

Abstract
We investigated the location of calcium entry sites and synaptic ribbons in the type-Mb goldfish bipolar neuron and the bullfrog saccular hair cell. Cells were loaded with a fast calcium indicator (Fluo-3 or Fluo-5F) and an excess of a high-affinity but slow Ca buffer (EGTA). The cell surface was imaged by evanescent field microscopy. Small fluorescent “hot spots” representing calcium entry sites appeared abruptly when a voltage step opened Ca channels and disappeared or dimmed abruptly when Ca channels closed. In bipolar cells, the fluorescence of hot spots tracked the calcium influx. Hair cells showed similar Ca hot spots. Synaptic ribbons or dense bodies were labeled by immunofluorescence with an antibody that recognizes the ribbon protein ribeye. The antibody labeled punctate structures beneath the plasma membrane. In both bipolar neurons and hair cells, the number of Ca entry sites was similar or identical to that of ribbons or dense bodies, consistent with the idea that calcium-channel clusters reside near ribbons, and that both mark active zones. In bipolar cells, the number of Ca entry sites and ribeye-positive fluorescent spots is also strikingly similar to that of exocytic active zones but significantly less than the number of total exocytic sites including solitary fusion events outside active zones. We suggest that in bipolar terminals, active zones, Ca entry sites, and synaptic ribbons all colocalize, but also that a significant number of vesicles can fuse outside active zones and, hence, independently of synaptic ribbons.
Keywords: calcium microdomains, ribeye, exocytosis, retina, bipolar cell, hair cell, evanescent field
Introduction
In some sensory synapses, presynaptic active zones are marked by electron-dense proteinaceous structures called ribbons or dense bodies. It is thought that the multitude of synaptic vesicles distributed throughout a terminal must first find their way to a relatively small number of ribbons beneath the plasma membrane (Bunt, 1971; Gray and Pease, 1971; Raviola and Gilula, 1975) before they can fuse with the membrane itself, although other regions of the plasma membrane may be much closer physically. Support for ribbon-specific exocytosis comes first from the finding that postsynaptic densities with their glutamate receptors (Matsubara et al., 1996; Morigiwa and Vardi, 1999; Qin and Pourcho, 1999; Ghosh et al., 2001) are located opposite ribbons. Second, electrophysiologic studies on goldfish bipolar neurons have shown that exocytosis slows greatly once a “readily releasable” pool of vesicles has been depleted, and that this pool is strikingly similar in size to the pool of vesicles attached to ribbons (von Gersdorff et al., 1996). Third, omega profiles are found primarily near dense bodies in electron tomographic reconstructions (Lenzi et al., 1999, 2002). Finally, direct observation has shown that vesicles fuse preferentially at a small number of active zones (Zenisek et al., 2000).
What makes exocytosis in ribbon synapses so site-specific? Perhaps only beneath ribbons does the Ca2+concentration rise high enough to trigger exocytosis. Indeed, exocytosis in bipolar cells requires calcium concentrations (Heidelberger et al., 1994) that are so high that they are expected only near open calcium channels. Electron micrographs show clusters of particles in the plasma membrane beneath synaptic ribbons (Raviola and Gilula, 1975; Raviola and Raviola, 1982) and dense bodies. In saccular hair cells, the particle clusters beneath dense bodies consist of Ca channels and Ca-activated K channels, and high-cytosolic Ca2+ concentrations do arise locally while the Ca channels open (Roberts et al., 1990). In addition, confocal microscopy has shown that Ca influx is localized to small regions of the plasma membrane in bullfrog saccular hair cells (Issa and Hudspeth, 1996) and in cochlear hair cells (Tucker and Fettiplace, 1995). However, in bipolar cells, local Ca influx has not been demonstrated. Indeed, it has been generally difficult to demonstrate that Ca influx is local at the micrometer or submicrometer level. Here, we used evanescent field-fluorescence microscopy to image calcium entry into goldfish bipolar cells and bullfrog saccular hair cells. The number of Ca entry sites is equal to that of ribbons as visualized by immunostaining.
A related question is how site-specific exocytosis is in bipolar neurons. The spatial distribution of single-vesicle fusion events does indicate preferential fusion at discrete active zones (Zenisek et al., 2000) that occur in numbers equal to those found here for ribeye-positive fluorescent spots and for Ca entry sites. In addition, however, solitary fusion events have also been observed outside active zones and account for approximately one-third of exocytic events during long stimuli (Zenisek et al., 2000). However, per unit area, their number is estimated here to be >100-fold lower than at active zones. Hence, fusion is indeed highly site-specific for active zones.
Materials and Methods
Cell and tissue preparation and electrical recording
Goldfish bipolar cells. Goldfish were decapitated, and eyes were removed and hemisected as described previously (Heidelberger and Matthews, 1992). To remove vitreous, eyecups were placed for 20 min in a solution of hyaluronidase (1100 U/ml) containing (in mm): 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1 MgCl2, 10 glucose, and 10 HEPES. Next, each retina was removed from the eyecups, cut into six to eight pieces, and digested in a papain solution (35 U/ml; Fluka, Buchs, Switzerland) for 40 min containing (in mm): 2.7 cysteine, 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1 MgCl2, 10 glucose, and 10 HEPES. Pieces of tissue were mechanically triturated using a fire-polished Pasteur pipette and plated to high-refractive index coverslips (n488 = 1.80; Plan Optik, Elsoff, Germany). Bipolar neurons were recognized by their unique morphology with large synaptic terminals [Zenisek et al. (2002), their Figs. 1 and 5] and used within 8 hr of removing the retina. For electrical recording and imaging, cells were placed in a recording solution containing (in mm): 120 NaCl, 2.5 CaCl2, 2.5 KCl, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4, with NaOH.
Cells were voltage-clamped using an EPC-9 amplifier (Heka Electronik, Lambrecht, Germany), running Pulse stimulus and acquisition software (Instrutech, Port Washington, NY). For whole-cell recording of bipolar cells, the patch pipette was placed on the cell body and the pipette solution contained (in mm): 120 Cs-glutamate, 4 Na2ATP, 0.5 GTP, 4 MgCl2, 10 TEA-Cl, pH 7.2, with CsOH, 10 EGTA, and 0.2 Fluo-5F pentapotassium salt (Molecular Probes, Eugene, OR).
Bullfrog hair cells
Hair cells were isolated from the sacculus of adult bullfrogs (Rana catesbeiana) as described previously (Assad and Corey, 1992). Briefly, the inner ears were removed in a low-calcium solution (LCS) containing (in mm): 110 NaCl, 2 KCl, 2 MgCl2–6 H2O, 3d-glucose, 10 HEPES, and 0.1 CaCl2. A small tear in the membrane above the sacculus was made, and tight junctions were disrupted with a 15 min incubation at room temperature (RT) in 1 mm EGTA in LCS. The saccular macula was then excised in LCS, and otoconia were removed. Isolated sacculi were then placed in papain solution (∼30 U/ml papain) (Fluka BioChemika, Ronkonkoma, NY) in double-distilled water containing 8.3 mml-cysteine for 5 min at room temperature. Sacculi were then transferred to DNase I solution (2 mg/ml LCS) for removal of otolithic membranes to expose hair cells. Cells were teased with an eyelash into a 60 μl droplet of LCS on a high-refractive index coverslip coated with concanavalin A (1 mg/ml LCS). After allowing several minutes for hair cells to settle, isolated cells were superfused with bath solution (Parsons et al., 1994) containing (in mm): 110 NaCl, 4 CaCl2, 2 KOH, 5 HEPES, and 3 d-glucose.
As with bipolar cells, hair cells were voltage-clamped using an EPC-9 amplifier with Pulse acquisition software. For electrical recordings, the pipette solution, adapted from Parsons et al. (1994), contained (in mm): 107.5 CsOH, 106 aspartate, 5 HEPES, 2 MgCl2, 0.078 CaCl2, 1 EGTA, and CsOH to bring pH to ∼7.4. Additional ingredients were added freshly before use (in mm): 10 EGTA, 10 TEA-Cl, 1.8 Na2ATP, and a 0.2 mm concentration of the Ca indicators Fluo-3, Fluo-5F, or Fluo-5N, all as pentaammonium salts (Molecular Probes). The osmolarity was adjusted to 240–250 mOsm. The external recording solution, adapted from Parsons et al. (1994), for hair cells was composed of (in mm): 110 NaCl, 4 or 17 CaCl2, 2 KOH, 5 HEPES, 3d-glucose, and NaOH to bring pH to ∼7.4.
Calcium imaging and data acquisition
Cells were viewed through an inverted microscope (Axiovert 135;Zeiss, Oberkochen, Germany) and modified for through-the-lens evanescent field illumination (Axelrod, 2001; Steyer and Almers, 2001) as described previously (Zenisek et al., 2002). A 488 nm wavelength beam from an argon laser (Coherent Inc., Santa Clara, CA) was applied by opening and closing a shutter for 1–5 sec periods of observation. The expanded beam was focused off-axis onto the back focal plane of a 1.65 NA objective (Apo ×100 O HR;Olympus Optical, Tokyo, Japan). It underwent total internal reflection at the interface between the coverglass and the cell and generated in the cell an evanescent field declining exponentially with distance from the interface, depending on the angle at which glass strikes the interface. For experiments with bipolar cells, we used the largest angle allowed by our objective (65.6–68°) as measured with a hemicylinder (Zenisek et al., 2002). For hair cells, a smaller angle (54o) was used. With this angle, the evanescent field is predicted to decline e-fold within 78.2 nm of the interface. Images were acquired with a fiber-coupled intensified frame-transfer CCD camera (I-Pentamax; Roper Scientific, Tucson, AZ) at frame rates between 20 and 50 Hz. Images were acquired and analyzed using Metamorph software (Universal Imaging Corporation, West Chester, PA).
