

Abstract
Endocannabinoids form a novel class of intercellular messengers, the functions of which include retrograde signaling in the brain and mediation or modulation of several types of synaptic plasticity. Yet, the signaling mechanisms and long-term effects of the stimulation of CB1 cannabinoid receptors (CB1-R) are poorly understood. We show that anandamide, 2-arachidonoyl-glycerol, and Δ9-tetrahydrocannabinol (THC) activated extracellular signal-regulated kinase (ERK) in hippocampal slices. In living mice, THC activated ERK in hippocampal neurons and induced its accumulation in the nuclei of pyramidal cells in CA1 and CA3. Both effects were attributable to stimulation of CB1-R and activation of MAP kinase/ERK kinase (MEK). In hippocampal slices, the stimulation of ERK was independent of phosphatidyl-inositol-3-kinase but was regulated by cAMP. The endocannabinoid-induced stimulation of ERK was lost in Fyn knock-out mice, in slices and in vivo, although it was insensitive to inhibitors of Src-family tyrosine kinases in vitro, suggesting a noncatalytic role of Fyn. Finally, the effects of cannabinoids on ERK activation were dependent on the activity of glutamate NMDA receptors in vivo, but not in hippocampal slices, indicating the existence of several pathways linking CB1-R to the ERK cascade. In vivo THC induced the expression of immediate-early genes products (c-Fos protein, Zif268, and BDNF mRNAs), and this induction was prevented by an inhibitor of MEK. The strong potential of cannabinoids for inducing long-term alterations in hippocampal neurons through the activation of the ERK pathway may be important for the physiological control of synaptic plasticity and for the general effects of THC in the context of drug abuse.
Keywords: hippocampus, cannabinoids, 2-AG, anandamide, CB1-R, THC, LPA, ERK, phosphorylation, Fyn, immediate-early genes, c-Fos, Zif268, BDNF, slices, rat, mouse
Introduction
The endocannabinoid system, which has recently emerged as a major player in the control of synaptic plasticity, is the pharmacological target of cannabis, the most widely used illicit drug of abuse. Cannabinoid receptors of the CB1 subtype (CB1-Rs) are highly expressed in the brain (Matsuda et al., 1990). They are the targets for endogenous ligands (endocannabinoids), including anandamide and 2-arachidonoyl glycerol (2-AG) (for review, see Di Marzo et al., 1998). In hippocampus, CB1-Rs are abundant and enriched in nerve terminals, especially those of a subpopulation of inhibitory interneurons (Tsou et al., 1998, 1999; Katona et al., 1999,2000). The production of 2-AG is increased after stimulation of Schaeffer collaterals (Stella et al., 1997). Massive activation of CB1-Rs decreases long-term potentiation (LTP) and depression (LTD) (Misner and Sullivan, 1999) and impairs short-term memory tasks associated with the firing of hippocampal neurons (Hampson and Deadwyler, 2000). Endocannabinoids play a major role in the modulation of synaptic transmission: they are released after depolarization of postsynaptic neurons and act backward on presynaptic CB1-Rs to suppress inhibitory neurotransmitter release (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001), a phenomenon called depolarization-induced suppression of inhibition. Thus, in physiological conditions the endocannabinoid system facilitates the induction of LTP (Carlson et al., 2002).
Despite the recent progress in understanding the actions of endocannabinoids on synaptic transmission, the signal transduction pathways regulated by Gi/o-coupled CB1-Rs in hippocampus are poorly characterized. Most of the data were obtained in non-neuronal cell lines, in which stimulation of CB1-Rs activates the extracellular-regulated kinase (ERK) subtype of mitogen-activated protein (MAP) kinases (Bouaboula et al., 1995a;Wartmann et al., 1995), leading to the expression of the immediate-early gene (IEG) Zif268 (also known asegr-1, NGFI-A, or Krox-24) (Bouaboula et al., 1995a). CB1-Rs are also coupled to the activation of protein kinase B/Akt (PKB) through the phosphatidylinositol 3 (PI3)-kinase pathway (Gomez del Pulgar et al., 2000, 2002). Little is known about the signaling pathways regulated by cannabinoids in the adult nervous system. Intraperitoneal injection of cannabinoid agonists increases the expression of IEGs, products including Zif268, c-Fos, and c-Jun, in rat forebrain (Mailleux et al., 1994; Glass and Dragunow, 1995), but the mechanism of these responses is not known. We have shown previously that cannabinoids augment tyrosine phosphorylation of the neuronal splice isoform of focal adhesion kinase (FAK) (Derkinderen et al., 1996, 2001b; Burgaya et al., 1997) and its association with the Src family tyrosine kinase Fyn (Derkinderen et al., 2001b). Cannabinoids also activate p38-MAPK, but not c-Jun N-terminal kinase, in hippocampal slices (Derkinderen et al., 2001a).
The aim of the present study was to determine whether cannabinoids could regulate ERK in hippocampus, to examine the signaling pathways involved, and to determine the role of this pathway in IEG expression. We report that stimulation of CB1-Rs activates the ERK cascade both in hippocampal slices and in vivo where it controls the expression of IEGs.
Materials and Methods
Reagents. Anandamide, cremophor El, Δ9-tetrahydrocannabinol (THC), diethyl pyrocarbonate, lysophosphatidic acid (LPA), protein A-Sepharose, and tetrodotoxin were purchased from Sigma (Saint Quentin Fallavier, France). 2-Arachidonoyl glycerol and WIN55212–2 were from Research Biochemicals (Saint Quentin Fallavier, France). CP55940 was from Pfizer. 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) was from Calbiochem (Meudon, France). LY290004 was from Biomol (Le Perray en Yvelines, France). PD98059 was from New England Biolabs (Ozyme, Orsay France). SR141716A was from Sanofi (Montpellier, France). U0126 and SL327 were kindly provided by Dr. James Trzaskos (DuPont Merck). Artificial CSF (ACSF) contained (in mm): 125 NaCl, 2.4 KCl, 0.83 MgCl2, 1.1 CaCl2, 0.5 KH2PO4, Na2SO4, 27 NaHCO3, 10 glucose, 11 HEPES, pH 7.4. Ca2+-free ACSF had the same composition except for MgCl2, which was 1.93 mm. The following commercially available phosphospecific antibodies were used for Western blotting: monoclonal anti-MAP kinase, activated (clone MAPK-YT, Sigma; 1:10,000), anti-diphospho-ERK (Promega; 1:5000), or anti-diphospho-ERK (New England Biolabs; diluted 1:1000), anti-phospho-Ser218-Ser222 MEK1/2 (New England Biolabs; diluted 1:1000). Mouse monoclonal antibodies (Zymed; 1:1000) or rabbit affinity-purified IgG (Upstate Biotechnology, Lake Placid, NY; 1 μg/10 ml) was used to detect the total amounts of ERK 1 and 2 proteins in homogenates. Monoclonal antibodies to phosphotyrosine (4G10) were purchased from Upstate Biotechnology (diluted 1:4000). For immunocytochemistry, polyclonal anti-phospho-Thr/Tyr-ERK antibodies (New England Biolabs; diluted 1:200) and polyclonal anti-c-Fos antibodies (Santa Cruz Biotechnology, Santa Cruz, CA; diluted 1:500) were used.
Rat and mice hippocampal slices. Rat hippocampal slices (300 μm thick) were prepared from young male Sprague Dawley rats (150–200 gm) with a McIlwain tissue chopper, as described previously (Siciliano et al., 1994). Briefly, slices were dissected in ice-cold, Ca2+-free ACSF and placed for 10 min in polypropylene tubes (three slices per tube) containing 1 ml of Ca2+-free ACSF at 35°C, equilibrated at pH 7.4 in O2/CO2 (95:5, v/v). They were then incubated at 35°C in 900 μl ACSF containing 1.1 mm Ca2+ and 1 μm TTX, for 45 min before pharmacological treatment. Treatment of control slices was performed by the addition of vehicle. TTX was added to prevent indirect effects attributable to neuronal firing and had no effects on tyrosine phosphorylation by itself (data not shown). At the end of the experiment, ACSF was aspirated, and slices were immediately frozen on dry ice and kept at −80°C. Hippocampal slices from wild-type, CB1-R knock-out (Ledent et al., 1999), and Fyn knock-out mice (Grant et al., 1992) were prepared as rat slices except for the use of a Vibratome instead of a McIllwain chopper.
