

Abstract
To identify molecular mechanisms that control activity-dependent gene expression in the CNS, we have characterized the factors that mediate activity-dependent transcription ofBDNF promoter III. We report the identification of a Ca2+-responsive E-box element, CaRE2, withinBDNF promoter III that binds upstream stimulatory factors 1 and 2 (USF1/2) and show that USFs are required for the activation of CaRE2-dependent transcription from BDNFpromoter III. We find that the transcriptional activity of the USFs is regulated by Ca2+-activated signaling pathways in neurons and that the USFs bind to the promoters of a number of neuronal activity-regulated genes in vivo. These results suggest a new function for the USFs in the regulation of activity-dependent transcription in neurons.
Keywords: upstream stimulatory factor, USF1, USF2, activity-dependent transcription, brain-derived neurotrophic factor, BDNF, calcium, neural plasticity, activity-dependent neural development
Introduction
Neural activity arising from sensory input induces the expression of new gene products that contribute to enduring adaptations in the CNS. These activity-dependent changes include the refinement of cortical circuitry during development (Katz and Shatz, 1996; Mao et al., 1999; Pallas, 2001; Sur and Leamey, 2001), the formation of long-term memories (Koenig and Lu, 1967; Nguyen et al., 1994), and the development of complex behaviors such as birdsong learning (Clayton, 1997). Neuronal activity and subsequent depolarization drive the influx of Ca2+ions via L-type voltage-sensitive Ca2+channels (L-VSCC) and the NMDA subtype of glutamate receptors, stimulating an intricate signaling network that regulates rapid transcriptional events at the promoters of downstream genes (Bito et al., 1997; Impey and Goodman, 2001; West et al., 2001). An especially interesting group of these activity-regulated genes encodes proteins that are expressed selectively in the nervous system and regulate synaptic maturation and function. Examples include the nicotinic acetylcholine receptor α7 subunit (nAchRα7) (Zhou et al., 2001), the major histocompatibility complex (MHC) class I genes (Corriveau et al., 1998; Huh et al., 2000), cyclooxygenase-2 (COX-2)/prostaglandin E synthase (PGES) (Yamagata et al., 1993), the neuropeptide preprotachykinin (Benson et al., 1994), and the neurotrophin brain-derived neurotrophic factor (BDNF). Investigating the regulatory mechanisms that control the transcription of these genes in neurons may provide important insights into activity-dependent neural development and synaptic plasticity.
To identify molecular mechanisms that mediate activity-dependent gene expression in neurons, we have studied the transcriptional regulation of BDNF. BDNF is highly expressed in neurons and plays important roles in neuronal survival (Bonni et al., 1999), cortical development (Ghosh, 1996), and synaptic plasticity (Poo, 2001). Transcription of theBDNF gene is stimulated dramatically by membrane depolarization in vitro (Ghosh et al., 1994) and by neural activity during kindling, long-term potentiation (LTP) induction, and visual associative learning (Ernfors et al., 1991; Patterson et al., 1992; Tokuyama et al., 2000). The BDNF gene comprises five exons, the first four of which are spliced alternatively to a single 3′ exon encoding the complete BDNF protein (Timmusk et al., 1993). Although the specific functions of the different BDNF transcripts are not yet clear, we have demonstrated previously that the promoter upstream of exon III is most responsive to membrane depolarization of cultured embryonic rat cortical neurons (Tao et al., 1998).
The cAMP/Ca2+-response element binding protein (CREB) is required for the activity-dependent transcription of a number of neuronal genes, including BDNF exon III (Shieh et al., 1998; Tao et al., 1998; Sasaki et al., 2000). However, activation of CREB alone is not sufficient to mediate the activity-dependent transcription of BDNF exon III. Mutations of BDNF promoter III 5′ to the CRE sequence severely reduce the responsiveness of the promoter to Ca2+influx (Shieh et al., 1998; Tao et al., 1998), indicating that there must exist additional transcription factors that cooperate with CREB to regulate BDNF promoter III in a Ca2+-dependent manner in neurons. We have conducted detailed mutagenesis of the region 5′ to the CRE inBDNF promoter III and find that it contains two distinct Ca2+-response elements (CaREs). In this report we show that one of these elements (CaRE2) is a Ca2+-responsive E-box. Using a yeast one-hybrid screen to identify factor(s) that bind this element, we found that upstream stimulatory factors 1 and 2 (USF1/2) bind toBDNF promoter III through the E-box sequence. The USFs are bound to BDNF promoter III in vivo, suggesting that they are likely to function as endogenous transcriptional regulators of BDNF expression in the brain. Consistent with a role for the USFs as regulators of Ca2+-dependent transcription of BDNF exon III, we observe that the transcriptional activity of the USFs is activated by Ca2+ influx into neurons. In addition to the evidence that USFs regulate BDNF expression, we demonstrate by chromatin immunoprecipitation that the USFs are bound in vivo to a number of other activity-regulated promoters. These data suggest a new function for the USFs as activity-dependent transcriptional regulators in the brain that play a role in orchestrating neural development and synaptic plasticity.
Materials and Methods
Plasmids. BDNF pIII(170)-Luc, EF-β-gal, pSG424 (Gal4 only), Gal4-Luc, Gal4-USF2, pSG5-USF2, pSG5-USF2DN, 3×UBE-Luc (pU3ML-Luc), CRE-Luc, CMV-A-USF1, and the control vectors pML-Luc and CMV were described previously (Sheng et al., 1991; Luo and Sawadogo, 1996a,b; Abdollah et al., 1997; Tao et al., 1998). TK-pRL was purchased from Promega (Madison, WI). We generated 2 bp substitutions of BDNF pIII(170)-Luc by QuickChange site-directed mutagenesis (Stratagene, La Jolla, CA) with a pair of complimentary 5′ oligos containing two random nucleotides at 2 bp intervals from nucleotides –54 to –41 in BDNFpromoter III.
Cell culture, transfection, stimulation, and luciferase assay.Cortical neurons from embryonic day 18 (E18) Long–Evans rats (Charles River, Wilmington, MA) and E16 C57/Black 6 mice were cultured as described (Tao et al., 1998). Neurons were transfected at 3 d in vitro (3 DIV) by the Ca2+ phosphate precipitation method (Xia et al., 1996). At 2 d after transfection the neurons were depolarized with 50 mm KCl (Tao et al., 1998) for 7–9 hr. Cell extracts were used for a luciferase reporter assay or a dual luciferase reporter assay if the cells were cotransfected with TK-pRL (Promega); 80 μl of extract was used for a β-galactosidase assay with ortho-nitrophenyl β-d-galactopyranoside as the substrate. The normalized luciferase activity was obtained by dividing thefirefly luciferase activity by either the renilla luciferase activity or the β-galactosidase activity. Each normalized value represents the average of at least three independent determinations, and the error bars indicate the SEM. L-VSCCs were blocked by the addition of 5 μm nimodipine (Roche Molecular Biochemicals, Indianapolis, IN), and NMDA receptors were blocked by 100 μmd-APV (Sigma, St. Louis, MO) for 5 min before depolarization. Glutamate receptors were activated by the addition of 20 μm glutamate.
Nuclear extracts and electrophoretic mobility shift assays.Nuclear extracts and electrophoretic mobility shift assays (EMSAs) were performed as described (Tao et al., 1998). The radiolabeled CaRE2 probe was synthesized by Klenow (3′→5′ exo−; New England Biolabs, Beverly, MA) with α-32P-dCTP (NEN Life Science, Boston, MA), using complementary oligonucleotides: 5′-GTG AGC TGT CAT ATG ATA CCT CCT CTG CCT C-3′ and 5′-GAG GCA GAG GAG G-3′ (Invitrogen, San Diego, CA). Wild-type and mutant unlabeled probes were synthesized by PCR with primers 5′-GGT AAT TCG TGC ACT AGA G-3′ and 5′-CGA GAG GGC TCC ACG CTG C-3′, using the wild-type or mutant BDNF pIII(170)-Luc vectors as templates. Anti-USF1 (sc-229X), anti-USF2 (sc-862X), and anti-c-Myc (sc-42X) antibodies for supershift were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Yeast one-hybrid screen. The yeast one-hybrid screen for CaRE2 binding proteins was performed by using the Matchmaker Yeast one-hybrid system (Clontech, Palo Alto, CA). Six repeats of the CaRE2 sequence (from nucleotides –56 to –39) were cloned upstream of the HIS3 gene. Then the plasmid was integrated into yeast genome to generate a yeast reporter strain, and the strain was transfected with a rat brain cDNA library containing ∼1 × 106 independent clones fused to the transcriptional activation domain of the yeast Gal4 protein (Clontech). Colonies that grew on minimal medium lacking histidine were selected, and the cDNA-containing plasmids were recovered and sequenced from these colonies.