Immunohistochemistry
Dissociated cells. Dissociated bipolar cells from retina of goldfish and hair cells from the inner ear of frogs were isolated as described above. The dissociated cells were fixed on coverslips with 2% formaldehyde at room temperature for 40 min. Cells were permeabilized in blocking buffer containing 3% bovine serum albumin (BSA), 10% normal goat serum (NGS), and 0.5% Triton X-100 in PBS at RT for 15 min followed by overnight incubation at 4°C with an antibody raised against C-terminal binding protein (CtBP1) fromXenopus laevis (diluted 1:200) in blocking buffer (antibody generously provided by Dr. Arie P. Otte, University of Amsterdam, Amsterdam, The Netherlands). The antibody recognizes CtBP2 (Sewalt et al., 1999), which has near-perfect sequence identity to the B-domain of ribeye, one of the components of the presynaptic ribbon (Schmitz et al., 2000). Cells were washed with washing buffer containing 0.2% BSA and 0.1% Triton X-100 in PBS four times (10 min each time) and then incubated with 1:1200 diluted Alexa Fluor 488 goat anti-rabbit IgG (heavy plus light) F(ab′)2 fragment conjugate (Molecular Probes) in blocking buffer at RT for 30 min. Cells were washed with washing buffer four times (10 min each time) and PBS four times (5 min each time). The coverslips were mounted on glass slides for observation. Cells were viewed by confocal microscopy (MRC 1000; Bio-Rad, Hercules, CA) with an S-Plan OlympusOptical 100 × 1.25 NA oil objective using a 0.5 μm iris. Both cell types were scanned by a confocal microscope with 0.5 μm z-step section, and three-dimensional images were reconstructed usingMetamorph software for localization of each ribbon or dense body. The number of ribbons and dense bodies was counted visually. Those at the bottom surface were determined in a 1-μm-thick optical section obtained by adding the two 0.5-μm-thick bottom-most sections. For the top surface, we added the two top-most sections.
In some experiments, the 0.1 ml of antiserum diluted 1:200 in blocking buffer was preincubated overnight (4°C) with 0.05 ml of Ni-NTA agarose beads (Qiagen, Hilden, Germany) coated with 0.4 ml of 0.01 mm His-tagged recombinant CtBP2 protein (kindly provided by Dr. Richard Goodman and Ngan Vo, Vollum Institute, Portland, OR). The beads were spun down, and the immunodepleted supernatant was used as described above for immunofluorescence staining of dissociated bipolar cells. As a control, the overnight incubation was performed with uncoated beads. The cells were viewed by epifluorescence on a Zeiss Axiovert microscope with a Planapo 100 × 1.4 NA objective, and images were taken with a cooled CCD camera (Micromax; Roper Scientific).
Immunofluorescence of retinal slices
Retinal tissue from goldfish was isolated and frozen in optimal cutting temperature-embedding medium (Sakura, Tokyo, Japan) on dry ice immediately. The cryostat sections with 15 μm thickness were made by using a microtome (2800 Frigocut; Reichert-Jung, Wetzlar, Germany). The glass slides with retinal sections were air-dried for 1 hr and stored in −30°C before use. Immunofluorescent staining of ribeye was performed as follows: Retinal sections were incubated with 1:200 Xenopus CtBP antibody diluted in blocking buffer (1% BSA, 10% NGS, and 0.2% Triton X-100 in PBS) at 4°C overnight. The slides were washed with buffer (0.5% BSA and 0.2% Triton X-100 in PBS) four times for 5 min. The sample was next incubated with 1:1200 goat anti-rabbit IgG Alexa Fluor 488 (Molecular Probes), diluted in blocking buffer for 30 min at room temperature, and washed four times for 20 min. Samples were mounted and observed by confocal microscopy with a 60× Nikon (Tokyo, Japan) oil objective.
Analysis
The average fluorescence in small circular regions was measured, and the fluorescence in a location outside the terminal “footprint” was subtracted as background. The difference was used in all additional analyses.
Measurements of leak conductance. Leak conductance was measured after each depolarization by applying a 15 mV hyperpolarization and measuring the current drop after allowing 100 msec for the capacitance transient to subside.
Unless otherwise noted, means are given as ±SE. All reagents are from Sigma (St. Louis, MO) and Aldrich (Milwaukee, WI) unless otherwise specified.
Locating Ca entry sites by using a high-affinity slow Ca buffer
When the internal Ca2+ concentration [Ca2+] is measured with a Ca indicator, the site of Ca2+ entry can be defined more sharply if the Ca indicator coexists with an abundance of a high-affinity Ca buffer such as BAPTA (Issa and Hudspeth, 1996). We show here that this approach is greatly enhanced by using a buffer that binds Ca2+ slowly, such as EGTA. Ignoring the effects of mobile endogenous calcium buffers, when a small quantity of Ca2+ is applied instantly and uniformly to the cytosol, the binding of calcium to indicator and buffer is governed by the following equations:
| Equation 1 |
| Equation 2 |
where [Ca-B] is the concentration of calcium buffer complex, [Ca] is the free calcium concentration,kon,1 is the binding rate constant,koff,1 is the unbinding rate constant for the buffer, [B] is the free calcium buffer, [F] is the concentration of free calcium indicator, [Ca-F] is the calcium-bound indicator, kon,2 is the binding rate constant, and koff,2 is the unbinding rate constant for the calcium indicator. On first entering the cell, calcium ions will predominantly bind to the calcium buffer with the highest on-rate. Assuming for the moment that Ca indicator and buffer are unoccupied before Ca entry, the fraction, R, of Ca ions binding to the Ca indicator rather than to the buffer is as follows:
| Equation 3 |
For Fluo-3, for instance, kon,1= 2.36 × 108m−1sec−1(Escobar et al., 1997), and a similar association rate constant may be assumed for the Ca indicator Fluo-5F (Naraghi, 1997). For EGTA, the on-rate is ∼100-fold slower (2.5 × 106m−1sec−1) (Naraghi, 1997). Hence with 10 mm EGTA and 0.2 mm Fluo-5F, Equation 3 predicts that Ca ions entering the cell are approximately twice as likely to bind to Fluo-5F than to EGTA. At equilibrium, calcium binding is determined by the equilibrium constants of the buffers as follows:
| Equation 4 |
| Equation 5 |
and Ca, initially bound to indicator, will relocate to EGTA. Assuming that the saturation is still negligible both for the indicator and buffer (total buffer, ∼[B]; total indicator, ∼[F]), the relative fraction of Ca still bound to indicator at equilibrium is as follows:
| Equation 6 |
where Kd,1 =koff,1/kon,1is the dissociation constant of the indicator andKd,2 is that for the buffer. WithKd = 2.3 μmfor Fluo-5F (Molecular Probes Data Sheet, Molecular Probes) and Kd = 150 nm for EGTA, pH 7.2, Equation 6 predicts that only 1 in 767 bound Ca ions is bound to indicator. Even if we allow the EGTA to be partly saturated, as it would be at a free calcium concentration of 1 μm, there are still 140 times more calcium ions bound to EGTA (Eqs. 4 and 5). Therefore, when traces of Ca indicator with a fast on-rate but low affinity coexist with an excess of Ca buffer with high affinity, the indicator will report the calcium ions that entered the cell most recently. These will be nearest the location of calcium entry.
The spatial definition of Ca entry sites thus imaged depends on the time the Ca remains bound to the indicator. From the on-rate of Fluo-5F and the dissociation constant, the off-rate can be estimated as 543 sec−1. Hence, a Ca-loaded indicator molecule has 1.8 msec to diffuse before losing its Ca and can cover a distance of 0.54 μm in the plane of the membrane if the Ca-indicator complex diffuses with a diffusion coefficient of 40 μm2/sec as in frog saccular hair cells (Issa and Hudspeth, 1996). Once Ca dissociates, the indicator greatly diminishes its fluorescence. Two-thirds of the Ca2+ will bind to another indicator molecule, and one-third will bind to EGTA and, hence, stop contributing to fluorescence. It will take several cycles of binding and unbinding from a Ca indicator before Ca finally translocates to EGTA. After four cycles of binding and unbinding, the average Ca has diffused 1.08 μm, and only 20% of Ca2+ remains bound to the indicator. Hence, the spatial definition of Ca entry sites with our mixture is expected to be in the micrometer range. Endogenous mobile calcium buffers may also confine the size of Ca spots. The cell types used in our studies have large concentrations of endogenous mobile calcium buffers (Roberts, 1993; Edmonds et al., 2000; Burrone et al., 2002). For example, tall thin hair cells contain ∼1.2 mmcalretinin, a protein that has five calcium-binding sites per molecule with association rate constants approximately fivefold slower than Fluo-5f and a Kd near 1 μm (Edmonds et al., 2000). With the high-concentration and fast-association rate constants of these buffers, calretinin would be expected to be sixfold more effective at binding calcium entering the cell than Fluo-5f. This will both reduce the calcium indicator signal surrounding calcium entry sites and limit the calcium indicator signal to one cycle of calcium binding and unbinding.
Results
Localized fluorescence changes during voltage steps in bipolar terminals
To image calcium entry sites in the giant-terminal retinal bipolar neuron of the goldfish (Carassius auratus), cells were loaded with a small quantity of calcium indicator and a high concentration of EGTA (10 mm) via a patch pipette. Cells showed dim featureless fluorescence at rest (Fig.1A), but two bright spots lit up when the membrane voltage was depolarized and calcium entered in discrete locations through voltage-gated calcium channels (Fig. 1B). When the cell was returned to its resting potential, the spots vanished (Fig. 1C; movies 1–4, available at www.jneurosci.org). The spots highlighted sites where Ca entered, because Ca2+ ions are expected to bind to the indicator in wherever they first entered the cell, and get absorbed by the excess EGTA buffer as they diffuse away from their entry site (see Materials and Methods). As in previous work on neutrophils by Omann and Axelrod (1996), we specifically looked at near-membrane calcium by taking advantage of the thin optical sectioning provided by evanescent field illumination. This technique excites fluorescence only in an ∼100 nm thin layer close to the plasma membrane in the footprint in which the cell adheres closely to the coverslip.
