

Abstract
Structural changes at synapses are associated with long-term facilitation (LTF) of synaptic transmission between sensory and motor neurons in Aplysia. We have cloned a cDNA encoding Aplysia adducin (ApADD), theAplysia homolog of mammalian adducins that are regulatory components of the membrane cytoskeleton. ApADD is recovered in the particulate fraction of nervous system extracts and is localized predominantly in the submembraneous region of Aplysianeurons. ApADD is phosphorylated in vitro by protein kinase C (PKC) at a site homologous to the in vivo PKC phosphorylation site in mammalian adducins. Phosphorylation of ApADD at this site is also detected in vivo in the intactAplysia nervous system and is increased 18 hr after serotonin-induced LTF. In contrast, there is no change in phosphorylation during short-term facilitation or 1 hr after initial LTF induction. Thus, ApADD is modulated specifically with later phases of LTF and provides an attractive candidate protein that contributes to structural changes accompanying long-lasting synaptic alteration.
Keywords: adducins, protein kinase C, long-term facilitation, cytoskeleton, synaptic plasticity, serotonin
Introduction
Morphological changes in the brain are associated with plastic processes leading to long-term memory and may be a critical mechanism for maintaining learning-associated synaptic changes. For example, long-term potentiation (LTP) leads to the formation of new spines on postsynaptic hippocampal dendrites and the appearance of multiple spine synapses between a single axon terminal and a dendrite (Engert and Bonhoeffer, 1999; Klintsova and Greenough, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999;Bonhoeffer and Yuste, 2002). In addition, synapse density is increased in the hippocampus of mice that are exposed to an enriched environment (Rampon et al., 2000). Finally, in Aplysia californica, the formation of long-term memory is accompanied by an increase in the number of synaptic active zones and presynaptic varicosities and by the growth of new presynaptic branches (Bailey and Chen, 1988; Bailey and Kandel, 1994; Wainwright et al., 2002).
Synaptic remodeling that accompanies learning requires alterations of the cytoskeleton at the site of growth, but the molecular mechanisms underlying those alterations have been difficult to identify. It is believed that the initial step in these structural changes involves a loss of cytoskeletal rigidity, allowing remodeling of synaptic contacts. At least two proteins have been implicated in this first step: (1) cell-adhesion molecules (CAMs) that link the plasma membrane to the extracellular matrix and are endocytosed during long-term facilitation (LTF) probably to be degraded (Bailey et al., 1992;Mayford et al., 1992) and (2) microtubule-associated protein 2 (MAP 2), which unbinds microtubules during phosphorylation, thus destabilizing the cytoskeleton (Audesirk et al., 1997; for review, see Sanchez et al., 2000). A particularly interesting set of cytoskeletal proteins are the adducins, which provide a link between signaling cascades and cell structure. Adducins cross-link actin filaments with spectrin, cap the barbed end of actin filaments, and bundle actin filaments (Gardner and Bennett, 1987; Mische et al., 1987; Kuhlman et al., 1996). Both phosphorylation of adducins by PKC and calcium/calmodulin binding inhibit the actin-capping and spectrin-recruitment properties of adducins (Gardner and Bennett, 1987; Kuhlman et al., 1996; Matsuoka et al., 1996, 1998). Mammalian adducins are encoded by three genes (α, β, and γ) (Joshi et al., 1991; Suriyapperuma et al., 2000), and αβ or αγ heterodimers appear to be the predominant active isoforms (Hughes and Bennett, 1995; Gilligan et al., 1999). Adducins have been implicated in structural change in numerous cell types (Waseem and Palfrey, 1988, 1990; Kaiser et al., 1989; Yue and Spradling, 1992; Fukata et al., 1999; Gilligan et al., 2002). They are expressed throughout the brain in regions that are rich in synaptic contacts (Seidel et al., 1995), and the phosphorylated form of adducin (phospho-adducin) is detected in the dendrites of hippocampal neurons (Matsuoka et al., 1998). Thus, the adducins represent a family of proteins that are ideal candidates to translate a complex interplay of signaling cascades into a localized change in cytoskeletal structure accompanying long-term synaptic plasticity.
Any morphological change in a neuron requires an alteration of the cytoskeletal architecture of the cell. To identify the cytoskeletal elements underlying such changes, we have taken advantage of a simple model system, the marine mollusk Aplysia, for which the neural correlates of memory have been extensively examined and can be studied in the intact nervous system. Sensitization of the gill and siphon withdrawal reflexes in Aplysia involves the facilitation of neurotransmission at identified synapses between sensory neurons (SNs) and motor neurons (MNs) (Frost et al., 1985). The cellular features of sensitization are mimicked in a reduced preparation of the Aplysia nervous system by the direct application of serotonin (5-HT), which is released in the CNS after sensitizing stimuli (Levenson et al., 1999; Marinesco and Carew, 2002). Sensitization, as well as synaptic facilitation induced by 5-HT, can exist in short-term, intermediate-term, and long-term forms (Carew et al., 1971; Pinsker et al., 1973; Emptage and Carew, 1993; Ghirardi et al., 1995; Sutton and Carew, 2000). In contrast to short-term facilitation (STF), LTF requires changes in gene expression and is associated with changes in synaptic structure (Montarolo et al., 1986;Castellucci et al., 1989; Bailey and Kandel, 1994; Martin et al., 1997). In this study, we first characterized Aplysia adducin (ApADD), the Aplysia homolog of mammalian adducins, and then analyzed how it is modulated during forms of synaptic plasticity that are cellular correlates of memory in Aplysia. ApADD is localized to the submembraneous compartment of neurons involved in synaptic facilitation and is phosphorylated by PKC in vitroand in vivo. Using the intact Aplysia nervous system and procedures shown previously to induce LTF (Montarolo et al., 1986; Emptage and Carew, 1993; Ghirardi et al., 1995), we found that phosphorylation of ApADD at a PKC consensus site is increased 18 hr after initial LTF induction. Our data suggest that ApADD contributes to later phases of LTF and may participate in structural changes during LTF that are a common feature of long-term synaptic plasticity and memory.
Materials and Methods
PCR homology screening. A PCR homology screen was performed using pools of inosine-containing oligonucleotides as primers and a λZAPII cDNA library from the Aplysia nervous system as a template (gift from Wayne Sossin, McGill University, Montreal, Canada). Oligonucleotides were designed using a multiple alignment ofDrosophila and mammalian adducin amino acid sequences. Amino acid regions with high similarity between adducin species were identified, and primers corresponding to two sequences (amino acids 309–315 and 386–392 of human α-adducin) were used successfully for PCR. The primer sequences were GA(AG) ACI (AG)TI GA(AG) GCI TT and GTI C(GT)(AG) TAI CCI (AG)I(AG) TT(AG) TC.
Cloning and plasmids. A λZAPII cDNA library was screened under high-stringency conditions with the 242 bp ApADD probe obtained via PCR (see above) (Sambrook et al., 1989). Positive plaques were replated and rescreened, and plaques that remained positive through three screenings were isolated. pBluescriptII phagemids were excised from the λZAPII vector in vivo with ExAssist helper phage, and plasmid inserts were sequenced. One clone contained a complete open reading frame encoding ApADD (GenBank accession numberAY191225).
Three vectors were constructed for the expression of recombinant polyhistidine-ApADD fusion proteins in bacteria. cDNAs, encoding full-length ApADD (FL; amino acids 1–701), the predicted N-terminal domain (NT) (amino acids 1–350), and the C-terminal domain of ApADD (CT) (amino acids 351–701) were amplified by PCR. PCR primers for the three cDNAs contained additional NdeI andBamHI sites for subcloning. All three cDNAs were subcloned first into a dT-tailed vector (TA Cloning Kit; Invitrogen, Carlsbad, CA) and after NdeI–BamHI restriction into the NdeI–BamHI cloning sites of the pET-19b vector. This vector allows bacterial expression of recombinant proteins with an N-terminal tag consisting of six histidine (His) residues.
Recombinant ApADD protein expression and preparation ofAplysia tissues homogenates. Polyhistidine-adducin constructs were transformed into Escherichia coli (BL21), and the expression of the recombinant His-ApADD fusion proteins was induced with isopropyl-β-d-thiogalactopyranoside (IPTG) (1 mm) according to the pET system manual (Novagen, Madison, WI). Recombinant proteins were purified from bacterial cultures under native conditions using the TALON Cell Thru metal affinity resin (Clontech User Manual; Clontech, Cambridge, UK).
Wild-caught adult Aplysia californica (∼150 gm; purchased from Marinus, Long Beach, CA) were anesthetized by injection of isotonic MgCl2 (∼100 ml/100 gm body weight). Ganglia and other tissues were removed immediately and homogenized in a glass homogenizer on ice in SDS sample buffer unless indicated otherwise.
Triton X-100 extraction of Aplysia ApADD.Pleural-pedal ganglia were rinsed twice and homogenized in cold PBS containing 5 mm EGTA and 4 mm Pefabloc (broad range protease inhibitor) in the presence of 0, 0.1, or 1% Triton X-100. Insoluble proteins in the particulate fraction were pelleted by centrifugation (100,000 ×g for 30 min) and resuspended in a volume equal to that of the soluble cytosolic fraction.