Immunocytochemistry. Anti-USF1 (C-20) and anti-USF2 (C-20) antibodies were purchased from Santa Cruz Biotechnology. Both antibodies were used at 1:400 for cell staining. The mouse TuJ1 anti-β-tubulin III antibody (Babco, Richmond, CA) was used at 1:300.
Chromatin immunoprecipitation assay. E18 rat cortical neurons (2 × 107 cells) at 5 DIV were treated with 1% formaldehyde at room temperature for 20 min. After two washes with 1× PBS, the cross-linked neurons were scraped off the plates. The neurons were pelleted, resuspended in 200 μl of lysis buffer, and processed for chromatin immunoprecipitation with the chromatin immunoprecipitation (ChIP) assay kit (Upstate Biotechnology, Lake Placid, NY) with the following modifications: (1) the cell lysates were sheared by sonication for a total of 100 sec at 20 sec per interval; (2) the protein A agarose/antibody/transcription factor complex was washed in the low-salt immune complex wash buffer twice, high-salt immune complex wash buffer twice, LiCl immune complex wash buffer twice, and then 1× TE three times; and (3) the chromatin fragments that were pulled down with specific antibodies were resuspended in 200 μl of H2O, 10 μl of which was used for PCR for 28–30 cycles. Anti-Bad (N-20), anti-c-Myc (C-33), and anti-Id (Z-8) antibodies were purchased from Santa Cruz Biotechnology. PCR primer sequences for BDNF exon III were 5′-GCG CGG AAT TCT GAT TCT GGT AAT-3′ and 5′-GAG AGG GCT CCA CGC TGC CTT GAC G-3′; forBDNF exon V were 5′-AAG TGT AAT CCC ATG GGT TAC ACG-3′ and 5′-CAG GAA GTG TCT ATC CTT ATG AAC CG-3′; for COX-2 promoter were 5′-CCT GCC CCT ATG GGT ATT ATG C-3′ and 5′-TTC GTG ACT GTG TCT TTC CGC-3′; for nAchRα7 promoter were 5′-ATT AAA CTG CAG GCG GGA CAG-3′ and 5′-GCG GCC AAG CTT GGC TAT-3′; for Nur77promoter were 5′-CCT GGT CGG TTA TTT CGG-3′ and 5′-AGC GCG GAT TGT TTG ATC-3′; for GAP-43 promoter were 5′-AGT GTG GAA GCA TAA ATG AGA TGT TTG-3′ and 5′-GGA GAT TTT GTG TGC AGT TGA TAA TTG-3′.
Quantitative real-time RT-PCR. RNA was prepared with the Absolutely RNA kit (Stratagene). Total RNA (1 μg) was used for reverse transcription with the First Strand Superscript II kit (Invitrogen). PCR was performed in an iCycler (Bio-Rad, Hercules, CA) with the use of SYBR-green (PE Applied Biosystems, Foster City, CA). Each independent sample was assayed in triplicate. The threshold cycle for each sample was chosen from the linear range and converted to a starting quantity by interpolation from a standard curve run on the same plate for each set of primers. The firefly luciferase mRNA levels were normalized for each well to cotransfected α-globin mRNA levels. Single PCR products were verified both by assessing that the melting temperature of the product had a single value and by viewing the PCR product on an agarose gel. The primer sequences forfirefly luciferase were 5′-GAG GTG AAC ATC ACG TAC GCG-3′ and 5′-AAG AGA GTT TTC ACT GCA TAC GAC G-3′ and forα-globin were 5′-CAA GAC CTA CTT CCC GCA CTT-3′ and 5′-GCT CAG GTC GAA GTG CGG-3′.
Results
An E-box sequence in BDNF promoter III is a Ca2+ response element
Previously, we found that 170 bp of the 5′ flanking sequence ofBDNF exon III is sufficient to activate reporter gene expression in response to membrane depolarization-induced Ca2+ influx via L-VSCCs in cultured embryonic rat cortical neurons (Tao et al., 1998), suggesting that key CaREs reside within this region. To identify these CaREs, we have made systematic deletions and mutations of the 170 bp of BDNFpromoter III and assessed the effects of these mutations on the Ca2+ inducibility of a luciferase reporter. Initially, we and others reported that both a CRE-like sequence bound by the transcription factor CREB as well as a discrete region 5′ to this element are required for induction of reporter gene expression (Shieh et al., 1998; Tao et al., 1998). With further mutagenesis we have determined that this distal regulatory region consists of two distinct elements. The most 5′ element (CaRE1) lies between nucleotides –73 to –64 relative to the transcriptional initiation site of BDNF exon III and is bound by a novel transcription factor, calcium-responsive transcription factor (CaRF), which regulates BDNF exon III transcription in a Ca2+- and neural-selective manner (Tao et al., 2002).
To characterize the element lying between CaRE1 and the CaRE3/CRE, we made two-nucleotide substitutions of the sequence between these elements in the context of the BDNF promoter III reporter gene construct. These mutant plasmids were transiently transfected into cultured cortical neurons, the cells were depolarized by exposure to elevated levels of KCl, and the induction of luciferase expression from the reporter gene was measured. As shown in Figure1a, transcription from the wild-type BDNF pIII(170)-Luc reporter gene was induced significantly in response to membrane depolarization. Most mutations of the nucleotide sequence between –52 and –43 bp 5′ to theBDNF exon III transcription initiation site severely reduced the ability of membrane depolarization to induce reporter gene expression, whereas mutations just outside this 10 bp region had little effect. These data identify the 10 bp nucleotide sequence from –52 to –43 bp relative to the BDNF exon III transcription initiation site as a critical Ca2+response element that we have named CaRE2.
Fig. 1.
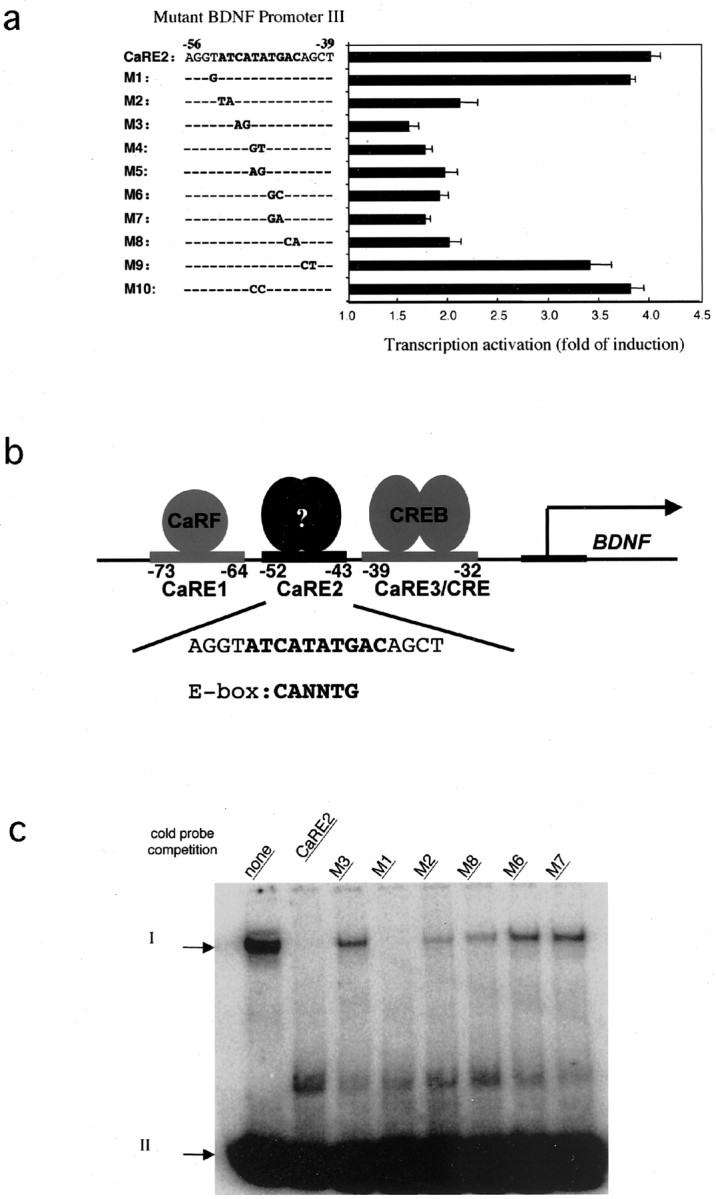
CaRE2 is a Ca2+-responsive E-box element in BDNF promoter III. a, Characterization of CaRE2. A luciferase reporter plasmid driven byBDNF promoter III with either wild-type or mutant sequences was transfected into E18 + 3 DIV rat cortical neurons. The fold of induction equals the ratio of normalized luciferase activity from stimulated cells to that from the unstimulated neurons. Thenumbering indicates the position relative to the first transcription start site for BDNF exon III. In the mutant sequences the letters indicate the mutated bases, and the dashed lines represent unchanged nucleotides.b, Schematic diagram of Ca2+ response elements (CaREs) in BDNF promoter III. The CaRE2 sequence (bolded) from BDNF promoter III is shown and compared with the canonical E-box sequence.c, Characterization of the CaRE2 binding protein(s). Shown is the specific interaction of CaRE2 and its binding protein(s) in neurons. Nuclear extracts from P1 rat brain were mixed with radiolabeled probes containing the CaRE2 sequence before electrophoresis in a native polyacrylamide gel. Unbound radiolabeled CaRE2 probes (arrow II) migrated faster than the ones retarded by the CaRE2 binding protein(s) (arrow I). The specificity of the interaction was determined by the ability of excess unlabeled mutant or wild-type CaRE2-containing probes to compete away the radiolabeled CaRE2 probes from the DNA/protein complex. The sequences for the mutant probes (M1–M3, M6–M8) are shown ina.