Fig. 1.
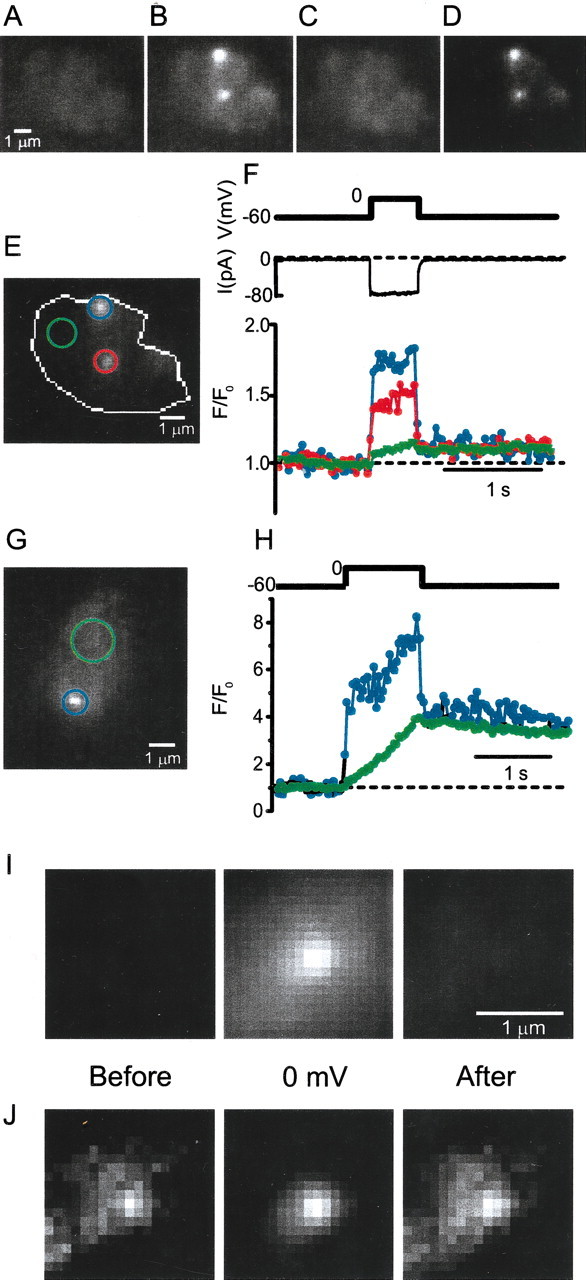
Fast and slow fluorescence changes in bipolar cell terminals loaded with a Ca indicator. Bipolar cells were loaded through the cell body with an internal solution containing 10 mmEGTA and 200 μm Fluo-5F (see movie 1). Images were taken under evanescent field illumination and show the footprint in which cells adhered tightly to the coverslip. A, Fluorescence collected for 1 sec before a 500 msec voltage step to 0 mV.B, Fluorescence collected during the 500 msec voltage step. C, Fluorescence collected during the 1 sec after return to −60 mV. D, Image of B after subtracting the average of images A andC. E, Image Dshowing regions used for monitoring fluorescence changes. White outline shows extent of cell footprint. F, Voltage step (top) and calcium current (middle); bottom shows the fluorescence in the three regions marked in E. Thetraces are color coded by region. For each region, intensity values were divided by their mean over the 1 sec before the voltage pulse. Insofar as the calcium concentration is uniform at the resting membrane potential, normalizing to initial fluorescence corrects for any local variations in cell footprint geometry or fluorophore concentration. G, As in B for another cell. H, As in F for the cell in G. Horizontal dotted lines inF (bottom) and H indicate initial fluorescence (F/F0 = 1). InI and J, eight 2 × 2 μm square regions from six cells were centered on fluorescent spots, as inB, that were selected to have a distance of >1 μm from the edge of the footprint. I, Light was collected for 500 msec before (left) and during a voltage step to 0 mV (middle). I, Right, Light was collected for a 500 msec interval starting 100 msec after the voltage was returned to the resting potential. The eight regions were superimposed and averaged. I, All images at same contrast. J, Contrast adjusted to take maximal advantage of the entire gray scale. Scale bar, 1 μm.
Figure 1D shows B subtracted by the average of C and A to better visualize the difference between the fluorescence when Ca channels were open and shut. Although fluorescence rose most dramatically in the two spots, it also increased elsewhere in the synaptic terminal. The number and location of hot spots stayed the same through up to 25 successive depolarizations over 15 min. In this and 16 similar experiments, such fluorescent hot spots populated the cell surface at an average of 0.112 ± 0.017 spots/μm−2.
To analyze the time course of fluorescence changes, we placed circular regions around each of the two spots and around a “quiet” region elsewhere in the cell, and plotted the fluorescence in each region against time (Fig. 1F). The resulting trace showed two kinetic components. A fast component rose and fell with the voltage pulse, and a slow component rose steadily during the pulse and fell only slowly thereafter. The slow component had approximately the same amplitude everywhere on the cell. It most likely reflects the bulk cytosolic calcium changes that are also observed using standard epifluorescence or confocal microscopy (Heidelberger and Matthews, 1992; Kobayashi and Tachibana, 1995; Neves and Lagnado, 1999; Zenisek and Matthews, 2000). In contrast, the fast component appeared and vanished as Ca channels opened and closed and was largest in the two discrete regions. In some cells, the rapid component was entirely confined to small bright spots (Fig. 1G,H). Hints of inhomogeneous fluorescence rises while calcium channels were open could also be seen under epifluorescence, but the fluorescent regions were larger and poorly defined (data not shown).
Although Ca entry sites stood out clearly only while Ca channels were open, close examination showed fluorescence at such sites also when most or all channels were closed. Figure 1I shows the average of eight spots before (left), during (middle), and after (right) a voltage step to 0 mV. As shown previously, a bright spot is seen while Ca channels were open. Enhancing the contrast from each of these images (Fig.1J) reveals that fluorescent spots also appeared before and after the voltage step, albeit dimly. The result can be explained if a small number of calcium channels remained open at −60 mV. Alternatively, the fluorescent spots at −60 mV may result from Ca-indicator binding to fixed structures beneath the plasma membrane (e.g., from ribbons, as in frog saccular hair cells in which indicator dye binds to dense bodies), the analog of synaptic ribbons (Issa and Hudspeth, 1994).
Do fluorescent hot spots result entirely from localized Ca indicator rather than localized Ca entry? If Ca entry were uniform, then fluorescence should rise by the same percentage everywhere on the plasma membrane. To test this idea, 2 μm circles were drawn around each hot spot and excised from the image of the entire footprint. The average fluorescence of the remaining image was taken as representative of plasma membrane outside hot spots. Fluorescence in hot spots was measured as averages in 1-μm-diameter circles. When Ca channels opened, fluorescence rose by 215 ± 45% in hot spots, more than four times as much as outside hot spots (46 ± 9%;n = 9 cells). Even this large difference is an underestimate if Ca2+ enters through Ca channel clusters of <1 μm in diameter. We conclude that local binding of Ca indicator cannot fully explain the intensity of fluorescent hot spots while Ca channels are open, and, therefore, that some or all Ca2+ enter locally.
Intensity of fluorescent spots depends on the magnitude of the calcium current
To investigate the relationship between fluorescence and calcium influx, cells were depolarized to different membrane potentials, and the rapid component of the fluorescence rise was measured. These measurements are demonstrated in Figure2. As a voltage step (Fig.2A, top) opened and closed Ca channels, the fluorescence of a hot spot (Fig. 2A,bottom, filled symbols) showed a rapid component that rose and fell with the voltage pulse as well as a slower component that remained even after the pulse. The contribution of the slow component (Fig. 2A, dashed line) was estimated by measuring the average fluorescence before and after the depolarization and connecting the two levels by a straight line. After subtraction of the slow component, the rapid component became a square pulse coincident with the voltage pulse (Fig. 2B). Its amplitude is taken to represent Ca influx through open Ca channels. The essentially constant amplitude indicates that Ca influx continued undiminished during the voltage pulse and did not decline, as might be expected if the Ca influx depleted calcium ions from the narrow space beneath the cell. In Figure2C, the amplitude of the fast component (Fig.2B, dashed line) is plotted against membrane potential. The relationship is similar or identical to that between Ca current and potential, consistent with the idea that the fast component tracks Ca influx.
Fig. 2.
