phenotype (A, B) is caused by decreased acetylcholine release from motor neurons.
Table 1.
Analysis of aldicarb paralysis
| Genotype (number of trials) | Transgene | 70 min | 110 min |
|---|---|---|---|
| WT (20) | None | 54 ± 4 | 99 ± 1 |
| WT (5) | acr-2∷egl-3 | 56 ± 4 | 100 ± 0 |
| WT (3) | combo∷egl-211-a | 55 ± 6 | 100 ± 0 |
| WT (2) | glr-1∷egl-21 | 55 ± 5 | 98 ± 3 |
| WT (2) | snb-1∷egl-21 | 54 ± 4 | 100 ± 0 |
| egl-3(nr2090) (13) | None | 17 ± 4 | 55 ± 5 |
| egl-3(nr2090) | unc-4∷egl-3 | 9 ± 3 | 54 ± 3 |
| egl-3(nr2090)(6) | acr-2∷egl-3 | 13 ± 5 | 56 ± 4 |
| egl-3(nu349) (5) | None | 10 ± 3 | 59 ± 2 |
| egl-3(nu349) (4) | unc-4∷egl-3 | 10 ± 3 | 60 ± 4 |
| egl-21(n476) (9) | None | 4 ± 2 | 47 ± 1 |
| egl-21(n576) (10) | None | 12 ± 4 | 69 ± 6 |
| egl-21(n476)(3) | glr-1∷egl-21 | 2 ± 2 | 42 ± 6 |
| egl-21(n476)(2) | combo∷egl-211-a | 3 ± 3 | 45 ± 5 |
| egl-21(n476) (4) | genomicegl-21 | 49 ± 4 | 98 ± 1 |
Fractions of animals paralyzed on 1 mm aldicarb after 70 and 110 min were measured as described in Materials and Methods. WT indicates wild-type animals. Values reported are means ± SE. Allegl-21 CPE transgenes used are GFP tagged.
combo∷egl-21 is a combination ofglr-1, unc-4, and acr-2 promoted transgenes.
The predicted egl-21 protein product, based on the sequence of egl-21 cDNAs, is 41% identical to human CPE (Fig.1B). Two residues that are known to be essential for CPE activity, S202 (Naggert et al., 1995; Fricker et al., 1996) and E300 (Qian et al., 1999), are conserved in EGL-21 (Fig.1B). Mammalian CPEs are produced initially as precursors with a short pro region (14–15 amino acids) that is cleaved, exposing the N terminus of mature CPE. Unlike human and rodent CPEs, EGL-21 does not appear to have a pro region with the predicted furin cleavage sequence (RRRRR), and it lacks the C-terminal membrane association domain (Fricker et al., 1990) (Fig. 1B). Like EGL-21, other CPEs also lack the furin cleavage sequence, including anglerfish and mollusc (Aplysia californica). The function of the pro region is unclear, because it is not required for folding (Manser et al., 1990; Varlamov and Fricker, 1996), sorting (Song and Fricker, 1997), or enzymatic activity (Manser et al., 1990;Parkinson, 1990).
unc-104 KIF1A is required for anterograde trafficking of dense-core vesicle precursors
The monomeric unc-104 KIF1A motor protein is required for anterograde movement of synaptic vesicle components in bothC. elegans and mouse (Hall and Hedgecock, 1991; Yonekawa et al., 1998). To determine whether unc-104 KIF1A also mediates anterograde transport of neuropeptide-containing vesicles, we compared the distribution of several dense-core vesicle components inunc-104 KIF1A mutants and wild-type animals. The distribution of egl-21 CPE was determined by immunostaining animals expressing a rescuing GFP-tagged EGL-21 with anti-GFP antibodies (Fig.2A–C). Immunostaining in wild-type animals showed that EGL-21:: GFP is expressed widely in the nervous system, with particularly strong expression in the neuronal processes of the nerve ring (Fig. 2A). EGL-21:: GFP was not expressed in any non-neuronal tissues. The egl-3 PC2 was visualized by immunostaining with anti-EGL-3 antibodies (Fig. 2D,E). Mature, fully processed FaRPs were immunostained with anti-Arg-Phe-NH2 antibodies (Fig.3A–D). In wild-type animals,egl-21 CPE (Fig. 2A), egl-3 PC2 (Fig. 2D), and FaRP (Fig. 3A) immunostaining were most concentrated in the nerve ring and other neuronal processes, whereas neuronal cell bodies had lower levels of expression. Immunostaining of egl-21 CPE (Fig.2B,C), egl-3 PC2 (Fig.2E), and FaRP (Fig. 3B,D) increased in neuronal cell bodies of unc-104 KIF1A mutants, whereas staining in axons was proportionately diminished. In addition, a broader expression pattern for all three of these antigens was observed inunc-104 KIF1A mutants compared with wild-type controls, because retention in the cell bodies enabled identification of previously undetected neurons. For example, in wild-type animals (Fig.3A,C), FaRP staining was observed in a total of 25–30 neurons (Schinkmann and Li, 1992), whereas inunc-104 KIF1A mutants (Fig. 3B,D), FaRP staining was observed in ∼82 neurons, including 39 ± 4 neurons in the nerve ring ganglia, 33 ± 3 motor neurons in the ventral cord, 8 ± 1 neurons in the lumbar ganglion, and 2 in the pre-anal ganglion. We found similar increases in the numbers of egl-3 PC2- andegl-21 CPE-expressing neurons in unc-104 KIF1A mutants. In particular, egl-21 CPE immunostaining was found in ∼100 cells in the head and several neurons in the tail ganglia (15 ± 3) and pre-anal ganglion (7 ± 1) and motor neurons of the ventral cord (38 ± 4). We identified a subset of theegl-21 CPE-expressing cells, including the following: the mechanosensory neurons ALM, AVM, and PVM; the interneurons BDU, SDQ; and the HSN egg-laying motor neurons. For an unc-104KIF1A cargo control, we tested localization of GFP-tagged synaptobrevin (SNB-1), expressed in the motor neurons. As reported previously (Nonet, 1999), we saw that GFP-SNB-1 in wild-type animals was expressed in a punctate synaptic pattern in the ventral cord (Fig. 3E) and became concentrated in neuronal cell bodies of unc-104 KIF1A mutants (Fig. 3F). Examination of GFP-SNB-1 expression in egl-3 PC2 andegl-21 CPE mutants showed no alterations in cell numbers or axon morphologies of ventral cord motor neurons (data not shown). The unc-104 KIF1A-dependent localization ofegl-21 CPE, egl-3 PC2, and FaRPs indicates that UNC-104 is the anterograde motor for dense-core vesicle precursors. Moreover, these results also demonstrate that a large fraction ofC. elegans neurons are likely to produce neuropeptides.
