

Abstract
Angelman syndrome (AS) is a disorder of human cognition characterized by severe mental retardation and epilepsy. Recently, a mouse model for AS (Ube3a maternal null mutation) was developed that displays deficits in both context-dependent learning and hippocampal long-term potentiation (LTP). In the present studies, we examined the molecular basis for these LTP and learning deficits. Mutant animals exhibited a significant increase in hippocampal phospho-calcium/calmodulin-dependent protein kinase II (CaMKII), specifically at sites Thr286 and Thr305, with no corresponding change in the levels of total CaMKII. In addition, mutants show a reduction in CaMKII activity, autophosphorylation capability, and total CaMKII associated with postsynaptic density. These findings are the first to implicate misregulation of CaMKII as a molecular cause for the neurobehavioral deficits in a human learning disorder.
Keywords: Angelman syndrome, calcium/calmodulin-dependent protein kinase II, long-term potentiation, postsynaptic density, protein phosphatase, autophosphorylation
Introduction
Angelman syndrome (AS) is a neurological disorder characterized by severe mental retardation, propensity for seizure (associated with a characteristic electroencephalogram), and an ataxic gait (Williams et al., 1995; Buoni et al., 1999; Laan et al., 1999). AS is estimated to occur in 1 of every 15,000–20,000 births and is most often caused by maternal chromosomal deletion of 15q11-q13; however, maternal uniparental disomy and imprinting center mutations also can cause AS (Clayton-Smith, 1993). In 1997, the Ube3a gene within the 15q11–13 chromosomal region was identified as the genetic locus for AS (Kishino et al., 1997; Matsuura et al., 1997; Sutcliffe et al., 1997).Ube3a codes for an E6-AP ubiquitin ligase, an enzyme involved in protein degradation through the ubiquitin-associated proteosome-mediated pathway. The E6-AP ubiquitin ligase is known to be involved in the degradation of four proteins, including the p53 tumor suppressor protein (Huibregtse et al., 1991), a human homolog to the yeast DNA repair protein Rad23 (Kumar et al., 1999), the multicopy maintenance protein 7 subunit involved in the initiation of DNA replication (Kuhne and Banks, 1998), and E6-AP, which is a target for itself (Nuber et al., 1998). Unfortunately, identification of these target proteins has yet to shed light on any possible mechanism underlying the etiology and learning and memory deficits of AS.
Since the identification of the AS locus, some progress has been made in understanding the genetic and biochemical mechanisms underlying this disorder. One important step forward in this endeavor was the development of a mouse model for AS (Jiang et al., 1998). This mouse model, generated by a maternal null mutation in the Ube3agene, revealed the characteristic subregion-specific paternal silencing of the Ube3a gene in the brain. Because of imprinting, theUbe3a gene product in the hippocampus and Purkinje cell layer of the cerebellum is derived primarily from the maternal copy of the gene. Importantly, the mouse model recapitulates the brain-specific imprinting of Ube3a in humans, and also the seizure, ataxic gait, and hippocampus-dependent abnormalities observed in human AS.
In undertaking these studies, we reasoned that understanding the deficits in hippocampal long-term potentiation (LTP) that we had observed previously in the AS mouse model should facilitate identifying the putative molecular mechanisms underlying the behavioral learning deficits in AS. Thus, in the present studies, we examined the hippocampal LTP deficits observed in the Ube3amaternal-deficient (m−/p+) mice and investigated the biochemical basis for these deficits.Ube3a maternal-deficient mice exhibited deficits in both NMDA receptor (NMDAR)-dependent and -independent LTP, suggesting that the source of the observed LTP deficits resides downstream of calcium influx. These results led us to examine calcium-dependent signal transduction pathways activated during LTP induction. We found aberrant autophosphorylation and diminished activity of hippocampal calcium/calmodulin-dependent protein (CaM) kinase II (CaMKII), without a change in total CaMKII concentrations. The change in the phosphorylation state of CaMKII was coupled to a significant reduction in CaMKII associated with postsynaptic density (PSD). In addition, we found that activity for the major phosphatases for CaMKII, protein phosphatase 1 (PP1) and PP2A, is significantly reduced in m−/p+ mice. Together, these results suggest that changes in CaMKII underlie the hippocampal LTP deficits. More importantly, these data indicate that alterations in calcium-dependent hippocampal CaMKII activation may play a role in the learning and memory deficits in the m−/p+ mutant mice and in humans with AS as well.
Materials and Methods
Hippocampal slice preparation and electrophysiology.Mutants with the Ube3a null mutation were developed as described previously (Jiang et al., 1998). Hippocampal slice preparation and electrophysiology were performed as described previously (Roberson and Sweatt, 1996). Briefly, hippocampal slices (400 μm) were maintained on an interface chamber and bathed in oxygenated artificial CSF (in mm: 125 NaCl, 2.5 KCl, 1.24 NaH2PO4, 25 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2) (1 ml/min) in an interface chamber maintained at either 25 or 32°C. Extracellular field recordings were made in area CA1 stratum radiatum by stimulation of the Schaffer collateral synapse. Stable baseline synaptic transmission was established for 20 min at an intensity of 40–50% of the maximum population EPSP (pEPSP) before LTP-inducing high-frequency stimulation (HFS). HFS consisted of one or three sets, each set consisting of two trains of 100 Hz stimulation for 1 sec, separated by 20 sec (NMDA-dependent LTP induction) or three trains of 200 Hz stimulation for 0.5 sec, separated by 2 min (NMDA-independent LTP induction). Stimulus intensity of the HFS was matched to the intensity used in the baseline recordings. Responses were monitored for 20 min before high-frequency stimulation was given to ensure a stable baseline. Measurements are shown as the average slope of the pEPSP from six individual traces and are standardized to baseline recordings.
Tissue preparation. Mouse brains were immediately removed and perfused in ice-cold saline (in mm: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d-glucose, 2 CaCl2, and 1 MgCl2, saturated with 95% O2–5% CO2, pH 7.4). Hippocampi were dissected and then homogenized in 2–3 ml of buffer (20 mm Tris-HCl, pH 7.5, 1 mm EGTA, 1 mmEDTA, 25 μg/ml aprotinin, 25 μg/ml leupeptin, 1 mmNa4P2O7, 500 μm phenylmethylsulfonyl fluoride, 4 mmpara-nitrophenylphosphate, and 1 mm sodium orthovanadate). Protein concentrations were determined using the BCA protein determination assay (BCA protein assay reagent; Pierce, Rockford, IL).
Western blot analysis. Hippocampal tissue homogenates were resolved on 10% SDS-polyacrylamide gels and transferred to Immobilon-P membranes. The membranes were blocked in 5% dry-milk solution for 1 hr. The primary antibody was diluted 1:3000 in the same 5% dry-milk solution. The membranes were incubated in the primary antibody for 1 hr at room temperature and then washed in Tween–TBS (TTBS) buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.05% Tween 20). The secondary antibody was diluted 1:30,000 in 5% dry-milk solution, and the membranes were incubated in the secondary antibody for 1 hr at room temperature. The membranes were again washed in TTBS before being developed using the enhanced chemiluminescence method (Amersham Pharmacia Biotech, Arlington Heights, IL). The bands of each Western blot were quantified with densitometry using a StudioScan desktop scanner and NIH Image software to determine the amount of each of the tested proteins.
Kinetic assays. CaMKII enzyme activity was measured by quantifying incorporation of [32P]PO4 into a synthetic peptide substrate as described by Roberson et al. (1996). Briefly, in vitro assays consisted of 5–10 μg of hippocampal homogenate protein resuspended in reaction buffer (in mm: 200 Tris, 5 EGTA, 10 EDTA, and 20 Na2PO4), 100 μm autocamtide peptide substrate, [γ-32P]ATP, 0.5 mm ATP, and H2O in a final assay volume of 50 μl. The reaction samples were incubated at 37°C for 20 min and terminated by addition of 25 μl of stop solution (in mm: 225 H3PO4 and 1 ATP). Two aliquots from each sample were transferred onto Whatman P-51 paper. After drying at room temperature, the filter papers were washed three times for 10 min in 0.25 mH3PO4 and once for 2 min in 95% (v/v) ethanol with gentle agitation. Chromatography papers were air dried before quantitation by liquid scintillation counting.