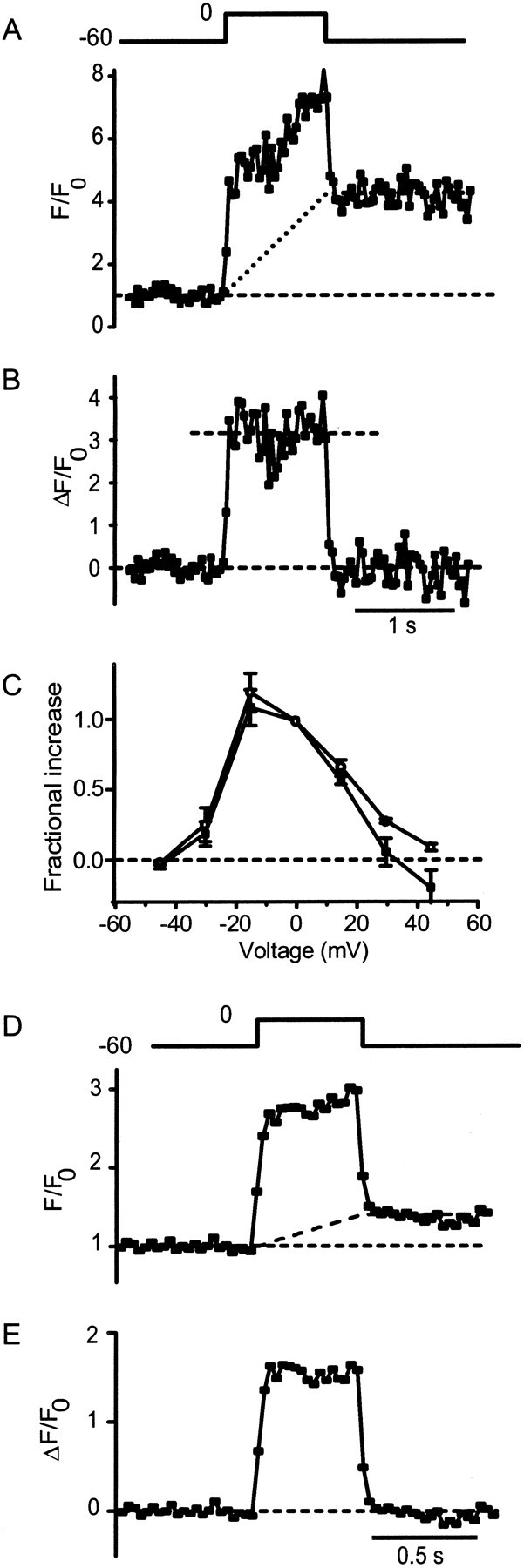
Fluorescence of spots follows the magnitude of calcium current. A, Top, Voltage pulse;bottom, fluorescence in a 1 μm circle encompassing a hot spot (filled symbols) as a function of time. The dotted line estimates the contribution from the slow component. This is the same experiment as that in Figure 1,G and H, in which fluorescence is expressed as F/F0, whereF0 is the fluorescence in the circle before depolarization (see Materials and Methods). B, Fluorescence trace in A after subtraction of thedotted line. It estimates the fast component, whose amplitude was measured as the average value during the voltage pulse (dashed line). C, Amplitude of the fast component determined as in B (dashed line) and plotted against membrane potential (open circles). Calcium current is the average inward current during depolarization (Fig.1F, bottom) after subtracting leak current (see Materials and Methods). For each cell, both calcium current and fluorescence amplitude were normalized to their values at 0 mV; the results were averaged (7 hot spots in 4 cells).D, Image of E as in A andB but in a cell loaded with the low-affinity Ca indicator Fluo-5N instead of Fluo-5F. An average oftraces from four depolarizations to 0 mV is shown.
One may wonder whether the square shape of the fast component arose from local saturation of the Ca indicator; however, this seems unlikely. First, the fluorescence at that location did rise throughout the voltage pulse, which is inconsistent with saturation (Fig.1A). Second, the amplitude of the fast component varied in proportion with the Ca current (Fig. 2C), indicating that even the fast component is not caused by saturated Ca indicator. Nonetheless, we tested Fluo-5N, a Ca indicator of nearly 40-fold lower affinity to Ca (dissociation constant, 90 μm; Molecular Probes Data Sheet). Both slow and fast components were observed (Fig.2D), and the fast component had a square shape (Fig. 2E).
The fast component varies in proportion to calcium current
We also tested whether the rapid component of the fluorescence change at hot spots tracks calcium influx. Bipolar cells were stepped to a series of different membrane potentials (Fig.3A), in which each generated different amounts of calcium current (Fig. 3B). If the rapid component at hot spots reflected local Ca entry, it should have the same time course as the calcium current (Ica). In contrast, changes in bulk cytoplasmic calcium concentration should follow the time integral of the calcium current (Fig. 3C, QCa). Figure 3D (filled circles) shows the fluorescence in a hot spot (Fig. 1G, red). Two kinetic components are apparent. A fast component rose and fell with the Ca current, whereas a slower component rose continuously and outlasted the voltage pulses. To determine the fast component in isolation, the time integral of the Ca current, QCa (Fig. 3C), was scaled to superimpose onto the fluorescence trace both before and after the series of voltage pulses. It was then subtracted from Figure3D, and the result is plotted in Figure 3F(left axis, open circles) along with the Ca current trace in Figure 3B after the trace was inverted and scaled (Fig. 3F, right axis, solid line). Within experimental error, the two traces show an identical time course that is consistent with the idea that the fast component of fluorescence at hot spots tracks Ca influx through open Ca channels. Similar results were seen for five spots in three cells.
Fig. 3.
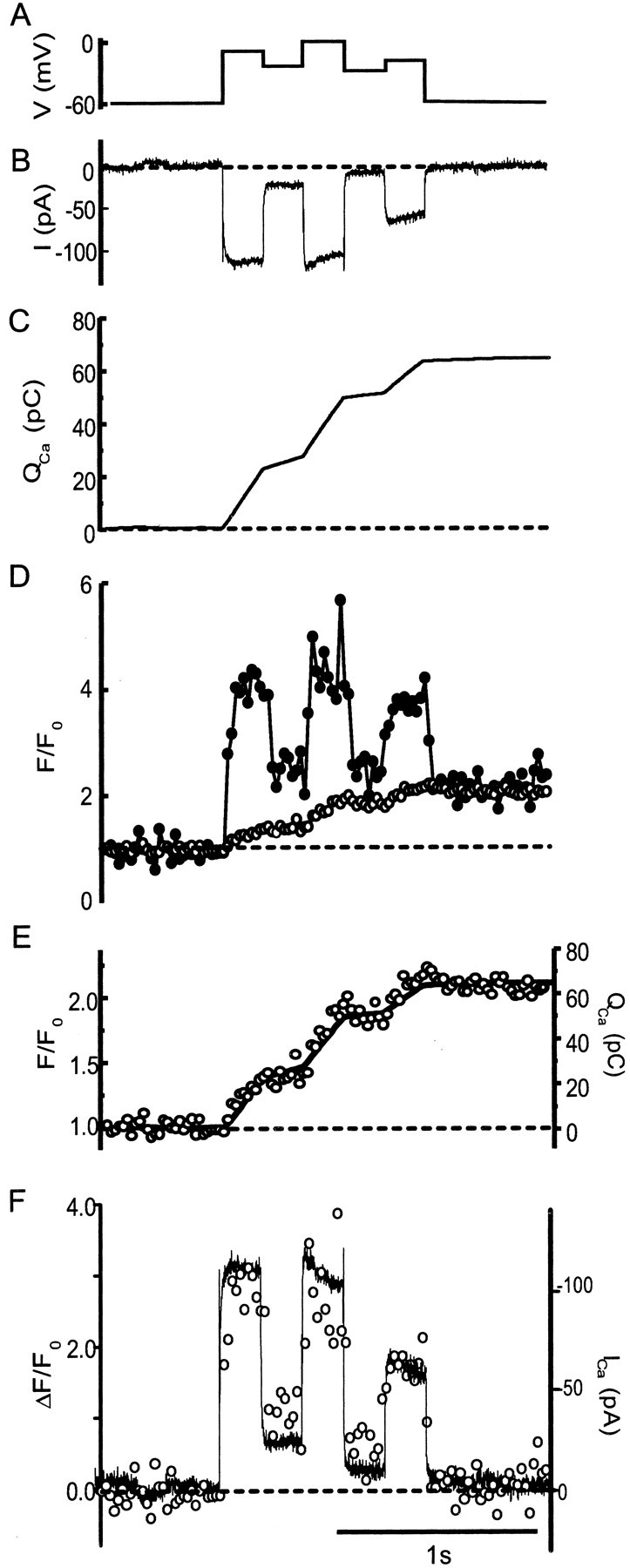
Fluorescence of hot spots as a component that tracks calcium current. A, B, Voltage (A) and leak subtracted (B) current. Because the cell was dialyzed with a Cs-glutamate-based internal solution designed to block potassium channels, the current reflects almost entirely calcium flux through L-type voltage-gated calcium channels (Heidelberger and Matthews, 1992; Tachibana et al., 1993). C, Time integral of the current inB (QCa). D, Fluorescence in a circle surrounding a hot spot (filled symbols) and in another circle in a quiet region of the cell (open symbols). Regions and cell are the same as in Figure 1, G and H. Fluorescence in the quiet region slowly increased while calcium entered the cell, whereas it responded more quickly to changes in calcium current in the hot spot. E, Fluorescence in the quiet region (open symbols) replotted with the integral of the calcium current (QCa) from C. F, Calcium current and fast component of fluorescence change in the hot spot. To estimate the fast fluorescence component, QCa was scaled and a constant was added so that values of the resulting trace were the same as the fluorescence of the hot spot (D,filled symbols) immediately before (averaged for 500 msec) and after (averaged for 500 msec) the series of voltage steps. The result was subtracted from the trace in D(filled circles). The open circles(left axis) show the result of this subtraction. The fluorescence rises and falls in proportion to the calcium current (right axis, solid line). Dashed lines indicate average values before depolarization.
Figure 3D also shows the fluorescence measured in a quiet region outside hot spots (open circles). Unlike the region encompassing the hot spots, the fluorescence in this region increased slowly and monotonically during the depolarization. The trace is magnified in Figure 3E (left ordinate) along with a scaled version of Figure 3C(right ordinate, solid line). Evidently the fluorescence of the quiet region in this cell tracked the integral of the Ca influx. At quiet regions, QCaaccounts for most of the fluorescence change, and in this example, QCa accounts for all of the fluorescence change.