Fig. 2.

Expression of egl-21 CPE andegl-3 PC2 in wild-type animals andunc-104 KIF1A mutants. Anti-GFP antibody was used to stain transgenic animals expressing a full-length rescuinggfp:: egl-21 genomic construct (A–C). A, The full-length rescuing gfp:: egl-21 translational fusion shows wide neural expression, including the cell bodies of neurons in the head and tail ganglia and axons in the ventral (VNC) and lateral nerve cords (LNC) (arrowheads). In addition, many axons in the nerve ring (NR) stained brightly.B, C, In an unc-104(e1265) KIF1A mutant, egl-21 CPE is localized to cell bodies. Reduction in unc-104 KIF1A-mediated trafficking ofegl-21 CPE out to axonal processes reveals a wider expression pattern including ∼100 head neurons (B) and the ventral cord motor neurons (C, arrows). D, Wild-type anti-egl-3 PC2 staining is primarily in nerve ring axons (NR), whereas cell bodies are weakly stained.E, In an unc-104(e1265) KIF1A mutant, anti-egl-3 PC2 staining is localized to cell bodies. Scale bars, 10 μm.
Fig. 3.

The unc-104 KIF1A motor is required for anterograde trafficking of FaRP-containing vesicles.A, Anti-FaRP immunostaining in wild-type animals was very bright in the nerve ring (NR) and ventral cord.B, Loss of unc-104 kinesin trafficking led to increased cell body staining. This revealed a wider expression pattern: more neurons in the head stained for FaRPs, including several in the retrovesicular ganglion (RVG) and lateral ganglion (LG) (RVG and LGin brackets). C, D, Staining in the ventral cord was predominant in neuronal processes for wild-type animals (C) but localized to cell bodies of ventral cord motor neurons in unc-104 mutants (D, arrows). E,F, Distribution of GFP-tagged synaptobrevin expressed in ventral cord motor neurons (using the acr-2 promoter) in wild-type (E) andunc-104(e1265) KIF1A mutant animals (F). Synaptobrevin is retained in the cell bodies of unc-104 KIF1A mutants (F), compared with wild-type controls (E). E, F,Arrowheads point to motor neuron cell bodies in the ventral cord. Scale bars, 10 μm.
FaRPs are substrates for processing by egl-3 PC2 andegl-21 CPE
The unc-104 KIF1A-dependent trafficking ofegl-21 CPE, egl-3 PC2, and FaRPs, combined with their similar expression patterns, suggested that FaRPs might be substrates of egl-21 CPEand egl-3 PC2. To determine whether egl-3 PC2 or egl-21 CPE mutants had decreased levels of processed FaRPs, we stained mutant animals with the anti-FaRP antibody. Because this antibody recognizes the C-terminal Arg-Phe-NH2 moiety, only FaRPs that had been processed previously by proprotein convertases, carboxypeptidases, and amidating enzymes should be detected (Marder et al., 1987). We found that FaRP immunostaining was decreased in many neurons in the nerve ring ganglia of egl-3 PC2 mutants (Fig.4B). By contrast, FaRP immunostaining was nearly eliminated in egl-21 CPE mutants (Fig. 4C), including ventral cord staining, compared with wild-type controls (Fig. 4A). Wild-type levels of FaRP staining were restored in both egl-3 PC2 andegl-21 CPE mutants by transgenes containing wild-type copies of these genes (Fig. 4D,E). When expression of egl-21 CPE was restored in a subset of cells (using the glr-1 promoter), FaRP staining was restored only in those cells expressing egl-21 CPE (Fig.4F). Thus, egl-3 PC2 and egl-21CPE activities were required to produce normal levels of FaRP staining, and egl-21 CPE is required in the FaRP-expressing cells. These results suggest that proFaRP precursors are processed byegl-3 PC2 and egl-21 CPE. In particular, it appears that loss of egl-21 CPE activity is likely to produce a severe decrease in the abundance of active endogenous FaRPs.