Autophosphorylation. CaMKII autophosphorylation assays were performed from whole hippocampal homogenates. Homogenates were thawed before use, and 15 μg of protein was used for each assay. Each 50 μl assay reaction was incubated on ice for 15 min with the addition of 10 μm ATP to allow the CaMKII enzyme to reach a state of autophosphorylation equilibrium. Reactions were initiated with the addition of 0.5 μCi of [γ-32P]ATP and 0.5 mm ATP with or without the 10 nm calmodulin and 1 mmCaCl2. Reactions were incubated at 32°C for 3 min and stopped with the addition of SDS-containing sample buffer. Samples were heated at 90°C for 1 min, and 25 μl was loaded onto a 7.5% SDS-polyacrylamide gel. After electrophoresis, gels were dried and placed on Biomax film overnight at −70°C in the presence of an intensifying screen. Band intensities were analyzed and quantified by densitometry using a Studioscan desktop scanner and NIH Image software.
Thr305 serum specificity. The synthetic peptides CRRKLKGAIL-pT-TMLATRN containing the phosphorylated threonine (corresponding to αCaMKII-Thr305) were coupled to keyhole limpet hemocyanin and injected into rabbits (Elgersma et al., 2002). The antisera were screened for recognition to activated CaMKII using purified CaMKII (Sigma, St. Louis, MO) subjected to a kinase reaction in the presence or absence of Ca2+ and calmodulin (see Fig.4A). No immunoreactivity was seen when blots were probed with P-Thr305 CaMKII serum to purified CaMKII samples incubated with PP1 (Sigma) after the autophosphorylation reaction (data not shown). Specificity to the P-Thr305 CaMKII site was tested by preincubation of the serum with either the synthetic peptide phosphorylated at Thr305 or a synthetic peptide lacking the Thr305phosphate. Precleared serum was used to probe blots from hippocampal homogenates that were subjected to a kinase phosphorylation reaction in the presence or absence of Ca2+ and calmodulin (see Fig. 4A). Because of the apparent high specificity of the serum exclusively to the P-Thr305 CaMKII site, the serum was not affinity purified and was directly used to probe blots of hippocampal homogenates from wild-type (WT) and m−/p+animals.
Fig. 4.

Increased phosphorylated Thr305CaMKII in m−/p+ mutants.A, Left blot, Western blot analysis showing that P-Thr305/306 CaMKII serum recognizes purified CaMKII activated with Ca2+/CaM, but not nonactivated CaMKII. Right blot, Specificity for the Thr305 autophosphorylation site was tested with autophosphorylation reactions with hippocampal homogenates from wild-type mice in the absence [control (Ctrl)] or presence (+Ca2+/CaM) of calcium and calmodulin. Control Western blots show an increase in immunoreactivity for activated CaMKII corresponding to the α and β subunits of CaMKII. Addition of antigen completely blocked recognition of phospho-Thr305/306 CaMKII in control and activated homogenates (peptide block). B,Top, Representative Western blot analysis of hippocampal homogenates probed for P-Thr305/306 CaMKII (right blot). The same blot was then stripped and reprobed for total αCaMKII (left blot).Bottom, Quantitative Western blot analysis shows no change in total αCaMKII but an increased Thr305αCaMKII phosphorylation in m−/p+ mutants in whole hippocampal homogenates [wild type (Wt), 100 ± 12, n = 7; m−/p+, 133.2 ± 3.3,n = 7; p = 0.02]. Data represent mean ± SEM. *p < 0.05.
Postsynaptic density isolation. PSD was isolated as described previously (Carlin et al., 1980). After decapitation, brains were removed and hippocampi were dissected and frozen on dry ice within 2 min. The speed of hippocampus isolation and flash freezing were optimized to reduce ischemia-induced CaMKII translocation to the PSD, as described previously by Suzuki et al. (1994). Hippocampi were homogenized by hand using a homogenization buffer without detergents (0.32 m sucrose, 1 mmNaHCO3, 1 mmMgCl2, and 0.5 mmCaCl2). The homogenates are then centrifuged at 1400 × g for 10 min. The resulting pellet is washed in the homogenization buffer and centrifuged at 710 ×g for 10 min. The supernatant from this spin is combined with the one from the first spin, and this sample is centrifuged at 13,800 × g for 10 min. The pellet from this spin is then resuspended in a second homogenization buffer (0.32m sucrose and 1 mmNaHCO3). This sample is then placed on top of a trilevel sucrose gradient (0.85, 1.0, and 1.4 msucrose). The sample is centrifuged through the gradient at 82,500 × g for 1 hr. The protein fraction between the 1.0 and 1.4m sucrose layers is removed. A Triton solution (1% v/v Triton X-100 in 0.32 m sucrose and 12 mm Tris-HCl, pH 8.1) and the second homogenization buffer are added to this fraction in equal amounts. This suspension is stirred gently and then centrifuged at 30,800 ×g for 20 min. The resulting pellet is resuspended in the second homogenization buffer and used in subsequent Western blotting experiments.
Immunohistochemistry/microscopy. Mice were perfused, and brains were harvested as described previously (Varga et al., 2000). Forty micrometer coronal hemisections were cut on a cryostat and placed in cryoprotectant until time of experiment. Sections were first washed three times for 10 min in TBS and then blocked for 2 hr (blocking buffer: 1% BSA, 5% NGS, and 0.3% Triton X-100 in TBS). Sections were then incubated in primary antibody for 24 hr (antibody buffer: 0.25% BSA, 5% NGS, and 0.3% Triton X-100 in TBS). Phospho-Thr286 CaMKII primary antibody (Promega, Madison, WI) was used at 1:500. Sections were again rinsed three times for 10 min with TBS and then incubated in fluorescent-labeled secondary antibody for 2 hr in the dark. Alexa Fluor 568 goat anti-rabbit IgG (Molecular Probes, Eugene, OR) was diluted to 1:500 in TBS. Sections were again rinsed three times for 10 min, mounted on Plus slides, and coverslipped with Supermount aqueous mounting medium (Biogenex). Confocal imaging was accomplished using a FluoView FV300 confocal laser scanning microscope on a BX50WI fixed-stage upright microscope equipped with a FV5-ZM stepper motor and FluoView software (OlympusOptical, Melville, NY). Fluorescent images were acquired via excitation with a krypton laser (568 nm line), a 510–550 nm bandpass emission filter set, and a 10× or 20× objective (OlympusOptical) using the confocal aperture suggested by the manufacturer. Kalman accumulation averaging of three or four was used. Maximum projection images were generated with the FluoView software. All figures were ultimately prepared using Photoshop software (Adobe Systems, San Jose, CA).
PP1/PP2A phosphatase assay. Phosphatase activity in whole hippocampal homogenates was measured using a serine–threonine phosphatase assay kit with a PP1/PP2A-specific phosphopeptide (K-R-pT-I-R-R) (Upstate Biotechnology, Lake Placid, NY). Free phosphate was detected after a 20 min incubation at 32°C using a colorimetric assay and compared with a freshly prepared phosphate standard. Specific activity was determined as the amount of picomoles of phosphate released per minute per microgram of protein. Data represent mean ± SEM. An analysis with a value ofp < 0.05 was considered to be statistically significant.
Data collection and statistical analysis. Hippocampal area CA1 pEPSPs were recorded using Axon 1320 Digidata data acquisition hardware operated by Axon pClamp 8.2 software. One-way ANOVA was used for multiple group data, and the paired Student's t test was used to evaluate differences in experimental control and mutant values. GraphPad Prism data analysis software was used for graph production, curve fitting, and statistical analysis. Data represent mean ± SEM. An analysis with a value of p < 0.05 was considered to be statistically significant.
Results
The AS mouse model is a powerful tool that can be effectively used to better understand the biochemical mechanisms involved in human maternal E6-AP deficiency. However, relating the genetic and biochemical abnormalities observed in a mouse model for AS to humans with AS has certain limitations. For instance, we have gained substantial insight into the brain region-specific maternal imprinting of Ube3a from investigation of the AS mouse model. This is consistent with our current knowledge that the human Ube3agene undergoes brain-specific imprinting (Rougeulle et al., 1997; Vu and Hoffman, 1997), but the exact brain regions for Ube3aimprinting in the human can only be inferred. However, there are many phenotypic and morphological abnormalities that are quite similar in the AS mouse model. For example, human newborns appear to be physically well formed, but by 12 months of age show signs of cranial growth declination and decreased brain size. Although not as prominent, the m−/p+ mice show similar changes in head size, with reduced brain weight at 18 d of age that persists into adulthood (Jiang et al., 1998). In addition, many other phenotypic characteristics of human AS are also seen in the AS mouse model, such as an ataxic gait, seizure, an abnormal electroencephalogram, and learning deficiencies. We reasoned that, by understanding the deficits in hippocampal synaptic function and molecular and biochemical changes in the AS mouse model, we could identify putative molecular mechanisms contributing to the cognitive deficits in human AS.