Characterization and cloning of the CaRE2 binding protein
The core of the CaRE2 sequence (ATCATATGAC) fits the consensus for an E-box element (CANNTG). E-box elements are bound by members of the basic helix-loop-helix (bHLH) family of transcription factors. However, analysis of the TRANSFAC database (http://www.cbil.upenn.edu/tess/index.html) indicated that none of the previously characterized factors favor the specific E-box sequence we had identified as CaRE2 in BDNF promoter III. Therefore, to identify the transcription factor(s) that mediate Ca2+-dependent expression ofBDNF exon III through CaRE2, we cloned and characterized the protein(s) that bind to this element.
Because BDNF is highly expressed in neurons, we asked whether there is a protein within the nucleus of cortical neurons that binds specifically to the CaRE2 sequence. Nuclear protein extracts were prepared from embryonic rat cortex, mixed with a radiolabeled probe encompassing the CaRE2 sequence, and then subjected to an EMSA. A protein in the cortical neuron nuclear extract was found to bind to and retard the mobility of the radiolabeled CaRE2 probe in a nondenaturing polyacrylamide gel (Fig. 1c). The association of this nuclear protein with CaRE2 was specific, because its binding to CaRE2 could be competed by the addition of an excess of unlabeled wild-type CaRE2 probe, but not with the addition of excess CaRE2 sequences that failed to support Ca2+-dependent induction in the context of the BDNF promoter III reporter gene (Fig.1a). This correlation between the ability of the neuronal nuclear protein to bind CaRE2 sequences and the ability of these CaRE2 sequences to drive Ca2+-dependent transcription of BDNF promoter III supports the hypothesis that there exists a protein in cortical neurons for which the interaction with CaRE2 is required for Ca2+-dependent induction ofBDNF promoter III transcription.
To identify the protein that regulates BDNF transcription through CaRE2, we used a yeast one-hybrid system to screen a rat brain cDNA library for CaRE2 binding protein(s). After screening 250,000 clones, we obtained three positive colonies. Protein extracts from these yeast contained a protein that bound to CaRE2 with the same specificity for wild-type and mutant CaRE2 sequences as the endogenous CaRE2 binding protein from neuronal nuclear extracts (data not shown). After sequencing of the brain cDNA recovered from these clones, we were surprised to find that in each case the expressed protein was the mammalian bHLH transcription factor USF1.
USF1 is a 43 kDa bHLH family transcription factor (Sawadogo et al., 1988; Gregor et al., 1990) originally purified as a cellular protein that regulates the adenovirus major late promoter (Carthew et al., 1985; Sawadogo and Roeder, 1985; Moncollin et al., 1986). A highly homologous family member, USF2, of 44 kDa also has been characterized (Sawadogo et al., 1988; Sirito et al., 1994). Both USFs are expressed ubiquitously (Sirito et al., 1994; Viollet et al., 1996) and bind to E-box elements as homo- or heterodimers (Gregor et al., 1990; Sirito et al., 1992). Preliminary characterization of the endogenous CaRE2 binding proteins from neurons had suggested that they were heat-stable proteins with molecular weights of ∼45 kDa (data not shown), consistent with the properties of the USFs. Although USF1 and USF2 are known to be expressed in brain (Sirito et al., 1994), neither protein has been implicated previously in activity-dependent transcription. Therefore, we conducted an additional series of experiments to determine whether the USFs contribute to Ca2+-regulated BDNF transcription.
USF1 and USF2 bind CaRE2
To examine whether the USFs bind CaRE2, we first asked whether the USFs are part of the CaRE2 binding complex that we observed by EMSA with cortical neuron nuclear extracts. The addition of anti-USF1 or USF2 antibodies to the CaRE2–neuronal nuclear protein complex caused an additional retardation of the complex by EMSA, suggesting that both USF1 and USF2 are in the CaRE2–protein complex (Fig.2a). In contrast, the addition of antibodies that recognize a closely related bHLH family transcription factor, c-Myc, had no effect on the CaRE2–protein complex. These results indicate that endogenous USF1 and USF2 are part of the complex of proteins present in neuronal extracts that bind to CaRE2.
Fig. 2.
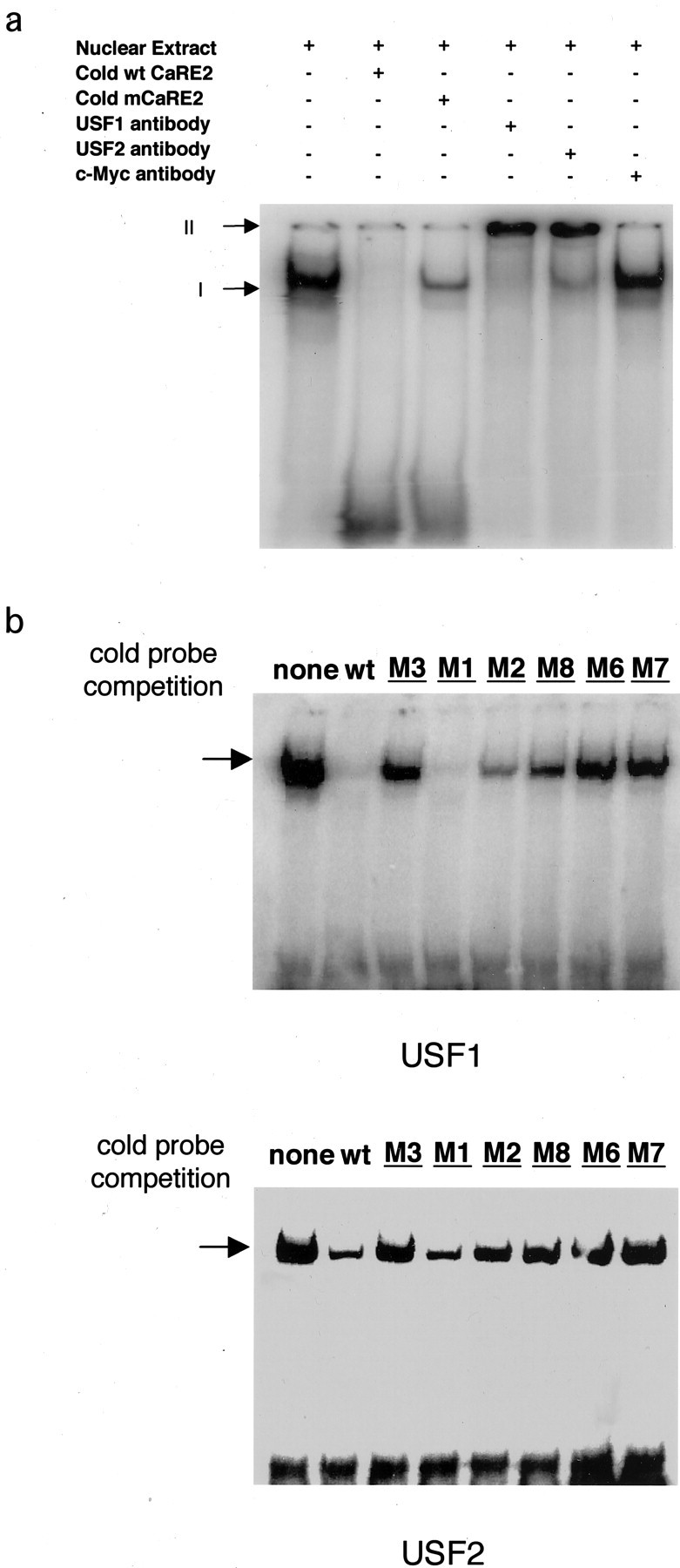
Upstream stimulatory factors (USFs) bind CaRE2. a, Endogenous USFs in neurons bind CaRE2. Nuclear extracts from P1 rat brain were incubated with excess unlabeled wild-type or mutant CaRE2 probes, anti-USF1 antibody, anti-USF2 antibody, or anti-c-Myc antibody on ice for 1 hr. Then radiolabeled CaRE2 probes were added to the mixture, which subsequently was subjected to EMSA. Arrow I marks the complex formed by the radiolabeled CaRE2 probes with the endogenous CaRE2 binding proteins. Arrow II indicates the supershifted complexes formed by anti-USF antibodies, the endogenous CaRE2 binding protein, and the radiolabeled CaRE2 probe. b, Recombinant USFs are sufficient to bind CaRE2. In vitro translated USF1 or USF2 proteins were mixed with radiolabeled CaRE2 probes and an excess of unlabeled wild-type or mutant CaRE2 probes and then subjected to EMSA. The sequences for the mutant CaRE2 probes are the same as those in Figure 1a. The arrow indicates the specifically retarded band.
