

Abstract
The central glutamatergic system has been implicated in the pathogenesis of neuropathic pain, and a highly active central glutamate transporter (GT) system regulates the uptake of endogenous glutamate. Here we demonstrate that both the expression and uptake activity of spinal GTs changed after chronic constriction nerve injury (CCI) and contributed to neuropathic pain behaviors in rats. CCI induced an initial GT upregulation up to at least postoperative day 5 primarily within the ipsilateral spinal cord dorsal horn, which was followed by a GT downregulation when examined on postoperative days 7 and 14 by Western blot and immunohistochemistry. Intrathecal administration of the tyrosine kinase receptor inhibitor K252a and the mitogen-activated protein kinase inhibitor PD98059 for postoperative days 1–4 reduced and nearly abolished the initial GT upregulation in CCI rats, respectively. Prevention of the CCI-induced GT upregulation by PD98059 resulted in exacerbated thermal hyperalgesia and mechanical allodynia reversible by the noncompetitive NMDA receptor antagonist MK-801, indicating that the initial GT upregulation hampered the development of neuropathic pain behaviors. Moreover, CCI significantly reduced glutamate uptake activity of spinal GTs when examined on postoperative day 5, which was prevented by riluzole (a positive GT activity regulator) given intrathecally twice a day for postoperative days 1–4. Consistently, riluzole attenuated and gradually reversed neuropathic pain behaviors when the 4 d riluzole treatment was given for postoperative days 1–4 and 5–8, respectively. These results indicate that changes in the expression and glutamate uptake activity of spinal GTs may play a critical role in both the induction and maintenance of neuropathic pain after nerve injury via the regulation of regional glutamate homeostasis, a new mechanism relevant to the pathogenesis of neuropathic pain.
Keywords: neuropathic pain, allodynia, hyperalgesia, glutamate transporter, nerve injury, riluzole, tyrosine kinase, mitogen-activated protein kinase
Introduction
Peripheral nerve injury could result in neuropathic pain syndromes, including hyperalgesia, allodynia, and spontaneous pain. Both peripheral and central mechanisms have been implicated in the pathogenesis of neuropathic pain (Wall et al., 1974;Devor, 1983; Woolf, 1983; Dubner, 1991; Mao et al., 1995; Woolf and Mannion, 1999), and dynamic interactions between peripheral and central mechanisms also have been indicated (Gracely et al., 1992; Mao et al., 1992b,c). An increase in neuronal excitability within the CNS may be initiated and maintained after excessive activation of central glutamate receptors by their endogenous ligands, namely glutamate and aspartate. To date, much effort and progress have been made in investigating the role of glutamate receptor activation and subsequent intracellular events in the pathogenesis of neuropathic pain (Dougherty and Willis, 1991; Dubner, 1991; Wilcox, 1991; Dougherty et al., 1992;Mao et al., 1992a,b, 1995; Yamamoto and Yaksh, 1992; Malmberg et al., 1997; Woolf and Mannion, 1999; Guo et al., 2002). Little has been known about the role of regulating endogenous ligands of glutamate receptors in the central mechanisms of neuropathic pain.
The homeostasis of extracellular glutamate, as well as aspartate, is regulated tightly by a highly active glutamate transporter (GT) system within the CNS (Robinson and Dowd, 1997; Semba and Wakuta, 1998;Mennerick et al., 1999; Jabaudon et al., 2000; Danbolt, 2001). Thus far, at least five Na+-dependent cell membrane GT proteins have been cloned, including EAAT1 [GLAST (glutamate/aspartate transporter)], EAAT2 (GLT-1), and EAAT3 (EAAC1) (Kanai and Hediger, 1992; Pines et al., 1992; Storck et al., 1992; Arriza et al., 1993; Tanaka, 1993; Fairman et al., 1995). GLAST and GLT-1 are expressed primarily in glial cells, whereas EAAC1 is the predominant neuronal GT (Robinson and Dowd, 1997; Danbolt, 2001). Both neuronal and glial GTs actively participate in a number of fundamental physiological functions, including synaptic plasticity, by regulating extracellular glutamate concentration (Mennerick et al., 1999; Lievens et al., 2000; Vorwerk et al., 2000; Trotti et al., 2001). Importantly, GTs regulate the duration and intensity of glutamate receptor activation during signal transduction. As such, GTs play a critical role in preventing overstimulation of glutamate receptors, a process that could trigger both neuroplastic changes and excitotoxic cascades under a variety of pathological conditions, including spinal cord and peripheral nerve injury (Mennerick et al., 1999; Lievens et al., 2000;Vorwerk et al., 2000; Trotti et al., 2001).
Thus spinal GTs could change after peripheral nerve injury and contribute to the pathogenesis of neuropathic pain, a pathological condition known to involve the central glutamatergic system. In the present study changes in the expression and glutamate uptake activity of spinal GTs and their functional relation to neuropathic pain behaviors were examined by using immunohistochemistry, Western blot,in vitro glutamate uptake assay, and pharmacological evaluation in a rat model induced by chronic constriction sciatic nerve injury (CCI). Previous studies have indicated that GT expression could be modulated by neurotrophic factors via the activation of tyrosine kinase (Trk) receptors and intracellular mitogen-activated protein kinases (MAPK) (Danbolt, 2001). Accordingly, the effects of the Trk receptor and MAPK activation on the regulation of GT expression and their relation to the development of neuropathic pain were examined in CCI rats by using K252a and PD98059, nonselective inhibitors for Trk receptors and MAPK, respectively. In addition, the role of modulating spinal GT glutamate uptake activity in the induction and maintenance of neuropathic pain was investigated by using riluzole, a positive regulator of GT uptake activity (Azbill et al., 2000; Mu et al., 2000), before or after the development of neuropathic pain in CCI rats.
Materials and Methods
Animal surgery and drugs
Adult male Sprague Dawley rats weighing 275–325 gm (Charles River Laboratories, Wilmington, MA) were used. The animal room was lighted artificially from 7:00 A.M. to 7:00 P.M. The surgical procedure was performed aseptically under halothane anesthesia (2%). CCI rats were produced by loosely ligating the common sciatic nerve according to the method of Bennett and Xie (1988). Briefly, on one side the rat's sciatic nerve was exposed in the mid-thigh, and four loose ligatures of 4.0 chromic gut were made around the dissected nerve with a 1.0–1.5 mm interval between each of them. Skin wound was closed with 6.0 nylon sutures. Sham rats were made by following the same surgical procedure except for nerve ligation. In some groups intrathecal catheter implantation was made under the same surgical condition, and a PE10 catheter was inserted into the lumbar enlargement according to the method described previously (Yaksh and Rudy, 1976). Those rats exhibiting postoperative neurological deficits or poor grooming were excluded from the experiments.
Riluzole [2-amino-6-(trifluoromethoxy)-benzothiazole] and MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo(a,b)cyclohepten-5,10-imine maleate] were purchased from Sigma (St. Louis, MO), and K252a and PD98059 were purchased from Calbiochem (La Jolla, CA). Riluzole, K252a, and PD98059 were dissolved in 10% DMSO diluted in normal saline (vehicle), and MK-801 was dissolved in normal saline. In those experiments in which riluzole, K252a, or PD98059 was used as a treatment, the nontreatment groups received vehicle as control. The experiments were conducted with the experimenters blinded to treatment conditions.
Behavioral tests
Animals were habituated to the test environment for 2 d consecutively before baseline testing. All animals were tested for thermal hyperalgesia and mechanical allodynia. For testing thermal hyperalgesia, we exposed the plantar surface of a rat's hindpaw to a beam of radiant heat through a transparent perplex glass surface (Hargreaves et al., 1988). The withdrawal latency was averaged from at least two trials separated by a 2 min interval, and the cutoff was set at 25 sec to avoid tissue damage.
For examining mechanical allodynia, we placed each rat on a metal mesh floor, covered it with a plastic box (15 × 15 ×18 cm), and allowed it 30 min to habituate. The mechanical stimulation resulting from the bending force of a von Frey filament was applied to the plantar surface of each hindpaw. Each trial consisted of five applications of a von Frey filament given every 4 sec, and the cutoff force was 20 gm. Brisk foot withdrawals (at least three times of five applications) in response to von Frey filament stimulation were considered positive. Depending on the initial response, subsequent filaments were applied in the order of either descending or ascending force to determine the threshold force (Tal and Bennett, 1994; Mao et al., 1997).
Statistical analysis of behavioral data
Data from the thermal hyperalgesia test were analyzed first by generating difference scores between two hindpaws (contralateral withdrawal latency minus ipsilateral withdrawal latency). As such, a higher difference score represents a higher degree of thermal hyperalgesia. The data from both thermal hyperalgesia and mechanical allodynia (threshold bending force) tests were analyzed by using two-way ANOVA repeated across testing time points to detect overall differences among treatment groups. Whenever applicable, the data also were examined by using two-way ANOVA repeated across treatment groups to examine overall differences among testing time points. In both cases, when significant main effects were observed, the Waller–DuncanK-ratio t test was performed to determine sources of differences. Differences were considered to be statistically significant at the level of α = 0.05.