Recovery of Ube3a mutant LTP
Numerous examples exist of differential responses of hippocampal CA1 LTP induction with variations in stimulus intensities, duration of stimulation, or changes in incubation temperatures (Krelstein et al., 1990; Muller and Lynch, 1990). A standard LTP-inducing stimulation consisting of a 1 sec, 100 Hz stimulation elicits a long-lasting increase in synaptic potentiation when slices are maintained at room temperature (25°C). Alternatively, maintaining slices at a higher temperature (30–32°C) and/or increasing the number of high-frequency trains of stimulation can produce a potentiation that is substantially higher in magnitude and longer lasting. Manipulating variables such as stimulus intensity, temperature, and number of stimulations is often used to determine the efficacy or potency of an applied drug or, as in this case, to evaluate the severity or penetrance of a mutant genotype.
Initial studies showing hippocampal CA1 LTP deficits in theUbe3am−/p+mutants (Jiang et al., 1998) were performed in an interface chamber maintained at 25°C and coupled with a modest LTP-inducing stimulus (consisting of two 1 sec trains of 100 Hz stimulation separated by 20 sec). As with any LTP deficit determined with a single LTP-induction protocol, it is unknown whether the LTP impairment in the m−/p+mutants is caused by the inability to reach an LTP induction threshold for a specific type of LTP, or alternatively, whether it reflects a more severe perturbation in a signal transduction cascade absolutely required for the induction of LTP. Therefore, we varied temperature and LTP-induction protocols to determine the nature of the LTP deficit in the Ube3a knock-out.
To test the possibility that the LTP deficits in m−/p+ mice were caused by an increase in the threshold of LTP induction, LTP was induced while maintaining hippocampal slices at increased temperatures and increasing the number of stimulating trains. In the first experiment, we maintained the interface chamber temperature at 32°C. As observed previously for LTP induced at 25°C, LTP induced at 32°C was essentially eliminated in AS mice (Fig.1A). In the next experiment, we combined the increase in temperature (32°C) with an increase in the number of stimulus trains delivered to the slice. Surprisingly, when the slice was maintained at 32°C and 100 Hz stimulation was increased from one to three sets of HFS, LTP induction was rescued in the Ube3a mutant (Fig. 1B). These results suggest that the LTP impairment inUbe3a-deficient mice is primarily attributable to an alteration in the threshold of synaptic stimulation necessary for LTP induction to occur.
Fig. 1.

The LTP deficit in m−/p+ mice is NMDA receptor dependent. Baseline synaptic responses of wild-type (m+/p+) (○) and maternal-deficient mutant (m−/p+) (●) animals were measured before application of LTP-inducing HFS (indicated byarrows) to hippocampal area CA1. A, Slices were maintained at 32°C, and LTP was induced with a single set of HFS consisting of two trains of 100 Hz stimulation for 1 sec separated by 20 sec. B, Slices were maintained at 32°C, and NMDA receptor-dependent LTP was induced with three sets of HFS, with each set separated by 10 min. C, NMDA receptor-independent LTP was induced with three sets of HFS consisting of 200 Hz stimulation for 1 sec separated by 4 min. The black bar represents the application of the NMDA receptor antagonist AP-5. All results are graphed as the percentage of potentiation standardized to the baseline recording. Dashed linesrepresent the 100% mark of baseline synaptic responses. Data represent mean ± SEM.
The induction of LTP is highly dependent on an influx of postsynaptic Ca2+. The ability to rescue LTP in m−/p+ mice with saturating HFS raises the question of whether the derangement of LTP induction was upstream (NMDA receptor activation) or downstream of Ca2+ influx. To help determine whether the increase in LTP threshold observed in m−/p+Ube3a mutants is caused by insufficient postsynaptic Ca2+ influx, slices were stimulated in a manner that allows calcium-dependent but NMDA receptor-independent LTP. Thus, LTP was induced with repeated very-high-frequency stimulation (200 Hz for 1 sec at 32°C) in the presence of the NMDA receptor antagonist AP-5. Using this protocol, a robust, long-lasting potentiation was induced in m+/p+controls, but Ube3am−/p+mutants exhibit a loss of potentiation by 30 min after the last stimulus (Fig. 1C). The inability of m−/p+mutants to achieve NMDA receptor-independent LTP induction suggests that at least one locus of the observed LTP deficit resides downstream of calcium influx.
Potential protein kinase targets
The absence of E6-AP ubiquitin ligase causes the accumulation of E6-AP targeted proteins (such as p53) in areas for which maternal gene imprinting is occurring, but no known LTP-associated protein has been identified as a target of E6-AP. Therefore, potential candidates for the observed physiologic deficits downstream of calcium influx could include any calcium-dependent, or alternatively, calcium-activated enzyme necessary for LTP induction (Fig.2A). Thus, in the next phase of our experiments, we focused on four calcium-regulated protein kinase families that have gained attention for playing prominent roles in LTP and learning and memory in the behaving animal: protein kinase C (PKC), cAMP-dependent protein kinase A (PKA), extracellular signal-regulated kinase (ERK), and CaMKII. Common characteristics make these attractive candidates for possible involvement in the m−/p+ mutant LTP and behavioral phenotype. For example, all four kinases are (1) activated by HFS in the hippocampus, (2) directly or indirectly affected by calcium influx into the postsynaptic neuron, (3) involved in the induction of hippocampal LTP, and (4) known to be necessary in mammalian learning. Because the m−/p+mutants are deficient for E6-AP ubiquitin ligase in the hippocampus, we predicted that there would be a significant increase in the relevant protein target of E6-AP in the mouse. Therefore, an examination was performed of the levels of a wide variety of specific members of these protein kinase families in the hippocampus of our m−/p+ mutant mice.
Fig. 2.