Phosphatase treatment of Aplysia pleural–pedal homogenates. Pleural–pedal ganglia were homogenized in 10 mm Tris-HCl, pH 7.5, in the presence of 4 mm Pefabloc. Mixtures for phosphatase and mock treatment contained (in mm): 20 MgCl2, 100 NaCl, and 0.5 EGTA. In addition, mock mixtures contained 20 mm NaF and 20 mm β-glycerophosphate, whereas phosphatase mixtures contained 2 U of calf intestinal phosphatase (Sigma, St. Louis, MO). Mixtures were added to 40 μg pleural–pedal homogenate and incubated at 37°C for 30 min. Reactions were terminated by the addition of SDS sample buffer.
In vitro phosphorylation of recombinant ApADD proteins with PKC. Purified recombinant His-ApADD fusion proteins (FL, NT, and CT; 10 μg each) were phosphorylated by incubation with 0.1 mm ATP, 5 mmMgCl2, 1 mm EGTA, 50 mm Tris-HCl, pH 7.5, and 2.5 μg/ml rat brain PKC (Calbiochem, La Jolla, CA) at room temperature for 2 hr. The reaction was terminated by the addition of SDS sample buffer.
Antisera. A phospho-adducin antiserum (Upstate Biotechnology, Lake Placid, NY) was raised against a phosphopeptide corresponding to amino acids 656–668 of human γ-adducin [KKFRTP(pS)FLKKNK]. General adducin antisera were raised in rabbits against synthetic peptides corresponding to various regions of human α- and β-adducins (Gilligan et al., 2002). These peptide sequences are conserved between mammalian and Aplysiaadducins. The antibody referred to as “adducin antibody” was affinity-purified using recombinant human α-adducin coupled to cyanogen bromide-activated Sepharose (Gilligan et al., 2002). This antibody recognizes human α-, β-, and γ-adducin via a C-terminal epitope. Where indicated, another antiserum was affinity-purified against synthetic peptides corresponding to mammalian adducin sequences. This antibody reacts with both N-terminal and C-terminal epitopes. In one experiment, an antibody was used that had been affinity-purified against human recombinant β-adducin (Gilligan et al., 1999).
Western blot analysis and immunocytochemistry. Homogenates of Aplysia ganglia (20 μg) were separated on 8–15% SDS-polyacrylamide gradient gels and transferred to nitrocellulose membranes. Membranes were blocked with 4% BSA and probed with adducin or phospho-adducin antibodies followed by protein A-HRP (BioRad, Hercules, CA). Adducin signals were visualized by enhanced chemiluminescence (Renaissance; NEN, Boston, MA).
Pleural–pedal ganglia were desheathed and fixed in 4% paraformaldehyde, PBS, and 20% sucrose overnight at 4°C. Ganglia were washed three times for 15 min in PBS, permeabilized for 1 hr in 4% Triton X-100/PBS, rinsed in PBS, and blocked in 2% normal goat serum for 1 hr at room temperature. Ganglia were incubated with adducin antibody affinity-purified against human recombinant α-adducin (0.5 mg/ml; 1:10000 in blocking medium) for 2 d at 4°C, washed three times for 15 min in PBS, and kept in goat anti-rabbit-Alexa-Fluor 488 (Molecular Probes, Eugene, OR) (1:100 in blocking medium) for 2.5 hr at room temperature. After three washes in PBS, ganglia were mounted in Citifluor, and immunoreactivity was visualized using a BioRad MRC 1020 confocal microscope with a krypton/argon laser. Images were collected using LaserSharp imaging software.
Ganglia preparation for 5-HT stimulation. Pleural–pedal ganglia were removed from Aplysia immediately after anesthesia and transferred to Sylgard-coated recording dishes containing a 1:1 mixture of isotonic MgCl2 and artificial sea water (ASW) containing (in mm): 460 NaCl, 55 MgCl2, 11 CaCl2, 10 KCl, and 10 Tris, pH 7.6, to prevent synaptic transmission during dissection. Ganglia were pinned down and desheathed so that somatic clusters of tail SNs in the pleural and tail MNs in the pedal ganglia were exposed. After dissection, preparations were continuously perfused at a rate of 5 ml/min at room temperature with ASW or 5-HT (50 μm) dissolved in ASW. In stimulation experiments, one of the paired pleural–pedal ganglia from each animal was used as an internal control and perfused with ASW only.
To investigate the modulation of ApADD after STF, preparations were stimulated with a single 5 min pulse of 5-HT. To analyze the modulation of ApADD after the induction of LTF, preparations received five spaced 5 min pulses of 5-HT (10 min between pulses).
Quantitation of ApADD and phospho-ApADD levels in Aplysiaganglia. The amount of protein, total ApADD, and phospho-ApADD in each sample was determined from densitometric scans of Coomassie-stained gels or Western blot analysis. Western blots were exposed for different time intervals, and multiple exposures were scanned and analyzed for each blot to ensure that the obtained signals were not saturated. For each ganglion sample, the ApADD or phospho-ApADD signal was normalized to the total protein content. Phospho-ApADD signals were also normalized to total adducin signals in the same samples as indicated. Finally, the means obtained for the control groups (unstimulated ganglia) were set to 1.
All data in the experiments on synaptic facilitation were normally distributed. Comparisons between control and 5-HT-treated ganglia were performed using Student's unpaired t test. The difference between mean values was regarded as statistically significant ifp < 0.05.
Results
Cloning of a cDNA encoding ApADD
We generated an alignment of available vertebrate and invertebrate adducin amino acid sequences to identify regions that are highly conserved between species. Conserved amino acid sequences were selected to design pools of degenerate inosine-containing oligonucleotides. These oligonucleotides were used as primers in a PCR homology screen with a cDNA library from the Aplysia nervous system as a template. One primer pair (corresponding to amino acids 309–315 and 386–392 of human α-adducin) amplified a 242 bp DNA fragment that was cloned and sequenced. The deduced amino acid sequence of this clone was 58% identical to human γ-adducin. Therefore, we used this 242 bp DNA fragment to screen an Aplysia nervous system cDNA library under high-stringency conditions. One of the clones identified in this screen contained an open reading frame encoding a putative polypeptide of 701 amino acids (calculated molecular weight: 77.4 kDa) (Fig.1A). The polypeptide shows sequence homology to Drosophila (43% identity, 62% similarity with the Drosophila adducin homolog R1) and mammalian (42% identity with human α-adducin) adducin and was named ApADD. Regions of high similarity to invertebrate and vertebrate adducins exist throughout the ApADD sequence (Fig.1A,B), but similarity is highest in the region corresponding to the N-terminal head domain of adducins (amino acids 1–336, 50% identity with human α-adducin). This region also shows the highest conservation between different mammalian adducins. Like other adducin family members, ApADD also contains a C-terminal highly basic domain with sequence similarity to the mammalian myristoylated alanine-rich C kinase substrate (MARCKS) protein (Joshi et al., 1991; Dong et al., 1995) (amino acids 675–685 in ApADD have 48% identity with the corresponding sequence in chicken MARCKS). A serine residue (Ser 668 of human γ-adducin) within the MARCKS-related domain was characterized previously as a major in vitro and in vivo PKC phosphorylation site for mammalian adducins (Dong et al., 1995; Matsuoka et al., 1998). This site is conserved in ApADD (FRMPSF, amino acids 683–688), suggesting that it may be a target for PKC phosphorylation.
Fig. 1.

ApADD resembles other adducin family members.A, An alignment of the deduced ApADD amino acid sequence with human, mouse, and Drosophila adducins was created using the Clustal Method (MegAlign; DNAStar, Madison, WI). Amino acids that are identical between species are boxed inblack. The arrowhead indicates thein vivo PKC phosphorylation site in the MARCKS domain of mammalian adducins. This site is conserved in ApADD and recognized by the phospho-adducin specific antibody. B, Comparison between ApADD and predicted polypeptides with adducin homology inC. elegans and S.pombe. A region from the N-terminal domain of ApADD was aligned with deduced amino acid sequences from C.elegans (cosmids F39C12 and F57F5) and S.pombe (GenBank accession numbers O42892 and Q9P5M9).
Like mammalian adducins, ApADD has a highly hydrophilic C-terminal domain
The strong similarity of ApADD to mammalian adducins suggests that ApADD has a similar domain structure with distinct N-terminal and C-terminal domains. We expressed full-length ApADD and polypeptides corresponding to its predicted N-terminal and C-terminal domains as fusion proteins with an N-terminal tag of six histidine residues in bacteria. The recombinant proteins were purified via nickel chelate chromatography (Fig.2A). The electrophoretic migration of the NT protein was consistent with the molecular weight calculated from its amino acid sequence (49 kDa). In contrast, the relative mobilities of FL and CT were higher than the molecular weight calculated from the amino acid sequences (FL, Mr 100 vs 80 kDa calculated; CT, Mr 50 vs 35 kDa calculated). This anomalous electrophoretic migration has also been observed for mammalian adducins and is most likely attributable to the strong hydrophilicity of the C-terminal domain (Joshi et al., 1991). The hydrophilicity profile of ApADD shows that the predicted C-terminal domain of ApADD (amino acids 335–701) is highly enriched in hydrophilic residues, whereas the N-terminal domain contains both hydrophilic and hydrophobic residues (Fig. 2B). Therefore, ApADD and mammalian adducins share a common hydrophilicity profile.