Western blot analysis. Slices were lysed by sonication in 1% SDS (v/v) containing 1 mm sodium orthovanadate at 100°C. Equal amounts of lysate slices (60 μg) were separated by SDS-PAGE (8 or 10%) before electrophoretic transfer onto nitrocellulose membrane (Hybond Pure, Amersham, Orsay, France). Membranes were blocked for 1 hr at room temperature in Tris-buffered saline (TBS) (100 mm NaCl, 10 mm Tris, pH 7.5) with 0.1% Tween 20 or 5% nonfat dry milk for the detection of phosphorylated and nonphosphorylated proteins, respectively. Membranes were incubated overnight at 4°C with the primary antibodies. Bound antibodies were detected with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies (Amersham; diluted 1:4000) and visualized by enhanced chemiluminescent detection (ECL, Amersham). When necessary, membranes were stripped in buffer [containing 100 mm glycine, pH 2.5, 200 mm NaCl, 0.1% Tween 20 (v/v) and 0.1% (v/v) β-mercaptoethanol] for 45 min at room temperature, followed by extensive washing in TBS before reblocking and reprobing. The relevant immunoreactive bands were quantified with laser-scanning densitometry using Scion Image software. To allow comparison between different autoradiographic films, the density of the bands was expressed as a percentage of the average of control (untreated) slices. The value of active diphospho-ERK2 (P-ERK2) was normalized to the amount of total ERK2 in the same sample and expressed as a percentage of controls. Statistical analysis was done by ANOVA followed byt test using Prism 3.02 software.
In vivo experiments. Male CD-1 mice (Charles River) weighing 20–22 gm were used. They were housed in cages in groups of five in a temperature-controlled room (21 ± 1°C) 1 week before the experiments were started. The mice were given access to food and water ad libitum and were maintained on a 12 hr light/dark cycle. Animal care was conducted in accordance with standard ethical guidelines (Guide for the Care and Use of Laboratory Animals, National Academy Press, 1996) and approved by the local ethics committee. THC was purchased as a 10 mg/ml solution in ethanol, which was diluted 1:100 in distilled water containing 5% ethanol and 5% cremophor El. A volume of 0.1 ml per 10 gm of body weight of this mixture containing 0.1 mg/ml THC was injected intraperitoneally. The CB1-R antagonist SR141716A (3 mg/kg) was dissolved in a solution of 10% ethanol, 10% cremophor El, and 80% distilled water, and injected intraperitoneally in a final volume of 0.2 ml per 10 gm of body weight 15 min before THC injection. SL327 (100 mg/kg in DMSO) or DMSO was injected intraperitoneally 1 hr before THC injection. At specified times after receiving THC (1 mg/kg, i.p.) brains were fixed by intracardiac perfusion of 4% paraformaldehyde (PFA) in 0.1mNa2HPO4/NaH2PO4buffer, pH 7.5, as described previously (Valjent et al., 2000). Brains were removed and postfixed in the same fixative solution overnight, sectioned (30 μm) on a Vibratome (Leitz), and then kept in a solution containing 30% ethylene glycol, 30% glycerol, 0.1m phosphate buffer, 0.1% diethyl pyrocarbonate at −20°C until processed for in situ hybridization or immunohistochemistry.
Immunohistochemistry. Detection of active ERKs or c-Fos proteins was performed as described previously (Atkins et al., 1998;Valjent et al., 2000). Briefly, free-floating sections were rinsed in TBS (0.25 M Tris, 0.5 M NaCl, pH 7.5), incubated for 5 min in TBS containing 3% H2O2 and 10% methanol, and rinsed three times 10 min each in TBS (0.1 mm NaF was included in all buffers and incubation solutions). After 15 min incubation in 0.2% Triton X-100 in TBS, sections were rinsed three times in TBS and incubated overnight with the primary antibody (see below) at 4°C. After three rinses in TBS, sections were incubated for 2 hr at room temperature with the secondary biotinylated antibody (anti-IgG) using a dilution twice that of the first antibody in TBS. After three rinses in TBS, sections were incubated overnight in avidin–biotin–peroxidase complex (ABC) solution (Vector Laboratories; final dilution 1:50). Sections were then washed two times in TBS and two times in TB (Tris 0.25 m, pH 7,5), 10 min each, placed in a solution of TB containing 0.1% 3–3′ diaminobenzidine (50 mg/100 ml), and developed by adding H2O2 (0.02%). After processing, tissue sections were mounted onto gelatin-coated slides and dehydrated through alcohol to xylene for light microscopic examination.
In situ hybridization. The antisense (complementary to cellular mRNA) probes were35S-radiolabeled riboprobes. For Zif268 and BDNF riboprobes, murine cDNA subclones were used. Zif268 insert corresponding to 1.6 kbp was linearized after HindIII digestion and transcribed with T7 RNA polymerase. The BDNF (327 bp) riboprobe was transcribed with T7 RNA polymerase after linearization with SmaI. Transcription reactions contained 1 μm [α35S]-UTP (1000 Ci/mmol; Isotopchim), 250 μmATP, CTP, GTP, and unlabeled UTP (10.5 μm) and were incubated at 39°C for 2 hr. After DNase I digestion, the labeled RNA was purified by phenol/chloroform/isoamyl alcohol (25:24:1) extraction and ethanol precipitation. Gel electrophoresis showed the transcripts to be predominantly full length. Free-floating sections were mounted on SuperFrost/plus slides (Menzel-Gläser) in RNase-free conditions. Once dried, mounted sections were rinsed in PBS and treated for 10 min with 0.1 m glycine in 0.1m Tris-HCl, pH 7.4. Sections were rinsed for 5 min at 37°C in 0.1 m Tris-HCl, pH 8, 50 mm EDTA, and treated for 15 min at 37°C with 1 mg/ml proteinase K in the same buffer. Before hybridization, sections were subjected to the following treatment: postfixation for 15 min in 4% PFA, 5 mm MgCl2 in PBS at room temperature, acetylation for 20 min in acetic anhydride/triethanolamine, pH 8, at room temperature, and stepwise dehydration in alcohol. The following hybridization solution was applied to sections, which were then covered with GelBond Film (FMC Bioproducts). The hybridization mixture contained 200 ng/ml (4 ng per section) of 35S-RNA probe in 20 mm Tris-HCl, pH 8, 300 mm NaCl, 5 mm EDTA, 10% dextran sulfate, 1× Denhardt's solution (0.02% Ficoll, 0.02% polyvinyl pyrolidone, 10 mg/ml BSA), 0.5 mg/ml Escherichia coli tRNA, 0.1 m dithiothreitol (DTT), and 50% formamide. Hybridization was performed at 60°C in humid chambers for 16 hr. After the GelBond coverslips were removed in 4× SSC (1× SSC is 0.15 m NaCl/0.015 mNa citrate), 10 mm DTT, the slides were washed in the same solution for 1 hr at room temperature and then in 50% formamide, 10 mm Tris-HCl, pH 8, 75 mm NaCl, 2.5 mm EDTA. Sections were treated with RNase A (20 μg/ml; Sigma) in 400 mm NaCl, 10 mmTris-HCl, pH 7.5, 50 mm EDTA for 1 hr at 37°C, and then rinsed for 15 min at 60°C in 2× SSC followed by 0.1× SSC. After dehydration, sections were air dried and exposed with Biomax MR films (Kodak) for 3 d.
Results
Cannabinoids activate ERK in the hippocampus
ERKs are present in brain and are particularly abundant in the hippocampus (Fiore et al., 1993; Flood et al., 1998). Anandamide (1 μm), 2-AG (1 μm), synthetic CB1-R agonists (1 μm CP 55940 and 100 μm WIN 55212–2), and THC increased the active form of ERK1 and ERK2 in hippocampal slices (Fig.1A,B). These effects were completely prevented by preincubation of slices with 100 μm SR141716A, a CB1-R antagonist (Fig.1A,B). As a control, we used lysophosphatidic acid, a lipidic intercellular messenger unrelated to cannabinoids and known to activate ERK in several cell lines (Kumagai et al., 1993). In hippocampal slices LPA (0.2 μm) increased ERK phosphorylation, and these effects were not blocked in the presence of SR141716A (Fig.1A,B). It should be noted that although ERK1 and ERK2 were found at similar levels in hippocampal slices, the signal was always much stronger for P-ERK2 than for P-ERK1, which was below the detection threshold in some experiments. However, when detected, the effects on ERK1 were qualitatively comparable with those on ERK2. Because 2-AG is the endocannabinoid produced in hippocampal slices after electrical stimulation (Stella et al., 1997), we studied the time course and time dependence of the effects of 2-AG on ERK phosphorylation. The activation of ERK2 phosphorylation by 2-AG was rapid (half-maximal effect at ∼1 min) (Fig. 1C) and concentration dependent (half-maximal effect at ∼20 nm) (Fig. 1D).
Fig. 1.