Western blot
Animals were killed after being exposed to a CO2 chamber. Fresh tissue samples taken from the L4 and L5 spinal cord dorsal horns were removed as quickly as possible by laminectomy at the lumbar region. The choice of examining the L4 and L5 segments was based on the consideration that these lumbar segments are the major contributor to the sciatic nerve and that previous studies have detected a high level of neurochemical and metabolic changes in these lumbar segments in CCI rats (Bennett et al., 1989; Kajander and Xu, 1995; Mao et al., 1995). Dorsal horns ipsilateral and contralateral to CCI were homogenized separately with a hand-held pellet pestle in a lysis buffer containing a mixture of phosphatase inhibitors (100×) and proteinase inhibitors (25×; Sigma). The quantification of protein contents was made by the Bradford method. Protein samples (40 μg) were separated on SDS-PAGE gel (4–15% gradient gel; Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride filters (Millipore, Bedford, MA). The filters were blocked with 3% milk and incubated overnight at 4°C with a polyclonal guinea pig anti-GLAST (1:2500) or rabbit anti-GLT-1 (1:2500; both fromChemicon, Temecula, CA) or rabbit anti-EAAC1 (1:2500; Alpha Diagnostics, San Antonio, TX) primary antibody. Then the blots were incubated for 1 hr at room temperature (RT) with a corresponding HRP-conjugated secondary antibody (1:3000; Amersham Biosciences, Arlington Heights, IL), visualized in ECL solution (PerkinElmer Life Sciences, Emeryville, CA) for 1 min, and exposed onto X-Omat film from Kodak (Rochester, NY) for 1–30 min. Finally, the blots were incubated in a stripping buffer (67.5 mm Tris, pH 6.8, 2% SDS, and 0.7% β-mercaptoethanol) for 30 min at 50°C and reprobed with a polyclonal rabbit anti-β-actin antibody (1:1000; Alpha Diagnostics) as loading controls.
The Western analysis was made in triplicate. All three primary antibodies have been used extensively in previous studies, and our Western blot bands showed the same band sizes as indicated in the antibody information sheets. The density of each specific band was measured with a computer-assisted imaging analysis system (IPLab software, Scanalytics, Fairfax, VA). There was no significant difference in the density of β-actin-loading control bands among groups. To compare the differences between control and treatment groups, we first normalized the density of each specific band against the density of the corresponding internal loading band. The percentage of change of the band density between treatment and control groups then was determined by using the equation: [(band density of a control group – band density of a treatment group)/band density of a control group] × 100%. Differences for each GT were compared by using Student's t test between two groups or by using one-way ANOVA for multiple groups repeated across time points, followed by Fisher's test.
Immunohistochemistry
Animals were anesthetized with halothane and perfused transcardially with 200 ml of normal saline, followed by 200–300 ml of 4% paraformaldehyde in 0.1 m phosphate buffer (PB). The lumbar spinal cords were dissected, postfixed for 1.5 hr, and transferred into 30% sucrose in 0.1 m PB for overnight. Tissues from both treatment and control groups then were mounted together in OCT compound and frozen on dry ice. Spinal cords (20 μm sections) were cut on a cryostat, mounted serially onto microscope slides, and stored at –80°C. For immunostaining, spinal cord sections were rinsed in 0.1 m PBS three times before being covered in 2% normal donkey serum containing 0.3% Triton X-100 for 1 hr at RT. The sections were incubated overnight at 4°C with a guinea pig anti-GLAST (1:4000), rabbit anti-GLT-1 (1:2000), or goat anti-EAAC1 (1:1000) (Chemicon) primary antibody in PBS with 0.1% Triton X-100. Then the sections were incubated in a corresponding FITC-conjugated secondary antibody (1:40; Jackson ImmunoResearch, West Grove, PA) for 1 hr at RT in 0.1m PBS containing 0.1% Triton X-100. Nonspecific bindings were examined by omitting the corresponding primary antibody during the incubation process. Four to six nonadjacent sections from the L4 and L5 segments were selected randomly, and the topographic distribution of GT changes within the spinal cord dorsal horn was observed and recorded with a CCD camera (Soft Imaging System, M∫nster, Germany).
In vitro glutamate uptake assay
An in vitro spinal synaptosome preparation was used to assess glutamate uptake activity of spinal GTs according to the previously published method (Mitrovic et al., 1999; Azbill et al., 2000; Mu et al., 2000). In brief, animals were killed after being exposed to a CO2 chamber, and fresh tissue samples from the L4 and L5spinal dorsal horns were removed as quickly as possible by laminectomy. Dorsal horns ipsilateral and contralateral to CCI were homogenized separately in an ice-cold buffer solution, pH 7.2, containing 0.32m sucrose plus (in μg/ml) 4 pepstatin, 5 aprotinin, 20 trypsin inhibitor, 4 leupeptin, and (in mm) 0.2 PMSF, 2 EDTA, 2 EGTA, and 20 HEPES. The homogenates were centrifuged at 1500 rpm for 10 min at 4°C, and the supernatant was collected. The remaining pellets were resuspended by using the same buffer solution and recentrifuged as above. Both supernatants were combined and again centrifuged at 13,000 rpm for 10 min at 4°C. The so-obtained synaptosomal pellets, which contain both neuronal and glial GTs (Mitrovic et al., 1999; Azbill et al., 2000; Mu et al., 2000), were suspended in 2 ml of Locke's buffer containing (in mm) 154 NaHCO3, 5.6 glucose, 5 HEPES, pH 7.2, and saturated with 95% O2/5% CO2. The protein concentration of final synaptosome pellets was measured by the Bradford method and was adjusted to 200 μg/ml in Locke's buffer. Glutamate uptake activity was determined by incubating the synaptosome preparation (in 100 μg protein content) with the solution containing [3H]l-glutamate (0.4 μCi/mmol; Amersham) at 37°C for 5 min. The reaction was terminated by filtering synaptosomes through a Whatman (Maidstone, UK) GF/C 2.4 cm filter presoaked in the same buffer solution. Then the filter was washed three times with ice-cold Locke's buffer (2 ml) and transferred to a vial containing scintillation mixture (10 ml; Fisher Scientific, Houston, TX). The radioactivity in the final samples was measured by a liquid scintillation counter (Bio-Rad). The basal uptake activity in counts per minute (cpm) was measured in the absence of any treatment. The percentage of change in glutamate uptake activity was calculated with the following equation: [(basal cpm without treatment – cpm with treatment)/(basal cpm without treatment)] × 100%. Differences between two groups were compared by using Student'st test.
Experimental design
Experiment 1: time course of changes in spinal GT expression after CCI. To determine whether the expression of spinal GTs changed after CCI, we used six groups of rats (n = 8–10/group for two separate sampling procedures: immunostaining and Western blot) to examine the time course of GT changes in CCI rats. Thus spinal cords from one group of CCI rats were harvested on postoperative day 1, 4, 7, or 14 after the behavioral tests. Each group of sham-operated or naive rats (n = 4–5/group) also was included to serve as control, and their spinal cords were removed on postoperative day 14 after the behavioral tests. These samples were used for the semiquantification of spinal GT changes (Western blot) and for the examination of the topographic distribution of such changes (immunostaining).
Experiment 2: modulation of GT expression by K252a or PD98059 and its relation to the development of neuropathic pain. The above time course experiment revealed an initial GT upregulation after CCI (see Results). Because Trk receptors and MAPK have been shown to modulate GT expression (Danbolt, 2001), the effect of blocking Trk receptors and MAPK on GT expression and its relation to the induction of neuropathic pain behaviors were examined in CCI rats by using intrathecal K252a, a nonselective Trk receptor inhibitor, or PD98059, an inhibitor primarily for the extracellular signal-regulated kinase (ERK) members of MAPK (Encinas et al., 1999; Pascual et al., 2001;Troller et al., 2001). Five groups of rats (n = 6–8/group) were used, which included (1) CCI + vehicle, (2) CCI + K252a (0.5 μg/10 μl), (3 and 4) CCI + PD98059 (1 μg/10 μl), and (5) sham + vehicle. The doses for K252a and PD98059 were based on previous and our pilot studies that showed reliable inhibition of Trk receptors and MAPK (Encinas et al., 1999; Pascual et al., 2001; Troller et al., 2001). Each treatment was given intrathecally twice a day for postoperative days 1–4 (groups 1, 2, 3, 5) or postoperative days 1–2 (group 4–PD98059), beginning immediately after the operation. Thermal hyperalgesia and mechanical allodynia were tested on postoperative days 1, 2, and 3 (all groups) as well as on day 5 (groups 1, 2, 3, 5 only) to examine the effect of K252a or PD98059 on behavioral changes. Spinal cords from these groups were harvested either on day 3 (group 4 only) or day 5 (groups 1, 2, 3, 5) to determine GT changes with Western blot. An additional group of CCI rats (n = 4) was treated with vehicle, and their spinal cords were taken on postoperative day 3 for comparing GT changes with the above group 4. Two groups of sham rats (n = 4/group) were included, and each received the same 4 d treatment regimen of K252a or PD98059 as that in CCI rats, with their spinal cords taken on postoperative day 5 to control for nonspecific effects of these agents on GT expression and behaviors.