egl-21 CPE and egl-3 PC2 regulate acetylcholine release at neuromuscular junctions
A shared phenotype of egl-21 CPE and egl-3PC2 is sluggish locomotion and a tendency to adopt a coiled posture during locomotion. In C. elegans, acetylcholine is the primary excitatory transmitter at the body wall neuromuscular junction (NMJ). To determine whether the locomotion defects in egl-21CPE and egl-3 PC2 mutants are associated with a change in synaptic transmission at the NMJ, we assayed steady-state release of acetylcholine at the NMJ by measuring the sensitivity of animals to the acetylcholinesterase inhibitor aldicarb (Fig.5A–C). Aldicarb enhances the effects of endogenously released acetylcholine by preventing acetylcholine breakdown, resulting in hypercontraction of body wall muscle and eventual paralysis in wild-type animals. Resistance to aldicarb (measured as delayed aldicarb-induced paralysis) is exhibited by mutants with defects in synaptic vesicle exocytosis and recycling (Nonet et al., 1993; Nguyen et al., 1995; Miller et al., 1996), as well as in mutants that are defective for modulation of acetylcholine release, e.g., egl-30 Gαq and egl-8 PLCβ (Miller et al., 1996; Lackner et al., 1999; Miller et al., 1999). We expected that neuropeptides might fit in this second class, having a modulatory role in synaptic transmission. We found that loss of function mutations in egl-21 CPE and egl-3 PC2 produced resistance to aldicarb (Fig. 5A,B, Table 1), suggesting that these mutants had decreased basal release of acetylcholine. Moreover, egl-21;egl-3 double mutants were more resistant than either single mutant (Fig. 5C). Expression of egl-21 CPE and egl-3 PC2 transgenes restored wild-type aldicarb sensitivity to egl-21 andegl-3 mutants, respectively (Fig. 5A,B). Rescue of the aldicarb sensitivity was obtained with transgenes containing the endogenous egl-21 or egl-3 promoters or using thesub-1 promoter, which has a pan-neuronal expression pattern. These results suggest that egl-21 CPE and egl-3 PC2 act in neurons to process endogenous neuropeptides that stimulate acetylcholine release at NMJs.
An alternative explanation for the aldicarb resistance observed in these mutants is that body wall muscles are less sensitive to acetylcholine. To test this possibility, we examined the sensitivity ofegl-21 CPE and egl-3 PC2 animals to levamisole, a nicotinic acetylcholine receptor agonist that directly activates the body muscle (Fig. 5D). We determined that loss ofegl-21 CPE had no effect on sensitivity to levamisole and that loss of egl-3 PC2 actually increased sensitivity. In contrast, animals lacking the unc-29 nicotinic acetylcholine receptor were resistant to levamisole paralysis. These results indicate that neuropeptides stimulate the release of acetylcholine from the motor neurons, and loss of neuropeptide processing in egl-21CPE and egl-3 PC2 mutant animals results in a decrease in steady-state acetylcholine release.
We next considered likely sites at which neuropeptides could be processed and released to facilitate acetylcholine release. Becauseegl-21 CPE and egl-3 PC2 were both expressed broadly in the nervous system (Fig. 2B–D) (Kass et al., 2001), we engineered vectors driving expression of these enzymes using several different promoters. The motor neurons were a likely site of neuropeptide processing and release, because this would directly affect the NMJ. Alternatively, interneurons, particularly the command neurons, which drive locomotion, could be the main site of neuropeptide processing. However, expression in a large number of the motor neurons (with the acr-2 orunc-4 promoters) or in the ventral cord interneurons (with the glr-1 promoter) failed to rescue the aldicarb sensitivity of egl-21 CPE and egl-3 PC2 mutants (Table 1) (data not shown). Furthermore, transgenes containing a combination of the above promoters also failed to rescue the aldicarb sensitivity of mutant animals (Table 1). Because the unc-4and acr-2 promoters are expressed in a large fraction (45 of 56) of ventral cord motor neurons, these results suggest that expression of egl-21 CPE and egl-3 PC2 in interneurons and motor neurons is not sufficient to facilitate acetylcholine release.
Discussion
Our results lead to four primary conclusions. First, theegl-21 gene encodes a protein that is very similar to vertebrate CPE. The egl-21 CPE is expressed in 60% of the nervous system, including interneurons, motor neurons, and sensory neurons. EGL-3, the C. elegans PC2 ortholog, is also broadly expressed in the nervous system (Kass et al., 2001). Taken together, these results suggest that a large fraction of the neurons in C. elegans use peptide neurotransmitters, which is consistent with the range of behavioral defects observed in mutants lacking these enzymes. Second, unc-104 KIF1A is required for anterograde transport of dense-core vesicle components. Third, FaRPs are processed by egl-3 PC2 and egl-21 CPE. Fourth,egl-3 PC2- and egl-21 CPE-processed peptides facilitate acetylcholine release from ventral cord NMJs.
Neuropeptides constitute a large, chemically diverse set of neurotransmitters proposed to play varied roles in physiology and behavior. Several factors have limited the analysis of neuropeptide functions. The vast number of neuropeptides limits the ability to systematically examine the functions of each peptide. For example, theC. elegans genome encodes 32 neuropeptide-like (nlp) genes, 23 FaRP (flp) genes, and 40 insulin-like (ins) genes (Duret et al., 1998; Gregoire et al., 1998; Li et al., 1999b; Kawano et al., 2000; Pierce et al., 2001). This could be an underestimate, because small genes are often missed by gene-predicting algorithms. Furthermore, each proneuropeptide gene encodes multiple peptides. Mutations in neuropeptide genes are rare. Finally, neuropeptides often have redundant functions. For example, seven different FaRPs have nearly identical effects on theDrosophila larval NMJ (Hewes et al., 1998). Thus, the observed phenotypes of a mutant lacking a single neuropeptide may underestimate the true range of its physiological functions.