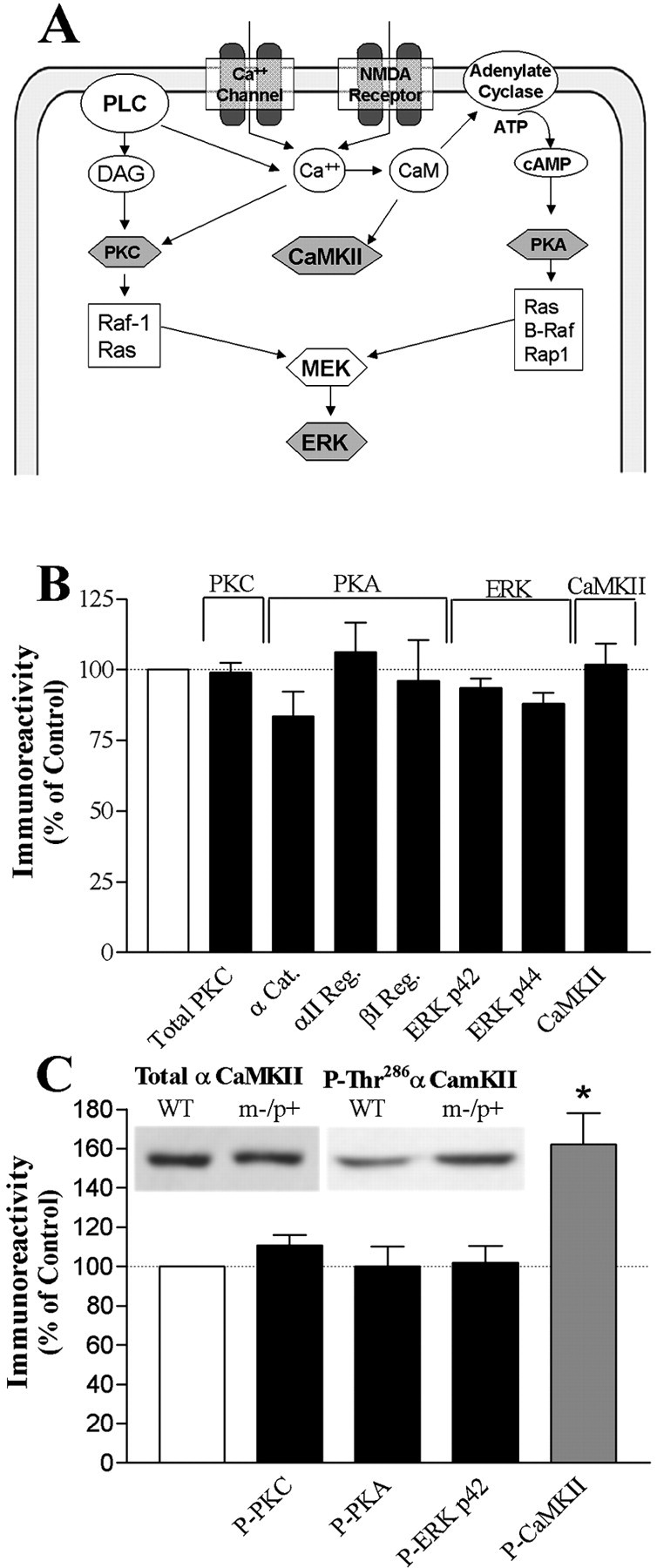
Kinase concentrations in m−/p+ mutant hippocampus.A, A schematic diagram of a postsynaptic neuron showing potential candidates (shaded hexagons) for LTP and learning disruption observed in E6-AP-deficient mice. B, Quantitative Western blot analysis on hippocampal homogenates from wild-type and m−/p+ mutants probed for total PKC, PKA [α catalytic (α Cat.), αII regulatory (αII Reg.), and βI regulatory (βI Reg.) subunits], ERK (p42 and p44 MAP kinase), and CaMKII. C, Top, Representative Western blot showing no change in total CaMKII but an increase in P-Thr286 αCaMKII in m−/p+ mouse hippocampal homogenates. Bottom, Quantitative Western blot analysis of hippocampal homogenates for protein levels of phospho-PKC, phospho-PKA, phospho-p42 ERK, and phospho-αCaMKII. A significant increase was detected in m−/p+mice only for total P-Thr286 αCaMKII (WT, 100 ± 7.1%, n = 10; m−/p+, 162 ± 16.2%,n = 10; p = 0.0024). Results shown are the percentage difference of m−/p+ protein levels compared with that of wild type. Dashed line represents control level of 100%. *p < 0.05. Data represent mean ± SEM. PLC, Phospholipase C; DAG, diacylglycerol; MEK, mitogen and extracellular signal regulated kinase.
PKC/PKA/ERK/CaMKII protein levels in m−/p+ mutants
Knock-out mouse models for PKC-β and PKC-γ make it evident that these specific isoforms likely play important roles in mammalian synaptic plasticity and memory (Abeliovich et al., 1993a,b; Weeber et al., 2000). Therefore, any ubiquitin ligase-dependent alterations in PKC isoform expression could have profound effects on LTP induction and learning. Levels of seven different PKC isoforms were unchanged in the hippocampus of Ube3a mutants (data not shown). To test whether slight changes in each isoform could result in a significant change in the total PKC complement, a broad-spectrum antibody for total PKC detection was used. We determined that the total PKC protein level in m−/p+homogenates was no different from that of controls (Fig.2B).
We were particularly interested in the levels of PKA, because ubiquitin-proteosome-dependent reductions in the regulatory subunit concentrations of PKA are required for long-term facilitation inAplysia as part of a mechanism by which autonomously active PKA is generated (Chain et al., 1999). In addition, PKA is known to be important in triggering the late phase of LTP and in long-term memory in mice (Chetkovich et al., 1991; Roberson and Sweatt, 1996; Abel et al., 1997). However, we found no difference in the α (catalytic) and αI or βII (regulatory) subunits of PKA in the hippocampus of AS mice (Fig. 2B). This suggests that E6-AP is not involved in PKA regulatory or catalytic subunit degradation, and that this site is not the locus of derangement in m−/p+mice.
It has only recently become clear that the ERK cascade is essential for the induction of NMDA receptor-dependent and -independent LTP and plays a critical role in mammalian learning (Adams and Sweatt, 2002). This prompted us to examine the protein levels of hippocampal p42 and p44 ERK levels in the m−/p+Ube3a mutant. Hippocampal homogenates from Ube3amutant animals probed for p42 and p44 ERK MAP (mitogen-activated protein) kinase immunoreactivity showed no change in protein levels compared with wild-type controls (Fig.2B).
CaMKII is particularly enriched in the brain and exhibits multifunctional roles in calcium-mediated signal transduction processes. CaMKII is known to be involved in synaptic plasticity and learning. Neuronal CaMKII is predominately composed of the α and β subunits, with sizes of 52 and 60 kDa, respectively. The regulation of CaMKII is complex, and is dependent on many factors, including calcium influx, subunit composition, binding to the postsynaptic density, calmodulin availability, and the autophosphorylation state of the holoenzyme. These characteristics make this enzyme especially susceptible to changes within the cellular milieu. However, no changes in total CaMKII concentrations were seen in AS mice (Fig.2B). Thus, the results on levels of specific PKC, PKA, ERK, and CaMKII isoforms strongly suggest that protein levels of several kinases known to be involved in synaptic plasticity and learning are normal in the AS hippocampus.
Hippocampal protein phosphorylation
Although an absence of significant changes in protein concentration were observed for PKC, PKA, ERK, and CaMKII, changes in the phosphorylated form of these kinases may not be evident by total protein determinations. The important regulatory actions of phosphorylation are well established, especially regarding the regulation of protein kinase activity. Such is the case in each of the kinases we investigated. Alterations in levels of phosphorylated protein kinases may underlie the disruption in LTP and learning in the AS mouse. Moreover, there are several examples of protein phosphorylation acting as a molecular switch to target a protein for ubiquitination and degradation through the proteosome pathway (Basu and Haldar, 2002; Zheng et al., 2002). Thus, phosphorylated kinases involved in LTP may be a target of E6-AP. To test this hypothesis, we examined the level of phosphorylated PKC, PKA, ERK, and CaMKII. No changes were seen in the phosphorylation levels of PKC, PKA, or ERK (P42) isolated from hippocampal homogenates of m−/p+ mice. However, a significant increase was detected in phospho-αCaMKII at the autophosphorylation site threonine 286 (Thr286) (Fig. 2C).
These data suggest that alterations in CaMKII activity, secondary to altered autophosphorylation, could contribute to the LTP and learning phenotype of the AS mouse. This observation becomes increasingly important as it has been widely demonstrated, using genetic and pharmacological manipulations, that normal CaMKII function is necessary for learning and memory. Moreover, the alteration in CaMKII autophosphorylation appears to be a specific and restricted phenomenon. Importantly, absence of E6-AP in the hippocampus of m−/p+ mice does not affect the overall phosphorylation state of any of the other proteins we tested. These results highlight the specific nature of the hippocampal derangement associated with the Ube3amaternal-deficient phenotype.
Autophosphorylation assays
To confirm the changes in αCaMKII autophosphorylation, we performed another experiment using a back-phosphorylation approach. Our rationale for this experiment is as follows: the result showing an increase in hippocampal P-Thr286 αCaMKII in our Ube3a maternal-deficient mice suggested that sites for additional autophosphorylation would no longer be available. Therefore, we performed an autophosphorylation assay in which CaMKII-activating concentrations of Ca2+and CaM were added to hippocampal homogenates, and the amount of subsequent additional autophosphorylation was quantified by assessing32P incorporation from [32P]ATP. Figure3A shows that the amount of Ca2+/CaM-induced CaMKII autophosphorylation was significantly reduced in hippocampal homogenates from our Ube3a-deficient mice. An important caveat to keep in mind when assessing these results is that this assay does not give an indication of which sites are autophosphorylated [i.e., the assay does not distinguish between autophosphorylation at threonine 286 versus threonine 305 or Ser314 (Patton et al., 1990) of the α catalytic subunit]. Nevertheless, this observation supports the Western blot analysis results showing that increased autophosphorylated αCaMKII is present in the Ube3a maternal-deficient mouse hippocampus.
Fig. 3.