Fig. 2.

Recombinant ApADD proteins are recognized by antibodies against mammalian adducins and show anomalous electrophoretic mobility. A, Full-length ApADD (amino acids 1–701) and its predicted N-terminal (amino acids 1–350) and C-terminal domains (amino acids 351–701) were expressed with an N-terminal tag of six histidine residues in bacteria. Recombinant proteins were purified from bacterial homogenates (H) via nickel chelate chromatography (F, flow-through from affinity column; E, eluate-containing recombinant proteins). B, Hydrophilicity profiles of human α-adducin and Aplysiaadducin ApADD. The profiles were generated using the Kyte-Doolittle algorithm within the program Protean (DNAStar). C, FL, NT, and CT ApADD were expressed with an N-terminal tag of six histidine residues in bacteria. Samples were taken from each bacterial culture immediately before the induction of expression with 1 mm IPTG (−) and 4 hr after induction (+). Homogenate from Aplysiapleural–pedal ganglia (Ap) was processed in parallel. For all samples, equal amounts of protein were separated by SDS-PAGE. Gels were stained with Coomassie Blue or blotted onto nitrocelluose and probed with adducin antibody that had been affinity-purified against α-adducin and reacts with C-terminal adducin epitopes, anti-adducin (CT), or with adducin antibody that had been purified against synthetic peptides and reacts with N-terminal and C-terminal adducin epitopes (anti-adducin, NT plus CT peptides).
ApADD is expressed in Aplysia nervous system
To gain tools for the analysis of ApADD function, we tested antibodies raised against synthetic peptides derived from human adducins for cross-reactivity with recombinant ApADD proteins. We used two different antibodies affinity-purified against recombinant human α-adducin [anti-adducin (CT)] or against synthetic peptides (anti-adducin, NT plus CT peptides) (Gilligan et al., 1999). Both antibodies cross-react with human and mouse α-, β-, and γ-adducin (D. Gilligan, unpublished observations).
The antibody affinity-purified against recombinant α-adducin reacts with FL and CT but not with NT ApADD (Fig. 2C,middle). This antibody also recognizes C-terminal epitopes in mammalian adducins (D. Gilligan, unpublished observations). The antibody affinity-purified against synthetic peptides recognizes FL, NT, and CT ApADD (Fig. 2C, bottom).
Both antibodies also strongly cross-react with a polypeptide of ∼100 kDa in extracts from Aplysia nervous system (Figs.2C, Ap,3A, adducin/Ap). This endogenous Aplysia polypeptide co-migrates with recombinant FL ApADD and with adducins detected in human red blood cell (RBC) ghosts (Figs. 2C, 3A). A comparable endogenous Aplysia protein is also detected with adducin antibody affinity-purified against human recombinant β-adducin (Fig.3A, β/Ap). This antibody reacts preferentially with mammalian β-adducin (D. Gilligan, unpublished observations). Therefore, we conclude that the antibodies against mammalian adducins cross-react with ApADD in Aplysia tissues.
Fig. 3.
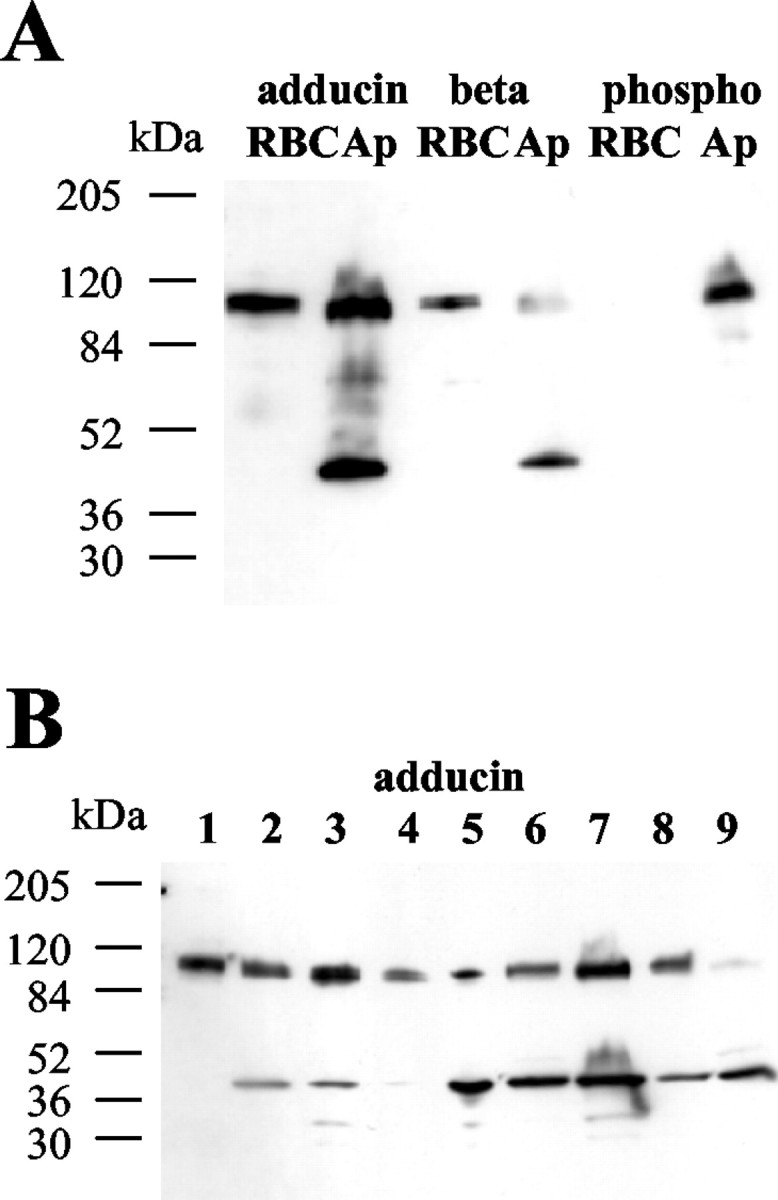
ApADD is expressed in Aplysiatissues. A, Western blot analysis of homogenates fromAplysia pleural–pedal ganglia. Human red blood cell (RBC) ghosts and pleural–pedal homogenates (Ap) were separated by SDS-PAGE, electroblotted, and probed with adducin antibody affinity-purified against human recombinant α-adducin (adducin) or human recombinant β-adducin (beta), or with the phospho-adducin specific antibody (phospho). B, ApADD expression in different Aplysia tissues. Human RBC ghosts (lane1), as well as homogenates fromAplysia pleural–pedal ganglia (lane 2), abdominal ganglia (lane 3), skin (lane 4), buccal mass (lane 5), penis (lane 6), heart (lane 7), hepatopancreas (lane 8), and body wall (lane 9) were separated by SDS-PAGE, electroblotted, and probed with anti-adducin antibody affinity-purified against human recombinant α-adducin.
All adducin antibodies also react with a polypeptide of a relative mobility of ∼50 kDa in Aplysia homogenates. This polypeptide is not detected in RBCs and may be a product of regulated proteolysis, an alternatively spliced form of ApADD, or the gene product of a different adducin gene in Aplysia. Regulated proteolysis and alternative splicing have been described for mammalian adducins (Lin et al., 1995; Gilligan et al., 1997; Sinard et al., 1998). However, we cannot entirely exclude the possibility that the 50 kDa polypeptide is an artifact that may have been generated during sample processing.
Western blot analysis of extracts from different Aplysiatissues shows that ApADD is expressed in skin, body wall, buccal mass, heart, hepatopancreas, and penis (Fig. 3B). This broad expression is consistent with the ubiquitous expression pattern found for mouse α- and γ-adducin (Gilligan et al., 1999).
ApADD is recovered in the particulate fraction of nervous system extracts and is localized in the submembraneous region of neurons
To analyze the subcellular distribution of ApADD, we generated particulate and cytosolic fractions of Aplysia nervous system extracts and performed Western blot analysis with adducin and phospho-adducin antibodies on these fractions. Approximately 95% of total and phosphorylated ApADD are recovered in the particulate fraction (Fig. 4). This is consistent with the localization of mammalian adducins to the cytoskeleton- and membrane-containing cellular components. To investigate additional similarities with mammalian adducins, we examined the extraction of ApADD from the particulate fraction with increasing concentrations of nonionic detergent (Triton X-100). After fractionation in the presence of 0.1% Triton X-100, a strong ApADD signal was detected in the soluble fraction (43% of total signal in soluble fraction). Extraction of ApADD was slightly increased in the presence of 1% Triton X-100 (54% of total adducin soluble) (Fig. 4). A partial extraction with nonionic detergent has also been described for mammalian adducins (Waseem and Palfrey, 1988). Phospho-ApADD shows extractability similar to ApADD. Treatment with 1% Triton X-100 leads to a slightly better extraction of phospho-ApADD compared with total ApADD (66% of total phospho-ApADD soluble; data not shown).