Our results suggest that characterizing mutants lacking particular proneuropeptide processing enzymes is an effective alternative strategy to analyze the physiological effects of neuropeptides. We have shown that mutants lacking either egl-3 PC2 (Kass et al., 2001) oregl-21 CPE have discrete behavioral defects that can be ascribed to changes in specific neural circuits. In the case ofegl-3 PC2, mutants have changes in sensitivity to mechanosensory stimuli, whereas both egl-3 PC2 andegl-21 CPE have diminished acetylcholine release at ventral cord NMJs. We estimate that candidate substrates processed byegl-3 PC2 and egl-21 CPE could include ∼200 unique neuropeptides, encoded by flp, nlp, and two ins genes (ins-1 and ins-18). Candidate substrates were identified by the presence of single and dibasic cleavage sites within predicted flp, nlp, andins genes. This may be an underestimate, because proprotein convertases have also been proposed to act at nonbasic cleavage sites (Che et al., 2001). Efforts to identify all of the neuropeptide-encoding genes in C. elegans and the expression pattern of each will provide useful information for identifying candidate neuropeptides that are responsible for the phenotypes observed in egl-21 CPE and egl-3 PC2 mutants (Li et al., 1999a,b; Nathoo et al., 2001). A similar strategy has also been used in Drosophila. Mutations in the DrosophilaPC2 (amontillado) and neuropeptide amidating enzyme (PHM) have been isolated (Siekhaus and Fuller, 1999;Jiang et al., 2000); however, in these cases, the homozygous mutants have an embryonic lethal phenotype. Recent work suggests that specific behavioral defects can be found when PHM expression is restored in a restricted subset of neurons with thegal4/UAS system (Taghert et al., 2001).
Processing by egl-3 PC2 and egl-21 CPE is required for producing biologically active neuropeptides and hormones that modulate several different neuronal circuits, indicated by the behavioral defects observed in mutants lacking these enzymes. In addition to egl-3 PC2, three other proprotein convertases are present in the C. elegans genome: kpc-1, bli-4/kpc-4, and aex-5/kpc-3 (Thacker and Rose, 2000). The bli-4/kpc-4 proprotein convertase has been shown to processes cuticular procollagens, and its expression pattern includes hypodermal tissue (Peters et al., 1991; Thacker et al., 1995). Theaex-5/kpc-3 proprotein convertase is predicted to be expressed only in muscle because it lies in the unc-54muscle myosin operon (Thacker and Rose, 2000). Thus, kpc-1is the most likely candidate to have some degree of functional overlap with egl-3 PC2, because a deletion was reported to have slightly uncoordinated locomotion, although its expression pattern has not been reported (Thacker and Rose, 2000). In addition to egl-21 CPE, two other predicted genes have significant similarity to carboxypeptidases; however, no mutants have been identified. The specific isoforms of the enzymes that process each neuropeptide are likely determined by the distinct expression patterns of the isoforms and by their substrate specificities. Moreover, it is also possible that different combinations of enzymes are used to process different neuropeptides. Some of our results are consistent with this idea. The egl-21 CPE;egl-3 PC2 double mutant had a more severe phenotype than either single mutant, suggesting that these enzymes are used in a combinatorial manner. For example, egl-3 PC2 and egl-21 CPE could be required for processing FaRPs in one set of neurons, whereas another proprotein convertase together with egl-21 CPE processes FaRPs in a distinct set of neurons. Thus, the egl-3PC2;egl-21 CPE double mutant would be predicted to have more severe defects than either single mutant.
Although egl-21 CPE may process multiple classes of proneuropeptides and other proproteins, it seems likely that lack of FaRP processing accounts for some of the behavioral defects seen inegl-3 PC2 and egl-21 CPE mutants. Our results demonstrate that egl-21 CPE mutants had significantly reduced levels of mature FaRPs. FaRPs have diverse physiological functions in both the CNS and the PNS throughout the animal kingdom (Raffa, 1988). Drosophila FaRPs enhance nerve-stimulated muscle contraction in a manner that is consistent with our findings (Hewes et al., 1998). The physiological effects of a fewC. elegans FaRPs have been tested in either C. elegans or Ascaris suum, a larger parasitic nematode (Maule et al., 1995; Marks et al., 1997, 1998, 1999, 2001; Rogers et al., 2001). To date, nematode FaRPs have been identified with inhibitory or excitatory effects on muscle contraction. In some cases, these effects are mediated by direct action on muscles, whereas in others the effects are dependent on synaptic input. Nonetheless, we expect that lack of other classes of neuropeptides also contributes to the phenotypes of egl-3 PC2 and egl-21 CPE.
Interestingly, egl-3 PC2 mutants had increased sensitivity to the acetylcholine agonist levamisole. We have seen a similar degree of levamisole hypersensitivity in some, but not all, aldicarb resistant mutants (Sieburth and Kaplan, unpublished observations). The increased responsiveness to levamisole could reflect a compensatory mechanism whereby muscle cells compensate for decreased acetylcholine secretion by increasing their responsiveness to acetylcholine. Further experiments will be required to determine the mechanisms underlying this effect.