To determine whether USF1 and USF2 are sufficient to bind to CaRE2 directly, we tested the ability of in vitro transcribed and translated USF1 and USF2 to bind to CaRE2 in an EMSA. As shown in Figure 2b, both USF1 and USF2 are capable of binding to CaRE2, as indicated by a retardation of the radiolabeled CaRE2 probe. Moreover, this binding shows the same specificity for CaRE2 sequences as we observed for the endogenous CaRE2 binding protein (Fig.1c). This correlation between the ability of USFs to bind CaRE2 sequences and the ability of these sequences to support activity-dependent transcription from BDNF promoter III suggests that USF binding to CaRE2 is relevant for activity-dependentBDNF exon III transcription in neurons.
USFs functionally regulate CaRE2-dependent Ca2+-inducible BDNF exon III transcription
To determine whether the USFs mediate Ca2+-inducible BDNF exon III transcription, we asked whether dominant-negative versions of USF block the activity-dependent induction of BDNF promoter III reporter gene transcription. Both USF1 and USF2 are expressed constitutively in the nuclei of cultured embryonic cortical neurons (Fig. 3a–h). To disrupt their function, we used a deletion mutant of USF2 (DN-USF2) that lacks the N-terminal transcriptional activation domain and effectively competes with endogenous USF1 and USF2 for binding to promoter E-boxes (Qyang et al., 1999) without activating USF-dependent transcription. Coexpression of DN-USF2 with the BDNF promoter III reporter gene resulted in a significant reduction of the activity-dependent induction of luciferase expression, whereas overexpression of wild-type USF2 had no effect (Fig. 3i). These data indicate that a nonfunctional USF bound to CaRE2 blocks activity-dependent BDNF exon III expression. To rule out the possibility that DN-USF2 was blocking BDNF transcription nonspecifically by occluding the binding of another bHLH protein to CaRE2, we examined the effect of a second dominant-negative USF construct that works via a different mechanism. A-USF is a dominant-negative USF construct that contains an acidic extension in its DNA binding domain and that, when overexpressed in neurons, dimerizes with both endogenous USF1 and USF2, thereby preventing these molecules from binding to CaRE2 (Qyang et al., 1999). Consistent with the effects of DN-USF2, overexpression of A-USF significantly inhibited membrane depolarization induction of theBDNF promoter III-driven reporter gene, whereas transfection of the empty plasmid vector alone had no effect (Fig.3j). In total, these data indicate that the USFs are required for activity-dependent transcription from BDNFpromoter III.
Fig. 3.
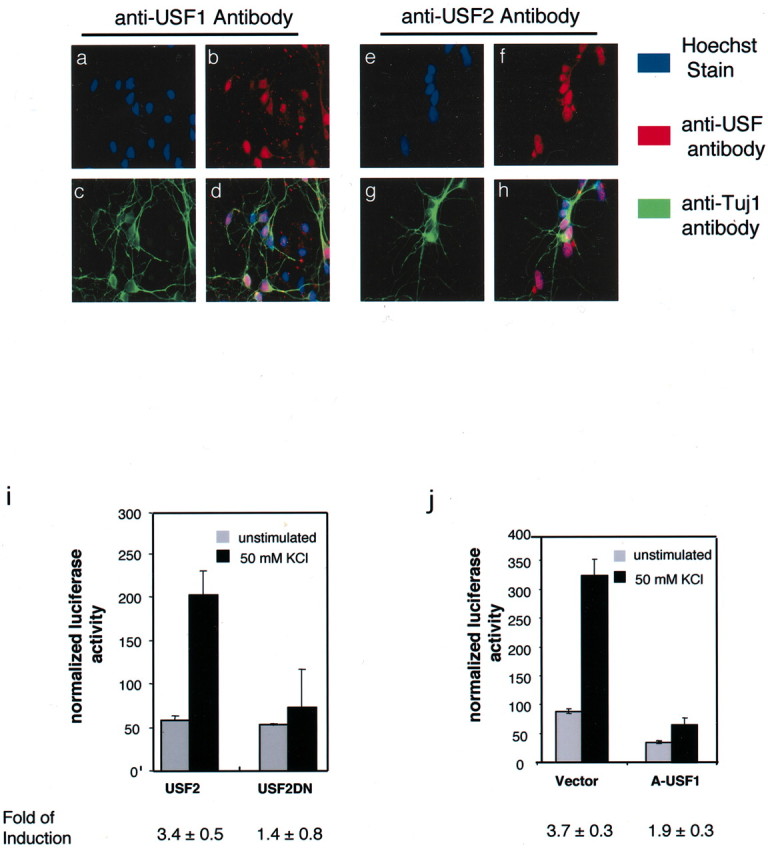
USFs mediate Ca2+-dependent activation of BDNF promoter III.a–h, USFs are expressed in the nucleus of cultured cortical neurons. E18 cortical cultures were grown for 5 DIV and then fixed and stained with the anti-USF1 or anti-USF2 antibodies (red; b, f) and an antibody against the neuronal marker β-tubulin III (anti-Tuj1, green; c, g). Nuclei were stained with the Hoechst dye (a, e). Nuclear USFs in Tuj1-positive cells appear in pink, with green marking the neuronal processes (d, h). i,j, Dominant-negative forms of USF block Ca2+-dependent activation of BDNFpromoter III. E18 + 3 DIV rat cortical cultures were transfected with the BDNF promoter III luciferase reporter and a mammalian vector expressing either the dominant-negative forms of USF (A-USF1 or USF2DN) or control vectors. A renilla luciferase reporter plasmid was cotransfected to normalize for transfection efficiency and sample handling.
Both USF1 and USF2 can regulate activity-dependent BDNFexon III transcription
The dominant-negative experiments suggest that USFs are required for Ca2+ regulation of BDNF transcription. However, both dominant negatives inhibit USF1 and USF2 and therefore do not distinguish whether one family member or both are the key regulators of Ca2+-dependent BDNF transcription. To ask whether either USF1 or USF2 is sufficient for activity-dependent BDNF exon III expression or whether both are required for the response, we cultured embryonic cortical neurons from either USF1 or USF2 null mice or their wild-type or heterozygous siblings. These cells were transfected with theBDNF reporter construct, and the induction of luciferase in response to depolarization was measured. We observed no significant difference in the membrane depolarization-mediated induction ofBDNF promoter III activity in any of the genotypes (Fig.4a). Neither did we observe significant alteration in the Ca2+responsiveness of BDNF promoter III in the USF1(−/−)/USF2(+/−) and USF1(+/−)/USF2(−/−) mice (data not shown). Taken together, these data suggest that both USF1 and USF2 are independently capable of mediating activity-dependent induction ofBDNF promoter III. Because the constitutive USF1/2 double knock-out mice die early in embryogenesis (Sirito et al., 1998), it is not possible at this stage to use these loss-of-function mutants to corroborate the role played by both USF1 and USF2 in BDNF transcription.
Fig. 4.

Endogenous USF1 and USF2 regulateBDNF promoter III in vivo.a, USF 1 and USF2 are both capable of regulating the Ca2+-inducible activation of BDNFpromoter III. E16 cortical neurons were obtained from USF1 and USF2 wild-type, heterozygous, or homozygous null mice and cultured in vitro for 3 d before transfection with theBDNF promoter III luciferase reporter. nrepresents the number of pups used for the measurement. The genotypes for the mice were determined by Southern blot analysis. Western blot analysis shows the relative amounts of USF1 and USF2 in nuclear extracts from USF1 and USF2 wild-type, heterozygous, and homozygous mice with antibodies specific to the USF1 and USF2 proteins.b, Endogenous USF1 and USF2 bind BDNFpromoter III in vivo. Cultured E18 + 5 DIV rat cortical neurons were treated with formaldehyde to cross-link DNA binding proteins to chromatin and then were subjected to chromatin immunoprecipitation with antibodies specific to USF1 and USF2 or a number of control antibodies. After reversing cross-links, we subjected eluted genomic DNA fragments to PCR with primers specific toBDNF promoter III or BDNF exon V. One percent of the input of the sheared chromatin before immunoprecipitation was used as a positive control for the PCR reaction. Negative controls include antibodies against c-Myc (a transcription factor), Bad (a cytoplasmic protein), Id (a bHLH protein lacking a DNA binding domain), or beads only.