Fig. 4.
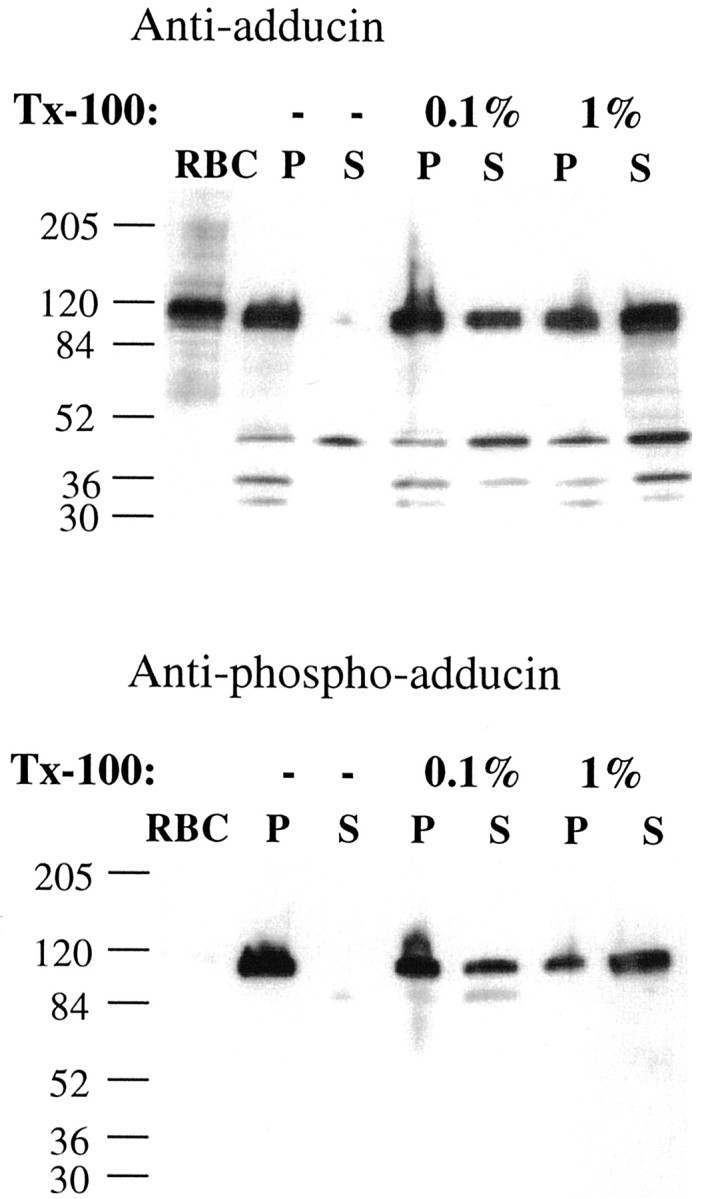
Subcellular distribution and extraction properties of ApADD. Crude particulate fractions (P) and cytosolic fractions (S) were prepared fromAplysia pleural–pedal ganglia in the absence (−) or presence of nonionic detergent (Triton X-100, 0.1 or 1%). Sample processing as described previously for Figure 3B.
To study further the cellular and subcellular localization of ApADD, we analyzed whole-mount preparations of pleural–pedal ganglia by immunofluorescence microscopy using affinity-purified adducin antibody. ApADD immunoreactivity was high in the majority of neuronal cell bodies but excluded from the nuclei (Fig.5A–C). Maximum signals were detected in a region just underneath the plasma membrane in all neurons observed (Fig. 5A,B, pleural SNs,C, pedal neurons). Only weak fluorescence was detected throughout the cytoplasmatic area of cells when adducin antibodies were replaced by nonimmune IgG or omitted entirely (data not shown). These results strongly support our hypothesis that ApADD is localized to the membrane cytoskeleton of Aplysia neurons.
Fig. 5.

ApADD immunoreactivity in pleural–pedal ganglia. ApADD immunoreactivity in pleural and pedal ganglia. Immunostaining was performed in whole-mount preparations from desheathed pleural and pedal ganglia, using adducin antiserum that had been affinity-purified against human recombinant α-adducin. The tail sensory neurons in the pleural ganglion show somatic ApADD immunoreactivity localized to the cytoplasm (A) and close to the plasma membrane (B, higher magnification). ApADD immunoreactivity is also found in neurons within the pedal ganglion (C). Scale bar, 100 μm.
ApADD is phosphorylated by PKC in vitro andin vivo
A 100 kDa polypeptide was also recognized in Aplysianervous system extracts by a phospho-adducin-specific antibody raised against a phospho-peptide corresponding to the major PKC-phosphorylation site in mammalian adducins (Fig. 3A). The sequence around this PKC phosphorylation site is highly conserved among mammalian adducins as well as in ApADD (Fig.1A).
To confirm that ApADD is in fact a PKC substrate, we performed in vitro phosphorylation of recombinant ApADD FL, NT, and CT polypeptides with purified active PKC from rat brain (Calbiochem, La Jolla, CA). We analyzed the phosphorylation of ApADD proteins by Western blot analysis with the phospho-adducin antibody. In the absence of PKC, none of the recombinant ApADD polypeptides was phosphorylated to a significant degree (−). Incubation with PKC (+) led to a strong phospho-signal for the FL and CT proteins but not to an increase in the signal for the NT protein (Fig. 6A). None of the three proteins were degraded during incubation with PKC because they remained detectable with adducin antibody affinity-purified against adducin peptides. This finding confirms that the C-terminal domain of ApADD is phosphorylated by PKC. Because PKC phosphorylation of ApADD is detectable with an antibody specific for the in vivo PKC site in mammalian adducins, we conclude that the corresponding site may also be phosphorylated by PKC inAplysia.
Fig. 6.

ApADD is phosphorylated by PKC. A, Recombinant ApADD proteins (FL, NT, andCT; see Fig. 2A) were phosphorylated in the absence (−) or presence (+) of rat brain PKCin vitro. Reactions were terminated by the addition of SDS sample buffer and samples were submitted to Western blot analysis with adducin antibody affinity-purified against adducin peptides or with phospho-adducin-specific antibody. B, Western blot analysis of pleural–pedal homogenates exposed to calf intestinal alkaline phosphatase (AP) or mock treatment (M). Samples were probed with phospho-adducin antibody and adducin antibody affinity-purified against human recombinant α-adducin.
We also tested the specificity of the phospho-adducin-specific antibody in Aplysia tissue extract. Treatment of tissue extract with alkaline phosphatase (AP) but not mock treatment (M) abolished the signal detected with the phospho-adducin antibody. Total ApADD as detected with adducin antibody remained unchanged (Fig.6B). This finding confirms that ApADD is phosphorylated at a PKC site in Aplysia nervous tissue and that the phospho-adducin antibody can be used to detect changes in the phosphorylation of this site.
PKC phosphorylation of ApADD is increased with multiple exposures to 5-HT that give rise to long-term facilitation of SN–MN synapses
To determine a possible role of ApADD in structural changes associated with synaptic plasticity, we examined ApADD expression and phosphorylation by PKC following procedures that induce STF and LTF. Facilitation was induced by direct application of 5-HT to the intact nervous system containing the presynaptic SN cell bodies and proximal synapses in the pleural ganglion and the SN–MN synapses and MN cell bodies in the pedal ganglion (Emptage and Carew, 1993; Mauelshagen et al.,1996; Sherff and Carew, 1999). For each animal, one ganglion pair was exposed to 5-HT, whereas the ganglia from the opposite side of the body served as an unstimulated control. Pleural and pedal ganglia were homogenized separately in SDS sample buffer, and total ApADD and phospho-ApADD levels in ganglia were analyzed by Western blotting.
A single 5 min pulse of 5-HT (50 μm), which induces STF (Ghirardi et al., 1995; Mauelshagen et al., 1996; Sutton and Carew, 2000), did not lead to any change in the PKC phosphorylation of ApADD in pleural or pedal ganglia immediately after the 5-HT pulse (Table1A). After five spaced pulses of 5-HT (5 min, 50 μm; 10 min between pulses), which induce LTF (Montarolo et al., 1986; Mauelshagen et al., 1996; Sherff and Carew, 1999), no change in phospho-ApADD or total ApADD levels was found at 1 hr after initial LTF induction (Fig.7B; Table 1B). Eighteen hours after the initial LTF induction, total ApADD levels were also not changed (Table 1C). In contrast, phospho-ApADD levels were selectively increased in pleural but not in pedal ganglia (Fig.7A,B; Table 1C) (unchanged phospho-adducin levels in the pedal ganglia were confirmed using Western blot exposures of various duration). However, changes at distal SN–MN synapses in the pedal ganglion may have occurred as well but may have been too small to be detected in pedal homogenate (see Discussion). In a subset of these experiments (n = 6; data not shown), the induction of LTF by 5-HT was confirmed with physiological recording (data kindly provided by C. M. Sherff, University of California, Irvine, CA). Because there was no difference in ApADD phosphorylation in this subset of experiments, these data were pooled with the total data set. These results suggest that ApADD phosphorylation at a PKC site may be increased in response to LTF induction in the presynaptic SNs. The observation that increased ApADD phosphorylation occurs at a late time point after initial LTF induction indicates that increased ApADD phosphorylation probably does not contribute to early but rather to late stages of LTF.