Cannabinoid agonists stimulate ERK phosphorylation in rat hippocampal slices. A, Rat hippocampal slices were incubated at 35°C, as described in Materials and Methods, for 50 min before the addition of vehicle (Control), 1 μm anandamide, 1 μm 2-AG, 1 μm CP 55940, 100 μm WIN 55212–2, 0.2 μm LPA, or 0.1 μm Δ9-THC for 5 min, in the absence or in the presence of 100 μm SR 141716A applied 30 min before. Slices were homogenized in SDS; 60 μg of protein per sample were subjected to immunoblot analysis using antibodies specific for the dually phosphorylated (active) forms of ERK1 and ERK2 (Blot P-ERK). After stripping, the membranes were reprobed with anti-ERK (Blot ERK) antibodies. B, For quantification the optical densities of P-ERK2-immunoreactive bands were measured, normalized to the optical densities of total ERK2 in the same samples, and expressed as percentages of controls. Data correspond to means ± SEM. Statistical analysis was done with ANOVA (F(13,24) = 22.8; p< 0.0001) followed by t test (treated vs control: ***p < 0.001, **p < 0.01; treated in the presence of SR141716A vs in its absence: °° p < 0.01, ° p < 0.05). C, D, Quantification of the effects of 2-AG on ERK2 active form: time course (drug concentration 1 μm) (C); concentration–response curve (treatment for 5 min) (D). Immunoreactivity was quantified by scanning densitometry using NIH image 1.62 software. Values are means ± SEM of four to eight independent experiments and are expressed as percentages of the maximal increase above unstimulated control values.
ERK is not activated by endocannabinoids in CB1 knock-out mice
Endocannabinoids have a potential for acting on other targets besides CB1-R, including CB2-R and possibly other receptors (Di Marzo et al., 2000; Al-Hayani et al., 2001). The effects of 2-AG and anandamide on ERK phosphorylation were mimicked by synthetic CB1-R agonists CP 55940 and WIN55212–2 and blocked by the specific CB1-R antagonist SR141716A (Fig. 1A,B); however, the required concentration of WIN55212–2 and SR141716A for complete effects was very high (100 μm). This could indicate a poor penetration of WIN55212–2 and SR141716A in slices, or the fact that the receptors involved in ERK activation were atypical. To assess the exact contribution of CB1-R to the effects of anandamide and 2-AG, we used hippocampal slices prepared from genetically altered mice lacking CB1-R (Ledent et al., 1999). In wild-type mice, anandamide and 2-AG increased ERK phosphorylation as in rats (Fig.2A,B). In contrast, endocannabinoids had no effect on ERK activation in CB1-R knock-out mice, although the expression levels of ERK1 and ERK2 were unchanged (Fig. 2A,B). The ERK pathway was normally functional in the hippocampus of CB1-R mutant mice because LPA was capable of activating ERKs to a similar level in knock-out and wild-type mice (Fig.2A,B). These results, combined with the pharmacological experiments, demonstrate that the effects of endocannabinoids on ERK in hippocampus are mediated through activation of the CB1-R.
Fig. 2.

The regulation of ERKs by endocannabinoids is absent in hippocampal slices from CB1-R knock-out mice.A, Hippocampal slices from wild-type (CB1-R +/+) and CB1-R knock-out (CB1-R −/−) mice were incubated at 35°C, as described in Materials and Methods, for 50 min before the addition of vehicle (Control), 1 μm anandamide, 1 μm 2-AG, or 0.2 μm LPA for 5 min. ERK phosphorylation was assayed as described in the legend to Figure 1. B, Quantification of the results for P-ERK2 as described in the legend to Figure 1. Data correspond to means ± SEM. Statistical analysis was done with ANOVA (anandamide: F(3,20) = 40.2,p < 0.0001; 2-AG and LPA:F(5,24) = 8.2, p < 0.0001) followed by t test (treated vs control: ***p < 0.001, **p < 0.01, *p < 0.05; treated in knock-out vs wild type: ° p < 0.05).
Endocannabinoids activate MEK and ERK independently of PI3-kinase
Activation of ERKs results from the phosphorylation of their activation loop, on a threonine and a tyrosine, by dual-specificity MEKs, MEK1 and MEK2. On the other hand, it has been shown that in some systems, activation of ERKs results from the inhibition of their dephosphorylation (Haneda et al., 1999). To determine the mechanism of action of endocannabinoids, we examined the phosphorylation state of MEK1/2 using a phosphospecific antibody reacting only with the active phosphorylated form of these enzymes (Fig. 3A). Anandamide and 2-AG increased the phosphorylation of MEK1/2. Moreover, the effects of endocannabinoids on ERK phosphorylation were completely inhibited by U0126 and partially by PD98059 (Fig.3B,C), two MEK inhibitors (Alessi et al., 1995; Favata et al., 1998). In contrast, both of these compounds fully antagonized the activation of ERKs by LPA (Fig.3B,C). The lower efficacy of PD98059 as compared with U0126 to inhibit ERK activation by endocannabinoids might indicate a preferential involvement of MEK2 in the activation of ERKs by cannabinoids, because PD98059 is 10-fold more active on MEK1 than on MEK2, whereas U0126 has the same potency on both kinases (Alessi et al., 1995; Favata et al., 1998).
Fig. 3.

Role of MEK in the effects of endocannabinoids in rat hippocampal slices. A, Rat hippocampal slices were incubated as described in Materials and Methods, for 50 min before the addition of vehicle (Control), 1 μmanandamide, or 1 μm 2-AG for 5 min. Homogenates (60 μg of protein per sample) were analyzed by immunoblotting with antibodies specific for the phosphorylated forms of MEK1/2. Quantification of P-MEK immunoreactivity (mean ± SEM): controls 100 ± 8, anandamide 857 ± 29, 2-AG 514 ± 57 (F(2,5) = 226, p < 0.001; t test treated vs control: p< 0.0001). B, Slices were treated with the same compounds as in A, or with 0.2 μm LPA, in the absence or in the presence of 50 μm PD98059 or 30 μm U0126, two MEK inhibitors. Homogenates were analyzed for active dually phosphorylated ERK by immunoblotting.C, Quantification of the results for P-ERK2 as described in the legend to Figure 1. Data correspond to mean ± SEM. Statistical analysis was done with ANOVA (F(11,45 = 7.4; p< 0.0001) followed by t test (treated vs control: ***p < 0.001; treated in the presence of MEK inhibitor vs in its absence: ° p < 0.05).
Multiple pathways lead from G-protein-coupled receptors to the activation of MEKs and ERKs (for review, see Derkinderen et al., 1999). In Chinese hamster ovary (CHO) cells transfected with CB1-R, wortmannin, a specific inhibitor of PI3-kinase, prevented the activation of ERK by cannabinoid agonists (Bouaboula et al., 1997). This observation was consistent with other reports in which activation of ERK by Gi-protein-coupled receptors required the activation of PI3-kinase (Hawes et al., 1996). Moreover, it has been shown that CB1-R is capable of activating PKB through a PI3-kinase-dependent pathway (Gomez del Pulgar et al., 2000, 2002). To test the role of PI3-kinase in the activation of ERK by endocannabinoids in hippocampal slices, we pretreated the slices with a specific inhibitor of this kinase, LY294002 (Vlahos et al., 1994). The effects on ERKs of 2-AG (Table 1) and anandamide (data not shown) were unaffected by the presence of LY294002. We verified that LY294002 was able to block insulin-induced activation of ERK as reported in cell lines (Shepherd et al., 1998) (data not shown). These results reveal that, in contrast to CB1-R-transfected CHO cells, the activation of ERKs by endocannabinoids in hippocampus does not require an active PI3-kinase.
Table 1.
Activation of ERK2 by 2-AG is independent of PI3-kinase and NMDA receptors in rat hippocampal slices
| Control | 2-AG | p | n | |
|---|---|---|---|---|
| Vehicle | 100 ± 28 | 800 ± 211 | <0.01 | 6 |
| LY294002 | 52 ± 8 | 626 ± 146 | <0.001 | 3 |
| p | NS | NS | ||
| Vehicle | 100 ± 17 | 900 ± 140 | <0.01 | 4 |
| MK801 | 200 ± 18 | 1100 ± 197 | <0.01 | 4 |
| p | <0.05 | NS |
Hippocampal slices were treated for 5 min with 1 μm2-AG, in the absence (Vehicle) or presence of 50 μmLY294002 (added 50 min before 2-AG), a PI3-kinase inhibitor, or, in a different series of experiments, in the absence (Vehicle) or presence of 20 μm MK801 (added 25 min before 2-AG). The homogenates were analyzed for the active dually phosphorylated form of ERK2 by immunoblotting and quantified as indicated in the legend to Figure 1. Statistical analysis was performed by ANOVA followed byt test.