What is the mechanism by which neuropeptides regulate the NMJ? Invertebrate studies have provided examples in which neuromodulators can exert both presynaptic and postsynaptic effects. Loss ofDrosophila calcium-activated protein for secretion (CAPS), a protein that has been proposed to promote priming of dense-core vesicles (Tandon et al., 1998), was reported to result in an accumulation of dense-core vesicles and a 50% decrease in evoked glutamate release at NMJs (Renden et al., 2001). aex-1, a novel C. elegans protein expressed in muscle, appears to regulate a retrograde signal at the NMJ to stimulate synaptic vesicle release from neurons (Doi and Iwasaki, 2002). In our case, the failure to produce mature egl-21 CPE- and egl-3PC2-processed peptides resulted in decreased acetylcholine secretion by ventral cord motor neurons, and this defect could be rescued by transgenes driving expression only in neurons. There are two mechanisms by which this could occur. We favor a straightforward model in which neuropeptides directly modulate acetylcholine release from motor neurons. However, our results do not exclude more complicated models in which neuropeptides act elsewhere to indirectly regulate acetylcholine release from motor neurons. In either case, our results show that neuropeptide processing is required in neurons. Finally, our results do not exclude the possibility that changes in acetylcholine release in these mutants are caused by failure to process non-neuropeptide substrates. Further experiments are needed to distinguish between these possibilities, including identification of relevant neuropeptides or hormones and the expression pattern of their receptors.
A search of the genome identified ∼130 genes encoding potential neuropeptide receptors (Bargmann, 1998; Nathoo et al., 2001), of which mutations have been isolated in one gene, npr-1, which encodes a receptor related to neuropeptide Y receptors (de Bono and Bargmann, 1998). On the other hand, knock-out mutations have been isolated in all 18 genes encoding heterotrimeric GTP-binding protein α-subunits (Mendel et al., 1995; Segalat et al., 1995; Brundage et al., 1996; Korswagen et al., 1997; Roayaie et al., 1998; Jansen et al., 1999), in two adenylyl cyclase genes (Berger et al., 1998; Korswagen et al., 1998; Moorman and Plasterk, 2002), and in one phospholipase Cβ gene (Lackner et al., 1999; Miller et al., 1999). We anticipate that further studies in C. elegans will be a productive strategy to define the behavioral impact of neuropeptides and to identify the downstream second messengers mediating these effects.
Footnotes
This work was supported by a research grant (NS32196) from the National Institutes of Health to J.K. T.C.J. is a Howard Hughes predoctoral fellow. We thank the following for advice, strains, and reagents: A. Fire, The C. elegans Genetic Stock Center, A. Coulson, J. Dittman, and L. Dreier. We also thank members of the Kaplan lab for comments on this manuscript.
Correspondence should be addressed to Dr. Joshua M. Kaplan, Department of Molecular Biology, Wellman 8, Massachusetts General Hospital, 50 Blossom Street, Boston, MA 02114. E-mail:kaplan@molbio.mgh.harvard.edu.
References
- 1.Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- 2.Berger A, Hart A, Kaplan J. G-α-s induced neurodegeneration in C. elegans. J Neurosci. 1998;18:2871–2880. doi: 10.1523/JNEUROSCI.18-08-02871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brundage L, Avery L, Katz A, Kim U-J, Mendel J, Sternberg P, Simon M. Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron. 1996;16:999–1009. doi: 10.1016/s0896-6273(00)80123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35:107–120. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- 6.Cain BM, Wang W, Beinfeld MC. Cholecystokinin (CCK) levels are greatly reduced in the brains but not the duodenums of Cpe(fat)/Cpe(fat) mice: a regional difference in the involvement of carboxypeptidase E (Cpe) in pro-CCK processing. Endocrinology. 1997;138:4034–4037. doi: 10.1210/endo.138.9.5490. [DOI] [PubMed] [Google Scholar]
- 7.Che FY, Yan L, Li H, Mzhavia N, Devi LA, Fricker LD. Identification of peptides from brain and pituitary of Cpe(fat)/Cpe(fat) mice. Proc Natl Acad Sci USA. 2001;98:9971–9976. doi: 10.1073/pnas.161542198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Bono M, Bargmann CI. Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell. 1998;94:679–689. doi: 10.1016/s0092-8674(00)81609-8. [DOI] [PubMed] [Google Scholar]
- 9.Doi M, Iwasaki K. Regulation of retrograde signaling at neuromuscular junctions by the novel C2 domain protein AEX-1. Neuron. 2002;33:249–259. doi: 10.1016/s0896-6273(01)00587-6. [DOI] [PubMed] [Google Scholar]
- 10.Duret L, Guex N, Peitsch MC, Bairoch A. New insulin-like proteins with atypical disulfide bond pattern characterized in Caenorhabditis elegans by comparative sequence analysis and homology modeling. Genome Res. 1998;8:348–353. doi: 10.1101/gr.8.4.348. [DOI] [PubMed] [Google Scholar]