Increased phosphorylated Thr286αCaMKII corresponds to reduced post hocautophosphorylation and kinase activity. A,Top, Representative autoradiograph from hippocampal homogenates corresponding to α and β catalytic subunits of CaMKII in the presence of [γ-32P]ATP before and after Ca2+/calmodulin stimulation. Bottom, Quantitative analysis of the total Ca2+/CaM-dependent CaMKII autophosphorylation in hippocampal homogenates of wild-type animals (■) or m−/p+ mutants (▪) shows a significant reduction in total CaMKII autophosphorylation after activation with calcium and calmodulin (wild type, 100 ± 5.5%,n = 6; m−/p+, 71 ± 8.9,n = 7; p = 0.002).Bars represent the percentage of quantified signal compared with that of wild types. B, Hippocampal homogenates from wild-type animals (■) or m−/p+ mutants (▪) were assayed for basal CaMKII activity and CaMKII activity after activation with Ca2+ and CaM. The m−/p+ mutants (n = 6) show no significant difference in basal phosphotransferase activity compared with wild-type controls (n = 7). Lower total activity after CaMKII activation in the presence of Ca2+ and CaM was seen in m−/p+ mutants (wild type, 0.33 ± 0.05, n = 6; m−/p+, 0.487 ± 0.04,n = 7; p = 0.048). Data represent mean ± SEM. N/D, Not detected;Wt, wild type. *p < 0.05.
In vitro CaMKII assay
Although the activity of CaMKII is highly sensitive to calcium influx, the activity of the enzyme and its sensitivity to successive increases in calcium concentrations are altered after autophosphorylation. Changes in the phosphorylation state of the α subunit of CaMKII can alter the kinetic properties of the holoenzyme, control translocation, and modify enzyme function, all without a detectable change in total CaMKII concentrations. Our observation of an altered phosphorylation state of CaMKII suggests a resulting change in CaMKII enzymatic activity. Specifically, a greater ratio of P-Thr286 αCaMKII to nonautophosphorylated αCaMKII enzyme would suggest an increased basal calcium/calmodulin-independent activity. The increase in autonomously active CaMKII complement should also translate into a reduced fold-increase in activity in response to activation with Ca2+ and CaM. We tested this hypothesis by performing in vitro CaMKII activity assays using hippocampal homogenates and comparing the amount of basal activity and Ca2+/CaM-stimulated activity between wild-type and m−/p+animals. The maternal-deficient mice showed a reduced, but not quite significant, basal CaMKII phosphotransferase activity (Fig.3B). Moreover, assays performed in the presence of calcium and calmodulin revealed a significant reduction in the phosphotransferase activity of the maternal-deficient mutants (Fig.3B). No differences are seen when the percentage of activation is determined (percentage change of basal vs activated phosphotransferase activity) (data not shown). These data demonstrate that the increases in the autophosphorylation state of CaMKII associated with E6-AP deficiency cause an overall reduction in CaMKII activity, most noticeably as a significant reduction in response to Ca2+ and CaM.
Thr305-αCaMKII autophosphorylation
Our in vitro CaMKII activity results were unexpected. We hypothesized that a greater P-Thr286-αCaMKII complement would translate into a greater overall basal CaMKII activity in the absence of Ca2+ and CaM. One explanation for these results is that a subset of CaMKII in the assay is in an inactive state. Previous in vitro experiments indicate that autophosphorylation of Thr305 and Thr306 sites on α and β CaMKII, respectively, occurs after Thr286-αCaMKII phosphorylation under some circumstances (Colbran and Soderling, 1990; Patton et al., 1990;Mukherji and Soderling, 1994). This can affect both the Ca2+/CaM-independent activity (Lou and Schulman, 1989) as well as the Ca2+/CaM-dependent activity (Kuret and Schulman, 1985; Hashimoto et al., 1987; Lickteig et al., 1988; Lou and Schulman, 1989; Mukherji and Soderling, 1994).
Therefore, we hypothesized that increased phosphorylation of Thr305 αCaMKII (P-Thr305 αCaMKII) was occurring in the AS mice as an explanation for the decreased basal enzyme activity despite the increase in Thr286-αCaMKII autophosphorylation. We tested this hypothesis using an antiserum raised against a peptide duplicate of the phosphorylated Thr305, Thr306 CaMKII site for the α and β CaMKII subunits, respectively (CRRKLKGAIL-pT-TMLATRN). It should be noted that, because of the high homology between the α and β subunits of CaMKII, the peptide used resulted in an antiserum that was able to recognize both the Thr305αCaMKII and Thr306 βCaMKII subunits (Fig. 4). Figure 4Ashows the specificity of the serum to the phosphorylated Thr305/306 CaMKII site. Purified CaMKII (Sigma) was placed in a kinase reaction mixture in the presence or absence of Ca2+ and CaM (described in Materials and Methods). Western blot analysis using the Thr305/306 CaMKII serum showed immunoreactivity specifically to the activated CaMKII. We then tested for specificity to the P-Thr305/306 CaMKII site using hippocampal homogenates from control wild-type mice. Again, homogenates were placed in a kinase reaction with or without Ca2+ and CaM, and Western blot analysis was performed using the Thr305/306 CaMKII serum. Both the α and β subunits of CaMKII were detected using the P-Thr305/306 CaMKII serum to probe hippocampal homogenates from wild-type animals (Fig.4A). The wild-type control samples (no peptide preincubation) show immunoreactivity corresponding to the α and β subunits of CaMKII, which is increased in the homogenate after activation with Ca2+ and CaM. Immunoreactivity is nearly absent if the Thr305/306 CaMKII serum is preincubated with a phosphorylated Thr305/306 peptide (Fig. 4A), and no change is seen in immunoreactivity if serum was preincubated with the nonphosphorylated synthetic peptide (data not shown). These results suggest that the Thr305/306 CaMKII serum is highly sensitive to the autophosphorylated species of CaMKII, is specific to the Thr305/306 CaMKII autophosphorylation site, and is reliable to test for changes in P-Thr305 αCaMKII in hippocampal extracts.
Using the Thr305/306 CaMKII serum, we found that mice maternally deficient for Ube3a showed a significant increase in Thr305 αCaMKII autophosphorylation in extracts of hippocampal homogenates (Fig.4B). Thus, the m−/p+mutants exhibit altered phosphorylation of αCaMKII at both sites of autophosphorylation, Thr286 (Fig.2C) and Thr305 (Fig.4B). The increase in P-Thr305 αCaMKII is consistent with the observed reduction in basal CaMKII activity in our in vitroassays and also with the loss of Ca2+/CaM-induced CaMKII activation, and indeed appears to be sufficient to explain the observation.
Postsynaptic density isolation
Recently, a T305D CaMKII-transgenic mouse was generated that mimics persistent inhibitory phosphorylation. This mutation is capable of decreasing the association of CaMKII with the PSD and can detrimentally affect LTP and learning (Elgersma et al., 2002). In contrast, blocking inhibitory phosphorylation increases CaMKII in the PSD. These remarkable results raise the interesting question of whether increased Thr305-phosphorylated αCaMKII in our m−/p+mice could result in decreased PSD-associated CaMKII. To test this hypothesis, we examined levels of total and autophosphorylated CaMKII from PSDs isolated from the hippocampus of wild-type and m−/p+ mice. We found that, in our m−/p+mutants, the total PSD-associated CaMKII was significantly reduced, to nearly one-half of that found in wild types (Fig.5A). These results suggest that in the AS mice, there are sufficient amounts of phosphorylated Thr305 αCaMKII to cause a considerable translocation of the CaMKII complement away from the PSD. To test further the phosphorylated state of the CaMKII present in the PSD, we probed for P-Thr286 and P-Thr305 CaMKII. Surprisingly, we found a 2.5-fold increase in P-Thr286 CaMKII but no change in P-Thr305 CaMKII (Fig.5B). These results support recent data showing that αCaMKII autophosphorylated at Thr305/306has lower affinity for the PSD than nonphosphorylated αCaMKII (Strack et al., 1997a; Shen et al., 2000).
Fig. 5.

PSD-associated CaMKII and autophosphorylated CaMKII. A, B, Top, Representative Western blot for PSD-associated total CaMKII (A) and P-Thr286 and P-Thr305 CaMKII (B).A, Bottom, PSD fractions isolated from the hippocampus of WT (■) or m−/p+ mutants (▪) show a reduction of ∼50% in total PSD-associated CaMKII (WT, 100.0 ± 4.2, n = 3; m−/p+, 54.6 ± 6.7,n = 4; p = 0.045).B, Bottom, P-Thr286CaMKII shows an ∼2.5-fold increase in the PSD fraction (WT, 100.0 ± 1.9, n = 3; m−/p+, 264.3 ± 36.4,n = 4; p = 0.012). P-Thr305 CaMKII levels in the PSD were detectable but showed no significant differences between wild-type and m−/p+ mice. Data represent mean ± SEM. *p < 0.05.