The experiments with USF null mice suggest that both USF1 and USF2 may regulate BDNF promoter III. To seek evidence that both USF1 and USF2 when expressed at physiological levels are bound to the endogenous CaRE2 element of BDNF promoter III in vivo, we used a ChIP assay to study the in vivooccupancy of BDNF promoter III by USF1 and USF2. After first cross-linking DNA-bound proteins to chromatin in neuronal cultures, we lysed the cells and sheared the chromatin to an average of 150 bp in length. Next we used specific antibodies against USF1 and USF2 to immunoprecipitate these proteins along with the bound chromatin. After extensive washing and reversal of the cross-linking, we used specific PCR primers to test for the presence of BDNF promoter sequences that coimmunoprecipitated with the USF proteins. We were able to detect BDNF promoter III sequences in the USF immunoprecipitates (Fig. 4b). However, BDNF exon V, which should have been sheared away from promoter III, was not found in the pellet, indicating that immunoprecipitation of promoter III was specific (Fig. 4b). To control for antibody specificity, we performed the immunoprecipitation with a number of control antibodies and found that neither promoter III nor exon V of the BDNFgene immunoprecipitated with any of the control antibodies (Fig.4b), although the anti-c-Myc antibody can precipitate its target promoters effectively in NIH 3T3 cells (data not shown). In total, these data strongly suggest that both USF1 and USF2 regulate transcription from promoter III of the endogenousBDNF gene in vivo.
USFs are activated by Ca2+ signals via L-VSCCs
The ability of USFs to regulate transcription of BDNFexon III through a Ca2+-responsive element suggested to us that the transcriptional activity of USFs might be regulated by Ca2+-activated signaling pathways in neurons. To isolate the activity of the USFs from that of other Ca2+-responsive transcription factors on BDNF promoter III, we studied the effects of calcium signaling pathways on transcription from a plasmid containing three copies of a consensus USF binding element in front of a luciferase reporter gene (3×UBE-Luc) (Qyang et al., 1999). Although a single USF element alone is not sufficient to confer a Ca2+ response in the context ofBDNF promoter III, many transcription factors that act cooperatively at complex promoters can drive transcription independently when their elements are present in multiple copies close to the start site of transcription of a luciferase reporter gene (Tao et al., 1998, 2002). Indeed, when transfected into cultured cortical neurons, membrane depolarization induced a significant increase in transcription from the 3×UBE-Luc reporter plasmid (Fig.5a), whereas no induction of transcription was seen from a plasmid containing the luciferase reporter but lacking the USF binding enhancer sequence. Blocking L-VSCCs with nimodipine completely inhibited depolarization-induced transcription of the UBE reporter gene. In contrast, blockade of the NMDA type of glutamate receptors with APV had little effect (Fig.5b). Furthermore, activation of glutamate receptors did not stimulate transcription from the USF reporter plasmid, although under these conditions glutamate effectively induced transcription from a CREB reporter gene (Fig. 5c). Taken together, these results suggest that Ca2+ influx via L-VSCCs regulates the transcriptional activation of the USFs in cortical neurons.
Fig. 5.

USFs are Ca2+-regulated transcription factors activated via L-type VSCCs. a, USF binding element (UBE) is regulated by membrane depolarization. Cultured E18 + 3 DIV rat cortical neurons were transfected with either a firefly luciferase reporter gene driven by three repeats of the consensus USF binding element (3×UBE) or the control vector. b,c, USFs are activated by Ca2+ influx via L-type VSCC. Cultured E18 + 3 DIV rat cortical neurons were transfected with either the 3×UBE-Luc reporter plasmid or CRE-Luc plasmids. At 2 d after transfection the cells were treated with nimodipine, APV, or the carrier solution before depolarization with 50 mm KCl or glutamate stimulation. After 8 hr of stimulation the cells were lysed, and the luciferase activities were measured.d, e, The transcriptional activity of USF2 is regulated by membrane depolarization. Cultured E18 + 3 DIV rat cortical neurons were transfected with a Gal4-luciferase reporter plasmid and either an expression plasmid for the Gal4 DNA binding domain alone (control) or the Gal4-DNA binding domain fused to the transcriptional activation domain of USF2.d, A renilla luciferase reporter plasmid was cotransfected as a control for transfection efficiency and sample handling. At 2 d after transfection the cells were depolarized with 50 mm KCl for 9 hr; then the cells were lysed, and the luciferase activities were measured. e, An α-globin expression vector was cotransfected to control for transfection efficiency and sample handling. At 2 d after transfection the neurons were depolarized with 50 mm KCl for 1 hr; then total RNA was purified. The total RNA was reverse transcribed into cDNA, and luciferase and α-globin cDNAs were measured by quantitative real-time PCR. f, The transcription activity of USF is regulated by Ca2+ influx via L-type VSCC. Cultured E18 + 3 DIV rat cortical neurons were cotransfected with Gal4-USF2 and Gal4-Luc plasmids. At 2 d after transfection the cells were treated with nimodipine, APV, or the carrier solution before depolarization with 50 mm KCl or glutamate stimulation. After 8 hr of stimulation the cells were lysed, and the luciferase activities were measured.
There are a number of mechanisms by which Ca2+ influx could regulate the USFs. In mast cells surface receptor activation leads to nuclear translocation of USF2 (Frenkel et al., 1998). However, when we expressed USF1 or USF2 fused to green fluorescent protein (GFP) in cultured cortical neurons, we observed nuclear localization for both proteins independent of membrane depolarization (data not shown). In addition, immunofluorescent staining with anti-USF1- and anti-USF2-specific antibodies and preparation of nuclear extracts from cultured cortical neurons indicate that both USF1 and USF2 are primarily nuclear under both unstimulated and membrane-depolarized conditions (Fig.3a–h; data not shown). A second possibility is that the USFs could undergo regulated binding to the CaRE2 element (Berger et al., 1998; Marmillot and Scovell, 1998; Cheung et al., 1999). However, we observed no change in the binding of the USFs to CaRE2 by EMSA, using nuclear extracts prepared from unstimulated or membrane-depolarized neurons (data not shown). In addition, using the ChIP assay, we found USF1 and USF2 bound to the endogenousBDNF promoter III under both unstimulated and membrane-depolarized conditions (data not shown), indicating that the DNA binding activities of USFs to BDNF promoter III are not regulated by Ca2+ influx into neurons.
Because Ca2+ influx does not appear to regulate the nuclear localization or DNA binding of the USFs, we considered the possibility that Ca2+-dependent signaling pathways might regulate the transcriptional activation domains of the USFs directly. To determine whether Ca2+ influx directly regulates the ability of the USFs to activate transcription, we tethered the transcriptional activation domain of USF2 to the DNA binding domain of the yeast transcription factor Gal4 (Luo and Sawadogo, 1996b). When it was transfected into cortical neurons along with a reporter gene containing the Gal4 upstream-activating sequence driving expression of luciferase (Gal4-Luc), membrane depolarization induced a significant increase in luciferase expression (Fig. 5d). This induction requires the activation domain(s) of USF2 because the control vector that contains no USF2 showed no increase in response to membrane depolarization (Fig. 5d). Similar to the results observed for induction of the 3×UBE-Luc, pretreatment with nimodipine completely blocked the activation of Gal4-USF2, whereas APV had no significant effect (Fig.5f). To evaluate whether Ca2+ induction of Gal4-USF2-dependent transcription occurs with a rapid time course that would be required for USFs to be Ca2+-responsive regulators of BDNF transcription, we used a quantitative real-time RT-PCR assay to assess the induction of luciferase mRNA after 1 hr of membrane depolarization. Under these conditions we observed a significant membrane depolarization-dependent induction of the activity of Gal4-USF2 (Fig. 5e). These data indicate that the ability of USFs to activate transcription is enhanced by Ca2+ signaling pathways in neurons and suggest that a rapid biochemical modification of either the USFs or critical interacting proteins mediates Ca2+ induction of USF-dependent transcription.