CCI rats (n = 4–5) treated with K252a, PD98059, or vehicle between postoperative days 1 and 4 as described above were given MK-801 (10 nmol, i.t.) at the end of the behavioral tests. Then the behavioral tests were repeated at 30 min after the MK-801 treatment to examine whether blocking NMDA receptors by MK-801 would reverse the effects of K252a or PD98059 on thermal hyperalgesia and mechanical allodynia.
Experiment 3: modulation of spinal GT uptake activity by riluzole and its relation to the induction and maintenance of neuropathic pain.To examine changes in spinal GT uptake activity and its modulation by riluzole (a positive regulator of GT activity) with an in vitro glutamate uptake assay, we used two groups each of CCI or sham rats (n = 4–5). CCI or sham rats were given either intrathecal vehicle or 10 μg of riluzole twice daily for postoperative days 1–4 beginning immediately after the operation. Spinal cord dorsal horn samples then were obtained on postoperative day 5 after the behavioral tests as described above. Differences in glutamate uptake activity were compared between sham and CCI groups treated with riluzole or vehicle. The basal glutamate uptake activity from additional naive rats (n = 3) also was obtained to control for possible changes in basal glutamate uptake activity in sham rats.
To examine whether pharmacological perturbation of GT uptake activity would regulate neuropathic pain behaviors in CCI rats, we gave riluzole intraperitoneally either before (pretreatment) or after (post-treatment) the development of neuropathic pain behaviors in CCI rats. Thus a total of six groups of rats (n = 6–8 group) was used, including (1) CCI + 1 mg/kg riluzole pretreatment, (2) CCI + 4 mg/kg riluzole pretreatment, (3) CCI + 1 mg/kg riluzole post-treatment, (4) CCI + 4 mg/kg riluzole post-treatment, (5) CCI + vehicle, and (6) sham + 4 mg/kg riluzole. The riluzole doses were chosen on the basis of previous studies that showed reliable changes in GT uptake activity under both in vitroand in vivo conditions (Azbill et al., 2000; Mu et al., 2000; Mao et al., 2002a,b). Both pretreatment and post-treatment regimens consisted of twice-daily injections for a 4 d period. The pretreatment regimen began immediately after the operation, whereas the post-treatment regimen began on postoperative day 5. The CCI + vehicle and sham + 4 mg/kg riluzole groups underwent the same treatment duration beginning on postoperative day 5. Behavioral tests were made on postoperative days 5, 8, and 10 in both treatment regimens.
Results
Changes in spinal GT expression after CCI
CCI induced significant changes in the expression of spinal GTs (EAAC1, GLAST, GLT-1) as revealed by Western blot (Fig.1). The GT changes occurred primarily within the spinal cord dorsal horn ipsilateral to CCI. There were no reliable differences in GT expression between CCI and sham rats on the contralateral spinal cord dorsal horn, nor were there differences in GT expression between sham and naive rats (p > 0.05). Further, the time course analysis showed a biphasic pattern of GT changes in CCI rats with an initial GT upregulation when examined on postoperative days 1 and 4, followed by a GT downregulation when examined on postoperative days 7 and 14 (Fig. 1). The β-actin level did not change after CCI, indicating a specific effect of CCI on the GT expression.
Fig. 1.

Time course of GT changes after CCI. CCI induced a biphasic change in the expression of EAAC1, GLAST, and GLT-1 within the spinal cord dorsal horn ipsilateral to CCI. There was an initial increase in GT expression when rats were examined on postoperative days 1 and 4, which was followed by a downregulation of all three GTs when rats were examined on postoperative days 7 and 14. Top, Western blot bands from representative CCI and sham rats showing postoperative changes in protein levels of EAAC1 (69 kDa), GLAST (67 kDa), and GLT-1 (67 kDa) within the ipsilateral spinal cord dorsal horn. β-Actin is a loading control. Bottom, Statistical analysis of relative density changes of Western blots between CCI and sham rats. The data are expressed as the percentage of changes in CCI rats as compared with sham controls.CCI-D1 to CCI-D14 refers to CCI rats on postoperative days 1–14. *p < 0.05 as compared with the sham control.
EAAC1
The EAAC1 level within the ipsilateral spinal cord dorsal horn was increased significantly on postoperative days 1 and 4 as compared with the sham group (Fig. 1). This increase in EAAC1 was followed by a decrease below the sham control level shown on postoperative days 7 and 14 (Fig. 1). The percentage of relative EAAC1 density on Western blots was 155, 125, 45, and 30% of the sham control on postoperative days 1, 4, 7, and 14 (ANOVA, F(4,17) = 4.02;p < 0.05), respectively.
GLAST
The pattern of postoperative GLAST expression was similar to that of EAAC1 (Fig. 1). As such, there was a significant increase over the sham control in GLAST expression within the ipsilateral spinal cord dorsal horn when it was examined on postoperative days 1 and 4. This initial increase was followed by a moderate decrease in the GLAST level when it was examined on postoperative day 7. On postoperative day 14 the GLAST level was at ∼41% of the sham control (ANOVA,F(4,17) = 3.89; p < 0.05).
GLT-1
An initial increase in GLT-1 expression as shown on postoperative days 1 and 4 also was followed by a decreased GLT-1 expression in CCI rats when it was examined on postoperative days 7 and 14 (Fig. 1). The percentage of relative GLT-1 density on Western blots from CCI rats was 141, 138, 69, and 48% of the sham control on postoperative days 1, 4, 7, and 14 (ANOVA, F(4,17) = 4.12;p < 0.05), respectively.
Topographic distribution of spinal GT changes in CCI rats
All three GTs were shown in the spinal cord dorsal horn in sham rats, indicating a basal level of GT immunostaining in this spinal region (Fig. 2). In particular, there was profound baseline immunostaining with EAAC1 and GLAST within laminas I–II of the spinal cord dorsal horn. In contrast, the immunostaining pattern of GLT-1 was rather diffuse across the spinal dorsal horn. In general, the pattern of spinal GT distribution is in agreement with the role of GTs in regulating extracellular glutamate, because the highest density of glutamate receptors also is located primarily within the superficial spinal cord dorsal horn (Petralia et al., 1994a,b; Kus et al., 1995; Bonnot et al., 1996; Kalb and Fox, 1997).
Fig. 2.

Topographic distribution of GT changes within the ipsilateral spinal cord dorsal horn. The photo micrographs taken from representative CCI and sham rats show an increase in EAAC1, GLAST, and GLT-1 in CCI rats (B, E, H) on postoperative day 4 within the ipsilateral spinal cord dorsal horn as compared with those of the sham control (A, D, G). All three GTs were downregulated when examined on postoperative day 14 (C, F, I). Note that EAAC1, GLAST, and GLT-1 are located primarily within the superficial laminas of the spinal cord dorsal horn. Scale bar, 100 μm.
Sham operation did not change the baseline immunostaining of all three GTs as compared with that of naive rats. Consistent with the semiquantitative GT changes revealed by Western blot (Fig. 1), CCI induced significant changes in GT immunostaining within the spinal cord dorsal horn ipsilateral to CCI that followed a similar biphasic pattern shown in the Western analysis (Fig. 2). In addition, the pattern of GT distribution within the spinal cord dorsal horn remained unchanged between sham and CCI rats. That is, the most significant change of GT immunostaining occurred primarily within laminas I–II after CCI (Fig.2). Thus both Western blot and immunostaining revealed consistent changes of GT expression in CCI rats, indicating that the ipsilateral superficial dorsal horn was the primary locus of GT changes at the spinal level after CCI.
Effects of K252a or PD98059 on GT upregulation
Intrathecal treatment with K252a (0.5 μg) or PD98059 (1 μg) in CCI rats significantly reduced the upregulation of EAAC1 (ANOVA,F(6,24) = 4.23; p < 0.05), GLAST (ANOVA, F(6,24) = 3.98;p < 0.05), and GLT-1 (ANOVA,F(6,24) = 4.12; p < 0.05) as compared with CCI rats treated with vehicle. The K252a or PD98059 treatment was initiated immediately after the surgical procedure and continued twice daily during the first several postoperative days. In particular, the PD98059 treatment nearly abolished the increased GT expression in CCI rats when examined on postoperative days 3 and 5 (Fig. 3;p < 0.05), whereas K252a partially reduced the GT expression when examined on postoperative day 5 (Fig. 3;p < 0.05). Of note is that the percentage of change of GT expression in vehicle-treated CCI rats did not differ significantly between postoperative days 3 and 5 (day 3: EAAC1, 153 ± 7.5%; GLAST, 132 ± 7.1%; GLT-1, 153 ± 7.8; day 5: EAAC1, 161 ± 7.1%; GLAST, 141 ± 6.7%; GLT-1, 165 ± 6.1; each p > 0.05), indicating that the CCI-induced GT upregulation was present up to at least postoperative day 5, and PD98059 effectively prevented the GT upregulation on both postoperative days 3 and 5. Neither K252a nor PD98059 alone significantly changed the baseline spinal GT expression (Fig. 3; p > 0.05), indicating a modulatory role of Trk receptors and MAPK in the upregulation of spinal GTs after CCI.