A caveat to these results arises because of the ability of CaMKII to rapidly translocate to the PSD after animals have been killed (Suzuki et al., 1994). Suzuki et al. (1994) have reported that, in rat frontal cortex, the amount of αCaMKII is 2.7, 4.0, 7.8, 8.6, or 9.4% of the total PSD protein at 0, 2, 5, 30, or 60 min after decapitation. To minimize the potential of postkilling CaMKII translocation, we rapidly removed and flash froze hippocampi within 2 min of decapitation. It is believed that an ischemia-induced chemical modification renders the enzyme insoluble and causes the association of CaMKII with the PSD; however, the exact mechanism underlying the translocation is unknown. Thus, it is difficult to link changes in PSD-associated CaMKII in mice deficient for hippocampal E6-AP with this phenomenon. Despite this caveat, our observations of an increased concentration of P-Thr286 αCaMKII in PSDs from maternal-deficient mice support findings by Elgersma et al. (2002) of the subsequent Thr305phosphorylation step promoting PSD dissociation. These observations also indicate that not all Thr286-phosphorylated CaMKII enzyme is also phosphorylated at Thr305, suggesting that the Thr286-autophosphorylated kinase does not automatically undergo subsequent phosphorylation at Thr305. Overall, these results are in remarkable agreement with the observations reported by Elgersma et al. (2002) and are consistent with our hypothesis of altered Thr305 autophosphorylation in the AS mouse model.
Immunohistochemistry
Immunohistochemical techniques were used to localize the changes seen in immunoreactivity to phosphorylated CaMKII at Thr286. Because we had seen a significant increase in Thr286-autophosphorylated CaMKII in our Western blot analysis of whole hippocampal homogenates (Fig. 2C) and in an isolated PSD fraction (Fig.5B), we used immunohistochemistry to determine whether there might be qualitative differences in the cellular distribution of hippocampal CaMKII in AS mice. As shown in Figure6, phosphorylated Thr286 CaMKII is localized in the wild-type mouse under basal conditions most strongly in the pyramidal cell layer of area CA1 as well as the granule cell layer of the dentate gyrus. The greatest increase in immunoreactivity is seen in the pyramidal cells in area CA3 (Fig.6E,F). Weaker immunoreactivity is seen in the dendritic layers of these cells, including the stratum oriens and stratum radiatum of areas CA1 and CA3 and the molecular layer of the dentate gyrus. The distribution of phosphorylated CaMKII at Thr286 in an Angelman mouse hippocampus appears qualitatively the same as that of a wild-type mouse, yet the total immunoreactivity is increased across all labeled areas. Overall, these data suggest that the increase in autophosphorylated CaMKII in AS mice is not specifically localized to one cellular subregion.
Fig. 6.

Increased immunoreactivity to phosphorylated CaMKII at Thr286 in Angelman mouse hippocampus. Phosphorylated CaMKII at Thr286 was detected immunohistochemically in the hippocampi of wild-type (A,C, E) and Angelman (B,D, F) mice. Increases in immunoreactivity are seen in the stratum pyramidale, stratum oriens, and stratum radiatum of CA1 and CA3 as well as the molecular layer and granule cell layer of the dentate gyrus. A,B, 100× magnification of the hippocampus.C, D, 200× magnification of area CA1.E, F, 200× magnification of area CA3.
Protein phosphatases PP1/PP2A
The observed increases in autophosphorylated CaMKII suggest that a causative candidate for this disruption would be a disruption of a CaMKII phosphatase. Previous investigation has revealed the necessity for phosphatase activity in the regulation of neuronal plasticity and memory formation (for review, see Winder and Sweatt, 2001). A group of these phosphatases, such as PP1 and PP2A, exhibit high CaMKII-specific phosphatase activity and are associated with LTP and learning (Strack et al., 1997b; Blitzer et al., 1998; Kasahara et al., 1999; Brown et al., 2000; Bennecib et al., 2001; Winder and Sweatt, 2001). Therefore, we sought to determine whether alterations in the levels of the major CaMKII phosphatases, PP1 and PP2A, were responsible for the increase in the P-Thr286 αCaMKII species. We found no significant difference in PP1 or PP2A total protein levels in hippocampal homogenates from m−/p+ mice (Fig. 7A). These results indicate that Ube3a deficiency does not affect protein levels of two known phosphatases associated in P-Thr286 αCaMKII dephosphorylation.
Fig. 7.

Decreased PP1/PP2A activity in m−/p+ mutants. A, Quantitative Western blot analysis showing no change in total protein levels of PP1 and PP2A from hippocampal homogenates of m−/p+ mutant animals compared with WT controls. The dashed line indicates wild-type protein levels. B, Phosphatase activity was measured for PP1/PP2A using a PP1/PP2A-specific phosphopeptide (K-R-pT-I-R-R). Phosphatase activity was significantly reduced in the m−/p+ mutants (▪) (0.659 ± 0.08 pmol · min−1 · μg−1protein; n = 5) compared with wild-type controls (■) (1.758 ± 0.27 pmol · min−1 · μg−1protein; n = 8; p = 0.009). Data represent mean ± SEM. *p < 0.05.
In a final series of experiments, PP1/PP2A phosphatase activity was assayed to determine whether the increase in CaMKII autophosphorylation was a result of alterations in phosphatase capacity. Phosphatase activity was measured using a PP1/PP2A-specific phosphopeptide (K-R-pT-I-R-R). Phosphatase activity using this assay system was found to be significantly reduced in the m−/p+mutants (0.659 ± 0.08 pmol · min−1 · μg−1protein; n = 5) compared with controls (1.758 ± 0.27 pmol · min−1 · μg−1protein; n = 8; p = 0.009) (Fig.7B). The prodigious 2.5-fold decrease in PP1/PP2A phosphorylase activity in the m−/p+mutants strongly suggests that the aberrant state of P-Thr286 and P-Thr305 αCaMKII phosphorylation is attributable to changes in the activity of one or both of these important phosphatases. This observation also suggests that the increase in Thr305 αCaMKII is attributable to both the increase in P-Thr286 αCaMKII, because phosphorylation at Thr286 precedes phosphorylation at Thr305 (Colbran and Soderling, 1990; Hanson and Schulman, 1992), and reduced PP1 and/or PP2A activity, which are known to dephosphorylate P-Thr305 αCaMKII in vitro(Patton et al., 1990).
Discussion
Consistent with the hypothesis that altered LTP parallels a learning deficit in mice and humans, we show that a mouse model for a human mental retardation syndrome displays pronounced hippocampal LTP deficits. Furthermore, the finding that CaMKII is perturbed, without changes in other learning-related kinases, suggests CaMKII as the site for the decreased plasticity and learning and memory defects. These results are consistent with the current literature implicating a role for CaMKII in learning, but extend this to include human learning as well. We reason that our mouse model reflects the mechanisms that are in place in children afflicted with AS. This is supported by our knowledge that the maternal imprinting of Ube3a occurs in both systems, and that this imprinting is restricted to the brain in both humans (Rougeulle et al., 1997; Vu and Hoffman, 1997) and mice (Jiang et al., 1998). Finally, the CaMKII-dependent LTP deficit appears to be caused by altered CaMKII regulation in the hippocampus, specifically because of alterations in the autophosphorylation state of the enzyme. These data thereby implicate CaMKII as being required for normal learning in humans.
Insights into the LTP defects in Ube3a maternal-deficient mice
The necessity of CaMKII for synaptic plasticity is well established. The induction of NMDA receptor-dependent LTP requires CaMKII activation in the postsynaptic neuron (Nicoll and Malenka, 1995;Lisman et al., 1997), and mice deficient for αCaMKII show deficits in hippocampal LTP (Silva et al., 1992; Hinds et al., 1998). The sites of CaMKII autophosphorylation are also important in LTP induction. Mutations of the Thr286 site to prevent autophosphorylation or, conversely, to produce a calcium-independent form of αCaMKII, result in LTP deficits for some types of LTP inducing stimulation (Giese et al., 1998). With the recent production of a knock-in mouse with mutations that either mimic or prevent inhibitory phosphorylation comes the first insight into the physiologic role of inhibitory autophosphorylation of CaMKII in synaptic plasticity and learning (Elgersma et al., 2002). These studies reveal that T305D mutants show a significant LTP deficit in studies using theta burst stimulation and 100 Hz, 1 sec HFS protocols. These results are especially exciting because we are investigating effects of both phospho-Thr286 and Thr305 CaMKII increases that, because of maternal imprinting of the Ube3a gene, are nearly exclusive to the hippocampus. We conclude that the derangements in CaMKII autophosphorylation at Thr286 and Thr305 contribute to the overall AS phenotype. Indeed, the results of Elgersma et al. (2002) suggest that the specific molecular change in phospho-Thr305 αCaMKII is sufficient to explain the LTP and learning deficits seen in the AS mouse model.