Table 1.
ApADD and phospho-ApADD levels in pleural and pedal ganglia after induction of STF or LTF
| Five minutes after 1 × 5-HT | ||
|---|---|---|
| 5-HT | Phospho-ADD/ADD | |
| Pleural ganglia | − | 1 ± 0.11 |
| + | 1.02 ± 0.11 | |
| Pedal ganglia | − | 1 ± 0.11 |
| + | 1.01 ± 0.09 | |
| One hour after 5 × 5-HT | |||||
|---|---|---|---|---|---|
| 5-HT | Total protein | ADD/protein | Phospho-ADD/ protein | Phospho-ADD/ ADD | |
| Pleural ganglia | − | 1 ± 0.03 | 1 ± 0.09 | 1 ± 0.12 | 1 ± 0.11 |
| + | 0.99 ± 0.03 | 1.07 ± 0.12 | 1.05 ± 0.12 | 1.02 ± 0.11 | |
| Pedal ganglia | − | 1 ± 0.03 | 1 ± 0.07 | 1 ± 0.12 | 1 ± 0.11 |
| + | 1.03 ± 0.02 | 0.98 ± 0.09 | 0.92 ± 0.07 | 1.01 ± 0.09 | |
| Eighteen hours after 5 × 5-HT | |||||
|---|---|---|---|---|---|
| 5-HT | Total protein | ADD/protein | Phospho-ADD/ protein | Phospho-ADD/ ADD | |
| Pleural ganglia | − | 1 ± 0.04 | 1 ± 0.06 | 1 ± 0.10 | 1 ± 0.11 |
| + | 1.03 ± 0.05 | 1.04 ± 0.06 | 1.37 ± 0.11* | 1.33 ± 0.11* | |
| Pedal ganglia | − | 1 ± 0.04 | 1 ± 0.05 | 1 ± 0.06 | 1 ± 0.04 |
| + | 1.06 ± 0.03 | 0.94 ± 0.04 | 0.95 ± 0.06 | 0.98 ± 0.05 | |
Pleural–pedal ganglia were stimulated and processed as described for Figure 7. Protein content (in LTF samples), total ApADD, and phospho-ApADD levels were quantified densitometrically from Coomassie blue stained gels and Western blot autoradiographs. ApADD and phospho-ApADD levels were normalized to the protein content of each sample. In addition, the phospho-ApADD level was also normalized to the ApADD level in the same sample, and the means obtained for control groups (−) were set to 1. Asterisks indicate means that are significantly different from the mean of the corresponding control group (p < 0.05).
Fig. 7.
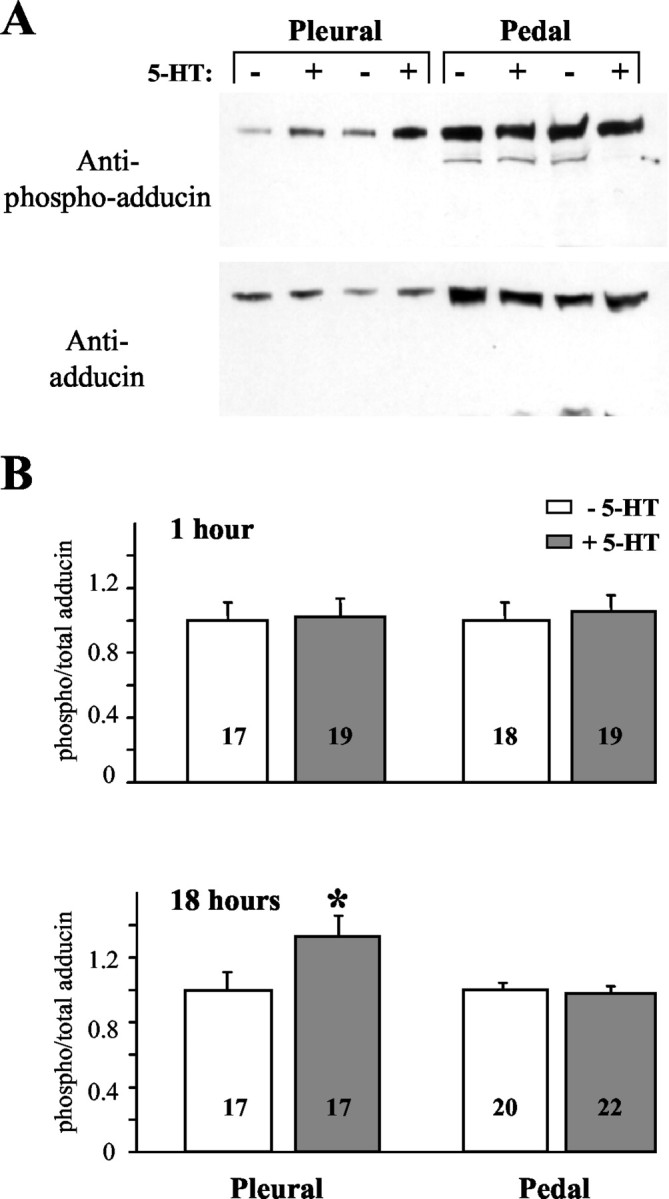
Adducin phosphorylation in pleural ganglia is increased 18 hr after initial LTF induction. Pleural–pedal ganglia were desheathed and continously perfused with ASW. One pleural–pedal complex from each animal served as a control and was perfused with ASW only. The pleural–pedal complex from the opposite body side received five spaced 5 min pulses of 5-HT (50 μm in ASW; 10 min between pulses) to induce LTF. Pleural and pedal ganglia were homogenized separately in SDS sample buffer 1 or 18 hr after 5-HT treatment. Sample processing was as described previously for Figure3B. A, Coomassie-stained gels and Western blots from samples taken 18 hr after 5-HT treatment (+) or control perfusion (−). B, ApADD and phospho-ApADD levels in samples were quantified densitometrically from Western blot autoradiographs. For each sample, phospho-ApADD levels were normalized to total ApADD levels. Numbers in the histograms indicate the number of samples in each group. Means were calculated for each treatment and control groups. The means for the control groups (unstimulated ganglia) were set to 1. Asterisks indicate means that are significantly different from the mean of the corresponding control group (p < 0.05).
Discussion
We have identified ApADD, an Aplysia homolog of mammalian cytoskeletal adducins. ApADD is enriched in the membrane skeletal region of neurons participating in synaptic facilitation and is phosphorylated by PKC in vitro and in vivo. In the intact Aplysia nervous system, ApADD phosphorylation at a PKC site is increased selectively 18 hr after multiple 5-HT exposures that give rise to LTF at tail SN–MN synapses (Emptage and Carew, 1993;Mauelshagen et al., 1996). These data lead us to propose a model in which ApADD phosphorylation serves both to destabilize the actin/spectrin membrane skeleton and permit increased actin polymerization (see below) (Fig. 8). Both steps could be critical for structural changes induced by LTF.
Fig. 8.

Model of the role of adducins in LTF.Top, Adducins are regulatory components of the membrane cytoskeleton. Bottom, Adducins are expressed in neurons, cross-link actin and spectrin in the cytoskeleton, and cap the fast-growing barbed end of actin filaments. Induction of LTF with 5-HT leads to the activation of PKC, an increase in ApADD phosphorylation at a PKC site, and a weakening of the interaction between ApADD and other cytoskeletal proteins. This chain of events may both promote destabilization of actin/spectrin complexes and increase actin polymerization. Both processes are critical for the initiation of structural changes accompanying LTF. After the completion of these changes, dephosphorylation of ApADD induces the stabilization of the newly remodeled synapse.
ApADD: a homolog of mammalian adducins
Regions of high similarity to mammalian and Drosophilaadducins are found throughout the entire ApADD deduced amino acid sequence, suggesting that the overall domain structure for these proteins is very similar. This similarity in structure is also supported by the observation that ApADD shows high hydrophilicity in the region corresponding to the highly hydrophilic C-terminal domain of mammalian adducins (Joshi et al., 1991). Two sites in mammalian adducins have been confirmed as in vivo phosphorylation sites that modulate adducin function. Phosphorylation of serine 726 in α-adducin by PKC inhibits its actin-capping activity and leads to a loss of the ability of adducin to recruit spectrin to the cytoskeleton (Matsuoka et al., 1998). In contrast, phosphorylation of threonine 445 by rho kinase enhances F-actin binding of adducin and regulates membrane ruffling and cell motility of Madin-Darby canine kidney epithelial cells (Kimura et al., 1998; Fukata et al., 1999). The region surrounding threonine 445 of α-adducin is highly conserved among mammalian adducins but less so in ApADD and other invertebrates. In contrast, the sequence surrounding serine 726 in the MARCKS-related domain is highly conserved between mammalian adducins, Drosophila hts R1, and ApADD. This suggests that phosphorylation by PKC may be a critical modulatory event for adducin function in different organisms, including Aplysia.