Counting synaptic ribbons in bipolar cells
In hair cells, Ca entry has been shown to colocalize with dense bodies, the equivalent of ribbons (Roberts et al., 1990; Issa and Hudspeth, 1996). If this is also true in bipolar cells, one can expect calcium entry sites and ribbons to occur in equal numbers. To immunostain synaptic ribbons, we took advantage of the finding that the ribbon protein ribeye and the protein CtBP2 are initiation variants from the same gene, and that the B-domain of ribeye contains nearly all of CtBP2. CtBP2 is an isoform of the transcriptional corepressor CtBP1. Because CtBP1 (formerly CtBP) (Sewalt et al., 1999) is 90% identical to CtBP2 in rats (Schmitz et al., 2000), we reasoned that an antibody against CtBP1 might also recognize ribeye. We first tested the antibody in cryosections of intact goldfish retina (Fig.4). Staining was strongest in the outer plexiform layer, in which the ribbons of photoreceptors are located. This is also found with other antibodies that label ribbon synapses (Balkema and Rizkalla, 1996; Muresan et al., 1999; Schmitz et al., 2000; Dick et al., 2001; Morgans, 2001). Weaker staining was present in the inner nuclear layer, in which the antibody recognized CtBP in nuclei, and in the inner plexiform layer, in which it presumably recognized ribeye in synaptic terminals.
Fig. 4.

Immunolocalization of CtBP/ribeye in goldfish retina. A, Section of fixed and frozen eyecups labeled with antibody for CtBP. Labels on the left indicate the layers of the retina (IPL, inner plexiform layer;OPL, outer plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer). The strongest immunoreactivity is found in the outer plexiform layer, which contains the photoreceptor ribbon synapses. Weaker staining is found in the inner plexiform layer containing the synaptic terminals of bipolar cells and in the inner nuclear layer containing the nuclei of bipolar cells. In the outer and inner plexiform layers, the antibody presumably labels the ribbon protein ribeye. B, C, Inner plexiform and outer plexiform layers at higher magnification. Staining is punctate, suggesting synaptic ribbons.
To examine this point in more detail, Figure5 shows the localization of CtBP and ribeye in single dissociated bipolar neurons. The antibody caused strong staining in the nucleus, where it presumably recognized CtBP. Punctate fluorescence (Fig. 5A, arrow) appeared in the synaptic terminal, where the antibody presumably recognized ribeye. In this epifluorescence micrograph, the punctate fluorescence of a few ribbons is superimposed on out-of-focus fluorescence from other ribbons. To test the specificity of staining, the serum was immunodepleted with recombinant His-tagged CtBP2 adsorbed to nickel beads. With immunodepleted serum, the staining is uniform, weak, and barely visible in Figure 5B.
Fig. 5.

Immunofluorescent localization of synaptic ribbons in bipolar cells. A chemically fixed single-dissociated bipolar cell is shown, immunostained with an antibody that recognizes CtBP1, CtBP2, and the B-domain of the ribbon protein ribeye. A, Active serum preincubated with uncoated nickel beads. Left, Bright field; right, fluorescence; arrowpoints to two ribbons. B, Image as inA but with serum immunodepleted by preincubation with nickel beads coated with recombinant His-tagged CtBP2.C–F, Confocal sections taken from the terminal of another bipolar cell, stained as described above except that the preincubation was omitted. Panels show the bottom-most section (C), two additional sections 1 μm (D) and 4 μm (E) higher, and the top-most section (F). Note that, except in the bottom and top sections, spots are found on the perimeter of the terminal, indicating that most or all staining is on the surface.
Similar results were obtained in other terminals. When serum was preincubated with beads lacking CtBP2, all of five cells showed punctate staining in the terminal and strong staining in the nucleus, but when beads were coated with CtBP2, all of eight terminals were devoid of punctate staining in the terminal and of nuclear stain. For a more detailed analysis, the staining in the terminal was examined by confocal microscopy. Strongly punctate staining was found, and it was located predominantly near the surface (Fig. 5C–F). We assume that the fluorescent dots represent ribbons. At the bottom-most plane, there were 0.10 ± 0.01 spots/μm−2 of cell surface, a number similar or identical to that of Ca entry sites (Table1). The result is consistent with the idea that each ribbon has a Ca entry site at its base, as has been inferred from electron microscopy in primate bipolar cells (Raviola and Raviola, 1982). The top-most plane had a similar surface density (0.09 ± 0.01 μm−2). The total number of spots was 25.4 ± 6.5 spots (mean ± SD) in 30 cells (mean diameter, 7.7 μm). The number is less than that found in two larger terminals examined by electron microscopy of intact retina (von Gersdorff et al., 1996) partly because of the size difference. A subset of synaptic terminals that had diameters >9 μm had an average of 35.7 ± 5.6 ribbons (mean diameter, 10.1 μm;n = 6 terminals). It is important to note that at the bottom-most plane of this subset of terminals, there were 0.09 ± 0.01 spots/μm−2, similar to the entire data set. Our results likely differ from the previous study partly because terminals in intact retina extend processes rich in ribbons, whereas the terminals in our dissociated cells were round. Some of the ribbon-bearing processes may have been lost during the dissociation procedure. Additionally, light microscopy may fail to distinguish between two closely adjacent ribbons. Because our calcium entry site determination is also done at the light level, the light level determination is relevant for our study.
Table 1.
Abundance of calcium spots, ribbons, and dense bodies in bipolar and hair cells
| Density (μm−2) | SE | Cells | |
|---|---|---|---|
| Bipolar cells | |||
| Calcium spots | 0.11 | 0.02 | 17 |
| Ribbons (bottom surface) | 0.10 | 0.01 | 30 |
| Ribbons (top surface) | 0.09 | 0.01 | 30 |
| Active zones | 0.10 | 0.03 | 7 |
| Total fusion sites | 0.24 | 0.04 | 7 |
| Hair cells | |||
| Calcium spots | 0.034 | 0.008 | 7 |
| Dense bodies | 0.048 | 0.008 | 8 |
Ca spots were counted from images as in Figures 1D and8D. CtBP-positive spots were counted from images that summed the bottom-most two images in stacks of serial confocal sections as in Figures 5C and 9B; the spots were considered to represent single ribbons in bipolar cells and dense bodies in hair cells. For bipolar cells, this table also shows the number of active zones of exocytosis (Zenisek et al., 2000) and the total number of fusion sites as determined in Figure 6. Data refer to the bottom surface of cells in which they adhere to the glass coverslip, except that CtBP-positive spots were counted in bipolar cells also on the top surface of the cell.
Active zones and fusion sites in bipolar cells
Most vesicles in bipolar cells undergo exocytosis at tightly clustered sites or active zones, defined as locations where two or more vesicles fuse within 300 nm of each other. On average, there were 0.10 ± 0.03 active zones per square micrometer (Zenisek et al., 2000), similar or identical to the number of ribbons and calcium entry sites observed here. In addition, however, solitary fusion events (“outliers”) occurred outside active zones, and we wanted to know whether or not they could be attributed to additional “reluctant” ribbons that were temporarily less active. Data from our previous work (Zenisek et al., 2000) were re-examined by selecting cells with >10 fusion events. Figure 6 shows an FM1–43-stained bipolar terminal. Single synaptic vesicles are not seen in this average of 500 video frames, but instead, dim and featureless fluorescence from residual dye in the plasma membrane indicates the footprint in which the terminal adhered tightly to the coverslip.Red dots indicate the location of all exocytic fusion events observed in this terminal.
Fig. 6.

Location of exocytic events in a bipolar neuron. Locations of all fusion events observed (red dots) were entered in the image of the footprint of the cell obtained by averaging 500 video frames. The 0.5-μm-diameter circles drawn are the minimum number required to include all fusion events.Blue circles indicate locations in which only one fusion event occurred; such fusions were classified as outliers in previous work (Zenisek et al., 2000). Green circles include more than one fusion event. All green circles except one (arrow) surrounded fusions occurring within 0.3 μm of each other; these qualified as active zones in previous work. Cells were analyzed only if we recorded at least 11 fusion events. The cell shown had relatively few fusions; most other cells had more (Zenisek et al., 2000). The two brightest spots remained unchanged for the entire 7 min of recordings and presumably represent stained debris.
Most events occurred within tight clusters of two or more separated by <300 nm (i.e., in active zones) (Fig. 6, green circles). Their presence suggests three ribbons in the footprint of this terminal. If the two outliers (Fig. 6, blue circles) also occurred at ribbons, how many additional ribbons were there? Because the longest axis of a bipolar cell ribbon is 0.5 μm (von Gersdorff et al., 1996), we assumed that fusions separated by larger distances occurred at different ribbons. The 0.5-μm-diameter circles drawn are the minimum number required to include all fusion events. There are three green circles surrounding active zones, a fourthgreen circle surrounding either a fourth active zone or closely spaced outliers, and two blue circles each including one outlier. In seven cells, an average of 0.24 ± 0.04/μm2 0.5-μm-diameter circles were required to enclose all fusion events. If all outliers occurred at ribbons, therefore, we would expect 0.24 ribbons per square micrometer. This number is significantly larger than the number of ribbons measured at the bottom surface of bipolar neurons, either as ribeye-positive spots (0.10 ± 0.01/μm2) or as Ca entry sites (0.11 ± 0.02/μm2; Table 1). We conclude that not all outlier fusion events occurred at ribbons.
If active zones occupy a circle of 0.5 μm diameter, then we can estimate the site specificity of fusion. Per square micrometer, there were 175 ± 32 times more fusions at active zones than elsewhere (n = 7 cells). An even higher multiple results if one assumes that the area occupied by docked vesicles beneath ribbons is smaller than that of a 0.5-μm-diameter circle. Although in percentage terms the number of outlier fusions may appear significant (36%) (Zenisek et al., 2000), it is small compared with that at active zones on a per-area basis. Hence, fusion at active zones in bipolar neurons is strongly site-specific.