Regulatory consequences of altered CaMKII phosphorylation
What are the potential effects associated with an increased Thr286 and Thr305 phosphorylation in CaMKII in m−/p+mutants? In a population of CaMKII molecules in which Thr286 and Thr305 are chronically phosphorylated, one would predict that the overall effect would be reduced basal activity, because of Thr305 phosphorylation, and insensitivity to increased Ca2+/CaM concentrations, because of the combination of Thr286 and Thr305 autophosphorylation. This scenario is in agreement with our in vitro assay results, which do not show an increase in basal activity in the absence of Ca2+/CaM, despite the presence of increased P-Thr286 αCaMKII. Also in agreement is the reduced activity in the presence of Ca2+/CaM, which could reflect both the Ca2+/CaM-insensitive Thr286 and inhibited Thr305 CaMKII complement. Moreover, this may also explain the hippocampal LTP deficit when using a modest LTP-inducing HFS protocol, but in which there is normal LTP with saturating HFS. A compromised, but not absent, CaMKII signal in CA1 may result in an inability of LTP induction with the application of a modest stimulus input. However, if an LTP-inducing protocol is robust, the result is sufficient CaMKII activation needed to trigger LTP induction.
Altered PSD-associated CaMKII
The potential regulatory problems involved with the aberrant autophosphorylated CaMKII are compounded in light of recent studies focusing on CaMKII autophosphorylation and enzyme translocation. In a series of elegant studies by Shen and colleagues (Shen and Meyer, 1999;Shen et al., 2002), it was found that the activation of NMDA receptors results in translocation of CaMKII from the cytosol to the postsynaptic density regions (Shen and Meyer, 1999). Also, HFS triggers a transient translocation of CaMKII from the cytosol to the PSD that is primarily dependent on the autophosphorylation state of the CaMKII at Thr286 (Shen et al., 2000). Results byElgersma et al. (2002) reveal that a T305D CaMKII mouse, which mimics persistent inhibitory phosphorylation, shows decreased association of CaMKII with the PSD. Conversely, a TT305/6VA CaMKII mutation results in greater PSD-associated CaMKII. Together, these results are consistent with published reports that P-Thr286αCaMKII is localized to the PSD, whereas P-Thr305 αCaMKII causes disassociation from the PSD. Our results suggest a derangement of this regulatory mechanism in our AS mouse model.
Our results show that the total pool of CaMKII in AS mice has more Thr286 and Thr305 autophosphorylation. However, the PSD-associated fraction of CaMKII shows increased Thr286 phosphorylation but not increased Thr305 phosphorylation. This supports the model that Thr286 phosphorylation promotes PSD association by binding to the NMDAR and is in agreement with the observation that association with the NMDAR effectively blocks inhibitory phosphorylation (Bayer et al., 2001) and that Thr305 phosphorylation promotes PSD dissociation (Elgersma et al., 2002). This situation does not preclude there being a population of unaffected CaMKII available for normal signaling and activation, supported by our saturating HFS protocol LTP results. The in vitro activity results showing CaMKII activation in the m−/p+mutants, albeit reduced total activation, is also consistent with this idea. Regardless, the alterations seen for both CaMKII autophosphorylated regulatory sites (Thr286 and Thr305) may represent a cumulative disruption in CaMKII signaling capability in the hippocampus. This may explain why the behavioral and LTP deficits in the E6-AP maternal-deficient mice are more pronounced than those in the transgenic mice described by Elgersma et al. (2002).
Protein phosphatase activity and altered autophosphorylated CaMKII
Our data support the hypothesis that CaMKII is not a target of E6-AP-dependent degradation but is indirectly affected by E6-AP deficiency, as observed by the significant increase in the P-Thr286 and P-Thr305 αCaMKII without an increase in total CaMKII complement. The considerable reduction in PP1/PP2A phosphatase activity suggests that the changes in CaMKII phosphorylation are a direct result of reduced protein phosphatase activity, specifically PP1 and/or PP2A. What may be the underlying mechanism of reduced PP1/PP2A activity? The observed alterations in P-Thr286 and P-Thr305 CaMKII may be caused by the endogenous alterations associated with hippocampal E6-AP deficiency, specifically an increase in proteins usually targeted by a now-absent ubiquitin ligase. For example, p53 is a known target of E6-AP and grossly accumulates in the hippocampus of m−/p+ mice, as well as in humans with AS. An interesting possibility arises from the identification of a p53-associated protein, designated p53 binding protein 2 (p53BP2), which can bind p53 and PP1 in a mutually exclusive manner (Helps et al., 1995). Very little is known about p53BP2, and its physiological importance in the CNS is unclear. However, it is known that p53BP2 can inhibit the phosphatase activity of PP1 at nanomolar concentrations. It is hypothesized that p53BP2 may be involved in the dephosphorylation and regulation of p53, but its effect on CaMKII phosphorylation is completely unknown at present. This raises the interesting possibility that the chronic presence and considerable increase in p53 in the imprinted areas of AS mice and humans can directly affect PP1 activity through p53BP2. The effect of p53BP2 may occur either through an upregulation of p53BP2 in response to p53 levels or through a change in the localization of p53, and subsequently p53BP2, throughout the neuron. An alternative hypothesis is that phosphorylation of copious p53 protein can cause phosphatase inhibition. This hypothesis is supported by recent research showing that inhibition of PP1 can occur after p53 phosphorylation in rat cardiomyocytes (Long et al., 2002). The actions of p53BP2 or p53 phosphorylation in Thr286 CaMKII dephosphorylation represent an exciting area of future research.
Conclusions
The association of the observed neurobehavioral features of AS is consistent with our current knowledge of the role of CaMKII in mammalian neural processes. The most important implication of our results relates to learning in humans. Various experimental data support the presupposition that the AS mouse model accurately reflects the condition in vivo in AS children. Our data support the hypothesis that the loss of hippocampal LTP underlies the learning deficit in humans. Finally, our observations support the hypothesis that the Ube3a maternal deficient-associated increase in the P-Thr286 and Thr305 CaMKII may underlie the deficits in synaptic plasticity and learning in our AS mouse model, and potentially underlie the etiology of human AS. This study is significant because it represents the first report that an alteration in CaMKII has the potential to cause a human disorder associated with a severe learning deficiency.
Footnotes
This work was supported by the cores of the Baylor College of Medicine Mental Retardation Research Center (HD24064). We thank Dr. Jonathan Levenson for constructive criticism and helpful discussion during the preparation of this manuscript.
Y.-H.J. and Y.E. contributed equally to this work.
Correspondence should be addressed to Dr. J. David Sweatt, Baylor College of Medicine, Division of Neuroscience, One Baylor Plaza, Houston, TX 77030. E-mail: jsweatt@bcm.tmc.edu.