Expression and localization of ApADD
Using antibodies generated against mammalian adducins, we found that ApADD is widely expressed in Aplysia. This result is again consistent with findings for mammalian α- and γ-adducins and for Drosophila adducin (Yue and Spradling, 1992; Seidel et al., 1995; Gilligan et al., 1999). In whole-mount preparations of pleural–pedal ganglia, we detected ApADD immunoreactivity in the majority of neurons observed. Staining was excluded from nuclei and was strongest in the submembraneous region of neurons, which is consistent with a localization of ApADD to the membrane cytoskeleton. Adducin immunoreactivity has also been detected in hippocampal neurons along the entire cell membrane, in the cytosol, and in dendrites (Matsuoka et al., 1998). In our preparation, staining was not visible in neurites but may have been too weak to be detected. It is possible that the access of the antibody to ApADD may be blocked intracellularly by cytoskeletal structures within the neurites themselves.
In accordance with a localization to the membrane skeleton, ApADD was primarily recovered in the particulate fraction of cells that contains the cytoskeleton and was partially extracted by nonionic detergent like mammalian adducins (Gardner and Bennett, 1986; Dong et al., 1995). These data suggest that ApADD is localized to the membrane skeleton ofAplysia neurons, including those neurons critically involved in synaptic facilitation at tail SN–MN synapses.
Functional activation of ApADD
Western blotting experiments using an antibody that was shown to be specific for the PKC phosphorylation site in mammalian adducins (Gilligan et al., 2002) strongly suggest that ApADD is phosphorylatedin vivo at a PKC site in its C-terminal domain. We found that this antibody recognizes polypeptides in the Aplysianervous system that co-migrate with recombinant ApADD and adducins from red blood cells. In addition, we showed that PKC phosphorylates recombinant proteins in vitro corresponding to ApADD and the predicted C-terminal domain of ApADD, whereas a protein corresponding to the predicted N-terminal domain of ApADD was not phosphorylated by PKC. These findings strongly support our hypothesis that ApADD is phosphorylated by PKC at a site in its C-terminal domain that corresponds to the in vivo phosphorylation site for PKC in mammalian adducins. The specificity of the phospho-adducin antibody for PKC phosphorylated ApADD is also supported by experiments on nervous system extract: phosphatase, but not mock treatment of extracts, led to loss of the signal obtained with the phospho-adducin antibody, whereas total ApADD as detected with mammalian adducin antibody remained unchanged. Although our data point to PKC being the kinase responsible for ApADD phosphorylation, we cannot entirely exclude the possibility that a different kinase is responsible for its phosphorylation at this site.
Both mammalian adducins and ApADD contain additional predicted consensus sequences for PKC phosphorylation. However, only one additional site in mammalian adducins (Ser 716 in human α-adducin, Ser703 in β-adducin) has been shown to be phosphorylated by PKCin vitro (Matsuoka et al., 1996). Because this site is not conserved in ApADD (or Drosophila adducin), the existence of additional PKC sites in ApADD is rather unlikely.
Interestingly, basal ApADD phosphorylation in the Aplysianervous system is higher than basal adducin phosphorylation in RBCs and platelets. High levels of phospho-adducin were also detected in unstimulated rat hippocampus and dentate gyrus (Matsuoka et al., 1998). A high basal level of adducin phosphorylation may be a typical feature of nervous tissue and could provide a higher degree of regulation of adducin function by both protein kinases and phosphatases.
We found that phosphorylation of ApADD at a PKC site in the intactAplysia nervous system was significantly increased 18 hr, but not 1 hr, after patterns of 5-HT exposure that induce LTF. Interestingly, we found no change in ApADD phosphorylation immediately after a single 5-HT pulse that induces STF. These findings suggest that PKC phosphorylation of ApADD plays a role in LTF and not in STF. The data also suggest that ApADD is not involved in the early steps of LTF induction but rather at a later time in LTF. We found that ApADD phosphorylation by PKC was increased in the pleural ganglia, which contain the presynaptic SN cell bodies and proximal SN synapses, but not in the pedal ganglia, which contain the SN–MN synapses and the postsynaptic MN cell bodies. Thus, our findings are consistent with the hypothesis that ApADD phosphorylation by PKC is increased in the presynaptic SNs that are critical for LTF.
PKC involvement in long-term memory
A role for PKC in intermediate and long-term forms of synaptic plasticity and memory has been described by several laboratories. For example, a persistently activated catalytic fragment of PKCzeta [persistently active catalytic fragment (PKM)-ζ] was recently shown to be necessary and sufficient for the maintenance of late-phase LTP in mammalian hippocampus (Ling et al., 2002). In Aplysia, a persistent activation of PKC was detected during intermediate-term sensitization (Sossin et al., 1994). More recently, it was shown that the induction of intermediate-term sensitization by a single tail shock requires the calpain-dependent proteolysis of PKC, yielding a persistently active PKM (Sutton et al., 2002). In addition, Drier et al. (2002) recently demonstrated that induction of a PKM transgene enhanced the formation of long-term memory for olfactory conditioning in Drosophila. Finally, a persistent activation of PKC in the time domain of intermediate- and long-term memory was also found for olfactory conditioning in honeybees (Gruenbaum and Mueller, 1998).
In Aplysia, a persistent activation of PKC was detected during intermediate-term but not long-term facilitation (Sossin et al., 1994). However, these experiments were conducted using a protocol for the induction of long-term facilitation that differed from ours (prolonged, 90 min, 5-HT application vs five spaced pulses in our experiments). Different modes of inducing long-lasting facilitation have been shown to lead to the recruitment of different signaling cascades in the sensory neurons of Aplysia (Sutton et al., 2001). In addition, differential activation of signaling pathways has also been observed in response to different modes of inducing long-term memory in both Drosophila and honeybee (Tully et al., 1990; Mueller, 1996). Therefore, although no PKC activation is detected during long-term facilitation induced by prolonged 5-HT application (Sossin et al., 1994), long-term PKC activation may well be induced by spaced applications. Moreover, Sossin et al. (1994) measured PKC activation as an increase in PKC activity in the particulate fraction. As these authors point out, this method will reveal only one possible mode of PKC activation; activation, for example, via translocation to the cytoskeleton, in which ApADD is localized, or to the nucleus, would not be detected. Interestingly, it has been shown recently in Aplysia that the induction of intermediate-term memory for sensitization by a single tail shock requires the calpain-dependent proteolysis of PKC, yielding a persistently active and soluble PKM (Sutton et al., 2001; Sutton and Carew, 2002).
We should also emphasize that the net phosphorylation state of ApADD will be determined by the competing activities of kinases (PKC) and phosphatases. Therefore, it is also possible that ApADD phosphorylation could be triggered initially by PKC activation in the intermediate-term facilitation (ITF) temporal domain, but could be maintained during LTF by the downregulation of one or more phosphatases acting on ApADD. Such a mechanism would allow for increased adducin phosphorylation in the LTF temporal domain without a concomitant activation of PKC above basal activity levels.
Several lines of evidence suggest that PKC, like adducins, is associated with the cytoskeleton. For example, vertebrate PKC-ε as well as both Aplysia PKC isoforms have been demonstrated to bind to actin (Nakhost et al., 1998; Prekeris et al., 1998). Moreover, Aplysia PKC Apl II was shown to colocalize with F-actin in neuronal growth cones (Nakhost et al., 1998). Therefore, we hypothesize that the interaction of PKC with actin filaments in the membrane cytoskeleton could bring PKC into the immediate vicinity of cytoskeletal substrates such as ApADD. These observations are thus consistent with the hypothesis that a form of PKC that is persistently activated during ITF and possibly LTF could mediate increased ApADD phosphorylation during LTF.
A possible role of ApADD in long-term synaptic plasticity
An important feature of long-term memory, LTP in the hippocampus, and LTF in Aplysia is its association with structural changes at specific synapses (Bailey and Chen, 1988; Glanzman et al., 1990; Chang et al., 1991; Bailey and Kandel, 1994; Engert and Bonhoeffer, 1999; Klintsova and Greenough, 1999; Maletic-Savatic et al., 1999; Bonhoeffer and Yuste, 2002). The activation of protein kinases and the induction of gene expression after learning or 5-HT treatment and the eventual structural changes themselves have been extensively investigated in Aplysia (Glanzman et al., 1990; Bailey and Kandel, 1994). However, much less is known about the connection between increased kinase activities and structural changes.