Fig. 3.

Modulation of GT expression by the Trk receptor inhibitor K252a or the MAPK inhibitor PD98059. The upregulation of EAAC1, GLAST, and GLT-1, respectively, was reduced and nearly abolished by intrathecal treatment with K252a (0.5 μg) and PD98059 (1 μg) given twice daily between postoperative days 1 and 4 beginning immediately after nerve ligation. Top, Western blots from representative CCI and sham rats with samples taken on postoperative day 3 (the C+P3 group) or day 5 (the remaining groups). Bottom, Statistical data showing GT expression in CCI rats treated with K252a (C+K), PD98059-day 3 (C+P3), PD98059-day 5 (C+P5), or vehicle (C+V) and in sham rats treated with vehicle (SHAM), K252a (S+K), or PD98059 (S+P). *p < 0.05 and **p < 0.01 as compared with the sham control; +p < 0.05 as compared with the C+V group. Note that the C+V group shows the bands from vehicle-treated CCI rats on day 5, because the percentage of change of GT expression in vehicle-treated CCI rats did not differ significantly between postoperative days 3 and 5 (see Results for details).
Effects of K252a or PD98059 on neuropathic pain behaviors: reversal by MK-801
The PD98059 treatment exacerbated the development of thermal hyperalgesia (Fig. 4A; ANOVA, F(3,23) = 4.76;p < 0.01) and mechanical allodynia (Fig.4B; ANOVA, F(3,23) = 3.88; p < 0.05) in these same CCI rats as compared with the vehicle-treated CCI rats. The exacerbation of neuropathic pain behaviors was manifested as a shortened onset of and an increased degree of thermal hyperalgesia and mechanical allodynia in CCI rats showing the blocked GT upregulation by PD98059 (Fig. 4). K252a or PD98059 alone did not affect baseline behaviors in sham rats. CCI rats treated with K252a did not show reliable exacerbation of thermal hyperalgesia or mechanical allodynia as compared with CCI rats treated with vehicle (Fig. 4; p > 0.05). Given that the GT upregulation was nearly abolished by PD98059 but only partially reduced by K252a (Fig. 3), these data indicate that the initial increase in GT expression played a significant role in hampering the development of neuropathic pain behaviors in CCI rats.
Fig. 4.

Modulation of thermal hyperalgesia and mechanical allodynia by PD98059. Thermal hyperalgesia (A) and mechanical allodynia (B) were exacerbated in CCI rats treated with PD98059 (1 μg, i.t.) twice daily between postoperative days 1 and 4 beginning immediately after the operation. This exacerbation was manifested as a shortened onset of and an enhanced degree of thermal hyperalgesia and mechanical allodynia in those CCI rats. K252a did not increase neuropathic pain behaviors significantly. *p < 0.05 and **p < 0.01 as compared with sham control;+p < 0.05 as compared with CCI rats treated with vehicle. Diff. score, Difference scores (contralateral minus ipsilateral withdrawal latency).
The noncompetitive NMDA receptor antagonist MK-801 reversed both thermal hyperalgesia and mechanical allodynia potentiated by PD98059. Thus a single intrathecal administration with 10 nmol of MK-801 on postoperative day 5 reversed thermal hyperalgesia (Fig.5, top) and mechanical allodynia (Fig. 5, bottom) in CCI rats treated with PD98059 (p < 0.05) when they were examined at 30 min after the MK-801 treatment. The same MK-801 dose also reversed thermal hyperalgesia and mechanical allodynia in CCI rats treated with vehicle or K252a. MK-801 alone did not change the baseline responses to thermal or mechanical stimulation. These results indicate that the effects of modulating GT expression by PD98059 on neuropathic pain behaviors were mediated at least in part via the activation of NMDA receptors in CCI rats.
Fig. 5.

MK-801 reversed thermal hyperalgesia and mechanical allodynia potentiated by PD98059. A single intrathecal administration of 10 nmol of MK-801 on postoperative day 5 reversed thermal hyperalgesia (top) and mechanical allodynia (bottom) in CCI rats treated with PD98059 (1 μg, i.t.) when they were examined at 30 min after the MK-801 treatment. The same dose of MK-801 also reversed thermal hyperalgesia and mechanical allodynia in CCI rats treated with vehicle or K252a. MK-801 alone did not change the baseline response to thermal or mechanical stimulation. *p < 0.05 and **p < 0.01 as compared with the corresponding baseline level. Pre-MKand After-MK refer to nociceptive responses before and at 30 min after the MK-801 treatment on postoperative day 5, respectively. For definition of Diff. score, see Figure4.
Changes in spinal glutamate uptake activity in CCI rats and its modulation by riluzole
There was a significant reduction of glutamate uptake activity (> 30% reduction) in the ipsilateral L4–L5 spinal cord dorsal horn sample of CCI rats as compared with that of sham rats when examined on postoperative day 5 (Fig. 6;p < 0.05). No reliable differences in glutamate uptake activity were observed between CCI and sham rats on the contralateral side of the spinal cord dorsal horn (Fig. 6; p > 0.05). Sham operation also did not affect basal glutamate uptake activity as compared with that of naive rats. However, glutamate uptake activity in the ipsilateral dorsal horn of CCI rats was enhanced on postoperative day 5 by intrathecal treatment with 10 μg of riluzole (a positive regulator of GT uptake activity), but not vehicle, given twice daily for postoperative days 1–4 beginning immediately after the operation (Fig. 6; p < 0.05). The 4 d riluzole treatment also moderately enhanced the glutamate uptake activity in the contralateral spinal cord dorsal horn of CCI rats as well as in both contralateral and ipsilateral spinal cord dorsal horn of sham rats (Fig. 6). These results indicate that (1) CCI reduced GT uptake activity primarily within the ipsilateral spinal cord dorsal horn and (2) the in vivo riluzole treatment was effective in enhancing spinal GT glutamate uptake activity.
Fig. 6.
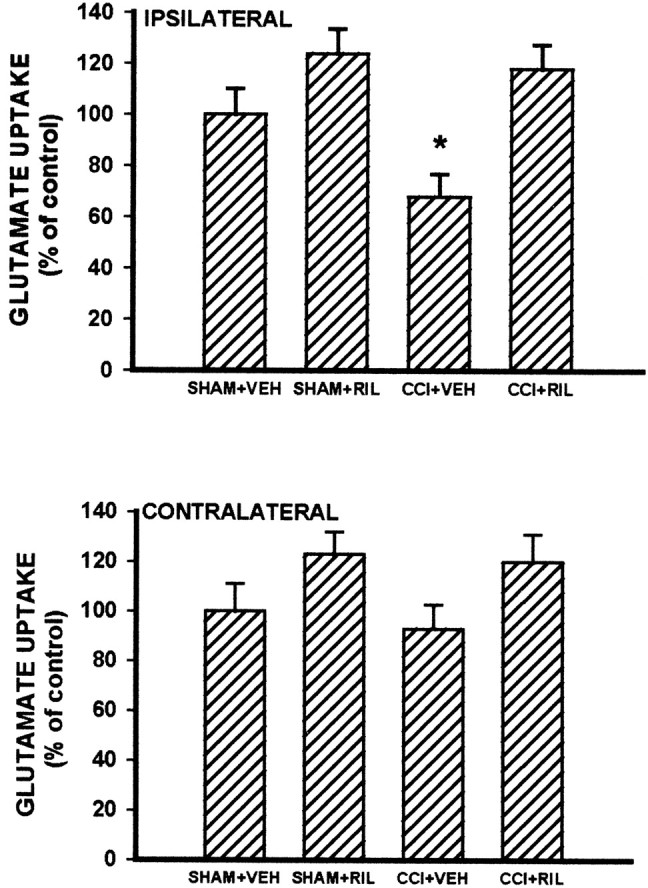
Changes in spinal glutamate uptake activity in CCI rats and its modulation by riluzole. CCI induced a significant reduction of glutamate uptake activity in the samples taken from the ipsilateral L4–L5 spinal cord dorsal horn as compared with that of sham rats when examined on postoperative day 5. When they were examined on postoperative day 5, glutamate uptake activity in CCI rats was enhanced by intrathecal treatment with 10 μg riluzole (CCI+RIL), but not vehicle (CCI+VEH), given twice daily for postoperative days 1–4 beginning immediately after the operation. Note that the 4 d riluzole treatment also moderately enhanced the glutamate uptake activity of the contralateral spinal cord dorsal horn in CCI rats and of bilateral spinal cord dorsal horn in sham rats (SHAM+RIL). *p < 0.05 as compared with the SHAM+VEH group.