USFs are general regulators of activity-dependent transcription
These experiments suggest that USFs are Ca2+-regulated transcription factors in neurons and raise the possibility that USFs may contribute to the inducible expression of activity-regulated genes in addition toBDNF. In fact, a number of genes characterized in vitro by EMSA and by reporter gene assays as USF target genes (Paterson et al., 1995; Howcroft et al., 1999) also are known to be induced by neuronal activity in vivo (Fig.6b), although the USFs previously have not been suggested to mediate this induction. We used the ChIP assay (described in Fig. 4b) to assess whether the USFs bind to the endogenous promoters of several of these activity-regulated genes in neurons. As shown in Figure 6a, anti-USF1 and anti-USF2 antibodies specifically precipitate the promoters for COX-2 and nAchRα7, which are known to be activity-regulated in vivo (Yamagata et al., 1993; Zhou et al., 2001) and to be USF target genes in vitro(Liu et al., 1999; Nagavarapu et al., 2001). Anti-USF antibodies also precipitated the promoter for the immediate early geneNur77, suggesting that Nur77 also may be a USF target gene. To determine the specificity of anti-USF antibodies in these ChIP experiments, we showed that these anti-USF antibodies do not pull down the GAP-43 promoter, which is neither activity-regulated nor a known USF target gene. Given that the transcriptional activity of the USFs is regulated by Ca2+ signaling in neurons and that USFs are bound to the endogenous promoters of a number of activity-inducible genes, these findings suggest that USF1 and USF2 may orchestrate the activity-regulated expression of a sizable group of neuronal genes that are important for neural development and synaptic plasticity.
Fig. 6.

USFs are general regulators of Ca2+-dependent gene transcription in the nervous system. a, USFs bind to the promoters of several activity-regulated neuronal genes in vivo. Cultured E18 + 5 DIV rat cortical neurons were treated with formaldehyde to cross-link DNA binding proteins to chromatin and were subjected to chromatin immunoprecipitation with antibodies specific to USF1 and USF2 or a number of control antibodies. After reversing the cross-linking, we subjected eluted genomic DNA fragments to PCR with primers specific for COX-2, Nur77,nAchRα7, and GAP-43 promoters.b, Neuronal activity-regulated genes that are known USF targets.
Discussion
USFs regulate activity-dependent transcription fromBDNF promoter III
In this study we have defined a new Ca2+-responsive E-box element, CaRE2 (ATCATATGAC), in BDNF promoter III. This element is required to confer Ca2+-responsive transcriptional activation of BDNF promoter III. Using a yeast one-hybrid screen, we identified the upstream stimulatory factors (USF1 and USF2) as CaRE2 binding proteins. Several lines of evidence indicate that the USFs are relevant regulators of activity-dependentBDNF transcription through CaRE2. We have shown that endogenous USF1 and USF2 from neuronal nuclear extracts bind to the CaRE2 sequence in vitro. The USFs are sufficient to bind directly to the CaRE2 sequence and only bind CaRE2 sequences that support activity-dependent transcription of the BDNFpromoter III luciferase reporter. Overexpression of dominant-negative forms of the USFs block activity-dependent transcription fromBDNF promoter III, suggesting that the transcriptional activity of the USFs is required for this induction. Finally, using a chromatin immunoprecipitation assay, we demonstrate that in neurons both USF1 and USF2 are bound to the CaRE2-containing region of promoter III in the endogenous BDNF gene. Either USF1 or USF2 appears to be sufficient to support activity-dependent BDNFexpression, because we observe that depolarization-induced increases inBDNF promoter III activity occur normally in mice null for either USF1 or USF2, as well as in USF1(−/−)/USF2(+/−) and USF1(−/+)/USF2(−/−) mice. Because the constitutive USF1/2 double knock-out mice exhibit early embryonic lethality (Sirito et al., 1998), loss-of-function studies that corroborate the role played by the USFs in BDNF transcription will await the generation of conditional or brain-specific USF1/2 double knock-out mice. Nonetheless, in total, our data strongly support a critical role for the USFs in the induction of activity-dependent transcription fromBDNF promoter III.
A new role for the USFs in the CNS
Although the USFs were among the first bHLH transcription factors to be identified and they were shown to be expressed in brain (Sirito et al., 1994), little was known about their function in the nervous system. Our data now suggest that these transcription factors may play an important role in the regulation of activity-dependent gene expression in neurons. We find that both USF1 and USF2 are expressed in the nuclei of neurons throughout the brain, including both the neocortex and the hippocampus (Fig. 5a; data not shown), brain regions in which neuronal activity induces transcription of a large set of genes. In these neurons both USF1 and USF2 are bound to the endogenous promoters of a number of activity-induced genes for which the products are important regulators of synaptic maturation and function, including BDNF, nAchRα7, andCOX-2. In addition, we find that USF1 and USF2 are bound to the promoter for the Nur-77 gene, an immediate early gene that encodes a transcription factor for which the expression is upregulated dramatically by neuronal activity.
On BDNF promoter III we have shown that the USFs cooperate with other transcription factors to regulate activity-dependent gene expression. We have identified three discrete Ca2+-response elements (CaRE1, CaRE2, and CaRE3/CRE) in BDNF promoter III that are required for the induction of exon III transcription in response to Ca2+ influx in neurons (Tao et al., 1998,2002). Mutation of any one of the CaREs effectively blocks the activity-dependent induction of BDNF exon III transcription, indicating that within the context of BDNF promoter III these three elements cooperatively promote transcription. However, when isolated from BDNF promoter III, each of these elements is independently inducible by neuronal activity, and each is bound by distinct transcription factors (CaRF, USF1/2, or CREB) for which the transcriptional activity can be modulated by Ca2+ signaling in neurons (Tao et al., 1998, 2002). These observations raise the question as to why all three factors are required to promote transcription in the context of the intact BDNF promoter III.
One possibility is that each of the BDNF regulatory factors serves a unique but essential role in regulating Ca2+ induction of BDNFtranscription. Like the induction of BDNF exon III expression itself, the activity of the CaRE1 binding protein CaRF is regulated in a Ca2+- and neural-selective manner, suggesting that this factor may confer stimulus and cell type selectivity with the expression of BDNF exon III (Tao et al., 2002). In contrast, the CaRE3/CRE binding protein CREB can be activated by a wide variety of stimuli, all of which lead to the phosphorylation of CREB at Ser133. However, recent studies have shown that Ca2+ influx into neurons induces phosphorylation of CREB at two additional sites, Ser142 and Ser143, both of which also are required for calcium-mediated CREB-dependent transcription in neurons (Kornhauser et al., 2002). Phosphorylation at these two additional sites appears to disrupt the interaction of CREB with CBP (Parker et al., 1999), raising the possibility that a factor other than CBP may mediate Ca2+ regulation of CREB-dependent transcription in neurons. USF activity together with CaRF and CREB may be required for the modification of chromatin structure and the recruitment of the basal transcriptional machinery to BDNFpromoter III, thereby facilitating effective Ca2+ regulation of BDNFpromoter III.
Interestingly, USF binding elements are found in tandem with CREB binding elements in a number of promoters (Cvekl et al., 1994; Durham et al., 1997; Kingsley-Kallesen et al., 1999), suggesting that cooperative activation of these two factors may be critical for the transcription of a number of Ca2+-inducible neuronal genes. A recent report suggests that the USFs may also be involved in the regulation ofBDNF promoter I (Tabuchi et al., 2002). Anti-USF antibodies were able to supershift a CRE binding complex on BDNFpromoter I, and a dominant-interfering form of USF (A-USF) was found to reduce moderately the calcium inducibility of BDNF promoter I. It remains to be determined whether the USFs directly interact withBDNF promoter I and whether USFs associate with this promoter in vivo as assessed by chromatin immunoprecipitation analysis. Because A-USF can act to sequester other USF interacting proteins such as E47 (Dear et al., 1997), the exact role of USF family members in BDNF promoter I regulation awaits further characterization. However, the potential involvement of USFs together with CREB in the regulation of alternativeBDNF promoters provides support for the idea that cooperation between these two calcium-responsive factors plays a role in regulating activity-dependent transcription in neurons.
Regulation of USF transcriptional activity
In addition to showing that CaRE2 is required for activity-dependent induction of BDNF promoter III, our findings suggest that the activity of USFs may be induced by calcium influx into neurons. As shown in Figure 5, membrane depolarization induced robust activation of USF-dependent transcription. This effect was blocked completely by L-type VSCCs-specific inhibitors but was unaffected by NMDA receptor inhibitors, suggesting that Ca2+ entry via L-type VSCCs mediates the activation of USF-dependent transcription in membrane-depolarized neurons. In contrast to membrane depolarization with elevated levels of KCl, the addition of glutamate to cultured neurons failed to activate USF-dependent transcription. Under these conditions glutamate induces significant Ca2+ influx through NMDA receptors and a modest amount of calcium influx through the L-type VSCC (Bading et al., 1995). The failure of glutamate to induce USF-dependent transcription under these conditions suggests that Ca2+ influx through the NMDA receptor may not activate USF activity. Glutamate-induced calcium influx through the L-type VSCC under these conditions appears not to be sufficient to activate the signaling molecules responsible for USF transcriptional activation. It is possible that the calcium signaling pathways activated by the NMDA receptors may be antagonistic to those activated by the L-VSCCs. Experiments designed to distinguish among these possibilities may yield insights into the integrative effect of calcium signals on activity-dependent neuronal gene expression.