Calcium entry sites in hair cells
Because dense bodies in frog saccular hair cells bind the calcium indicator Fluo-3 (Issa and Hudspeth, 1994), they fluoresce even when Ca channels are shut. Hence, Ca indicator fluorescence can be used to locate dense bodies. Figure 7 shows a hair cell loaded with calcium indicator imaged under differential interference contrast and under evanescent field fluorescence. Like previous authors (Roberts et al., 1990; Issa and Hudspeth, 1994, 1996), we concentrated on the basal half of the cell that carries most active zones (Fig. 7B, white outline). Two small fluorescent spots are readily seen in the basal portion (Fig.7B). Hair cells were imaged before (Fig.8A), during (B), and after (C) voltage pulses that opened Ca channels. During the voltage step, the two fluorescent spots brightened dramatically (Fig. 8B) without spreading significantly (Fig. 8D). The result resembles those on bipolar cells, except that while Ca channels are shut, the Ca entry sites are more clearly visible in hair cells. The fluorescence in hot spots showed both fast and slow kinetic components (see movies 1–4), whereas only a slow component appeared in quiet regions of the cell (Fig. 8G, compare green andred traces). In all eight cells, all 19 Fluo-3-stained regions showed a rapid rise and fall of fluorescence as the plasma membrane was depolarized and then repolarized. The fluorescence changes outside these regions were slow and gradual. To count calcium entry sites, cells were divided into basal (Fig. 7B, white outline) and apical (gray outline) regions. As expected for dense bodies (Roberts et al., 1990), most calcium entry sites (19 of 21) were found in the basal half of the cell. They populated the plasma membrane there at a density of 0.034 ± 0.008 (n = 7) sites per square micrometer.
Fig. 7.
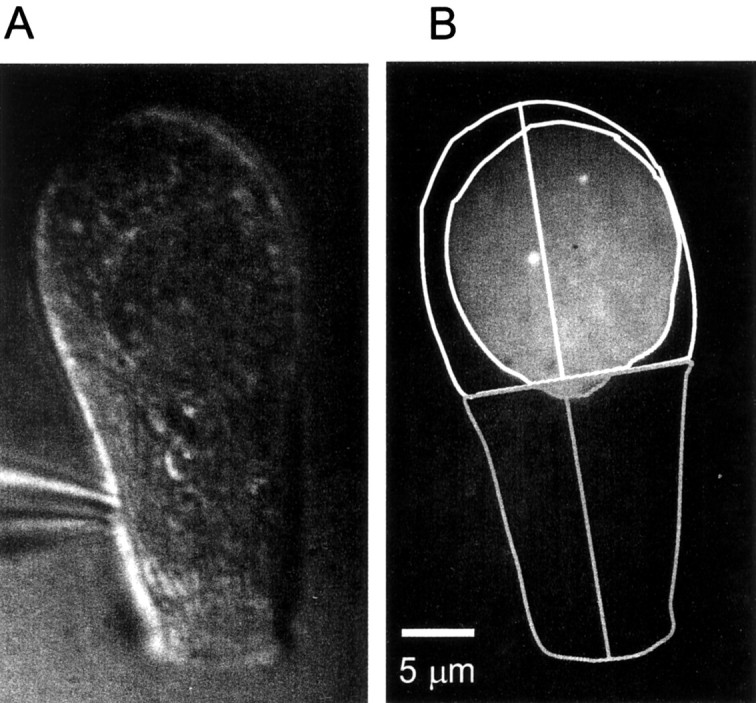
Fluo-3 labels dense bodies in frog saccular hair cells. A, Differential interference contrast image of a frog vestibular hair cell loaded with Fluo-3 and 10 mm EGTA via a patch pipette (bottom left). B, Evanescent field fluorescence image with outline of the cell under differential interference contrast (A). To define apical and basal regions, a line was drawn down the long axis of the cell and bisected in the middle. The apical half is enclosed by thegray outline; it was not used in additional analyses. The white outline surrounds the basal half. A secondnear-circular outline within indicates the area of the footprint. Two bright spots in the cell are readily seen; they mark the locations of two dense bodies (Issa and Hudspeth, 1996).
Fig. 8.
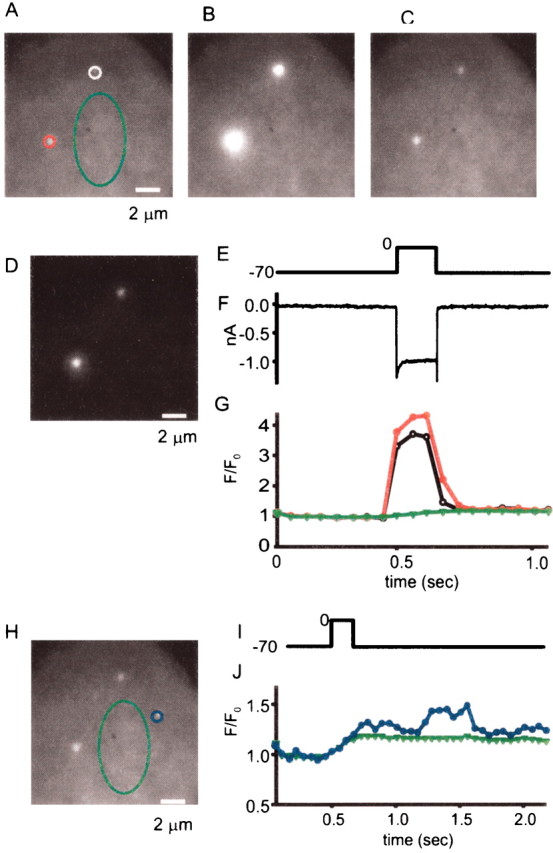
Localized calcium entry in frog hair cells.A–C, Basal portion of a hair cell before, during, and after a voltage pulse from −70 to 0 mV. Exposures are for 150 msec inA and C and for 150 msec inB. In hair cells, fluorescent spots are clearly visible even in the absence of a depolarizing stimulus, and they brighten dramatically when Ca channels open and seem to broaden; this is caused by contrast saturation. When B is printed at diminished brightness, fluorescence is tightly localized to the two bright spots (D). E, Voltage (in millivolts). F, Calcium current. G, Fluorescence in the three regions outlined in A, plotted against time. Traces are color-coded to indicate the regions to which they apply. As in Figure 1, fluorescence for each region is normalized by the average value during 350 msec before the voltage pulse. H, Average of three 50 msec exposures taken ∼1 sec after the voltage pulse. Green region is the same as in A; blue region shows a local increase in Ca. I, Fluorescence ingreen and blue regions plotted against time. Note that the blue trace shows a fluorescence increase occurring after Ca channels were shut by returning the membrane potential to −70 mV.
In addition to the fluorescence changes at dense bodies, hair cells also exhibited small discrete locations in which fluorescence fluctuated independently of the cell stimulus. Figure8H–J shows an example. In a small region circled inblue (Fig. 8H), the fluorescence showed a transient increase starting ∼500 msec after the cell had been returned to −70 mV (Fig. 8J, blue symbols). Similar fluorescence fluctuations were observed in eight of nine hair cells. Because these fluorescence “flickers” persist even in the absence of significant calcium fluxes, they may represent calcium release from internal stores. Indeed, the existence of internal calcium stores suitable for Ca release has been suggested for both cochlear (Shigemoto and Ohmori, 1991; Sridhar et al., 1997; Fuchs, 2002; Kennedy and Meech, 2002) and vestibular (Yoshida et al., 1994) hair cells.
CtBP staining of hair cell-dense bodies
Figure 9 shows immunofluorescence images from a hair cell labeled with anti-CtBP. Bright punctate fluorescence is readily seen near the top or bottom surfaces. Fluorescent spots were concentrated in the basal region, with no staining found in the stereocilia (data not shown). Hair cells contained 23.9 ± 3.0 spots (mean ± SD), similar to the number of dense bodies estimated from electron microscopy (18.6 ± 2.3) (Roberts et al., 1990). Similar to what has been reported for dense bodies in electron micrographs (73%) (Roberts et al., 1990), 79.1% of the fluorescent spots in nine cells were found within the basal half of the cell. Figure 9B–D shows individual confocal sections. Spots occupy locations throughout the bottom surface (Fig. 9B) but populate only the periphery in sections near the cell equator. The staining pattern is consistent with most or all fluorescent spots located on the cell surface. We counted the number of CtBP-positive spots in the bottom-most confocal sections of the basal half of cells. Their average number was similar to that of Ca hot spots (Table 1).
Fig. 9.

Immunofluorescence staining of dense bodies in frog hair cells. A, Maximum fluorescence at each pixel location in all 25 confocal sections through an anti-CtBP-labeled hair cell. Fluorescent spots representing dense bodies are mostly found in the basal half of the cell. The nucleus did not stain prominently in hair cells. B, Bottom-most confocal section of the cell.C, D, Two sections taken near the middle of the cell. Note that CtBP spots in middle sections are found on the cell periphery, as expected for synaptic ribbons.
Discussion
We show here that Ca entry sites can be imaged with submicrometer definition in neurons or hair cells loaded with calcium indicator dyes and high concentrations of EGTA. In cells that are so loaded, two kinetic components of fluorescence change resulted when Ca channels were opened by a voltage pulse (Zenisek and Matthews, 1995). The faster component rose and fell with the calcium current, whereas the slower component followed the integral of the calcium current. The rapid component was seen most strikingly in discrete regions on the footprint of the cell, causing these regions to brighten during the depolarization in proportion to the calcium current. We provide evidence that the fast component quantitatively tracks Ca influx through open Ca channels. Interestingly, the fast component at hot spots was maintained during a 1 sec pulse and did not diminish, as it might if Ca were depleted in the space beneath the terminal.