References
- 1.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Modified hippocampal long-term potentiation in PKC-γ-mutant mice. Cell. 1993a;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- 3.Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKC-γ mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993b;75:1263–1271. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- 4.Adams JP, Sweatt JD. Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 5.Basu A, Haldar S. Signal-induced site specific phosphorylation targets Bcl2 to the proteasome pathway. Int J Oncol. 2002;21:597–601. [PubMed] [Google Scholar]
- 6.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 7.Bennecib M, Gong C, Grundke-Iqbal I, Iqbal K. Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser262/356. FEBS Lett. 2001;490:15–22. doi: 10.1016/s0014-5793(01)02127-5. [DOI] [PubMed] [Google Scholar]
- 8.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 9.Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM. Long-term potentiation induced by theta frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci. 2000;20:7880–7887. doi: 10.1523/JNEUROSCI.20-21-07880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buoni S, Grosso S, Pucci L, Fois A. Diagnosis of Angelman syndrome: clinical and EEG criteria. Brain Dev. 1999;21:296–302. doi: 10.1016/s0387-7604(99)00007-8. [DOI] [PubMed] [Google Scholar]
- 11.Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22:147–156. doi: 10.1016/s0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- 13.Chetkovich DM, Gray R, Johnston D, Sweatt JD. N-Methyl-d-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proc Natl Acad Sci USA. 1991;88:6467–6471. doi: 10.1073/pnas.88.15.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clayton-Smith J. Clinical research on Angelman syndrome in the United Kingdom: observations on 82 affected individuals. Am J Med Genet. 1993;46:12–15. doi: 10.1002/ajmg.1320460105. [DOI] [PubMed] [Google Scholar]
- 15.Colbran RJ, Soderling TR. Calcium/calmodulin-independent autophosphorylation sites of calcium/calmodulin-dependent protein kinase II. Studies on the effect of phosphorylation of threonine 305/306 and serine 314 on calmodulin binding using synthetic peptides. J Biol Chem. 1990;265:11213–11219. [PubMed] [Google Scholar]
- 16.Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM, Giese KP, Silva AJ. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron. 2002;36:493–505. doi: 10.1016/s0896-6273(02)01007-3. [DOI] [PubMed] [Google Scholar]
- 17.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the α-calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 18.Hanson PI, Schulman H. Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J Biol Chem. 1992;267:17216–17224. [PubMed] [Google Scholar]
- 19.Hashimoto Y, Schworer CM, Colbran RJ, Soderling TR. Autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. Effects on total and Ca2+-independent activities and kinetic parameters. J Biol Chem. 1987;262:8051–8055. [PubMed] [Google Scholar]
- 20.Helps NR, Barker HM, Elledge SJ, Cohen PT. Protein phosphatase 1 interacts with p53BP2, a protein which binds to the tumour suppressor p53. FEBS Lett. 1995;377:295–300. doi: 10.1016/0014-5793(95)01347-4. [DOI] [PubMed] [Google Scholar]
- 21.Hinds HL, Tonegawa S, Malinow R. CA1 long-term potentiation is diminished but present in hippocampal slices from αCaMKII mutant mice. Learn Mem. 1998;5:344–354. [PMC free article] [PubMed] [Google Scholar]
- 22.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 24.Kasahara J, Fukunaga K, Miyamoto E. Differential effects of a calcineurin inhibitor on glutamate-induced phosphorylation of Ca2+/calmodulin-dependent protein kinases in cultured rat hippocampal neurons. J Biol Chem. 1999;274:9061–9067. doi: 10.1074/jbc.274.13.9061. [DOI] [PubMed] [Google Scholar]
- 25. Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15 1997. 70 73[Erratum (1997) 15:411]. [DOI] [PubMed] [Google Scholar]
- 26.Krelstein MS, Thomas MP, Horowitz JM. Thermal effects on long-term potentiation in the hamster hippocampus. Brain Res. 1990;520:115–122. doi: 10.1016/0006-8993(90)91696-e. [DOI] [PubMed] [Google Scholar]
- 27.Kuhne C, Banks L. E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J Biol Chem. 1998;273:34302–34309. doi: 10.1074/jbc.273.51.34302. [DOI] [PubMed] [Google Scholar]
- 28.Kumar S, Talis AL, Howley PM. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J Biol Chem. 1999;274:18785–18792. doi: 10.1074/jbc.274.26.18785. [DOI] [PubMed] [Google Scholar]
- 29.Kuret J, Schulman H. Mechanism of autophosphorylation of the multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 1985;260:6427–6433. [PubMed] [Google Scholar]
- 30.Laan LA, v Haeringen A, Brouwer OF. Angelman syndrome: a review of clinical and genetic aspects. Clin Neurol Neurosurg. 1999;101:161–170. doi: 10.1016/s0303-8467(99)00030-x. [DOI] [PubMed] [Google Scholar]
- 31.Lickteig R, Shenolikar S, Denner L, Kelly PT. Regulation of Ca2+/calmodulin-dependent protein kinase II by Ca2+/calmodulin-independent autophosphorylation. J Biol Chem. 1988;263:19232–19239. [PubMed] [Google Scholar]
- 32.Lisman J, Malenka RC, Nicoll RA, Malinow R. Learning mechanisms: the case for CaM-KII. Science. 1997;276:2001–2002. doi: 10.1126/science.276.5321.2001. [DOI] [PubMed] [Google Scholar]
- 33.Long X, Wu G, Gaa ST, Rogers TB. Inhibition of protein phosphatase-1 is linked to phosphorylation of p53 and apoptosis. Apoptosis. 2002;7:31–39. doi: 10.1023/a:1013508811252. [DOI] [PubMed] [Google Scholar]
- 34.Lou LL, Schulman H. Distinct autophosphorylation sites sequentially produce autonomy and inhibition of the multifunctional Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1989;9:2020–2032. doi: 10.1523/JNEUROSCI.09-06-02020.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 36.Mukherji S, Soderling TR. Regulation of Ca2+/calmodulin-dependent protein kinase II by inter- and intrasubunit-catalyzed autophosphorylations. J Biol Chem. 1994;269:13744–13747. [PubMed] [Google Scholar]
- 37.Muller D, Lynch G. Synaptic modulation of N-methyl-d-aspartate receptor mediated responses in hippocampus. Synapse. 1990;5:94–103. doi: 10.1002/syn.890050203. [DOI] [PubMed] [Google Scholar]
- 38.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 39.Nuber U, Schwarz SE, Scheffner M. The ubiquitin-protein ligase E6-associated protein (E6-AP) serves as its own substrate. Eur J Biochem. 1998;254:643–649. doi: 10.1046/j.1432-1327.1998.2540643.x. [DOI] [PubMed] [Google Scholar]
- 40.Patton BL, Miller SG, Kennedy MB. Activation of type II calcium/calmodulin-dependent protein kinase by Ca2+/calmodulin is inhibited by autophosphorylation of threonine within the calmodulin-binding domain. J Biol Chem. 1990;265:11204–11212. [PubMed] [Google Scholar]
- 41.Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- 42.Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–15. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- 43.Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 44.Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- 45.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 46.Strack S, Choi S, Lovinger DM, Colbran RJ. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J Biol Chem. 1997a;272:13467–13470. doi: 10.1074/jbc.272.21.13467. [DOI] [PubMed] [Google Scholar]
- 47.Strack S, Westphal RS, Colbran RJ, Ebner FF, Wadzinski BE. Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res. 1997b;49:15–28. doi: 10.1016/s0169-328x(97)00117-4. [DOI] [PubMed] [Google Scholar]
- 48.Sutcliffe JS, Jiang YH, Galijaard RJ, Matsuura T, Fang P, Kubota T, Christian SL, Bressler J, Cattanach B, Ledbetter DH, Beaudet AL. The E6-AP ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region [letter]. Genome Res. 1997;7:368–377. doi: 10.1101/gr.7.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki T, Okumura-Noji K, Tanaka R, Tada T. Rapid translocation of cytosolic Ca2+/calmodulin-dependent protein kinase II into postsynaptic density after decapitation. J Neurochem. 1994;63:1529–1537. doi: 10.1046/j.1471-4159.1994.63041529.x. [DOI] [PubMed] [Google Scholar]
- 50.Varga AW, Anderson AE, Adams JP, Vogel H, Sweatt JD. Input-specific immunolocalization of differentially phosphorylated Kv4.2 in the mouse brain. Learn Mem. 2000;7:321–332. doi: 10.1101/lm.35300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17:12–13. doi: 10.1038/ng0997-12. [DOI] [PubMed] [Google Scholar]
- 52.Weeber EJ, Atkins CM, Selcher JC, Varga AW, Mirnikjoo B, Paylor R, Leitges M, Sweatt JD. A role for the β isoform of protein kinase C in fear conditioning. J Neurosci. 2000;20:5906–5914. doi: 10.1523/JNEUROSCI.20-16-05906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams CA, Angelman H, Clayton-Smith J, Driscoll DJ, Hendrickson JE, Knoll JH, Magenis RE, Schinzel A, Wagstaff J, Whidden EM, Zori RT. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet. 1995;56:237–238. doi: 10.1002/ajmg.1320560224. [DOI] [PubMed] [Google Scholar]
- 54.Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- 55.Zheng YJ, Furukawa T, Ogura T, Tajimi K, Inagaki N. M-phase specific expression and phosphorylation-dependent ubiquitination of the ClC-2 channel. J Biol Chem. 2002;277:32268–32273. doi: 10.1074/jbc.M202105200. [DOI] [PubMed] [Google Scholar]