Role of cAMP in the control of ERK activation by cannabinoids in hippocampal slices
Stimulation of CB1-R inhibits adenylyl cyclase, thus decreasing cAMP levels (Howlett, 1995). Depending on the cell type and experimental conditions, cAMP can either stimulate or inhibit the ERK pathway, through various signaling pathways (Stork and Schmitt, 2002). In various non-neuronal cells, cAMP prevents the activation of ERK by mitogens, whereas in neuronal cells such as PC12, an increase in the intracellular concentration of cAMP activates the ERK pathway. Because we had demonstrated previously that cAMP was involved in the effects of endocannabinoids on protein tyrosine phosphorylation in hippocampal slices (Derkinderen et al., 1996, 2001b), we examined its role in ERK regulation. We treated hippocampal slices with 8-Bromo (Br)-cAMP, a cell-permeant analog of cAMP, or forskolin, a stimulator of adenylyl cyclase, for different periods of time. Treatment of slices with these compounds for <30 min induced an increase in ERK phosphorylation, in agreement with previous reports (Impey et al., 1998, 1999). In contrast, when slices were treated for periods ranging from 30 to 45 min, ERK phosphorylation dramatically decreased as compared with controls (Fig.4A and unpublished results). A 45 min pretreatment with 8-Br-cAMP completely prevented the activation of ERK by endocannabinoids (Fig.4B,C), whereas LPA was still capable of activating ERKs in these conditions (data not shown). Because a major and prolonged effect of cAMP is to activate cAMP-dependent protein kinase (PKA), this result prompted us to test whether inhibition of PKA could participate in ERK activation. We used two unrelated PKA inhibitors: Rp-cAMPS, which prevents cAMP binding to the regulatory subunit of the kinase, and H-89, an inhibitor of the catalytic subunit of PKA. Remarkably, both compounds stimulated ERK activity in hippocampal slices (Fig.4B,C). Taken together, these results underline the complex effects of cAMP on ERK phosphorylation in hippocampus, revealing a time-dependent combination of stimulatory and inhibitory effects. They also suggest that a decrease in cAMP levels and, consequently, in PKA activity, may participate in the effects of CB1-R on the ERK pathway in hippocampus.
Fig. 4.

Role of cAMP in the effects of endocannabinoids on ERK phosphorylation. A, Rat hippocampal slices were incubated with forskolin (50 μm) for the indicated period of time. Homogenates were analyzed for active dually phosphorylated ERK by immunoblotting. B, Rat hippocampal slices were incubated as described in Materials and Methods, in the presence or in the absence of 4 mm 8-Br-cAMP for 45 min before the addition of vehicle (Control), 1 μmanandamide, or 2-AG for 5 min. In other experiments, slices were incubated in the presence of the PKA inhibitors H-89 (100 μm) or Rp-cAMPS (1 mm) for 20 min. Homogenates were analyzed for active dually phosphorylated ERK by immunoblotting as described in the legend to Figure 1.C, Quantification of the results for P-ERK2 as described in the legend to Figure 1. Data correspond to mean ± SEM. Statistical analysis was done with ANOVA (8-Br-cAMP:F(5,18) = 14.5; H89 and RpcAMPs:F(2,7) = 16.1; p < 0.01) followed by t test (treated vs control: ***p < 0.001, **p < 0.01, *p < 0.05; treated in the presence of 8-Br-cAMP vs in its absence: °° p < 0.01, ° p < 0.05).
Role of Fyn in the effects of endocannabinoids
We have shown previously that stimulation of CB1-Rs in hippocampus activates a protein tyrosine phosphorylation pathway, involving neuronal isoforms of FAK and an associated protein, p130-Cas (Derkinderen et al., 1996, 2001b). In cell lines, after autophosphorylation on Tyr-397, FAK recruits Src-family tyrosine kinases, including Src and Fyn, which in turn phosphorylate multiple residues in the kinase domain and C-terminal region of FAK, as well as in p130-Cas (Girault et al., 1999; Schaller, 2001). Our recent results showed that, in hippocampal slices, endocannabinoids increase the association of Fyn, but not Src, with FAK (Derkinderen et al., 2001b). In several cell types, recruitment of Src-family kinases by FAK has been shown to result in the activation of ERK through various signaling mechanisms (Girault et al., 1999; Schaller, 2001). Moreover, other pathways involving Src-family kinases can activate ERKs downstream of G-protein-coupled receptors, independently of FAK (Luttrell et al., 1996). To determine the role of Src-family kinases in the effects of endocannabinoids, we used PP2, a compound that inhibits potently the catalytic activity of this group of kinases (Hanke et al., 1996). As reported previously (Derkinderen et al., 1996), endocannabinoids increased tyrosine phosphorylation of a number of proteins in hippocampal slices (Fig. 5A). Pretreatment of slices with PP2 dramatically decreased the basal tyrosine phosphorylation of most proteins and abolished the effects of endocannabinoids (Fig. 5A), demonstrating that they are almost completely accounted for by Src-family kinases. However, despite the massive inhibition of protein tyrosine phosphorylation by PP2, the activation of ERK by endocannabinoids was still present (Fig.5A).
Fig. 5.

Role of Src-family kinases in the effects of endocannabinoids in rat hippocampal slices. A, Rat hippocampal slices were incubated as described in Materials and Methods, in the presence or absence of 5 μm PP2 for 45 min before the addition of vehicle (Control), 1 μm anandamide, or 2-AG for 5 min. Homogenates were analyzed by immunoblotting with antibodies specific for anti-phospho-tyrosine (Blot P-Tyr), active dually phosphorylated ERK (Blot P-ERK), or total ERK (Blot ERK). The optical densities of P-ERK2 were as follows: in the absence of PP2: control 100 ± 43, anandamide 767 ± 231, and 2-AG 413 ± 92; in the presence of PP2: control 89 ± 28, anandamide 1358 ± 280, and 2-AG 861 ± 191 (F(5,14) = 7.1,p < 0.001; followed by t test,p < 0.05 for endocannabinoid-treated vs control in both groups). B, Hippocampal slices from wild-type (Fyn +/+) and Fyn knock-out (Fyn −/−) mice were incubated for 50 min before the addition of either vehicle (Control) or 1 μm2-AG for 5 min. The optical densities of P-ERK2 were as follows: in Fyn +/+ slices: control 100 ± 8, 2-AG 1198 ± 351; in Fyn −/− slices: control 115 ± 23, 2-AG 108 ± 63 (F(3,8) = 9.3, p < 0.01; followed by t test, p < 0.05, 2-AG vs control in wild-type slices, and p < 0.05, 2-AG in knock-out vs control slices).
Because the endocannabinoids specifically increase the association of Fyn with FAK (Derkinderen et al., 2001b), we examined whether the effects of endocannabinoids on ERK were altered in Fyn knock-out mice. In these mice the effects of 2-AG on protein tyrosine phosphorylation were reduced dramatically (Fig. 5B), confirming the critical role of Fyn in tyrosine phosphorylation in hippocampus (Grant et al., 1995) and its specific involvement in the action of cannabinoids (Derkinderen et al., 2001b). Moreover, endocannabinoids were no longer capable of stimulating ERK (Fig. 5B), showing that Fyn is required for this effect. It is important to note that the levels of ERK (Fig. 5B) and CB1-R (Derkinderen et al., 2001b) were not altered in these mice. Altogether these results strongly suggest that Fyn in hippocampus is necessary for the activation of ERK by CB1-Rs, but that this requirement is independent from acute Fyn catalytic activity.
THC, a cannabinoid agonist, activates ERKs in the hippocampusin vivo
Our results demonstrated that endocannabinoids activate the ERK pathway in hippocampal slices. To determine whether this effect could be also induced by cannabinoids in vivo, we used THC, the cannabinoid agonist widely abused by humans, which has potent effects when administrated at the periphery. As demonstrated above, THC was a powerful stimulator of ERKs in hippocampal slices (Fig. 1). We analyzed ERK activation in hippocampus by immunocytochemistry with a phosphorylation state-specific antibody after injection of Δ9-THC (1 mg/kg, i.p.). A strong P-ERK-like immunoreactivity was detected in the pyramidal cell layers of CA1 (Fig.6A) and CA3 (Fig.6B) 10 min after THC injection (number of positive cells per section, mean ± SEM: vehicle 30 ± 4 vs THC 125 ± 7; n = 4 mice; t test;p < 0.01). This effect was transient because the number of P-ERK-positive cells decreased rapidly: 57 ± 13 at 20 min, 31 ± 8 at 30 min, and 18 ± 4 at 60 min (n = 4 mice per time point; no significant difference with controls). No significant effect was observed in dentate gyrus (data not shown). Analysis at a higher magnification indicated that ERK was activated in the soma and dendrites of many neurons, although only some principal neurons were labeled in response to THC (Fig.6A,B). In CA1, P-ERK-immunoreactivity was detected in pyramidal cells, whereas in CA3, both pyramidal and nonpyramidal cells were labeled with a dense plexiform dendritic network extending into the stratum radiatum. The activation of ERK in response to THC was prevented in CB1-R knock-out mice (Fig. 6C) as well as by pretreatment with SR141716A (3 mg/kg, i.p.) (Fig. 6D), demonstrating that THC activates ERK by acting on CB1-R. Because a dramatic alteration in the response to stimulation of CB1-R was observed in hippocampal slices of Fyn−/− mice, we examined whether the response to THC was also hampered in vivo in these animals. In these mutant mice, no P-ERK immunoreactivity was detected after injection of THC (Fig.6E), revealing that Fyn is necessary for the activation of ERK.