Because the transcriptional activity of a Gal4-USF2 fusion protein also can be regulated rapidly by calcium signaling pathways in neurons, we postulate that a post-translational modification of either USFs or components of the transcriptional machinery that USFs bring to the promoters of Ca2+-responsive genes may mediate Ca2+ induction of USF-dependent transcription. Phosphorylation is a common means of regulating the activity of transcription factors, and USF1 has been reported to be a phosphoprotein in HeLa cells (Galibert et al., 1997). In melanocytes UV stress induction of the phosphorylation of USF1 by p38 MAP kinase is required for UV-induced expression of Tyrosinase (Galibert et al., 2001). Our preliminary data from two-dimensional isoelectric focusing experiments suggest that, when overexpressed in neurons, both USF1 and USF2 exist as multiple species with distinct isoelectric focusing points, consistent with the phosphorylation of the USFs at multiple sites (data not shown). Several kinases are known to be activated in neurons in response to calcium influx via L-VSCCs, including the MAP kinases Erk1/2 and p38, the calcium-calmodulin kinases II and IV, and protein kinase A (Mao et al., 1999; Shaywitz and Greenberg, 1999; Dolmetsch et al., 2001). However, whether phosphorylation of USFs or USF-associated factors is regulated by neuronal activity remains to be determined.
One mechanism by which neuronal activity might control the function of the USFs is by regulating the interaction between USFs and components of the basal transcription machinery. The USFs have been shown to interact physically with TAFII55, and via this interaction USFs can recruit the TATA-box binding complex TFIID (Workman et al., 1990; Reach et al., 1991; Kokubo et al., 1993; Chiang and Roeder, 1995). The USFs also have been shown to interact with TFII-I, a factor highly expressed in brain that is thought to mediate transcription from TATA-less promoters via binding to an Inr element (Roy et al., 1991, 1997;Cheriyath et al., 1998). BDNF promoter III is a TATA-less promoter that contains an Inr element near the transcriptional initiation site (Timmusk et al., 1993), suggesting that BDNFpromoter III could be regulated by a TFII-I-dependent mechanism. Future studies aimed at elucidating the molecular events that control the recruitment of these and other basal transcription factors to activity-dependent promoters such as BDNF promoter III may help to identify the mechanism by which USFs mediate Ca2+-dependent transcription in neurons.
Footnotes
We acknowledge the gracious support of the F. M. Kirby Foundation to the Division of Neuroscience (M.E.G.), Mental Retardation Research Center Grant HD18655 (M.E.G.), and National Institutes of Health Grants NS28829-07 (M.E.G.) and CA79579 (M.S.). This work also was supported by a Howard Hughes Medical Institute Predoctoral Fellowship (W.G.C.) and an American Cancer Society Postdoctoral Fellowship (A.E.W.). We thank Dr. Mario Sirito for help and advice on the USF knock-out mice and Pieter Dikkes and Ruth Ann-Pimental for technical assistance. We also thank Dr. Eric Griffith, Dr. Chris Cowan, Dr. Ricardo Dolmetsch, Dr. Janine Zieg, Paul L. Greer, Elizabeth Nigh, and members of the Greenberg laboratory for helpful discussions and critical reading of this manuscript.
Correspondence should be addressed to Dr. Michael E. Greenberg, Division of Neuroscience, Children's Hospital and Harvard Medical School, 300 Longwood Avenue, Boston, MA 02115. E-mail:michael.greenberg@tch.harvard.edu.
X. Tao's present address: Curis Incorporated, Cambridge, MA 02138.
References
- 1.Abdollah S, Macias-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2–Smad4 complex formation and signaling. J Biol Chem. 1997;272:27678–27685. doi: 10.1074/jbc.272.44.27678. [DOI] [PubMed] [Google Scholar]
- 2.Bading H, Segal MM, Sucher NJ, Dudek H, Lipton SA, Greenberg ME. N-methyl-d-aspartate receptors are critical for mediating the effects of glutamate on intracellular calcium concentration and immediate early gene expression in cultured hippocampal neurons. Neuroscience. 1995;64:653–664. doi: 10.1016/0306-4522(94)00462-e. [DOI] [PubMed] [Google Scholar]
- 3.Benson DL, Huntsman MM, Jones EG. Activity-dependent changes in GAD and preprotachykinin mRNAs in visual cortex of adult monkeys. Cereb Cortex. 1994;4:40–51. doi: 10.1093/cercor/4.1.40. [DOI] [PubMed] [Google Scholar]
- 4.Berger A, Cultaro CM, Segal S, Spiegel S. The potent lipid mitogen sphingosylphosphocholine activates the DNA binding activity of upstream stimulating factor (USF), a basic helix-loop-helix zipper protein. Biochim Biophys Acta. 1998;1390:225–236. doi: 10.1016/s0005-2760(97)00180-x. [DOI] [PubMed] [Google Scholar]
- 5.Bito H, Deisseroth K, Tsien RW. Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- 6.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 7.Carthew RW, Chodosh LA, Sharp PA. An RNA polymerase II transcription factor binds to an upstream element in the adenovirus major late promoter. Cell. 1985;43:439–448. doi: 10.1016/0092-8674(85)90174-6. [DOI] [PubMed] [Google Scholar]
- 8.Cheriyath V, Novina CD, Roy AL. TFII-I regulates Vβ promoter activity through an initiator element. Mol Cell Biol. 1998;18:4444–4454. doi: 10.1128/mcb.18.8.4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung E, Mayr P, Coda-Zabetta F, Woodman PG, Boam DS. DNA-binding activity of the transcription factor upstream stimulatory factor-1 (USF-1) is regulated by cyclin-dependent phosphorylation. Biochem J. 1999;344[Pt 1]:145–152. [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang CM, Roeder RG. Cloning of an intrinsic human TFIID subunit that interacts with multiple transcriptional activators. Science. 1995;267:531–536. doi: 10.1126/science.7824954. [DOI] [PubMed] [Google Scholar]
- 11.Clayton DF. Role of gene regulation in song circuit development and song learning. J Neurobiol. 1997;33:549–571. [PubMed] [Google Scholar]
- 12.Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- 13.Cvekl A, Sax CM, Bresnick EH, Piatigorsky J. A complex array of positive and negative elements regulates the chicken α A-crystallin gene: involvement of Pax-6, USF, CREB and/or CREM, and AP-1 proteins. Mol Cell Biol. 1994;14:7363–7376. doi: 10.1128/mcb.14.11.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dear TN, Hainzl T, Follo M, Nehls M, Wilmore H, Matena K, Boehm T. Identification of interaction partners for the basic helix-loop-helix protein E47. Oncogene. 1997;14:891–898. doi: 10.1038/sj.onc.1200912. [DOI] [PubMed] [Google Scholar]
- 15.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel–calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 16.Durham PL, Sharma RV, Russo AF. Repression of the calcitonin gene-related peptide promoter by 5-HT1 receptor activation. J Neurosci. 1997;17:9545–9553. doi: 10.1523/JNEUROSCI.17-24-09545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- 18.Frenkel S, Kay G, Nechushtan H, Razin E. Nuclear translocation of upstream stimulating factor 2 (USF2) in activated mast cells: a possible role in their survival. J Immunol. 1998;161:2881–2887. [PubMed] [Google Scholar]
- 19.Galibert MD, Boucontet L, Goding CR, Meo T. Recognition of the E-C4 element from the C4 complement gene promoter by the upstream stimulatory factor-1 transcription factor. J Immunol. 1997;159:6176–6183. [PubMed] [Google Scholar]
- 20.Galibert MD, Carreira S, Goding CR. The USF-1 transcription factor is a novel target for the stress-responsive p38 kinase and mediates UV-induced Tyrosinase expression. EMBO J. 2001;20:5022–5031. doi: 10.1093/emboj/20.17.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh A. Cortical development: with an eye on neurotrophins. Curr Biol. 1996;6:130–133. doi: 10.1016/s0960-9822(02)00442-6. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 23.Gregor PD, Sawadogo M, Roeder RG. The adenovirus major late transcription factor USF is a member of the helix-loop-helix group of regulatory proteins and binds to DNA as a dimer. Genes Dev. 1990;4:1730–1740. doi: 10.1101/gad.4.10.1730. [DOI] [PubMed] [Google Scholar]