Effects of riluzole on the induction of neuropathic pain behaviors
Both thermal hyperalgesia and mechanical allodynia were observable on postoperative day 5 and continued on postoperative days 8 and10 in vehicle-treated CCI rats (Fig.7A,B). Intraperitoneal treatment with 1 or 4 mg/kg riluzole, given twice daily for the first 4 postoperative days beginning immediately after the operation, significantly reduced thermal hyperalgesia (Fig. 7A; ANOVA,F(3,22) = 5.12; p < 0.01) and mechanical allodynia (Fig. 7B; ANOVA,F(3,22) = 3.98; p < 0.05) as compared with vehicle-treated CCI rats. The effects of riluzole on thermal hyperalgesia and mechanical allodynia also were observed in CCI rats receiving a similar postoperative riluzole treatment regimen except that riluzole (10 μg) was given intrathecally (data not shown), suggesting a spinal site of riluzole action.
Fig. 7.

Effects of riluzole on the induction of thermal hyperalgesia and mechanical allodynia after CCI. Intraperitoneal treatment with 1 or 4 mg/kg riluzole (RIL-1 orRIL-4) twice daily for postoperative days 1–4 beginning immediately after the operation significantly reduced the development of thermal hyperalgesia (A) and mechanical allodynia (B). Note that the effects of riluzole on thermal hyperalgesia and mechanical allodynia lasted at least several days after its discontinuation on postoperative day 5, indicating a significant role of regulating GT uptake activity in the development of neuropathic pain behaviors in CCI rats. The effect of riluzole on thermal hyperalgesia was dose-dependent. *p < 0.05 and **p < 0.01 as compared with CCI rats treated with vehicle (CCI+VEH); +p < 0.05 as compared with the low dose (1 mg/kg) riluzole group. ForDiff. score, see Figure 4.
There are several noteworthy features concerning the riluzole effects. (1) When tested on postoperative day 5, thermal nociceptive threshold was not statistically different between riluzole-treated CCI rats and sham rats, indicating a preventive effect of riluzole on the development of thermal hyperalgesia. This riluzole effect lasted at least several days after its discontinuation on postoperative day 5 (Fig. 7A). (2) In contrast to the effects of riluzole on thermal hyperalgesia, mechanical allodynia was not reduced significantly on postoperative day 5 in CCI rats treated with riluzole (Fig. 7B). Instead, mechanical allodynia was reduced significantly in riluzole-treated CCI rats when tested on postoperative days 8 and 10 after the riluzole treatment had been discontinued on postoperative day 5 (Fig. 7B). The effects of riluzole on both thermal hyperalgesia and mechanical allodynia indicate a critical role of regulating GT uptake activity during the early stage of nerve injury in modulating the development of neuropathic pain behaviors in CCI rats. (3) There was a dose–response effect on thermal hyperalgesia from the riluzole treatment (Fig. 7A; 4 mg/kg > 1 mg/kg > vehicle; p < 0.05). (4) Riluzole itself produced a mild but statistically insignificant increase in thermal and mechanical nociceptive thresholds in hindpaws contralateral to CCI. Moreover, riluzole at the current doses did not produce sedative effects or decreased locomotor activity (e.g., impaired motor coordination, a loss of righting reflex), indicating a specific effect of riluzole on neuropathic pain behaviors in CCI rats.
Effects of riluzole on the maintenance of neuropathic pain behaviors
Riluzole also gradually reversed thermal hyperalgesia and mechanical allodynia in CCI rats when the treatment began on postoperative day 5. Thus riluzole (1 or 4 mg/kg, i.p.), given twice daily for 4 d beginning on postoperative day 5, significantly reduced thermal hyperalgesia (Fig.8A; ANOVA,F(3,21) = 4.23; p < 0.05) and mechanical allodynia (Fig. 8B; ANOVA,F(3,21) = 3.76; p < 0.05) as compared with vehicle-treated CCI rats when examined on postoperative days 8 and 10. Similar to the dose–response effect of riluzole on the induction of thermal hyperalgesia, CCI rats receiving 4 mg/kg riluzole resulted in more reduction of thermal hyperalgesia as compared with those treated with 1 mg/kg riluzole (Fig.8A; p < 0.05).
Fig. 8.

Effects of riluzole on the maintenance of thermal hyperalgesia and mechanical allodynia after CCI. Intraperitoneal treatment with 1 or 4 mg/kg riluzole (RIL-1 orRIL-4) twice daily for 4 d beginning on postoperative day 5 gradually reversed thermal hyperalgesia (A) and mechanical allodynia (B) when rats were examined on postoperative days 8 and 10. *p < 0.05 and **p < 0.01 as compared with CCI rats treated with vehicle (CCI+VEH); +p < 0.05 as compared with the low dose (1 mg/kg) riluzole group.Arrows indicate the beginning of the riluzole treatment on postoperative day 5. For Diff. score, see Figure4.
Discussion
The present findings demonstrate that both expression and glutamate uptake activity of spinal neuronal and glial GTs are altered after peripheral nerve injury, which contributes to neuropathic pain behaviors in CCI rats. First, spinal GT expression showed a biphasic change, with an initial upregulation followed by a downregulation after CCI. Second, prevention of this initial GT upregulation after CCI by the MAPK inhibitor PD98059 resulted in exacerbated thermal hyperalgesia and mechanical allodynia that were reversible by the noncompetitive NMDA receptor antagonist MK-801. Third, preventing CCI-induced reduction of spinal GT glutamate uptake activity by the positive GT activity regulator riluzole attenuated thermal hyperalgesia and mechanical allodynia when riluzole was given in a 4 d treatment regimen beginning either immediately after the operation or on postoperative day 5. These results provide, for the first time, lines of evidence indicating that spinal GTs may play a critical role in both induction and maintenance of neuropathic pain induced by peripheral nerve injury.
Overall consideration on data interpretation
Glutamate is a major excitatory amino acid neurotransmitter within the CNS participating in important physiological functions such as synaptic plasticity and cognitive awareness. Maintaining a low extracellular glutamate concentration is key to preventing glutamate neurotoxicity that may occur under many pathological conditions (Robinson and Dowd, 1997; Danbolt, 2001). Because the clearance of extracellular glutamate via glutamate metabolism or diffusion is virtually negligible (Robinson and Dowd, 1997; Danbolt, 2001) and inhibition of GT activity leads to extracellular glutamate build-ups within seconds (Jabaudon et al., 1999, 2000), regulation of extracellular glutamate is performed primarily by glutamate uptake via an efficient, high-capacity GT system (Robinson and Dowd, 1997;Danbolt, 2001). Among the identified cell membrane GT proteins (Kanai and Hediger, 1992; Pines et al., 1992; Storck et al., 1992; Arriza et al., 1993; Tanaka, 1993; Fairman et al., 1995), EAAC1, GLAST, and GLT-1 are particularly relevant for regulating glutamate uptake in broad CNS regions (Danbolt, 2001). Because the role of other cell membrane GTs (EAAT4 and EAAT5) remains unclear, EAAC1, GLAST, and GLT-1 were investigated specifically in the present experiments. Although GTs also regulate aspartate uptake, only glutamate will be mentioned in the following to simplify the discussion.
Several lines of evidence support a critical role of regulating GT expression and uptake activity in maintaining in vivoglutamate homeostasis as indicated in in vitro studies (Semba and Wakuta, 1998; Azbill et al., 2000; Jabaudon et al., 2000;Lievens et al., 2000; Matthews et al., 2000; Mu et al., 2000). First, in a previous study downregulation of spinal GTs induced by chronic morphine administration resulted in the exacerbated response to exogenous glutamate (Mao et al., 2002b). Second, perturbation of GT activity by using either a GT inhibitor (l-trans-pyrrolidine-2-4-dicarboxylate) or activator (riluzole) modulated abnormal pain sensitivity in morphine-tolerant rats that was mediated by NMDA receptors (Mao et al., 2002a,b). Third, mechanical allodynia induced by exogenous prostaglandin (PG) E2, PGF2α, NMDA, or AMPA was modulated by the GT inhibitor d,l-threo-β-benzyloxyaspartate (Minami et al., 2001). Fourth, in the present study the noncompetitive NMDA receptor antagonist MK-801 also effectively reversed thermal hyperalgesia and mechanical allodynia that were potentiated by preventing CCI-induced GT upregulation resulting from the PD98059 treatment. Moreover, riluzole both enhanced spinal GT uptake activity and effectively reduced thermal hyperalgesia and mechanical allodynia in CCI rats. Thus the effects of altered expression and glutamate uptake activity of spinal GTs on neuropathic pain behaviors in CCI rats are likely to be mediated via changes in regional glutamate homeostasis and at least in part via the subsequent activation of NMDA receptors.