Fig. 6.
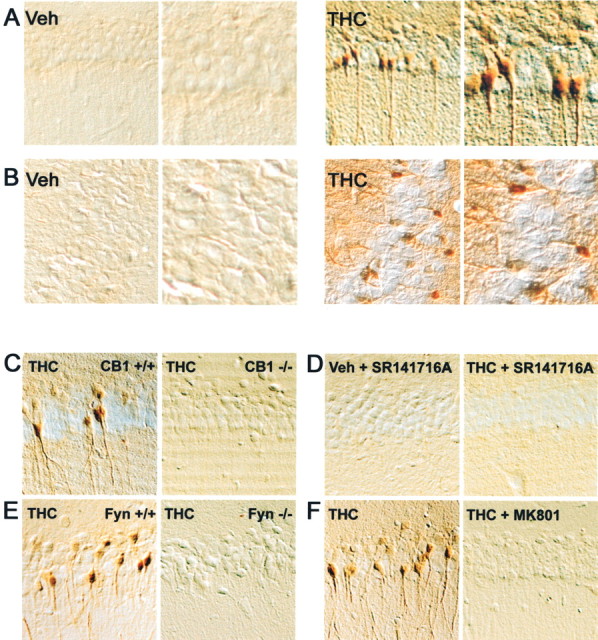
THC activates ERK in hippocampus in vivo by stimulating CB1 receptors. Mice were injected with vehicle (Veh) or THC (1 mg/kg, i.p.) 10 min before they were killed. Active ERK1 and ERK2 were detected by peroxidase immunocytochemistry in the hippocampus, using antibodies against the doubly phosphorylated protein. Results in CA1 (A) and CA3 (B) regions are shown. The right panel corresponds to a higher magnification of the THC-treated sections. In THC-treated mice, immunoreactive cells are mostly present in the pyramidal cell layer of CA1 and CA3. A strong labeling is visible in the cytoplasm (including the dendrites) and the nucleus.C, THC failed to activate ERK in CA1 of CB1 −/− mice, whereas it was active in CB1 +/+ matched controls. D, In the presence of the CB1-R antagonist, SR141716A (3 mg/kg) injected alone (Veh + SR141716A), or 15 min before THC injection (THC + SR141716A), no activation of ERK was observed in CA1. E, THC failed to activate ERK in CA1 of Fyn −/− mice, whereas its effects were present in Fyn +/+ matched controls.F, Stimulation of ERK phosphorylation by THC was abolished by the NMDA receptor antagonist MK801 (0.1 mg/kg), injected 15 min before THC. The results obtained in CB1 −/−, SR141716A-treated, Fyn −/−, and MK801-treated mice in CA3 (data not shown) were similar to those illustrated here in CA1.
In hippocampus, most CB1-Rs are located on GABA nerve terminals (Tsou et al., 1998, 1999; Katona et al., 1999, 2000), and their stimulation is capable of enhancing the activation of pyramidal cells by glutamatergic inputs through a decrease in GABA inhibitory tone (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Therefore, we tested the contribution of glutamate neurotransmission in the effects of THC on the ERK pathway. In mice pretreated with MK801 (0.1 mg/kg, i.p.), a noncompetitive antagonist of NMDA receptors that crosses the blood–brain barrier, THC did not increase P-ERK immunoreactivity in hippocampal neurons (Fig. 6F). This result suggests that the effects of CB1-R on ERK activation in vivo either are mediated by an increase in NMDA receptor stimulation or require the concomitant activation of these receptors. To determine whether the activation of the ERK pathway by cannabinoids in hippocampus was exclusively a consequence of an increased sensitivity to excitatory inputs, we examined the contribution of glutamate in the effects of endocannabinoids in hippocampal slices. In this preparation, MK801 had a small stimulatory effect on ERK phosphorylation by itself but did not prevent ERK activation by either 2-AG (Table 1) or THC (data not shown). In contrast, the stimulatory effect of glutamate on ERK phosphorylation was prevented in the presence of MK801 (data not shown). These results indicate that ERK activation by cannabinoids may be achieved by several mechanisms, one of which depends on NMDA receptors and appears predominant in vivo.
Role of ERK in the regulation of gene expression by THCin vivo
The strong accumulation of P-ERK in nuclei (Fig.7A) suggested its possible role in gene regulation via phosphorylation of transcription factors. To examine the consequences of ERK stimulation by THC in vivo we used SL327, a drug that crosses the blood–brain barrier and prevents the activation of ERK by inhibiting MEK (Atkins et al., 1998; Valjent et al., 2000). This drug, closely related to U0126, which inhibited completely ERK phosphorylation in hippocampal slices (Fig.3), had a dramatic effect on P-ERK immunoreactivity in hippocampal neurons and prevented its enhancement after THC treatment (Fig.7B). ERK phosphorylates and activates transcription factors of the ternary complex factor family, including Elk-1, that bind to the serum response element (SRE) (Whitmarsh and Davis, 1996; Valjent et al., 2001a). Moreover, ERK regulates genes that contain the cAMP/Ca2+ response element (CRE) in their promoter, possibly by phosphorylating and activating MAPK-activated protein kinase 2, which phosphorylates cAMP response element-binding protein (CREB) (Impey et al., 1999). Therefore, we examined the role of ERK in the induction of IEGs by THC, focusing our attention on c-Fos and Zif268, which contain both SRE and CRE in their promoter, and the expression of which is known to increase in response to cannabinoids in neurons (Mailleux et al., 1994; Glass and Dragunow, 1995). We analyzed c-Fos protein by immunohistochemistry, with a specific antibody that did not recognize Fos-related antigens (Fig.7C), and we analyzed Zif268 mRNA by in situ hybridization (Fig. 7D). THC increased the number and immunoreactivity of c-Fos-expressing neurons in CA1 (Fig. 7C) and in CA3 (data not shown). Zif268 mRNA levels were increased in CA1 and CA3 but not in the dentate gyrus (Fig. 7D,F). Both effects were prevented by injection of SL327 before THC (Fig.7C,D,F).
Fig. 7.
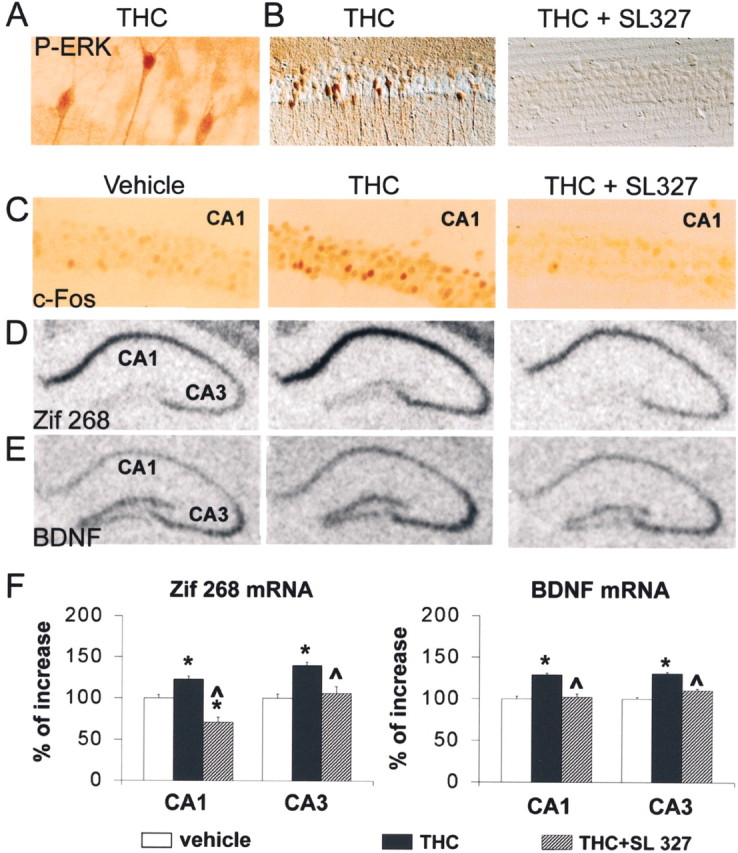
ERK-dependent induction of immediate-early genes by THC in hippocampus in vivo. A, Mice were injected with 1 mg/kg THC 10 min before they were killed, as in Figure 6A, High magnification of a peroxidase-labeled section shows the nuclear staining for dually phosphorylated ERK. B, The effects of THC (middle panel) were prevented in the CA1 region of mice injected with SL327 (100 mg/kg) 60 min before THC (right panel). Results shown are representative of four to six animals for each group. C, c-Fos immunoreactivity (peroxidase reaction) was examined in CA1 of mice injected with either vehicle (Veh) or 1 mg/kg THC 60 min before they were killed. SL327 (100 mg/kg) was injected 60 min before THC.D, Zif268 mRNA expression was analyzed by in situ hybridization in mouse hippocampus 1 hr after injection of vehicle, THC (1 mg/kg, i.p.), or THC and SL327 (100 mg/kg, 60 min before THC). Note the increased hybridization signals in the CA1 and CA3 regions. E, BDNF mRNA expression was analyzed byin situ hybridization in mouse hippocampus 1 hr after injection of vehicle, THC (1 mg/kg, i.p.), or THC and SL327 (100 mg/kg, 60 min before THC). F, Signals for mRNA hybridization were quantified using an image analyzer for six animals for each treatment. Statistical analyses used one-way ANOVA followed by apost hoc comparison with Newman–Keuls test; *p < 0.001 when comparing THC-treated mice with control mice; ^ p < 0.001 when comparing SL + THC with THC alone (n = 6 mice per group).