In Aplysia, an early step in the initiation of structural changes involves the internalization of the cell-adhesion molecule ApCAM in sensory neurons (Bailey et al., 1992; Mayford et al., 1992). This process is thought to lead to a loss of cell–cell contacts and may remove inhibition for synaptic growth. A second critical event in structural changes during LTF seems to be actin polymerization, which contributes to new growth at SN–MN synapses (Hatada et al., 2000). Adducins could contribute to structural changes associated with LTF and long-term memory in two ways. First, increased phosphorylation at a PKC site decreases the ability of adducins to recruit spectrin to actin filaments (Matsuoka et al., 1998). Therefore, increased phosphorylation of ApADD by PKC could lead to loss of structural rigidity in the membrane cytoskeleton and thereby permit structural changes associated with LTF to occur. After the internalization of ApCAM from the SN membrane, this could provide another permissive step for the initiation of structural changes during LTF. Second, adducins cap the fast-growing (barbed) end of actin filaments and thereby block the elongation of actin filaments (Kuhlman et al., 1996). This actin-capping activity is also inhibited by PKC phosphorylation of adducins (Matsuoka et al., 1998). Thus, increased PKC phosphorylation of ApADD during LTF might lead to a reduction of actin capping and could thereby allow increased actin polymerization, which is critical for structural changes associated with LTF.
Collectively, our data lead us to propose the following model for ApADD function in LTF (Fig. 8). Induction of LTF leads to a persistent activation of PKC in the intermediate-term (and perhaps long-term) temporal domains and, as a consequence, leads to increased phosphorylation of ApADD. This process modulates the interaction of ApADD with cytoskeletal components and increases both the flexibility of the membrane cytoskeleton and actin polymerization. Both of these processes could be necessary for structural changes to occur. After the completion of structural changes, dephosphorylation leads to a reassociation of ApADD with the membrane cytoskeleton and to an increase in the capping of actin filaments. Both processes would promote the stabilization of the newly remodeled synaptic structure.
In conclusion, we have identified a mechanism for rapid and reversible induction of cytoskeletal modification that may contribute to early stages of structural remodeling in synaptic modifications accompanying long-term changes in synaptic strength. These changes could be an important component of the ultrastructural remodeling underlying long-term memory.
Footnotes
This work was supported by National Institutes of Health Grant DKHL, 55005-01 (D.M.G.), Simone and Cino Del Luca and Institut National de la Santé et de la Recherche Médicale Fellowships (S.M.), Feodor-Lynen Fellowship from the Humboldt Society (L.M.G.), and National Science Foundation Grant IBN 004-9013 (T.J.C.). We thank Angela Purcell, Joanna Schaffhausen, Carolyn Sherff, and Michael Sutton for helpful comments on a previous version of this manuscript.
Correspondence should be addressed to Thomas J. Carew, Department of Neurobiology and Behavior, Center for Learning and Memory, 2205 BioSciII, University of California, Irvine, Irvine, CA 92697-4550. E-mail: tcarew@uci.edu.
References
- 1.Audesirk G, Cabell L, Kern M. Modulation of neurite branching by protein phosphorylation in cultured rat hippocampal neurons. Dev Brain Res. 1997;102:247–260. doi: 10.1016/s0165-3806(97)00100-4. [DOI] [PubMed] [Google Scholar]
- 2.Bailey CH, Chen M. Long-term memory in Aplysia modulates the total number of varicosities of single identified sensory neurons. Proc Natl Acad Sci USA. 1988;85:2373–2377. doi: 10.1073/pnas.85.7.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey CH, Kandel ER. Structural changes underlying long-term memory storage in Aplysia: a molecular perspective. Semin Neurosci. 1994;6:35–44. doi: 10.1016/s1040-7952(06)80034-0. [DOI] [PubMed] [Google Scholar]
- 4.Bailey CH, Chen M, Keller F, Kandel ER. Serotonin-mediated endocytosis of apCAM: an early step of learning-related synaptic growth in Aplysia. Science. 1992;256:645–649. doi: 10.1126/science.1585177. [DOI] [PubMed] [Google Scholar]
- 5.Bonhoeffer T, Yuste R. Spine motility: phenomenology, mechanisms, and function. Neuron. 2002;35:1019–1027. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 6.Carew TC, Castellucci VF, Kandel ER. An analysis of dishabituation and sensitization of the gill-withdrawal reflex in Aplysia. Int J Neurosci. 1971;2:79–98. doi: 10.3109/00207457109146995. [DOI] [PubMed] [Google Scholar]
- 7.Castellucci VF, Blumenfeld H, Goelet P, Kandel ER. Inhibitor of protein synthesis blocks long-term behavioral sensitization in the isolated gill-withdrawal reflex of Aplysia. J Neurobiol. 1989;20:1–9. doi: 10.1002/neu.480200102. [DOI] [PubMed] [Google Scholar]
- 8.Chang PL, Isaacs KR, Greenough WT. Synapse formation occurs in association with the induction of long-term potentiation in two-year-old rat hippocampus in vitro. Neurobiol Aging. 1991;12:517–522. doi: 10.1016/0197-4580(91)90082-u. [DOI] [PubMed] [Google Scholar]
- 9.Dong L, Chapline C, Mousseau B, Fowler L, Ramsay K, Stevens JL, Jaken S. 35H, a sequence isolated as a protein kinase C binding protein, is a novel member of the adducin family. J Biol Chem. 1995;270:25534–25540. doi: 10.1074/jbc.270.43.25534. [DOI] [PubMed] [Google Scholar]
- 10.Drier EA, Tello MK, Cowan M, Wu P, Blace N, Sacktor TC, Yin JCP. Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat Neurosci. 2002;5:316–324. doi: 10.1038/nn820. [DOI] [PubMed] [Google Scholar]
- 11.Emptage N, Carew TC. Long-term synaptic facilitation in the absence of short-term facilitation in Aplysia neurons. Science. 1993;262:253–256. doi: 10.1126/science.8211146. [DOI] [PubMed] [Google Scholar]
- 12.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 13.Frost WN, Castellucci VF, Hawkins RD, Kandel ER. Monosynaptic connections from the sensory neurons of the gill- and siphon-withdrawal reflex in Aplysia participate in the storage of long-term memory for sensitization. Proc Natl Acad Sci USA. 1985;82:8266–8269. doi: 10.1073/pnas.82.23.8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, Matsura Y, Kaibuchi K. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J Cell Biol. 1999;145:347–361. doi: 10.1083/jcb.145.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardner K, Bennett V. A new erythrocyte membrane-associated protein with calmodulin binding activity. J Biol Chem. 1986;261:1339–1348. [PubMed] [Google Scholar]
- 16.Gardner K, Bennett V. Modulation of spectrin-actin assembly by erythrocyte adducin. Nature. 1987;328:359–362. doi: 10.1038/328359a0. [DOI] [PubMed] [Google Scholar]
- 17.Ghirardi M, Montarolo PG, Kandel ER. A novel intermediate stage in the transition between short- and long-term facilitation in the sensory to motor neurons synapse of Aplysia. Neuron. 1995;14:413–420. doi: 10.1016/0896-6273(95)90297-x. [DOI] [PubMed] [Google Scholar]
- 18.Gilligan DM, Lozovatsky L, Silberfein A. Organization of the human beta adducin gene (ADD2). Genomics. 1997;43:141–148. doi: 10.1006/geno.1997.4802. [DOI] [PubMed] [Google Scholar]
- 19.Gilligan DM, Lozovatsky L, Gwynn B, Brugnara C, Narla M, Peters LL. Targeted disruption of the beta adducin gene causes spherocytosis in mice. Proc Natl Acad Sci USA. 1999;96:10717–10722. doi: 10.1073/pnas.96.19.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilligan DM, Sarid R, Weese J. Adducin in platelets: activation-induced phosphorylation by PKC and proteolysis by calpain. Blood. 2002;99:2418–2426. doi: 10.1182/blood.v99.7.2418. [DOI] [PubMed] [Google Scholar]
- 21.Glanzman DL, Kandel ER, Schacher S. Target-dependent structural changes accompanying long-term synaptic facilitation in Aplysia neurons. Science. 1990;249:4200–4213. doi: 10.1126/science.2389145. [DOI] [PubMed] [Google Scholar]
- 22.Gruenbaum L, Mueller U. Induction of a specific olfactory memory leads to a long-lasting activation of protein kinase C in the antennal lobe of the honeybee. J Neurosci. 1998;18:4384–4392. doi: 10.1523/JNEUROSCI.18-11-04384.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatada Y, Wu F, Sun Z-Y, Schacher S, Goldberg DJ. Presynaptic morphological changes associated with long-term synaptic facilitation are triggered by actin polymerization at preexisting varicosities. J Neurosci. 2000;20:1–5. doi: 10.1523/JNEUROSCI.20-13-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes CA, Bennett V. Adducin: a physical model with implications for function in assembly of spectrin-actin complexes. J Biol Chem. 1995;270:18990–18996. doi: 10.1074/jbc.270.32.18990. [DOI] [PubMed] [Google Scholar]