Previous electrophysiologic studies have demonstrated domains of high [Ca2+] that presumably surround Ca entry sites, first in hair cells (Roberts et al., 1990) and then in culturedXenopus motor neurons (Yazejian et al., 1997) and retinal bipolar neurons (Sakaba et al., 1997). Localized Ca entry sites were also demonstrated with calcium-dependent luminescent proteins at the squid giant synapse (Llinas et al., 1992, with Ca indicator dyes and confocal microscopy in frog (Issa and Hudspeth, 1996) and turtle (Tucker and Fettiplace, 1995) hair cells, and, finally, by “confocal spot” microscopy in Xenopus motor neurons (DiGregorio et al., 1999). In comparison with the previous work, the method described here was optimized for imaging the Ca entry across the plasma membrane or the release of Ca2+ from internal stores near the plasma membrane. We counted Ca entry sites presumably representing clusters of voltage-gated Ca channels. In both bipolar neurons and frog saccular hair cells, their number is similar or identical to that of ribbons or dense bodies marked by other means.
Surface density of synaptic ribbons
Frog saccular hair cells contain fixed, subplasmalemmal structures that stain with the Ca indicator dye Fluo-3 and appear as fluorescent spots (Issa and Hudspeth, 1994). Direct comparison of confocal fluorescence microscopy with Fluo-3 and electron micrographs from the same cells has shown that nearly all dense bodies are associated with fluorescent spots, and that >80% of the fluorescent spots are associated with dense bodies (Issa and Hudspeth, 1994). When Ca channels are opened with voltage pulses, the fluorescent structures light up as the Ca indicator dye binds Ca. The findings by Issa and Hudspeth (1994, 1996) provide direct support for the idea that Ca channels in hair cells congregate beneath dense bodies, as suggested previously in a study combining electron microscopy with electrophysiology (Roberts et al., 1990). Our findings are in excellent and quantitative agreement with their work. Like Issa and Hudspeth (1994), we find that the Ca indicator dye Fluo-3 labels stationary structures in hair cells that brighten dramatically when voltage pulses open Ca channels. These structures lie closely apposed to the plasma membrane as they appear under evanescent field illumination. Their number is nearly identical to that found by Issa and Hudspeth (1994,1996). We add a third way to image dense bodies, namely by immunostaining with an antibody that recognizes the B-domain of the synaptic ribbon protein ribeye. Ribeye-positive spots were seen to populate the cell surface at a density similar or identical to that of Ca entry sites. The previous results and those reported here leave little doubt that Ca entry sites are reliable markers of dense bodies in hair cells.
Dense bodies in hair cells are the analog of ribbons in other sensory neurons, including retinal bipolar cells. We found that, like hair cells, bipolar cells have a small number of micrometer- or submicrometer-sized Ca entry sites (Fig. 1I). As in hair cells, these sites remain visible, although dimly, even at potentials at which most or all Ca channels are shut. In analogy with hair cells, we suggest that they mark fixed subplasmalemmal structures beneath Ca entry sites that represent ribbons. That they are harder to see may be related to the volume of ribbons, which is smaller in bipolar cells [see electron micrographs in von Gersdorff et al. (1996)] than in saccular hair cells (Lenzi et al., 1999). Moreover, the number of calcium entry sites is identical to the number of ribeye-positive spots. We conclude that, as in hair cells, Ca-sensitive spots and ribeye mark synaptic ribbons. In turn, the numbers of Ca entry sites and ribeye-positive spots in bipolar cells are closely similar to that of active zones found by imaging single exocytic events. The agreement is strong evidence that Ca entry sites, ribeye-positive spots, and exocytic active zones all mark the structure recognized in electron microscopy as ribbons. At the light microscopic level, therefore, there are 0.1 ribbons per square micrometer in terminals of dissociated goldfish bipolar neurons.
Site-specificity of exocytosis at active zones
When imaging single exocytic fusion events in bipolar cells, it appeared that approximately one-third occurred outside active zones during 500 msec to 1 sec depolarizations to 0 mV (Zenisek et al., 2000). If all such events occurred at reluctant ribbons, there would need to be twice as many ribbons as those found by immunostaining. We do not believe that our immunofluorescence measurements are in error by that amount, and instead conclude that synaptic vesicles fuse also at sites that are remote from ribbons, similar to what has been suggested in hair cells (Beutner et al., 2001; Lenzi et al., 2002). Fusion by outliers may help contribute to a puzzling discrepancy between experiments aimed at defining the number of most rapidly releasable vesicles that can be released within milliseconds of stimulation. When exocytosis is stimulated by Ca entry through voltage-gated Ca channels, only ∼1100 vesicles per terminal can be released that fast, causing a capacitance increase of 30 fF. This number of vesicles is equal to that found morphologically to be both tethered to ribbons and docked at the plasma membrane (Mennerick and Matthews, 1996). In contrast, when stimulation by flash photolysis of caged Ca2+ raises [Ca2+] uniformly throughout the cell, there is also a pool of vesicles released in milliseconds, but it is 2.5-fold to fivefold higher (Heidelberger et al., 1994; Heidelberger, 1998). Qualitatively, such discrepancy is predicted by our results. Opening voltage-gated Ca channels would favor vesicles at active zones in which Ca channels are congregated at the highest concentration. In contrast, flash photolysis would raise cytosolic [Ca2+] equally near outliers and cause rapid release of both outliers and vesicles at active zones.
Can fusions outside ribbons occur physiologically? Synaptic vesicles docking at a distance from ribbons face a disadvantage, because they would also be far from the main sites of Ca entry. However, it is not certain that the plasma membrane outside active zones is completely devoid of Ca channels. When tracking the fluorescence of Ca indicator, we often, but not always, found a small fast component also in remote regions (Fig. 1E,F). This component may result from remote channels, and influx through such channels may trigger exocytosis of outliers during prolonged periods of activation. One may question whether sparse and remote channels can raise [Ca2+] sufficiently to trigger exocytosis, because little or no exocytosis was observed at <10 μm [Ca2+] byHeidelberger et al. (1994). However, others have also reported evidence for exocytosis at significantly lower [Ca2+] (Lagnado et al., 1996; Rouze and Schwartz, 1998).
It remains to be seen to what extent outliers fuse physiologically and what role such fusions might play. There may be extrasynaptic glutamate receptors to report glutamate release outside active zones. In mammals, immunofluorescence studies have shown glutamate receptors away from ribbons in the inner plexiform layer (Grunert et al., 2002). Such extrasynaptic glutamate receptors may be activated both by glutamate spillover from active zones (Matsui et al., 1998; Higgs and Lukasiewicz, 1999; Chen and Diamond, 2002) and by glutamate released from outliers.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants MH60600 and DK44239. V.D. is supported by NIH Training Grant DK07680. We thank Dr. Peter Gillespie for advice on hair cells, Dr. Trillium Blackmer for her helpful comments on this manuscript, and Drs. Richard Goodman and Arie Otte for reagents.
Correspondence should be addressed to Wolfhard Almers, Vollum Institute, 31 Southwest Sam Jackson Park Road, Portland, OR 97201. E-mail: almersw@ohsu.edu.
References
- 1.Assad JA, Corey DP. An active motor model for adaptation by vertebrate hair cells. J Neurosci. 1992;12:3291–3309. doi: 10.1523/JNEUROSCI.12-09-03291.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Axelrod D. Selective imaging of surface fluorescence with very high aperture microscope objectives. J Biomed Opt. 2001;6:6–13. doi: 10.1117/1.1335689. [DOI] [PubMed] [Google Scholar]
- 3.Balkema GW, Rizkalla R. Ultrastructural localization of a synaptic ribbon protein recognized by antibody B16. J Neurocytol. 1996;25:565–571. doi: 10.1007/BF02284824. [DOI] [PubMed] [Google Scholar]
- 4.Beutner D, Voets T, Neher E, Moser T. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 2001;29:681–690. doi: 10.1016/s0896-6273(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 5.Bunt AH. Enzymatic digestion of synaptic ribbons in amphibian retinal photoreceptors. Brain Res. 1971;25:571–577. doi: 10.1016/0006-8993(71)90461-6. [DOI] [PubMed] [Google Scholar]
- 6.Burrone J, Neves G, Gomis A, Cooke A, Lagnado L. Endogenous calcium buffers regulate fast exocytosis in the synaptic terminal of retinal bipolar cells. Neuron. 2002;33:101–112. doi: 10.1016/s0896-6273(01)00565-7. [DOI] [PubMed] [Google Scholar]
- 7.Chen S, Diamond JS. Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci. 2002;22:2165–2173. doi: 10.1523/JNEUROSCI.22-06-02165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dick O, Hack I, Altrock WD, Garner CC, Gundelfinger ED, Brandstatter JH. Localization of the presynaptic cytomatrix protein Piccolo at ribbon and conventional synapses in the rat retina: comparison with Bassoon. J Comp Neurol. 2001;439:224–234. doi: 10.1002/cne.1344. [DOI] [PubMed] [Google Scholar]
- 9.DiGregorio DA, Peskoff A, Vergara JL. Measurement of action potential-induced presynaptic calcium domains at a cultured neuromuscular junction. J Neurosci. 1999;19:7846–7859. doi: 10.1523/JNEUROSCI.19-18-07846.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edmonds B, Reyes R, Schwaller B, Roberts WM. Calretinin modifies presynaptic calcium signaling in frog saccular hair cells. Nat Neurosci. 2000;3:786–790. doi: 10.1038/77687. [DOI] [PubMed] [Google Scholar]