We also examined the effects of THC on the expression of BDNF, a growth factor that behaves as an IEG, transcribed in response to bursts of neuronal activity through its CREB-binding sites (Shieh et al., 1998;Tao et al., 1998), and plays an important role in hippocampal synaptic plasticity (Kang and Schuman, 1995; Hartmann et al., 2001; Patterson et al., 2001; Ying et al., 2002). We found a significant upregulation of BDNF mRNA levels in CA1 and CA3 1 hr after injection of THC (Fig.7E,F). This increase in BDNF mRNA was prevented by pretreatment with SL327 (Fig.7E,F). Thus, acute injection of THC can induce BDNF mRNA transcription through the ERK signaling pathway.
Discussion
Regulation of ERKs by cannabinoids in hippocampus
The present study shows that cannabinoids activate ERKs in hippocampus and in slices incubated in vitro, as well as in living mice. In slices, 2-AG, an endocannabinoid produced in hippocampus in response to stimulation of Schaeffer collaterals (Stella et al., 1997), increased ERK phosphorylation rapidly and at low concentrations. This effect was likewise achieved with other cannabinoids, including synthetic agonists and THC. Injection of THC in mice was also able to induce ERK activation in the hippocampus. The effects of THC on ERK activation were mediated by CB1-Rs because they were prevented by pretreatment with SR141716A, a specific CB1-R antagonist, and they were not observed in CB1-R knock-out mice. In the hippocampus, the neurons that expressed the strongest P-ERK immunoreactivity after in vivo administration of THC were pyramidal cells in CA1 and CA3. In contrast, CB1-R-immunoreactive neurons are located in all subfields in hippocampus and are mostly interneurons, with a large predominance of cholecystokinin-positive cells (Pettit et al., 1998; Katona et al., 1999; Tsou et al., 1999). In fact, CB1-Rs are highly concentrated in nerve terminals, and endocannabinoids have recently been shown to mediate retrograde signaling at hippocampal synapses (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Although we cannot formally rule out a contribution of CB1-Rs in principal cells, which express low levels of CB1-R mRNA (Marsicano and Lutz, 1999), THC is likely to act on pyramidal cells indirectly by modulating inputs at the presynaptic level. Because CB1-Rs are mostly located on GABA terminals, they could modulate indirectly the sensitivity to excitatory inputs by decreasing the inhibitory tone on pyramidal neurons (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Stimulation of NMDA glutamate receptors is capable of activating the ERK pathway in vitro and in vivo (Valjent et al., 2001a). Interestingly, however, the contribution of NMDA glutamate receptors in the effects of THC appeared different in vitro and in vivo. In living mice, MK801, an NMDA receptor antagonist, prevented the effects of THC on the ERK pathway, suggesting that THC effects on ERK in vivoeither were mediated by an increased stimulation or sensitivity of NMDA receptors or required the concomitant activity of these receptors.
Signaling pathways involved in ERK regulation by CB1-Rs
Although the effects of in vivo CB1-R stimulation were sensitive to a NMDA receptor inhibitor, a strong stimulation of ERK phosphorylation was observed in hippocampal slices in the presence of TTX, which prevents neuronal firing. This effect was also resistant to MK801, arguing in favor of a direct stimulation of the ERK pathway by CB1-Rs in this preparation. It should be pointed out that the precise cellular localization of the activation of ERK by CB1-Rs in hippocampal slices is not known. Nevertheless, the mechanism by which stimulation of CB1-Rs led to ERK activation in hippocampal slices appears to be different from what has been reported in transfected cells. Studies in CHO cells supported a role for βγ rather than for αi subunits in CB1-R–ERK coupling (Bouaboula et al., 1995a,b). Activation of ERKs in cell lines by Gi-coupled receptors can be mediated through recruitment of PI3-kinase by βγ subunits, independently of the inhibition of adenylyl cyclase (Hawes et al., 1996; Lopez-Ilasaca et al., 1997). However, in hippocampal slices, the cannabinoid-induced activation of ERKs was insensitive to the PI3-kinase inhibitor LY294002, strongly arguing against a role of the βγ subunits/PI3-kinase pathway in this effect. Other pathways linking G-protein-coupled receptors to MAP kinases involve tyrosine kinases of the Src family (Luttrell et al., 1997). For example, PP2, a potent inhibitor of these kinases, blocked the effects of various extracellular messengers on the activation of ERK in CHO cells (Igishi and Gutkind, 1998). We have shown previously that stimulation of CB1-Rs increases the tyrosine phosphorylation of the neuronal isoform of FAK in hippocampus and its association with Fyn (Derkinderen et al., 1996,2001b). Thus, FAK provides a possible link for the activation of ERK through recruitment of Src-family kinases or PI3-kinase (Girault et al., 1999; Schaller, 2001). However, the activation of ERK by CB1-Rs in hippocampal slices cannot be accounted for by these known pathways linking FAK to ERK, because it was resistant to inhibitors of PI3-kinase and Src-family kinases. It was particularly striking that PP2 did not prevent ERK activation in hippocampal slices, despite its dramatic effect on protein tyrosine phosphorylation. Yet, the Src-family kinase Fyn appeared to be critical for the activation of ERK by CB1-Rs because this activation was absent in Fyn mutant mice, bothin vitro and in vivo. Although we verified that CB1-R and ERK levels were unaltered in Fyn−/− mice, we cannot rule out the possibility that the absence of Fyn throughout development impaired indirectly the signaling pathways at other levels. An alternative explanation of our findings is that stimulation of CB1-Rs leads to the recruitment of Fyn and to the activation of ERK by a mechanism independent of Fyn catalytic activity. In support of this hypothesis, the closely related tyrosine kinase Src is known to exert some of its biological effects independently of its tyrosine kinase activity (Kaplan et al., 1995; Schwartzberg et al., 1997).
The role of cAMP in ERK regulation in hippocampus
Several additional pathways have been described that link G-protein-coupled receptors to ERK activation and could account for the effects of endocannabinoids (Marinissen and Gutkind, 2001). It is well established that CB1-Rs inhibit adenylyl cyclase, including in hippocampus (Bidaut-Russell et al., 1990), and our observations reveal that ERK activation by cannabinoids was closely associated with cAMP regulation. The effects of endocannabinoids on ERK were prevented by pretreatment of slices with cAMP analogs, suggesting that their mechanism of action may involve an inhibition of cAMP production. Similar effects of cAMP on the activation of ERK induced by anandamide have been reported in a human breast cancer cell line (Melck et al., 1999). The role of cAMP in the effects of CB1-Rs in hippocampus was further supported by the fact that two different inhibitors of PKA, acting on the regulatory or the catalytic subunit of PKA, also activated ERK in hippocampal slices. These effects of cAMP are reminiscent of those reported previously for FAK (Derkinderen et al., 1996, 2001b). Interestingly, a negative effect of cAMP on both FAK- and ERK-dependent responses has also been observed in a completely different context, when adherent NIH3T3 cells were placed in suspension (Howe and Juliano, 2000). These effects of cAMP might appear paradoxical in light of the known stimulation of ERK by cAMP in hippocampal slices (Impey et al., 1998, 1999). Indeed, we confirmed in our experimental conditions that 8-Br-cAMP as well as forskolin were also capable of activating ERK, in agreement with these previous reports. These stimulatory effects lasted for a short period of time and were followed by a decrease in ERK phosphorylation and its insensitivity to endocannabinoids. The effects of cAMP on ERK in hippocampus are complex, similar to what has been reported in other cell types (Stork and Schmitt, 2002). cAMP appears to exert two opposing effects on the ERK pathway in hippocampus, a stimulatory effect that may be partly independent of PKA and, as indicated by our findings, an inhibitory effect mediated by PKA. Thus, in hippocampal slices, stimulation of CB1-Rs could be a critical modulator of cAMP signaling, by opposing some of its effects and enhancing some others. Further work will have to determine whether these apparently opposing effects take place at different cellular or subcellular locations.