- 11.Feany MB, Quinn WG. A neuropeptide gene defined by the Drosophila memory mutant amnesiac. Science. 1995;268:869–873. doi: 10.1126/science.7754370. [DOI] [PubMed] [Google Scholar]
- 12.Fricker LD, Das B, Angeletti RH. Identification of the pH-dependent membrane anchor of carboxypeptidase E (EC 3.4.17.10). J Biol Chem. 1990;265:2476–2482. [PubMed] [Google Scholar]
- 13.Fricker LD, Berman YL, Leiter EH, Devi LA. Carboxypeptidase E activity is deficient in mice with the fat mutation. Effect on peptide processing. J Biol Chem. 1996;271:30619–30624. doi: 10.1074/jbc.271.48.30619. [DOI] [PubMed] [Google Scholar]
- 14.Friis-Hansen L, Lacourse KA, Samuelson LC, Holst JJ. Attenuated processing of proglucagon and glucagon-like peptide-1 in carboxypeptidase E-deficient mice. J Endocrinol. 2001;169:595–602. doi: 10.1677/joe.0.1690595. [DOI] [PubMed] [Google Scholar]
- 15.Furuta M, Yano H, Zhou A, Rouille Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci USA. 1997;94:6646–6651. doi: 10.1073/pnas.94.13.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gregoire FM, Chomiki N, Kachinskas D, Warden CH. Cloning and developmental regulation of a novel member of the insulin-like gene family in Caenorhabditis elegans. Biochem Biophys Res Commun. 1998;249:385–390. doi: 10.1006/bbrc.1998.9164. [DOI] [PubMed] [Google Scholar]
- 17.Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- 18.Hewes RS, Snowdeal EC, Saitoe M, 3rd, Taghert PH. Functional redundancy of FMRFamide-related peptides at the Drosophila larval neuromuscular junction. J Neurosci. 1998;18:7138–7151. doi: 10.1523/JNEUROSCI.18-18-07138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jansen G, Thijssen KL, Werner P, van der Horst M, Hazendonk E, Plasterk RH. The complete family of genes encoding G proteins of Caenorhabditis elegans. Nat Genet. 1999;21:414–419. doi: 10.1038/7753. [DOI] [PubMed] [Google Scholar]
- 20.Jiang N, Kolhekar AS, Jacobs PS, Mains RE, Eipper BA, Taghert PH. PHM is required for normal developmental transitions and for biosynthesis of secretory peptides in Drosophila. Dev Biol. 2000;226:118–136. doi: 10.1006/dbio.2000.9832. [DOI] [PubMed] [Google Scholar]
- 21.Kass J, Jacob TC, Kim P, Kaplan JM. The EGL-3 proprotein convertase regulates mechanosensory responses of Caenorhabditis elegans. J Neurosci. 2001;21:9265–9272. doi: 10.1523/JNEUROSCI.21-23-09265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawano T, Ito Y, Ishiguro M, Takuwa K, Nakajima T, Kimura Y. Molecular cloning and characterization of a new Insulin/IGF-like peptide of the nematode Caenorhabditis elegans. Biochem Biophys Res Commun. 2000;273:431–436. doi: 10.1006/bbrc.2000.2971. [DOI] [PubMed] [Google Scholar]
- 23.Korswagen H, Park J-H, Ohshima Y, Plasterk R. An activating mutation in a C. elegans Gs protein induces neural degeneration. Genes Dev. 1997;11:1493–1503. doi: 10.1101/gad.11.12.1493. [DOI] [PubMed] [Google Scholar]
- 24.Korswagen HC, van der Linden AM, Plasterk RH. G protein hyperactivation of the Caenorhabditis elegans adenylyl cyclase SGS-1 induces neuronal degeneration. EMBO J. 1998;17:5059–5065. doi: 10.1093/emboj/17.17.5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- 26.Lacourse KA, Friis-Hansen L, Rehfeld JF, Samuelson LC. Disturbed progastrin processing in carboxypeptidase E-deficient fat mice. FEBS Lett. 1997;416:45–50. doi: 10.1016/s0014-5793(97)01164-2. [DOI] [PubMed] [Google Scholar]
- 27.Lacourse KA, Friis-Hansen L, Samuelson LC, Rehfeld JF. Altered processing of procholecystokinin in carboxypeptidase E-deficient fat mice: differential synthesis in neurons and endocrine cells. FEBS Lett. 1998;436:61–66. doi: 10.1016/s0014-5793(98)01099-0. [DOI] [PubMed] [Google Scholar]
- 28.Li C, Chalfie M. Organogenesis in C. elegans: positioning of neurons and muscles in the egg-laying system. Neuron. 1990;4:681–695. doi: 10.1016/0896-6273(90)90195-l. [DOI] [PubMed] [Google Scholar]
- 29.Li C, Kim K, Nelson LS. FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. Brain Res. 1999a;848:26–34. doi: 10.1016/s0006-8993(99)01972-1. [DOI] [PubMed] [Google Scholar]
- 30.Li C, Nelson LS, Kim K, Nathoo A, Hart AC. Neuropeptide gene families in the nematode Caenorhabditis elegans. Ann NY Acad Sci. 1999b;897:239–252. doi: 10.1111/j.1749-6632.1999.tb07895.x. [DOI] [PubMed] [Google Scholar]
- 31.Manser E, Fernandez D, Loo L, Goh PY, Monfries C, Hall C, Lim L. Human carboxypeptidase E. Isolation and characterization of the cDNA, sequence conservation, expression and processing in vitro. Biochem J. 1990;267:517–525. doi: 10.1042/bj2670517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marder E, Calabrese RL, Nusbaum MP, Trimmer B. Distribution and partial characterization of FMRFamide-like peptides in the stomatogastric nervous systems of the rock crab, Cancer borealis, and the spiny lobster, Panulirus interruptus. J Comp Neurol. 1987;259:150–163. doi: 10.1002/cne.902590111. [DOI] [PubMed] [Google Scholar]