It should be pointed out that riluzole may have neuroprotective and anticonvulsive effects in part via its possible effects on glutamate release (Cheramy et al., 1992; Martin et al., 1993; Doble, 1996; Hammer et al., 1999). More recently, however, riluzole has been shown to be a positive regulator of GT uptake activity that potently increases spinal glutamate uptake under both in vitro and in vivoconditions at the dose range compatible to that used in the present study (Azbill et al., 2000; Mu et al., 2000). This also was supported by our present data indicating the modulatory effects of riluzole on spinal GT uptake activity of both CCI and sham rats. A caveat is that the effects of riluzole on regulating glutamate uptake are nonselective between neuronal and glial GTs. At present there is no reliable GT uptake activity activator selective for neuronal or glial GTs, although MS-153 has been proposed to be a potential candidate for glial GTs (Nakagawa et al., 2001). Thus the effects of riluzole on neuropathic pain behaviors in CCI rats should be considered to involve the regulation of glutamate uptake activity of both neuronal and glial GTs.
Possible mechanisms of GT changes after CCI
An interesting observation from the present study is that CCI-induced a biphasic change in GT expression primarily within the ipsilateral superficial spinal cord dorsal horn. All three GTs under examination showed an initial increase in their expression after CCI, which lasted up to at least postoperative day 5 and was followed by a GT downregulation when examined on postoperative days 7 and 14. The time course of this late phase GT downregulation is similar to that of changes in neurotransmitters and receptors within the spinal cord dorsal horn of CCI rats (Stevens et al., 1991; Kajander and Xu, 1995). Thus a plausible explanation for the GT downregulation is a loss of primary afferents resulting from CCI (Bennett et al., 1989), given that neuronal GTs are located primarily at presynaptic sites (Robinson and Dowd, 1997; Danbolt, 2001). Likewise, degenerative changes within the spinal cord might be attributable to, at least in part, the downregulation of glial GTs after their initial reactive phase after CCI. These possibilities merit future investigations. Conceivably, this late phase GT downregulation would be contributory to persistent neuropathic pain behaviors in CCI rats demonstrated both in the present and previous studies (Bennett and Xie, 1988; Mao et al., 1992a,c).
Several cellular and intracellular mechanisms may be implicated in the upregulation of spinal GTs during the early stage after CCI. For instance, nerve injury-induced glutamate release from the primary afferents may serve as a positive feedback on GT expression (Danbolt, 2001). Alternatively, Trk receptors and MAPK may play an active role in GT upregulation as well (Danbolt, 2001). Trk receptors include at least subtypes A, B, and C, which can be activated by nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3, respectively (Hagemann and Blank, 2001; Ji and Woolf, 2001). Expression of neurotrophic factors in the dorsal root ganglion and spinal cord is upregulated dramatically under pathological conditions (Coffey et al., 1997; Li et al., 1999; Oyesiku et al., 1999; Ha et al., 2001), and such neurotrophic factors can be released into the spinal cord after peripheral nerve injury (Deng et al., 2000; Walker et al., 2001). Activation of Trk receptors in turn initiates downstream cascades, including activation of MAPK (Encinas et al., 1999; Hagemann and Blank, 2001). Activation of MAPK enhances gene expression for a variety of protein elements (Gegelashvili et al., 1997; Hagemann and Blank, 2001;Williams et al., 2001; Ji et al., 2002) and indeed has been shown to induce the expression of both GLT-1 mRNA and protein in astrocytes (Gegelashvili et al., 2000; Zelenaia et al., 2000). Thus the Trk receptor and MAPK pathway may participate in GT upregulation in CCI rats.
Our findings indicate that the initial GT upregulation indeed was reduced significantly in CCT rats receiving the 4 d treatment with K252a (a nonselective Trk receptor inhibitor) or PD98059 (an inhibitor for ERK-MAPK) beginning immediately after the operation. Of significance is that a much greater reduction of GT expression was observed in CCI rats treated with PD98059 than with K252a. This is consistent with the possible role of MAPK as an intracellular downstream effector for a variety of upstream regulatory mechanisms (Hagemann and Blank, 2001; Ji and Woolf, 2001; Williams et al., 2001). That is, because the Trk receptor activation is likely to be one of those upstream mechanisms, its inhibition leads to a partial prevention of GT upregulation in comparison with the effects of the MAPK inhibition. Accordingly, prevention of the GT upregulation by PD98059, but not the partial GT reduction by K252a, resulted in the exacerbation of thermal hyperalgesia and mechanical allodynia in CCI rats. The functional implication is that the initial GT upregulation after CCI may serve as a protective mechanism to minimize adverse consequences from nerve injury-induced glutamate overstimulation. Therefore, although the initial GT upregulation alone is insufficient to prevent the development of neuropathic pain in CCI rats, it plays a critical role in hampering the induction of neuropathic pain after CCI, as indicated by the exacerbated hyperalgesia and allodynia in the absence of this initial GT upregulation. Of note is that a nonselective Trk receptor inhibitor was used in this study because of the lack of selective subtype Trk receptor inhibitors at present. In addition, because the ERK-MAPK inhibitor PD98059 was used in this study, future studies may examine whether other members of the MAPK family (e.g., p38 MAPK) are also contributory to this process.
Besides the altered GT expression after CCI, changes in GT glutamate uptake activity also play a significant role in regulating regional glutamate homeostasis and neuropathic pain behaviors in CCI rats. Previous studies that used the electrophysiological method have shown that inhibition of GT activity may not prolong significantly the single stimulus-induced excitatory postsynaptic glutamate-mediated currents, but it does so if the stimulus is repetitive and excessive (Overstreet et al., 1999), a condition that is encountered after peripheral nerve injury (Wall et al., 1974; Seltzer et al., 1991). In this regard, enhancing GT uptake activity by the positive GT activity regulator riluzole would be expected to reduce glutamate excitation regardless of changes in GT expression. This hypothesis is supported strongly by the present findings that riluzole reduced thermal hyperalgesia and mechanical allodynia when given during either the induction or maintenance phase of neuropathic pain behaviors in CCI rats. That is, even when the GT expression was reduced after the initial stage of CCI, an enhanced GT uptake activity by riluzole, as demonstrated in ourin vitro glutamate uptake assay, was able to offset significantly the effects of GT downregulation on neuropathic pain behaviors in CCI rats. Similar effects of regulating GT uptake activity have been observed on the development of morphine tolerance (Nakagawa et al., 2001; Mao et al., 2002a,b), a process that has been shown to share certain common glutamate-mediated cellular and intracellular mechanisms (Mao et al., 1994, 1995; Ossipov et al., 1995).
Contributions to the central mechanisms of neuropathic pain
Neuropathic pain is perhaps the most challenging chronic pain condition. Both peripheral and central mechanisms have been proposed to explain clinical features of neuropathic pain. With regard to the peripheral mechanisms, abnormal expression or distribution of certain sodium channels and spontaneous ectopic discharges are some of the proposed mechanisms likely to be contributory to the development of neuropathic pain (Wall et al., 1974; Devor, 1983; Porreca et al., 1999;Waxman et al., 1999; Gold et al., 2003). For over a decade the central glutamatergic system has been a major focus of research interest on the central mechanisms of neuropathic pain. Basic research from this field has generated enormous information that has contributed major progress in understanding the neural mechanisms of neuropathic pain. To date, however, studies on the role of the central glutamatergic system in neuropathic pain have been focused mainly on the modulation of glutamate receptors and the associated intracellular events (Dougherty and Willis, 1991; Dubner, 1991; Wilcox, 1991; Dougherty et al., 1992;Mao et al., 1992a,b, 1995; Yamamoto and Yaksh, 1992; Malmberg et al., 1997; Woolf and Mannion, 1999; Guo et al., 2002).
The present findings suggest a new mechanism of nerve injury-induced central changes that modulate regional glutamate availability by regulating glutamate uptake capability via spinal GTs. Changes in regional glutamate uptake capability could be attributable to an altered expression of GTs at the spinal and/or supraspinal loci, a decreased GT uptake activity, or both after peripheral nerve injury as seen in CCI rats. Conceivably, increased regional glutamate availability secondary to GT changes would result in persistent and enhanced activation of glutamate receptors leading to increased neuronal excitability and possibly neurotoxic consequences. Such a process could take place independently of the modulation of glutamate receptors and the related intracellular changes and/or reinforce the previously proposed glutamate-mediated cellular and intracellular changes, further implicating the central glutamatergic system in the pathogenesis of neuropathic pain.
Regulation of regional glutamate uptake has been linked critically to the prevention of glutamate neurotoxicity as well as the pathogenesis of neurological disorders, including brain ischemia, epilepsy, amyotrophic lateral sclerosis, spinal cord injury, and Alzheimer's disease (Mennerick et al., 1999; Lievens et al., 2000; Vorwerk et al., 2000; Bigini et al., 2001; Trotti et al., 2001; Vera-Portocarrero et al., 2002). The present findings suggest a new strategy for treating neuropathic pain by reducing regional glutamate availability and preventing glutamate overexcitation via an enhanced GT system. The potential clinical implications of such a strategy merit future investigations with the development of more specific GT regulators and improved clinical trial methodologies (Hammer et al., 1999; Galer et al., 2000).