- 24.Howcroft TK, Murphy C, Weissman JD, Huber SJ, Sawadogo M, Singer DS. Upstream stimulatory factor regulates major histocompatibility complex class I gene expression: the U2ΔE4 splice variant abrogates E-box activity. Mol Cell Biol. 1999;19:4788–4797. doi: 10.1128/mcb.19.7.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Impey S, Goodman RH. CREB signaling—timing is everything. Sci STKE. 2001;2001:E1. doi: 10.1126/stke.2001.82.pe1. [DOI] [PubMed] [Google Scholar]
- 27.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 28.Kingsley-Kallesen ML, Kelly D, Rizzino A. Transcriptional regulation of the transforming growth factor-β2 promoter by cAMP-responsive element-binding protein (CREB) and activating transcription factor-1 (ATF-1) is modulated by protein kinases and the coactivators p300 and CREB-binding protein. J Biol Chem. 1999;274:34020–34028. doi: 10.1074/jbc.274.48.34020. [DOI] [PubMed] [Google Scholar]
- 29.Koenig H, Lu C. RNA and protein synthesis in relation to neural function. A study of the neurobiological activity of actinomycin D in cat spinal cord. Trans Am Neurol Assoc. 1967;92:250–252. [PubMed] [Google Scholar]
- 30.Kokubo T, Takada R, Yamashita S, Gong DW, Roeder RG, Horikoshi M, Nakatani Y. Identification of TFIID components required for transcriptional activation by upstream stimulatory factor. J Biol Chem. 1993;268:17554–17558. [PubMed] [Google Scholar]
- 31.Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron. 2002;34:221–233. doi: 10.1016/s0896-6273(02)00655-4. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Antaya M, Boerboom D, Lussier JG, Silversides DW, Sirois J. The delayed activation of the prostaglandin G/H synthase-2 promoter in bovine granulosa cells is associated with down-regulation of truncated upstream stimulatory factor-2. J Biol Chem. 1999;274:35037–35045. doi: 10.1074/jbc.274.49.35037. [DOI] [PubMed] [Google Scholar]
- 33.Luo X, Sawadogo M. Antiproliferative properties of the USF family of helix-loop-helix transcription factors. Proc Natl Acad Sci USA. 1996a;93:1308–1313. doi: 10.1073/pnas.93.3.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo X, Sawadogo M. Functional domains of the transcription factor USF2: atypical nuclear localization signals and context-dependent transcriptional activation domains. Mol Cell Biol. 1996b;16:1367–1375. doi: 10.1128/mcb.16.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- 36.Marmillot P, Scovell W. Enhancement of transcription factor, USF, binding to the adenovirus major late promoter: effect of dithiothreitol and high mobility group protein-1. Biochim Biophys Acta. 1998;1395:228–236. doi: 10.1016/s0167-4781(97)00153-x. [DOI] [PubMed] [Google Scholar]
- 37.Moncollin V, Miyamoto NG, Zheng XM, Egly JM. Purification of a factor specific for the upstream element of the adenovirus-2 major late promoter. EMBO J. 1986;5:2577–2584. doi: 10.1002/j.1460-2075.1986.tb04537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagavarapu U, Danthi S, Boyd RT. Characterization of a rat neuronal nicotinic acetylcholine receptor α7 promoter. J Biol Chem. 2001;276:16749–16757. doi: 10.1074/jbc.M009712200. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 40.Pallas SL. Intrinsic and extrinsic factors that shape neocortical specification. Trends Neurosci. 2001;24:417–423. doi: 10.1016/s0166-2236(00)01853-1. [DOI] [PubMed] [Google Scholar]
- 41.Parker D, Rivera M, Zor T, Henrion-Caude A, Radhakrishnan I, Kumar A, Shapiro LH, Wright PE, Montminy M, Brindle PK. Role of secondary structure in discrimination between constitutive and inducible activators. Mol Cell Biol. 1999;19:5601–5607. doi: 10.1128/mcb.19.8.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paterson JM, Morrison CF, Mendelson SC, McAllister J, Quinn JP. An upstream stimulatory factor (USF) binding motif is critical for rat preprotachykinin-A promoter activity in PC12 cells. Biochem J. 1995;310:401–406. doi: 10.1042/bj3100401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 44.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 45.Qyang Y, Luo X, Lu T, Ismail PM, Krylov D, Vinson C, Sawadogo M. Cell type-dependent activity of the ubiquitous transcription factor USF in cellular proliferation and transcriptional activation. Mol Cell Biol. 1999;19:1508–1517. doi: 10.1128/mcb.19.2.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reach M, Xu LX, Young CS. Transcription from the adenovirus major late promoter uses redundant activating elements. EMBO J. 1991;10:3439–3446. doi: 10.1002/j.1460-2075.1991.tb04908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy AL, Meisterernst M, Pognonec P, Roeder RG. Cooperative interaction of an initiator-binding transcription initiation factor and the helix-loop-helix activator USF. Nature. 1991;354:245–248. doi: 10.1038/354245a0. [DOI] [PubMed] [Google Scholar]
- 48.Roy AL, Du H, Gregor PD, Novina CD, Martinez E, Roeder RG. Cloning of an Inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J. 1997;16:7091–7104. doi: 10.1093/emboj/16.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sasaki M, Gonzalez-Zulueta M, Huang H, Herring WJ, Ahn S, Ginty DD, Dawson VL, Dawson TM. Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc Natl Acad Sci USA. 2000;97:8617–8622. doi: 10.1073/pnas.97.15.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sawadogo M, Roeder RG. Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region. Cell. 1985;43:165–175. doi: 10.1016/0092-8674(85)90021-2. [DOI] [PubMed] [Google Scholar]
- 51.Sawadogo M, Van Dyke MW, Gregor PD, Roeder RG. Multiple forms of the human gene-specific transcription factor USF. I. Complete purification and identification of USF from HeLa cell nuclei. J Biol Chem. 1988;263:11985–11993. [PubMed] [Google Scholar]
- 52.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 53.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 54.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 55.Sirito M, Walker S, Lin Q, Kozlowski MT, Klein WH, Sawadogo M. Members of the USF family of helix-loop-helix proteins bind DNA as homo- as well as heterodimers. Gene Expr. 1992;2:231–240. [PMC free article] [PubMed] [Google Scholar]
- 56.Sirito M, Lin Q, Maity T, Sawadogo M. Ubiquitous expression of the 43- and 44-kDa forms of transcription factor USF in mammalian cells. Nucleic Acids Res. 1994;22:427–433. doi: 10.1093/nar/22.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sirito M, Lin Q, Deng JM, Behringer RR, Sawadogo M. Overlapping roles and asymmetrical cross-regulation of the USF proteins in mice. Proc Natl Acad Sci USA. 1998;95:3758–3763. doi: 10.1073/pnas.95.7.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sur M, Leamey CA. Development and plasticity of cortical areas and networks. Nat Rev Neurosci. 2001;2:251–262. doi: 10.1038/35067562. [DOI] [PubMed] [Google Scholar]
- 59.Tabuchi A, Sakaya H, Kisukeda T, Fushiki H, Tsuda M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J Biol Chem. 2002;277:35920–35931. doi: 10.1074/jbc.M204784200. [DOI] [PubMed] [Google Scholar]
- 60.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 61.Tao X, West AE, Chen WG, Corfas G, Greenberg ME. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33:383–395. doi: 10.1016/s0896-6273(01)00561-x. [DOI] [PubMed] [Google Scholar]
- 62.Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- 63.Tokuyama W, Okuno H, Hashimoto T, Xin Li Y, Miyashita Y. BDNF upregulation during declarative memory formation in monkey inferior temporal cortex. Nat Neurosci. 2000;3:1134–1142. doi: 10.1038/80655. [DOI] [PubMed] [Google Scholar]
- 64.Viollet B, Lefrancois-Martinez AM, Henrion A, Kahn A, Raymondjean M, Martinez A. Immunochemical characterization and transacting properties of upstream stimulatory factor isoforms. J Biol Chem. 1996;271:1405–1415. doi: 10.1074/jbc.271.3.1405. [DOI] [PubMed] [Google Scholar]
- 65.West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Workman JL, Roeder RG, Kingston RE. An upstream transcription factor, USF (MLTF), facilitates the formation of preinitiation complexes during in vitro chromatin assembly. EMBO J. 1990;9:1299–1308. doi: 10.1002/j.1460-2075.1990.tb08239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 69.Zhou Y, Deneris E, Zigmond RE. Nicotinic acetylcholine receptor subunit proteins α7 and β4 decrease in the superior cervical ganglion after axotomy. J Neurobiol. 2001;46:178–192. doi: 10.1002/1097-4695(20010215)46:3<178::aid-neu1001>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]