- 33.Marks NJ, Maule AG, Geary TG, Thompson DP, Davis JP, Halton DW, Verhaert P, Shaw C. APEASPFIRFamide, a novel FMRFamide-related decapeptide from Caenorhabditis elegans: structure and myoactivity. Biochem Biophys Res Commun. 1997;231:591–595. doi: 10.1006/bbrc.1997.6155. [DOI] [PubMed] [Google Scholar]
- 34.Marks NJ, Maule AG, Geary TG, Thompson DP, Li C, Halton DW, Shaw C. KSAYMRFamide (PF3/AF8) is present in the free-living nematode, Caenorhabditis elegans. Biochem Biophys Res Commun. 1998;248:422–425. doi: 10.1006/bbrc.1998.8982. [DOI] [PubMed] [Google Scholar]
- 35.Marks NJ, Maule AG, Li C, Nelson LS, Thompson DP, Alexander-Bowman S, Geary TG, Halton DW, Verhaert P, Shaw C. Isolation, pharmacology and gene organization of KPSFVRFamide: a neuropeptide from Caenorhabditis elegans. Biochem Biophys Res Commun. 1999;254:222–230. doi: 10.1006/bbrc.1998.9920. [DOI] [PubMed] [Google Scholar]
- 36.Marks NJ, Shaw C, Halton DW, Thompson DP, Geary TG, Li C, Maule AG. Isolation and preliminary biological assessment of AADGAPLIRFamide and SVPGVLRFamide from Caenorhabditis elegans. Biochem Biophys Res Commun. 2001;286:1170–1176. doi: 10.1006/bbrc.2001.5524. [DOI] [PubMed] [Google Scholar]
- 37.Maule AG, Geary TG, Bowman JW, Marks NJ, Blair KL, Halton DW, Shaw C, Thompson DP. Inhibitory effects of nematode FMRFamide-related peptides (FaRPs) on muscle strips from Ascaris suum. Invert Neurosci. 1995;1:255–265. doi: 10.1007/BF02211027. [DOI] [PubMed] [Google Scholar]
- 38.Mendel J, Korswagen H, Liu K, Hadju-Cronin Y, Simon M, Plasterk R, Sternberg P. Participation of the protein Go in multiple aspects of behavior in C. elegans. Science. 1995;267:1652–1655. doi: 10.1126/science.7886455. [DOI] [PubMed] [Google Scholar]
- 39.Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci USA. 1996;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller KG, Emerson MD, Rand JB. Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron. 1999;24:323–333. doi: 10.1016/s0896-6273(00)80847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore MS, DeZazzo J, Luk AY, Tully T, Singh CM, Heberlein U. Ethanol intoxication in Drosophila: genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell. 1998;93:997–1007. doi: 10.1016/s0092-8674(00)81205-2. [DOI] [PubMed] [Google Scholar]
- 42.Moorman C, Plasterk RH. Functional Characterization of the adenylyl cyclase gene sgs-1 by analysis of a mutational spectrum in Caenorhabditis elegans. Genetics. 2002;161:133–142. doi: 10.1093/genetics/161.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naggert JK, Fricker LD, Varlamov O, Nishina PM, Rouille Y, Steiner DF, Carroll RJ, Paigen BJ, Leiter EH. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet. 1995;10:135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- 44.Nathoo AN, Moeller RA, Westlund BA, Hart AC. Identification of neuropeptide-like protein gene families in Caenorhabditis elegans and other species. Proc Natl Acad Sci USA. 2001;98:14000–14005. doi: 10.1073/pnas.241231298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson LS, Rosoff ML, Li C. Disruption of a neuropeptide gene, flp-1, causes multiple behavioral defects in Caenorhabditis elegans. Science. 1998;281:1686–1690. doi: 10.1126/science.281.5383.1686. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen M, Alfonso A, Johnson CD, Rand JB. Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics. 1995;140:527–535. doi: 10.1093/genetics/140.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nonet ML. Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J Neurosci Methods. 1999;89:33–40. doi: 10.1016/s0165-0270(99)00031-x. [DOI] [PubMed] [Google Scholar]
- 48.Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- 49.Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nurrish S, Segalat L, Kaplan JM. Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron. 1999;24:231–242. doi: 10.1016/s0896-6273(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 51.Parkinson D. Two soluble forms of bovine carboxypeptidase H have different NH2-terminal sequences. J Biol Chem. 1990;265:17101–17105. [PubMed] [Google Scholar]
- 52.Peters K, McDowall J, Rose AM. Mutations in the bli-4 (I) locus of Caenorhabditis elegans disrupt both adult cuticle and early larval development. Genetics. 1991;129:95–102. doi: 10.1093/genetics/129.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pierce SB, Costa M, Wisotzkey R, Devadhar S, Homburger SA, Buchman AR, Ferguson KC, Heller J, Platt DM, Pasquinelli AA, Liu LX, Doberstein SK, Ruvkun G. Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 2001;15:672–686. doi: 10.1101/gad.867301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian Y, Varlamov O, Fricker LD. Glu300 of rat carboxypeptidase E is essential for enzymatic activity but not substrate binding or routing to the regulated secretory pathway. J Biol Chem. 1999;274:11582–11586. doi: 10.1074/jbc.274.17.11582. [DOI] [PubMed] [Google Scholar]