- 11.Escobar AL, Velez P, Kim AM, Cifuentes F, Fill M, Vergara JL. Kinetic properties of DM-nitrophen and calcium indicators: rapid transient response to flash photolysis. Pflügers Arch. 1997;434:615–631. doi: 10.1007/s004240050444. [DOI] [PubMed] [Google Scholar]
- 12.Fuchs P. The synaptic physiology of cochlear hair cells. Audiol Neurootol. 2002;7:40–44. doi: 10.1159/000046862. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh KK, Haverkamp S, Wassle H. Glutamate receptors in the rod pathway of the mammalian retina. J Neurosci. 2001;21:8636–8647. doi: 10.1523/JNEUROSCI.21-21-08636.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gray EG, Pease HL. On understanding the organization of the retinal receptor synapses. Brain Res. 1971;35:1–15. doi: 10.1016/0006-8993(71)90591-9. [DOI] [PubMed] [Google Scholar]
- 15.Grunert U, Haverkamp S, Fletcher EL, Wassle H. Synaptic distribution of ionotropic glutamate receptors in the inner plexiform layer of the primate retina. J Comp Neurol. 2002;447:138–151. doi: 10.1002/cne.10220. [DOI] [PubMed] [Google Scholar]
- 16.Heidelberger R. Adenosine triphosphate and the late steps in calcium-dependent exocytosis at a ribbon synapse. J Gen Physiol. 1998;111:225–241. doi: 10.1085/jgp.111.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heidelberger R, Matthews G. Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol (Lond) 1992;447:235–256. doi: 10.1113/jphysiol.1992.sp019000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- 19.Higgs MH, Lukasiewicz PD. Glutamate uptake limits synaptic excitation of retinal ganglion cells. J Neurosci. 1999;19:3691–3700. doi: 10.1523/JNEUROSCI.19-10-03691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Issa NP, Hudspeth AJ. Clustering of Ca2+ channels and Ca2+-activated K+ channels at fluorescently labeled presynaptic active zones of hair cells. Proc Natl Acad Sci USA. 1994;91:7578–7582. doi: 10.1073/pnas.91.16.7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Issa NP, Hudspeth AJ. The entry and clearance of Ca2+ at individual presynaptic active zones of hair cells from the bullfrog's sacculus. Proc Natl Acad Sci USA. 1996;93:9527–9532. doi: 10.1073/pnas.93.18.9527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy HJ, Meech RW. Fast Ca2+ signals at mouse inner hair cell synapse: a role for Ca2+-induced Ca2+ release. J Physiol (Lond) 2002;539:15–23. doi: 10.1113/jphysiol.2001.013171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi K, Tachibana M. Ca2+ regulation in the presynaptic terminals of goldfish retinal bipolar cells. J Physiol (Lond) 1995;483:79–94. doi: 10.1113/jphysiol.1995.sp020569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagnado L, Gomis A, Job C. Continuous vesicle cycling in the synaptic terminal of retinal bipolar cells. Neuron. 1996;17:957–967. doi: 10.1016/s0896-6273(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 25.Lenzi D, Runyeon JW, Crum J, Ellisman MH, Roberts WM. Synaptic vesicle populations in saccular hair cells reconstructed by electron tomography. J Neurosci. 1999;19:119–132. doi: 10.1523/JNEUROSCI.19-01-00119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenzi D, Crum J, Ellisman MH, Roberts WM. Depolarization redistributes synaptic membrane and creates a gradient of vesicles on the synaptic body at a ribbon synapse. Neuron. 2002;36:649–659. doi: 10.1016/s0896-6273(02)01025-5. [DOI] [PubMed] [Google Scholar]
- 27.Llinas R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a presynaptic terminal. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- 28.Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP. Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci. 1996;16:4457–4467. doi: 10.1523/JNEUROSCI.16-14-04457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsui K, Hosoi N, Tachibana M. Excitatory synaptic transmission in the inner retina: paired recordings of bipolar cells and neurons of the ganglion cell layer. J Neurosci. 1998;18:4500–4510. doi: 10.1523/JNEUROSCI.18-12-04500.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mennerick S, Matthews G. Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron. 1996;17:1241–1249. doi: 10.1016/s0896-6273(00)80254-8. [DOI] [PubMed] [Google Scholar]
- 31.Morgans CW. Localization of the alpha(1F) calcium channel subunit in the rat retina. Invest Ophthalmol Vis Sci. 2001;42:2414–2418. [PubMed] [Google Scholar]
- 32.Morigiwa K, Vardi N. Differential expression of ionotropic glutamate receptor subunits in the outer retina. J Comp Neurol. 1999;405:173–184. doi: 10.1002/(sici)1096-9861(19990308)405:2<173::aid-cne3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 33.Muresan V, Lyass A, Schnapp BJ. The kinesin motor KIF3A is a component of the presynaptic ribbon in vertebrate photoreceptors. J Neurosci. 1999;19:1027–1037. doi: 10.1523/JNEUROSCI.19-03-01027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- 35.Neves G, Lagnado L. The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J Physiol (Lond) 1999;515:181–202. doi: 10.1111/j.1469-7793.1999.181ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Omann GM, Axelrod D. Membrane-proximal calcium transients in stimulated neutrophils detected by total internal reflection fluorescence. Biophys J. 1996;71:2885–2891. doi: 10.1016/S0006-3495(96)79485-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsons TD, Lenzi D, Almers W, Roberts WM. Calcium-triggered exocytosis and endocytosis in an isolated presynaptic cell: capacitance measurements in saccular hair cells. Neuron. 1994;13:875–883. doi: 10.1016/0896-6273(94)90253-4. [DOI] [PubMed] [Google Scholar]
- 38.Qin P, Pourcho RG. Localization of AMPA-selective glutamate receptor subunits in the cat retina: a light- and electron-microscopic study. Vis Neurosci. 1999;16:169–177. doi: 10.1017/s0952523899161121. [DOI] [PubMed] [Google Scholar]
- 39.Raviola E, Gilula NB. Intramembrane organization of specialized contacts in the outer plexiform layer of the retina. A freeze-fracture study in monkeys and rabbits. J Cell Biol. 1975;65:192–222. doi: 10.1083/jcb.65.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raviola E, Raviola G. Structure of the synaptic membranes in the inner plexiform layer of the retina: a freeze-fracture study in monkeys and rabbits. J Comp Neurol. 1982;209:233–248. doi: 10.1002/cne.902090303. [DOI] [PubMed] [Google Scholar]
- 41.Roberts WM. Spatial calcium buffering in saccular hair cells. Nature. 1993;363:74–76. doi: 10.1038/363074a0. [DOI] [PubMed] [Google Scholar]
- 42.Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rouze NC, Schwartz EA. Continuous and transient vesicle cycling at a ribbon synapse. J Neurosci. 1998;18:8614–8624. doi: 10.1523/JNEUROSCI.18-21-08614.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakaba T, Ishikane H, Tachibana M. Ca2+-activated K+ current at presynaptic terminals of goldfish retinal bipolar cells. Neurosci Res. 1997;27:219–228. doi: 10.1016/s0168-0102(97)01155-3. [DOI] [PubMed] [Google Scholar]
- 45.Schmitz F, Konigstorfer A, Sudhof TC. RIBEYE, a component of synaptic ribbons: a protein's journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28:857–872. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 46.Sewalt RG, Gunster MJ, van der Vlag J, Satijn DP, Otte AP. C-terminal binding protein is a transcriptional repressor that interacts with a specific class of vertebrate Polycomb proteins. Mol Cell Biol. 1999;19:777–787. doi: 10.1128/mcb.19.1.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shigemoto T, Ohmori H. Muscarinic receptor hyperpolarizes cochlear hair cells of chick by activating Ca2+-activated K+ channels. J Physiol (Lond) 1991;442:669–690. doi: 10.1113/jphysiol.1991.sp018814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sridhar TS, Brown MC, Sewell WF. Unique postsynaptic signaling at the hair cell efferent synapse permits calcium to evoke changes on two time scales. J Neurosci. 1997;17:428–437. doi: 10.1523/JNEUROSCI.17-01-00428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steyer JA, Almers W. A real-time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol. 2001;2:268–275. doi: 10.1038/35067069. [DOI] [PubMed] [Google Scholar]
- 50.Tacchibana M, Okada T, Arimura T, Kobayashi K, Piccolino M. Dihydropyrdine-sensitive calcium current mediates neurotransmitter release from bipolar cells of the goldfish retina. J Neurosci. 1993;12:2898–2909. doi: 10.1523/JNEUROSCI.13-07-02898.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tucker T, Fettiplace R. Confocal imaging of calcium microdomains and calcium extrusion in turtle hair cells. Neuron. 1995;15:1323–1335. doi: 10.1016/0896-6273(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 52.von Gersdorff H, Vardi E, Matthews G, Sterling P. Evidence that vesicles on the synaptic ribbon of retinal bipolar neurons can be rapidly released. Neuron. 1996;16:1221–1227. doi: 10.1016/s0896-6273(00)80148-8. [DOI] [PubMed] [Google Scholar]
- 53.Yazejian B, DiGregorio DA, Vergara JL, Poage RE, Meriney SD, Grinnell AD. Direct measurements of presynaptic calcium and calcium-activated potassium currents regulating neurotransmitter release at cultured Xenopus nerve-muscle synapses. J Neurosci. 1997;17:2990–3001. doi: 10.1523/JNEUROSCI.17-09-02990.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida N, Shigemoto T, Sugai T, Ohmori H. The role of inositol trisphosphate on ACh-induced outward currents in bullfrog saccular hair cells. Brain Res. 1994;644:90–100. doi: 10.1016/0006-8993(94)90351-4. [DOI] [PubMed] [Google Scholar]
- 55.Zenisek D, Matthews G. Calcium recovery following depolarization in a synaptic nerve terminal. Biophys J. 1995;68:A397. [Google Scholar]
- 56.Zenisek D, Matthews G. The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron. 2000;25:229–237. doi: 10.1016/s0896-6273(00)80885-5. [DOI] [PubMed] [Google Scholar]
- 57.Zenisek D, Steyer JA, Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- 58.Zenisek D, Steyer JA, Feldman ME, Almers W. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron. 2002;35:1–20. doi: 10.1016/s0896-6273(02)00896-6. [DOI] [PubMed] [Google Scholar]