Footnotes
This work was supported by National Institutes of Health Grant R01DA08835 to J.M. We thank the Neural Plasticity Research Group at the Massachusetts General Hospital for generous technical support.
Correspondence should be addressed to Dr. Jianren Mao, Massachusetts General Hospital Pain Center, WACC 324, Department of Anesthesia and Critical Care, Massachusetts General Hospital, Harvard Medical School, 15 Parkman Street, Boston, MA 02114. E-mail: jmao@partners.org.
References
- 1.Arriza JL, Kavanaugh MP, Fairman WA, Wu YN, Murdoch GH, North RA, Amara SG. Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J Biol Chem. 1993;268:15329–15332. [PubMed] [Google Scholar]
- 2.Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 3.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 4.Bennett GJ, Kajander KC, Sahara Y, Iadarola MJ, Sugimoto T. Neurochemical and anatomical changes in the dorsal horn of rats with an experimental painful peripheral neuropathy. In: Cervero F, Bennett GJ, Headley PM, editors. Proceedings of sensory information in the superficial dorsal horn of the spinal cord. Plenum; New York: 1989. pp. 463–471. [Google Scholar]
- 5.Bigini P, Bastone A, Mennini T. Glutamate transporters in the spinal cord of the wobbler mouse. NeuroReport. 2001;12:1815–1820. doi: 10.1097/00001756-200107030-00011. [DOI] [PubMed] [Google Scholar]
- 6.Bonnot A, Corio M, Tramu G, Viala D. Immunocytochemical distribution of ionotropic glutamate receptor subunits in the spinal cord of the rabbit. J Chem Neuroanat. 1996;11:267–278. doi: 10.1016/s0891-0618(96)00173-1. [DOI] [PubMed] [Google Scholar]
- 7.Cheramy A, Barbeito L, Godeheu G, Glowinski J. Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci Lett. 1992;147:209–212. doi: 10.1016/0304-3940(92)90597-z. [DOI] [PubMed] [Google Scholar]
- 8.Coffey ET, Akerman KEO, Courtney MJ. Brain-derived neurotrophic factor induces a rapid upregulation of synaptophysin and tau protein via the neurotrophin receptor TrkB in rat cerebellar granule cells. Neurosci Lett. 1997;227:177–180. doi: 10.1016/s0304-3940(97)00335-2. [DOI] [PubMed] [Google Scholar]
- 9.Danbolt NC. Glutamate uptake. Prog Neurol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 10.Deng YS, Zhong JH, Zhou XF. Effects of endogenous neurotrophins on sympathetic sprouting in the dorsal root ganglia and allodynia following spinal nerve injury. Exp Neurol. 2000;164:344–350. doi: 10.1006/exnr.2000.7432. [DOI] [PubMed] [Google Scholar]
- 11.Devor M. Nerve pathophysiology and mechanisms of pain in causalgia. J Auton Nerv Syst. 1983;7:371–384. doi: 10.1016/0165-1838(83)90090-5. [DOI] [PubMed] [Google Scholar]
- 12.Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:S233–S241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- 13.Dougherty PM, Willis WD. Modification of the responses of primate spinothalamic neurons to mechanical stimulation by excitatory amino acids and an N-methyl-d-aspartate antagonist. Brain Res. 1991;542:15–22. doi: 10.1016/0006-8993(91)90991-4. [DOI] [PubMed] [Google Scholar]
- 14.Dougherty PM, Palecek J, Paleckova V, Sorkin LS, Willis WD. The role of NMDA and non-NMDA excitatory amino acid receptors in the excitation of primate spinothalamic tract neurons by mechanical, chemical, thermal, and electrical stimuli. J Neurosci. 1992;12:3025–3041. doi: 10.1523/JNEUROSCI.12-08-03025.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubner R. Neuronal plasticity and pain following peripheral tissue inflammation or nerve injury. In: Bond M, Charlton E, Woolf CJ, editors. Proceedings of the 5th World Congress on pain. Pain research and clinical management, Vol 5. Elsevier; Amsterdam: 1991. pp. 263–276. [Google Scholar]
- 16.Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem. 1999;73:1409–1421. doi: 10.1046/j.1471-4159.1999.0731409.x. [DOI] [PubMed] [Google Scholar]
- 17.Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 18.Galer BS, Twilling TL, Harle J, Cluff RS, Friedman E, Rowbotham MC. Lack of efficacy of riluzole in the treatment of peripheral neuropathic pain conditions. Neurology. 2000;55:971–975. doi: 10.1212/wnl.55.7.971. [DOI] [PubMed] [Google Scholar]
- 19.Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- 20.Gegelashvili G, Dehnes Y, Danbolt NC, Schousboe A. The high affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signaling mechanisms. Neurochem Int. 2000;37:163–170. doi: 10.1016/s0197-0186(00)00019-x. [DOI] [PubMed] [Google Scholar]
- 21.Gold MS, Weinreich D, Kim CH, Wang R, Treanor J, Porreca F, Lai J. Redistribution of NaV1.8 in uninjured axons enables neuropathic pain. J Neurosci. 2003;23:158–166. doi: 10.1523/JNEUROSCI.23-01-00158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gracely RH, Lynch SA, Bennett GJ. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 1992;51:175–194. doi: 10.1016/0304-3959(92)90259-E. [DOI] [PubMed] [Google Scholar]
- 23.Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci. 2002;22:6208–6217. doi: 10.1523/JNEUROSCI.22-14-06208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ha SO, Kim JK, Hong HS, Kim DS, Cho HJ. Expression of brain-derived neurotrophin factor in rat dorsal root ganglia, spinal cord, and gracile nuclei in experimental models of neuropathic pain. Neuroscience. 2001;107:301–309. doi: 10.1016/s0306-4522(01)00353-0. [DOI] [PubMed] [Google Scholar]
- 25.Hagemann C, Blank JL. The ups and downs of MEK kinase interactions. Cell Signal. 2001;13:863–875. doi: 10.1016/s0898-6568(01)00220-0. [DOI] [PubMed] [Google Scholar]
- 26.Hammer NA, Lilleso J, Pedersen JL, Kehlet H. Effect of riluzole on acute pain and hyperalgesia in humans. Br J Anaesth. 1999;82:718–722. doi: 10.1093/bja/82.5.718. [DOI] [PubMed] [Google Scholar]
- 27.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 28.Jabaudon D, Shimamoto K, Yasuda-KAmatani Y, Scanziani M, Gahwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA. 1999;96:8733–8738. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jabaudon D, Scanziani M, Gahwiler BH, Gerber U. Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci USA. 2000;97:5610–5615. doi: 10.1073/pnas.97.10.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- 31.Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kajander KC, Xu JX. Quantitative evaluation of calcitonin gene-related peptide and substance P levels in rat spinal cord following peripheral nerve injury. Neurosci Lett. 1995;186:84–88. doi: 10.1016/0304-3940(95)11294-7. [DOI] [PubMed] [Google Scholar]
- 33.Kalb RG, Fox AJ. Synchronized overproduction of AMPA, kainate, and NMDA glutamate receptors during human spinal cord development. J Comp Neurol. 1997;384:200–210. doi: 10.1002/(sici)1096-9861(19970728)384:2<200::aid-cne3>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 34.Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 35.Kus L, Saxon D, Beitz AJ. NMDA R1 mRNA distribution in motor and thalamic-projecting sensory neurons in the rat spinal cord and brain stem. Neurosci Lett. 1995;196:201–204. doi: 10.1016/0304-3940(95)11878-z. [DOI] [PubMed] [Google Scholar]
- 36.Li WP, Xian C, Rush RA, Zhou XF. Upregulation of brain-derived neurotrophic factor and neuropeptide Y in the dorsal ascending sensory pathway following sciatic nerve injury in rat. Neurosci Lett. 1999;260:49–52. doi: 10.1016/s0304-3940(98)00958-6. [DOI] [PubMed] [Google Scholar]
- 37.Lievens JC, Bernal F, Forni C, Mahy N, Kerkerian-Le Goff L. Characterization of striatal lesions produced by glutamate uptake alteration: cell death, reactive gliosis, and changes in GLT-1 and GADD45 mRNA expression. Glia. 2000;29:222–232. doi: 10.1002/(sici)1098-1136(20000201)29:3<222::aid-glia4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 38.Malmberg AB, Chen C, Tonegawa S, Basbaum AI. Preserved acute pain and reduced neuropathic pain in mice lacking PKCγ. Science. 1997;278:279–283. doi: 10.1126/science.278.5336.279. [DOI] [PubMed] [Google Scholar]
- 39.Mao J, Mayer DJ, Hayes RL, Lu J, Price DD. Differential roles of NMDA and non-NMDA receptor activation in induction and maintenance of thermal hyperalgesia in rats with painful peripheral mononeuropathy. Brain Res. 1992a;598:271–278. doi: 10.1016/0006-8993(92)90193-d. [DOI] [PubMed] [Google Scholar]