Functional implications of the regulation of ERK and immediate-early genes by cannabinoids in hippocampus
Our findings have two types of implications concerning the possible physiological function of the endocannabinoid system in hippocampus and the effects of THC in the context of drug abuse. Recent work from several laboratories has unveiled the important role of the endocannabinoid system in hippocampus. Although massive stimulation of CB1-Rs inhibits several forms of synaptic plasticity in hippocampus, in vivo and in vitro, including LTP and LTD in CA1 neurons (Collins et al., 1995; Terranova et al., 1995;Misner and Sullivan, 1999), it appears that focal stimulation of these receptors has more subtle effects. By decreasing locally the GABAergic inhibition of hippocampal neurons (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001), endocannabinoids facilitate the induction of LTP (Carlson et al., 2002). In addition to these direct effects on synaptic transmission, our results show that stimulation of CB1-Rs has potent effects on signaling pathways that are known to be critical for long-term synaptic plasticity. Stimulation of CB1-Rs activates ERK, a protein kinase known to be critical in several forms of synaptic plasticity and in learning and memory (English and Sweatt, 1997; Atkins et al., 1998). The ERK pathway appears essential for some forms of LTP in hippocampus (Winder et al., 1999) and for the regulation of gene expression in the context of synaptic plasticity (Impey et al., 1999). Remarkably, the activation of ERK by THC was absent in the hippocampus of mice lacking the tyrosine kinase Fyn, which display altered synaptic plasticity (Grant et al., 1992).
CB1-R-mediated stimulation of ERK was responsible for the induction of Zif268, a transcription factor essential for transition from short- to long-term synaptic plasticity and for the expression of long-term memories (Jones et al., 2001). Moreover, we demonstrate that THC also induced the expression of BDNF, a neurotrophic factor that increases synaptic efficiency (Kang and Schuman, 1995; Patterson et al., 2001;Ying et al., 2002). Thus, stimulation of CB1-Rs is by itself capable of activating signaling cascades known to be important for synaptic plasticity. Localized increases in endocannabinoid production could facilitate local synaptic efficiency not only by inhibiting presynaptic release of GABA but also by activating signaling pathways important for long-term modifications.
In contrast to this possible physiological role of the local activation of CB1-Rs, their general pharmacological stimulation in the whole tissue clearly has a negative effect on plasticity (Collins et al., 1995; Terranova et al., 1995; Misner and Sullivan, 1999; Bohme et al., 2000). The effects of THC on ERK phosphorylation in mice, reported here, were observed at doses (1 mg/kg) that are low for experimental studies in animals and may correspond to the amounts of THC absorbed by human subjects during heavy intoxication (Barnett et al., 1985). This dose of THC has minimal aversive effects in mice, and, given after a priming injection in the home cage, is capable of inducing conditioned place preference (Valjent and Maldonado, 2000). It should be pointed out that general administration of THC also stimulates the ERK pathway in other brain regions, including the dorsal striatum and nucleus accumbens (Valjent et al., 2001b). In these brain regions, however, the activation of ERK was entirely dependent on activation of dopamine D1 receptors, whereas in hippocampus the effects of THC were still observed in the presence of SCH23390, a potent blocker of D1 receptors (our unpublished observations). Thus, the present study demonstrates that THC can activate ERK in brain by several pathways, and in regions beyond those usually thought to be the major targets of drugs of abuse. Therefore it will be important to determine whether effects of THC on ERK in hippocampus and their consequences on gene expression may participate in the cognitive and memory impairment reported in heavy cannabis users (Pope et al., 2001; Solowij et al., 2002).
P.D. and E.V. contributed equally to this work.
P.D. was supported by a Poste d'accueil Institut National de la Santé et de la Recherche Médicale. This work was supported in part by grants from Mission Interministerielle de Lutte contre les Drogues et la Toxicomanie, Human Frontier Science Programme Organization, Fondation pour la Recherche Médicale, Fondation Schlumberger pour l'Enseignement et la Recherche, and Action Concertée Incitative (Biologie du Développement et Physiologie Intégrative). Prof. Seth Grant (University of Edinburgh) is gratefully acknowledged for providing some of the Fyn knock-out mice used in this study.
Correspondence should be addressed to Dr. Jean-Antoine Girault, Institut National de la Santé et de la Recherche Médicale U536, Institut du Fer à Moulin, 17 rue du Fer à Moulin, 75005 Paris, France. E-mail:girault@ifm.inserm.fr.
References
- 1.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 2.Al-Hayani A, Wease KN, Ross RA, Pertwee RG, Davies SN. The endogenous cannabinoid anandamide activates vanilloid receptors in the rat hippocampal slice. Neuropharmacology. 2001;41:1000–1005. doi: 10.1016/s0028-3908(01)00145-9. [DOI] [PubMed] [Google Scholar]
- 3.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 4.Barnett G, Licko V, Thompson T. Behavioral pharmacokinetics of marijuana. Psychopharmacology. 1985;85:51–56. doi: 10.1007/BF00427321. [DOI] [PubMed] [Google Scholar]
- 5.Bidaut-Russell M, Devane WA, Howlett AC. Cannabinoid receptors and modulation of cyclic AMP accumulation in the rat brain. J Neurochem. 1990;55:21–26. doi: 10.1111/j.1471-4159.1990.tb08815.x. [DOI] [PubMed] [Google Scholar]
- 6.Bohme GA, Laville M, Ledent C, Parmentier M, Imperato A. Enhanced long-term potentiation in mice lacking cannabinoid CB1 receptors. Neuroscience. 2000;95:5–7. doi: 10.1016/s0306-4522(99)00483-2. [DOI] [PubMed] [Google Scholar]
- 7.Bouaboula M, Bourrié B, Rinaldi-Carmona M, Shire D, Le Fur G, Casellas P. Stimulation of cannabinoid receptor CB1 induces krox-24 expression in human astrocytoma cells. J Biol Chem. 1995a;270:13973–13980. doi: 10.1074/jbc.270.23.13973. [DOI] [PubMed] [Google Scholar]
- 8.Bouaboula M, Poinot-Chazel C, Bourrié B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G, Casellas P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995b;312:637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1: evidence for a new model of receptor/ligand interactions. J Biol Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 10.Burgaya F, Toutant M, Studler JM, Costa A, Le Bert M, Gelman M, Girault JA. Alternatively spliced focal adhesion kinase in rat brain with increased autophosphorylation activity. J Biol Chem. 1997;272:28720–28725. doi: 10.1074/jbc.272.45.28720. [DOI] [PubMed] [Google Scholar]
- 11.Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- 12.Collins DR, Pertwee RG, Davies SN. Prevention by the cannabinoid antagonist, SR141716A, of cannabinoid-mediated blockade of long-term potentiation in the rat hippocampal slice. Br J Pharmacol. 1995;115:869–870. doi: 10.1111/j.1476-5381.1995.tb15889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derkinderen P, Toutant M, Burgaya F, Le Bert M, Siciliano JC, De Franciscis V, Gelman M, Girault JA. Regulation of a neuronal form of focal adhesion kinase by anandamide. Science. 1996;273:1719–1722. doi: 10.1126/science.273.5282.1719. [DOI] [PubMed] [Google Scholar]
- 14.Derkinderen P, Enslen H, Girault JA. The ERK/MAP-kinase cascade in the nervous system. NeuroReport. 1999;10:R24–34. [PubMed] [Google Scholar]
- 15.Derkinderen P, Ledent C, Parmentier M, Girault JA. Cannabinoids activate p38 mitogen-activated protein kinases through CB1 receptors in hippocampus. J Neurochem. 2001a;77:957–960. doi: 10.1046/j.1471-4159.2001.00333.x. [DOI] [PubMed] [Google Scholar]
- 16.Derkinderen P, Toutant M, Kadaré G, Ledent C, Parmentier M, Girault JA. Dual role of Fyn in the regulation of FAK+6,7 by cannabinoids in hippocampus. J Biol Chem. 2001b;276:38289–38296. doi: 10.1074/jbc.M105630200. [DOI] [PubMed] [Google Scholar]
- 17.Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- 18.Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, Zimmer A, Martin BR. Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem. 2000;75:2434–2444. doi: 10.1046/j.1471-4159.2000.0752434.x. [DOI] [PubMed] [Google Scholar]
- 19.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 20.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 21.Fiore RS, Bayer VE, Pelech SL, Posada J, Cooper JA, Baraban JM. p42 mitogen-activated protein kinase in brain: prominent localization in neuronal cell bodies and dendrites. Neuroscience. 1993;55:463–472. doi: 10.1016/0306-4522(93)90516-i. [DOI] [PubMed] [Google Scholar]