- 25.Joshi R, Gilligan DM, Otto E, McLaughlin T, Bennett V. Primary structure and domain organization of human alpha and beta adducin. J Cell Biol. 1991;115:665–675. doi: 10.1083/jcb.115.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaiser HW, O'Keefe E, Bennett V. Adducin: Ca++-dependent association with sites of cell-cell contact. J Cell Biol. 1989;109:557–569. doi: 10.1083/jcb.109.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura K, Fukata Y, Matsuoka Y, Bennet V, Matsuura Y, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of the association of adducin with actin filaments by rho-associated kinase (Rho-kinase) and myosin phosphatase. J Biol Chem. 1998;273:5542–5548. doi: 10.1074/jbc.273.10.5542. [DOI] [PubMed] [Google Scholar]
- 28.Klintsova AY, Greenough WT. Synaptic plasticity in cortical systems. Curr Opin Neurobiol. 1999;9:203–208. doi: 10.1016/s0959-4388(99)80028-2. [DOI] [PubMed] [Google Scholar]
- 29.Kuhlman PA, Hughes CA, Bennett V, Fowler VM. A new function for adducin: calcium/calmodulin-regulated capping of the barbed ends of actin filaments. J Biol Chem. 1996;271:7986–7991. doi: 10.1074/jbc.271.14.7986. [DOI] [PubMed] [Google Scholar]
- 30.Levenson J, Byrne JH, Eskin A. Levels of serotonin in the hemolymph of Aplysia are modulated by light/dark cycles and sensitization training. J Neurosci. 1999;19:8094–8103. doi: 10.1523/JNEUROSCI.19-18-08094.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin B, Nasir J, McDonald H, Graham R, Rommens JM, Goldberg YP, Hayden MR. Genomic organization of the human alpha-adducin gene and its alternatively spliced isoforms. Genomics. 1995;25:93–99. doi: 10.1016/0888-7543(95)80113-z. [DOI] [PubMed] [Google Scholar]
- 32.Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- 33.Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1860–1861. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 34.Marinesco S, Carew TJ. Serotonin release evoked by tail-nerve stimulation in the central nervous system of Aplysia: characterization and relationship to heterosynaptic plasticity. J Neurosci. 2002;22:2229–2312. doi: 10.1523/JNEUROSCI.22-06-02299.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin KC, Casadio A, Zhu HEY, Rose JC, Chen M, Bailey CH, Kandel ER. Synapse-specific, long-term facilitation of Aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- 36.Matsuoka Y, Hughes CA, Bennett V. Adducin regulation. Definition of the calmodulin-binding domain and sites of phosphorylation by protein kinases A and C. J Biol Chem. 1996;271:25157–25166. doi: 10.1074/jbc.271.41.25157. [DOI] [PubMed] [Google Scholar]
- 37.Matsuoka Y, Li X, Bennett V. Adducin is an in vivo substrate for protein kinase C: phosphorylation in the MARCKS-related domain inhibits activity in promoting spectrin-actin complexes and occurs in many cells, including dendritic spines of neurons. J Cell Biol. 1998;142:485–497. doi: 10.1083/jcb.142.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mauelshagen J, Parker G, Carew TC. Dynamics of induction and expression of long-term synaptic facilitation in Aplysia. J Neurosci. 1996;16:7099–7108. doi: 10.1523/JNEUROSCI.16-22-07099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayford M, Barzilai A, Keller F, Schacher S, Kandel ER. Modulation of a NCAM-related adhesion molecule with long-term synaptic plasticity in Aplysia. Science. 1992;256:638–644. doi: 10.1126/science.1585176. [DOI] [PubMed] [Google Scholar]
- 40.Mische S, Mooseker M, Morrow J. Erythrocyte adducin: a calmodulin regulated actin-binding protein that stimulates spectrin-actin binding. J Cell Biol. 1987;105:2837–2849. doi: 10.1083/jcb.105.6.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–1254. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- 42.Mueller U. Inhibition of nitric oxide synthase impairs a distinct form of long-term memory in the honeybee, Apis mellifera. Neuron. 1996;16:541–549. doi: 10.1016/s0896-6273(00)80073-2. [DOI] [PubMed] [Google Scholar]
- 43.Nakhost A, Forscher P, Sossin WS. Binding of protein kinase C isoforms to actin in Aplysia. J Neurochem. 1998;71:1221–1231. doi: 10.1046/j.1471-4159.1998.71031221.x. [DOI] [PubMed] [Google Scholar]
- 44.Pinsker HM, Hening WA, Carew TJ, Kandel ER. Long-term sensitization of a defensive withdrawal reflex in Aplysia. Science. 1973;182:1039–1042. doi: 10.1126/science.182.4116.1039. [DOI] [PubMed] [Google Scholar]
- 45.Prekeris R, Hernandez RM, ayhew MW, White MK, Terrain DM. Molecular analysis of the interactions between protein kinase C- and filamentous actin. J Biol Chem. 1998;273:26790–26798. doi: 10.1074/jbc.273.41.26790. [DOI] [PubMed] [Google Scholar]
- 46.Rampon C, Tang YP, Goddhouse J, Shimizu E, Kyin M, Tsien JZ. Enrichement induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci. 2000;3:205–206. doi: 10.1038/72945. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. Cold Spring Harbor Laboratory; Plainview, NY: 1989. [Google Scholar]
- 48.Sanchez C, Diaz-Nido J, Avila J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol. 2000;61:133–168. doi: 10.1016/s0301-0082(99)00046-5. [DOI] [PubMed] [Google Scholar]
- 49.Seidel B, Zuschratter W, Wex H, Garner CC, Gundelfinger ED. Spatial and sub-cellular localization of the membrane cytoskeleton-associated protein alpha-adducin in the rat brain. Brain Res. 1995;700:13–24. doi: 10.1016/0006-8993(95)00962-p. [DOI] [PubMed] [Google Scholar]
- 50.Sherff CM, Carew TJ. Coincident induction of long-term facilitation in Aplysia: cooperativity between cell bodies and remote synapses. Science. 1999;285:1911–1914. doi: 10.1126/science.285.5435.1911. [DOI] [PubMed] [Google Scholar]
- 51.Sinard JH, Stewart GW, Stabach PR, Argent AC, Gilligan DM, Morrow JS. Utilization of an 86 bp exon generates a novel adducin isoform (beta4) lacking the MARCKS homology domain. Biochim Biophys Acta. 1998;1396:57–66. doi: 10.1016/s0167-4781(97)00167-x. [DOI] [PubMed] [Google Scholar]
- 52.Sossin WS, Sacktor TC, Schwartz JH. Persistent activation of protein kinase C during the development of long-term facilitation in Aplysia. Learn Mem. 1994;1:189–202. [PubMed] [Google Scholar]
- 53.Suriyapperuma S, Lozovatsky L, Ciciotte SL, Peters LL, Gilligan DM. The murine adducin gene family: alternative splicing and chromosomal localization. Mamm Genome. 2000;11:16–23. doi: 10.1007/s003350010004. [DOI] [PubMed] [Google Scholar]
- 54.Sutton MA, Carew TJ. Parallel molecular pathways mediate expression of distinct forms of intermediate-term facilitation at tail sensory-motor synapses in Aplysia. Neuron. 2000;26:219–231. doi: 10.1016/s0896-6273(00)81152-6. [DOI] [PubMed] [Google Scholar]
- 55.Sutton MA, Carew TJ. Behavioral, cellular and molecular analysis of memory in Aplysia I: Intermediate-term memory. Integ Comp Biol. 2002;42:725–735. doi: 10.1093/icb/42.4.725. [DOI] [PubMed] [Google Scholar]
- 56.Sutton MA, Bagnall MW, Carew TJ. Molecular mechanisms of site-specific memory for sensitization in Aplysia. Soc Neurosci Abstr. 2001;965:12. [Google Scholar]
- 57.Toni N, Buchs PA, Nikenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- 58.Tully T, Boynton S, Brandes C, Dura JM, Mihalek R, Preat T, Villella A. Genetic dissection of memory formation in Drosophila melanogaster. Cold Spring Harb Symp Quant Biol. 1990;55:203–211. doi: 10.1101/sqb.1990.055.01.022. [DOI] [PubMed] [Google Scholar]
- 59.Wainwright ML, Zhang H, Byrne JH, Cleary LJ. Localized neuronal outgrowth induced by long-term sensitization training in Aplysia. J Neurosci. 2002;22:4132–4141. doi: 10.1523/JNEUROSCI.22-10-04132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waseem A, Palfrey HC. Erythrocyte adducin. Eur J Biochem. 1988;178:563–573. doi: 10.1111/j.1432-1033.1988.tb14483.x. [DOI] [PubMed] [Google Scholar]
- 61.Waseem A, Palfrey HC. Identification and protein kinase C-dependent phosphorylation of alpha-adducin in human fibroblasts. J Cell Sci. 1990;96:93–98. doi: 10.1242/jcs.96.1.93. [DOI] [PubMed] [Google Scholar]
- 62.Yue L, Spradling AC. hu-li tai shao, a gene required for ring canal formation during Drosophila oogenesis, encodes a homolog of adducin. Genes Dev. 1992;6:2443–2454. doi: 10.1101/gad.6.12b.2443. [DOI] [PubMed] [Google Scholar]