- 40.Mao J, Price DD, Hayes RL, Lu J, Mayer DJ. Intrathecal GM1 ganglioside and local nerve anesthesia reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res. 1992b;584:28–35. doi: 10.1016/0006-8993(92)90874-9. [DOI] [PubMed] [Google Scholar]
- 41.Mao J, Price DD, Mayer DJ, Lu J, Hayes RL. Intrathecal MK-801 and local nerve anesthesia synergistically reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res. 1992c;576:254–262. doi: 10.1016/0006-8993(92)90688-6. [DOI] [PubMed] [Google Scholar]
- 42.Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and opiate tolerance: a current view of their possible interactions. Pain. 1995;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- 44.Mao J, Price DD, Zhu J, Lu J, Mayer DJ. The inhibition of nitric oxide-activated poly(ADP-ribose) synthetase attenuates transsynaptic alteration of spinal cord dorsal horn neurons and neuropathic pain in the rat. Pain. 1997;72:355–366. doi: 10.1016/s0304-3959(97)00063-8. [DOI] [PubMed] [Google Scholar]
- 45.Mao J, Sung B, Ji RR, Lim G. Neuronal apoptosis associated with morphine tolerance: evidence for an opioid-induced neurotoxic mechanism. J Neurosci. 2002a;22:7650–7661. doi: 10.1523/JNEUROSCI.22-17-07650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci. 2002b;22:8312–8323. doi: 10.1523/JNEUROSCI.22-18-08312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin D, Thompson MA, Nadle JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250:473–476. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- 48.Matthews CC, Zielke HR, Wollack JB, Fishman PS. Enzymatic degradation protects from glutamate excitotoxicity. J Neurochem. 2000;75:1045–1052. doi: 10.1046/j.1471-4159.2000.0751045.x. [DOI] [PubMed] [Google Scholar]
- 49.Mennerick S, Shen W, Xu W, Benz A, Tanaka K, Shimamoto K, Isenberg KE, Krause JE, Zorumski CF. Substrate turnover by transporters curtails synaptic glutamate transients. J Neurosci. 1999;19:9242–9251. doi: 10.1523/JNEUROSCI.19-21-09242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Minami T, Matsumura S, Okuda-Ashitaka E, Shimamoto K, Sakimura K, Mishina M, Mori H, Ito S. Characterization of the glutamatergic system for induction and maintenance of allodynia. Brain Res. 2001;895:178–185. doi: 10.1016/s0006-8993(01)02069-8. [DOI] [PubMed] [Google Scholar]
- 51.Mitrovic AD, Maddison JE, Johnson GAR. Influence of the oestrous cycle on l-glutamate and l-aspartate transport in rat brain synaptosomes. Neurochem Int. 1999;34:101–108. doi: 10.1016/s0197-0186(98)00066-7. [DOI] [PubMed] [Google Scholar]
- 52.Mu X, Azbill RD, Springer JE. Riluzole improves measures of oxidative stress following traumatic spinal cord injury. Brain Res. 2000;870:66–72. doi: 10.1016/s0006-8993(00)02402-1. [DOI] [PubMed] [Google Scholar]
- 53.Nakagawa T, Ozawa T, Shige K, Yamamoto R, Minami M, Satoh M. Inhibition of morphine tolerance and dependence by MS-153, a glutamate transporter activator. Eur J Pharmacol. 2001;419:39–45. doi: 10.1016/s0014-2999(01)00965-7. [DOI] [PubMed] [Google Scholar]
- 54.Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci Lett. 1995;199:87–90. doi: 10.1016/0304-3940(95)12022-v. [DOI] [PubMed] [Google Scholar]
- 55.Overstreet LS, Kinney GA, Liu YB, Billups D, Slate NT. Glutamate transporters contribute to the time course of synaptic transmission in cerebellar granule cells. J Neurosci. 1999;19:9663–9673. doi: 10.1523/JNEUROSCI.19-21-09663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oyesiku NM, Evans CO, Houston S, Darrell RS, Smith JS, Fulop ZL, Dixon CE, Stein DG. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999;833:161–172. doi: 10.1016/s0006-8993(99)01501-2. [DOI] [PubMed] [Google Scholar]
- 57.Pascual M, Climent E, Guerri C. BDNF induces glutamate release in cerebrocortical nerve terminals and in cortical astrocytes. NeuroReport. 2001;12:2673–2677. doi: 10.1097/00001756-200108280-00017. [DOI] [PubMed] [Google Scholar]
- 58.Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci. 1994a;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci. 1994b;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain l-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- 61.Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert D, Sanggameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PNS/SNS and NaN/SNS2, in a rat model of chronic pain. Proc Natl Acad Sci USA. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson MB, Dowd LA. Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Adv Pharmacol. 1997;37:69–115. doi: 10.1016/s1054-3589(08)60948-5. [DOI] [PubMed] [Google Scholar]
- 63.Seltzer Z, Cohn S, Ginzgurg R, Berlin B. Modulation of neuropathic pain behavior in rats by spinal disinhibition and NMDA receptor blockade of injury discharge. Pain. 1991;45:69–75. doi: 10.1016/0304-3959(91)90166-U. [DOI] [PubMed] [Google Scholar]
- 64.Semba J, Wakuta MS. Regional differences in the effects of glutamate uptake inhibitor trans-pyrrolidine-2,4-bicarboxylic acid on extracellular amino acids and dopamine in rat brain: an in vivo microdialysis study. Gen Pharmacol. 1998;31:399–404. doi: 10.1016/s0306-3623(98)00047-0. [DOI] [PubMed] [Google Scholar]
- 65.Stevens CW, Kajander KC, Bennett GJ, Seybold VS. Bilateral and differential changes in spinal cord mu, delta, and kappa opioid binding in rats with a painful, unilateral neuropathy. Pain. 1991;46:315–326. doi: 10.1016/0304-3959(91)90114-D. [DOI] [PubMed] [Google Scholar]
- 66.Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from the rat brain. Proc Natl Acad Sci USA. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:275–382. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka K. Expression cloning of a rat glutamate transporter. Neurosci Res. 1993;16:149–153. doi: 10.1016/0168-0102(93)90082-2. [DOI] [PubMed] [Google Scholar]
- 69.Troller U, Zeidman R, Svensson K, Larsson C. A PKCβ isoform mediates phorbol ester-induced activation of Erk1/2 and expression of neuronal differentiation genes in neuroblastoma cells. FEBS Lett. 2001;508:126–130. doi: 10.1016/s0014-5793(01)03043-5. [DOI] [PubMed] [Google Scholar]
- 70.Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown RHJ, Hediger MA. Amyotrophic lateral sclerosis-linked glutamate transporter mutation has impaired glutamate clearance capacity. J Biol Chem. 2001;276:576–582. doi: 10.1074/jbc.M003779200. [DOI] [PubMed] [Google Scholar]
- 71.Vera-Portocarrero LP, Mills CD, Ye Z, Fullwood SD, McAdoo DJ, Hulsebosch CE, Westlund KN. Rapid changes in expression of glutamate transporters after spinal cord injury. Brain Res. 2002;927:104–110. doi: 10.1016/s0006-8993(01)03329-7. [DOI] [PubMed] [Google Scholar]
- 72.Vorwerk CK, Naskar R, Schuettauf F, Quinto K, Zurakowski D, Gochenauer G, Robinson, Mackler SA, Dreyer EB. Depression of retinal glutamate transporter function leads to elevated intravitreal glutamate levels and ganglion cell death. Invest Ophthalmol Vis Sci. 2000;41:3615–3621. [PubMed] [Google Scholar]
- 73.Walker SM, Mitchell VA, White DM, Rush RA, Duggan AW. Release of immunoreactive brain-derived neurotrophic factor in the spinal cord of the rat following sciatic nerve transection. Brain Res. 2001;899:240–247. doi: 10.1016/s0006-8993(01)02259-4. [DOI] [PubMed] [Google Scholar]
- 74.Wall PD, Waxman SG, Basbaum AI. Ongoing activity in peripheral nerve: injury discharge. Exp Neurol. 1974;45:576–589. doi: 10.1016/0014-4886(74)90163-0. [DOI] [PubMed] [Google Scholar]
- 75.Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci USA. 1999;96:7635–7639. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilcox G. Excitatory neurotransmitters and pain. In: Bond MR, Charlton JE, Woolf CJ, editors. Proceedings of the 6th World Congress on pain. Elsevier; New York: 1991. pp. 97–114. [Google Scholar]
- 77.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 78.Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 79.Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- 80.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 81.Yamamoto T, Yaksh TL. Spinal pharmacology of thermal hyperalgesia induced by constriction injury of sciatic nerve. Excitatory amino acid antagonists. Pain. 1992;49:121–128. doi: 10.1016/0304-3959(92)90198-K. [DOI] [PubMed] [Google Scholar]
- 82.Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-κB. Mol Pharmacol. 2000;57:667–678. doi: 10.1124/mol.57.4.667. [DOI] [PubMed] [Google Scholar]