- 22.Flood DG, Finn JP, Walton KM, Dionne CA, Contreras PC, Miller MS, Bhat RV. Immunolocalization of the mitogen-activated protein kinases p42MAPK and JNK1, and their regulatory kinases MEK1 and MEK4, in adult rat central nervous system. J Comp Neurol. 1998;398:373–392. doi: 10.1002/(sici)1096-9861(19980831)398:3<373::aid-cne6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 23.Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M. FAK and PYK2/CAK in the nervous system, a link between neuronal activity, plasticity and survival? Trends Neurosci. 1999;22:257–263. doi: 10.1016/s0166-2236(98)01358-7. [DOI] [PubMed] [Google Scholar]
- 24.Glass M, Dragunow M. Induction of the Krox 24 transcription factor in striosomes by a cannabinoid agonist. NeuroReport. 1995;6:241–244. doi: 10.1097/00001756-199501000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Gomez del Pulgar T, Velasco G, Guzman M. The CB1 cannabinoid receptor is coupled to the activation of protein kinase B/Akt. Biochem J. 2000;347:369–373. doi: 10.1042/0264-6021:3470369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez del Pulgar T, De Ceballos ML, Guzman M, Velasco G. Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2002;277:36527–36533. doi: 10.1074/jbc.M205797200. [DOI] [PubMed] [Google Scholar]
- 27.Grant SGN, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- 28.Grant SGN, Karl KA, Kiebler MA, Kandel ER. Focal adhesion kinase in the brain: novel subcellular localization and specific regulation by Fyn tyrosine kinase in mutant mice. Genes Dev. 1995;9:1909–1921. doi: 10.1101/gad.9.15.1909. [DOI] [PubMed] [Google Scholar]
- 29.Hampson RE, Deadwyler SA. Cannabinoids reveal the necessity of hippocampal neural encoding for short-term memory in rats. J Neurosci. 2000;20:8932–8942. doi: 10.1523/JNEUROSCI.20-23-08932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haneda M, Sugimoto T, Kikkawa R. Mitogen-activated protein kinase phosphatase: a negative regulator of the mitogen-activated protein kinase cascade. Eur J Pharmacol. 1999;365:1–7. doi: 10.1016/s0014-2999(98)00857-7. [DOI] [PubMed] [Google Scholar]
- 31.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok K, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor: study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 32.Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawes BE, Luttrell LM, Van Biesen T, Lefkowitz RJ. Phosphatidylinositol 3-kinase is an early intermediate in the Gβgamma-mediated mitogen-activated protein kinase signaling pathway. J Biol Chem. 1996;271:12133–12136. doi: 10.1074/jbc.271.21.12133. [DOI] [PubMed] [Google Scholar]
- 34.Howe AK, Juliano RL. Regulation of anchorage-dependent signal transduction by protein kinase A and p21-activated kinase. Nat Cell Biol. 2000;2:593–600. doi: 10.1038/35023536. [DOI] [PubMed] [Google Scholar]
- 35.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 36.Igishi T, Gutkind JS. Tyrosine kinases of the Src family participate in signaling to MAP kinase from both Gq, and Gi-coupled receptors. Biochem Biophys Res Commun. 1998;244:5–10. doi: 10.1006/bbrc.1998.8208. [DOI] [PubMed] [Google Scholar]
- 37.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 38.Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- 39.Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- 40.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- 42.Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katona I, Sperlagh B, Magloczky Z, Santha E, Kofalvi A, Czirjak S, Mackie K, Vizi ES, Freund TF. GABAergic interneurons are the targets of cannabinoid actions in the human hippocampus. Neuroscience. 2000;100:797–804. doi: 10.1016/s0306-4522(00)00286-4. [DOI] [PubMed] [Google Scholar]
- 44.Kumagai N, Morii N, Fujisawa K, Yoshimasa T, Nakao K, Narumiya S. Lysophosphatidic acid induces tyrosine phosphorylation and activation of MAP-kinase and focal adhesion kinase in cultured Swiss 3T3 cells. FEBS Lett. 1993;329:273–276. doi: 10.1016/0014-5793(93)80236-n. [DOI] [PubMed] [Google Scholar]
- 45.Ledent C, Valverde O, Cossu C, Petitet F, Aubert LF, Beslot F, Böhme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- 46.Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 47.Luttrell LM, Hawes BE, Van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gβgamma subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 48.Luttrell LM, Van Biesen T, Hawes BE, Koch WJ, Krueger KM, Touhara K, Lefkowitz RJ. G-protein-coupled receptors and their regulation: activation of the MAP kinase signaling pathway by G-protein-coupled receptors. Adv Second Messenger Phosphoprotein Res. 1997;31:263–277. [PubMed] [Google Scholar]
- 49.Mailleux P, Verslype M, Preud'homme X, Vanderhaeghen J-J. Activation of multiple transcription factor genes by tetrahydrocannabinol in rat forebrain. NeuroReport. 1994;5:1265–1268. doi: 10.1097/00001756-199406020-00028. [DOI] [PubMed] [Google Scholar]
- 50.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci. 2001;22:368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 51.Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- 52.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 53.Melck D, Rueda D, Galve-Roperh I, De Petrocellis L, Guzman M, Di Marzo V. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999;463:235–240. doi: 10.1016/s0014-5793(99)01639-7. [DOI] [PubMed] [Google Scholar]
- 54.Misner DL, Sullivan JM. Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci. 1999;19:6795–6805. doi: 10.1523/JNEUROSCI.19-16-06795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 56.Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- 57.Pettit DA, Harrison MP, Olson JM, Spencer RF, Cabral GA. Immunohistochemical localization of the neural cannabinoid receptor in rat brain. J Neurosci Res. 1998;51:391–402. doi: 10.1002/(SICI)1097-4547(19980201)51:3<391::AID-JNR12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 58.Pope HG, Jr, Gruber AJ, Hudson JI, Huestis MA, Yurgelun-Todd D. Neuropsychological performance in long-term cannabis users. Arch Gen Psychiatry. 2001;58:909–915. doi: 10.1001/archpsyc.58.10.909. [DOI] [PubMed] [Google Scholar]
- 59.Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta. 2001;1540:1–21. doi: 10.1016/s0167-4889(01)00123-9. [DOI] [PubMed] [Google Scholar]
- 60.Schwartzberg PL, Xing LP, Hoffmann O, Lowell CA, Garrett L, Boyce BF, Varmus HE. Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src−/− mutant mice. Genes Dev. 1997;11:2835–2844. doi: 10.1101/gad.11.21.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signaling. Biochem J. 1998;333:471–490. doi: 10.1042/bj3330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 63.Siciliano JC, Gelman M, Girault JA. Depolarization and neurotransmitters increase neuronal protein tyrosine phosphorylation. J Neurochem. 1994;62:950–959. doi: 10.1046/j.1471-4159.1994.62030950.x. [DOI] [PubMed] [Google Scholar]
- 64.Solowij N, Stephens RS, Roffman RA, Babor T, Kadden R, Miller M, Christiansen K, McRee B, Vendetti J. Cognitive functioning of long-term heavy cannabis users seeking treatment. JAMA. 2002;287:1123–1131. doi: 10.1001/jama.287.9.1123. [DOI] [PubMed] [Google Scholar]
- 65.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 66.Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 67.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 68.Terranova JP, Michaud JC, Le Fur G, Soubrié P. Inhibition of long-term potentiation in rat hippocampal slices by anandamide and WIN55212–2: reversal by SR141716 A, a selective antagonist of CB1 cannabinoid receptors. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:576–579. doi: 10.1007/BF00169393. [DOI] [PubMed] [Google Scholar]
- 69.Tsou K, Nogueron MI, Muthian S, Sañudo-Peña MC, Hillard CJ, Deutsch DG, Walker JM. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–140. doi: 10.1016/s0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- 70.Tsou K, Mackie K, Sanudo-Pena MC, Walker JM. Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience. 1999;93:969–975. doi: 10.1016/s0306-4522(99)00086-x. [DOI] [PubMed] [Google Scholar]
- 71.Valjent E, Maldonado R. A behavioural model to reveal place preference to delta 9- tetrahydrocannabinol in mice. Psychopharmacology (Berl) 2000;147:436–438. doi: 10.1007/s002130050013. [DOI] [PubMed] [Google Scholar]
- 72.Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valjent E, Caboche J, Vanhoutte P. Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: a molecular substrate for learning and memory? Mol Neurobiol. 2001a;23:83–99. doi: 10.1385/MN:23:2-3:083. [DOI] [PubMed] [Google Scholar]
- 74.Valjent E, Pages C, Rogard M, Besson MJ, Maldonado R, Caboche J. Delta 9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur J Neurosci. 2001b;14:342–352. doi: 10.1046/j.0953-816x.2001.01652.x. [DOI] [PubMed] [Google Scholar]
- 75.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase,2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 76.Wartmann M, Campbell D, Subramanian A, Burstein SH, Davis RJ. The MAP kinase signal transduction pathway is activated by the endogenous cannabinoid anandamide. FEBS Lett. 1995;359:133–136. doi: 10.1016/0014-5793(95)00027-7. [DOI] [PubMed] [Google Scholar]
- 77.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 78.Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 79.Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- 80.Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]