- 55.Raffa RB. The action of FMRFamide (Phe-Met-Arg-Phe-NH2) and related peptides on mammals. Peptides. 1988;9:915–922. doi: 10.1016/0196-9781(88)90141-6. [DOI] [PubMed] [Google Scholar]
- 56.Renden R, Berwin B, Davis W, Ann K, Chin CT, Kreber R, Ganetzky B, Martin TF, Broadie K. Drosophila CAPS is an essential gene that regulates dense-core vesicle release and synaptic vesicle fusion. Neuron. 2001;31:421–437. doi: 10.1016/s0896-6273(01)00382-8. [DOI] [PubMed] [Google Scholar]
- 57.Renn SC, Park JH, Rosbash M, Hall JC, Taghert PH. A pdf neuropeptide gene mutation and ablation of PDF neurons each cause severe abnormalities of behavioral circadian rhythms in Drosophila. Cell. 1999;99:791–802. doi: 10.1016/s0092-8674(00)81676-1. [DOI] [PubMed] [Google Scholar]
- 58.Roayaie K, Crump JG, Sagasti A, Bargmann CI. The G alpha protein ODR-3 mediates olfactory and nociceptive function and controls cilium morphogenesis in C. elegans olfactory neurons. Neuron. 1998;20:55–67. doi: 10.1016/s0896-6273(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 59.Rogers CM, Franks CJ, Walker RJ, Burke JF, Holden-Dye L. Regulation of the pharynx of Caenorhabditis elegans by 5-HT, octopamine, and FMRFamide-like neuropeptides. J Neurobiol. 2001;49:235–244. doi: 10.1002/neu.1078. [DOI] [PubMed] [Google Scholar]
- 60.Rovere C, Viale A, Nahon J, Kitabgi P. Impaired processing of brain proneurotensin and promelanin-concentrating hormone in obese fat/fat mice. Endocrinology. 1996;137:2954–2958. doi: 10.1210/endo.137.7.8770919. [DOI] [PubMed] [Google Scholar]
- 61.Schinkmann K, Li C. Localization of FMRFamide-like peptides in Caenorhabditis elegans. J Comp Neurol. 1992;316:251–260. doi: 10.1002/cne.903160209. [DOI] [PubMed] [Google Scholar]
- 62.Segalat L, Elkes D, Kaplan J. Go modulation of serotonin-controlled behaviors in C. elegans. Science. 1995;267:1648–1651. doi: 10.1126/science.7886454. [DOI] [PubMed] [Google Scholar]
- 63.Settle SH, Jr, Green MM, Burtis KC. The silver gene of Drosophila melanogaster encodes multiple carboxypeptidases similar to mammalian prohormone-processing enzymes. Proc Natl Acad Sci USA. 1995;92:9470–9474. doi: 10.1073/pnas.92.21.9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siekhaus DE, Fuller RS. A role for amontillado, the Drosophila homolog of the neuropeptide precursor processing protease PC2, in triggering hatching behavior. J Neurosci. 1999;19:6942–6954. doi: 10.1523/JNEUROSCI.19-16-06942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song L, Fricker LD. The pro region is not required for the expression or intracellular routing of carboxypeptidase E. Biochem J. 1997;323:265–271. doi: 10.1042/bj3230265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taghert PH, Hewes RS, Park JH, O'Brien MA, Han M, Peck ME. Multiple amidated neuropeptides are required for normal circadian locomotor rhythms in Drosophila. J Neurosci. 2001;21:6673–6686. doi: 10.1523/JNEUROSCI.21-17-06673.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tandon A, Bannykh S, Kowalchyk JA, Banerjee A, Martin TF, Balch WE. Differential regulation of exocytosis by calcium and CAPS in semi-intact synaptosomes. Neuron. 1998;21:147–154. doi: 10.1016/s0896-6273(00)80522-x. [DOI] [PubMed] [Google Scholar]
- 68.Thacker C, Rose AM. A look at the Caenorhabditis elegans Kex2/Subtilisin-like proprotein convertase family. BioEssays. 2000;22:545–553. doi: 10.1002/(SICI)1521-1878(200006)22:6<545::AID-BIES7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 69.Thacker C, Peters K, Srayko M, Rose AM. The bli-4 locus of Caenorhabditis elegans encodes structurally distinct kex2/subtilisin-like endoproteases essential for early development and adult morphology. Genes Dev. 1995;9:956–971. doi: 10.1101/gad.9.8.956. [DOI] [PubMed] [Google Scholar]
- 70.Trent C, Tsuing N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Udupi V, Gomez P, Song L, Varlamov O, Reed JT, Leiter EH, Fricker LD, Greeley GH. Effect of carboxypeptidase E deficiency on progastrin processing and gastrin messenger ribonucleic acid expression in mice with the fat mutation. Endocrinology. 1997;138:1959–1963. doi: 10.1210/endo.138.5.5113. [DOI] [PubMed] [Google Scholar]
- 72.Varlamov O, Fricker LD. The C-terminal region of carboxypeptidase E involved in membrane binding is distinct from the region involved with intracellular routing. J Biol Chem. 1996;271:6077–6083. doi: 10.1074/jbc.271.11.6077. [DOI] [PubMed] [Google Scholar]
- 73.Westphal CH, Muller L, Zhou A, Zhu X, Bonner-Weir S, Schambelan M, Steiner DF, Lindberg I, Leder P. The neuroendocrine protein 7B2 is required for peptide hormone processing in vivo and provides a novel mechanism for pituitary Cushing's disease. Cell. 1999;96:689–700. doi: 10.1016/s0092-8674(00)80579-6. [DOI] [PubMed] [Google Scholar]
- 74.Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol. 1998;141:431–441. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]