

Abstract
At GABAergic synapses, GABA receptors form high-density clusters opposite GABA release sites. Whether GABA release per se plays a role in the formation of GABA receptor clusters remains uncertain. To address this question in vivo, we characterized GABA receptor clustering in the nematode Caenorhabditis elegans. In C. elegans, body wall muscles receive excitatory inputs from cholinergic motor neurons and inhibitory inputs from GABAergic neurons. Using immunohistochemistry and green fluorescent protein-tagged proteins, we observed that the muscle GABA receptor UNC-49 is precisely clustered opposite GABA release sites. During development, these clusters appear slightly after the detection of presynaptic vesicles. If motor axons are mislocalized as in unc-5 mutants, GABA receptors cluster opposite ectopic axons at GABA release sites. Together, these data imply that a motor neuron-derived factor is instructing GABA receptor clustering. Presynaptic localization of this clustering activity requires the neuronal kinesin UNC-104, suggesting that release of GABA from synaptic vesicles may represent the clustering signal. However,unc-25 mutants do not synthesize GABA but do cluster postsynaptic GABA receptors indistinguishably from the wild type. Therefore, at GABAergic neuromuscular junctions, GABA receptor clustering requires nerve–muscle interaction but not GABA neurotransmission.
Keywords: GABA receptor, synaptogenesis, neuromuscular junction, C. elegans, receptor clustering, activity-dependent mechanism
Introduction
In the cerebral cortex, a single pyramidal cell receives thousands of inputs from distinct classes of excitatory and inhibitory neurons. At each of these synapses, appropriate neurotransmitter receptors are clustered opposite the site of neurotransmitter release. How does the postsynaptic cell cluster receptors to match the presynaptic input? One possibility is that the neurotransmitter itself acts as a trans-synaptic signal to cluster or stabilize the appropriate receptors opposite neurotransmitter release sites. The release of neurotransmitter by embryonic growth cones before the formation of synaptic contacts (Young and Poo, 1983) is consistent with such a hypothesis. Despite the appeal of this model, molecular evidence demonstrates that it is not a general mechanism for receptor clustering (for review, see Craig and Boudin, 2001). For example, at the vertebrate neuromuscular junction, acetylcholine receptors cluster in response to agrin, an extracellular matrix protein secreted by motor neurons (for review, see Ruegg and Bixby, 1998; Sanes and Lichtman, 2001). In contrast, choline acetyltransferase mutant mice that do not synthesize any acetylcholine do form neuromuscular synapses that contain high-density clusters of acetylcholine receptors (Misgeld et al., 2002).
However, the role of neurotransmitter in the differentiation of inhibitory synapses remains unclear. In cultured spinal cord neurons, chronic application of the glycine receptor blocker strychnine inhibits the formation of glycine receptor clusters (Kirsch and Betz, 1998; Levi et al., 1998). These results suggest that the activation of the receptors by the neurotransmitter glycine is required to initiate or stabilize glycine receptor clusters. However, the effect of strychnine might be attributable to a block of glycine receptor recycling after endocytosis, resulting in a subsequent depletion of receptors at postsynaptic domains (Rasmussen et al., 2002). In contrast to the glycine receptor clustering studies, initial reports indicate that GABAA receptor expression and clustering do not require neurotransmitter release. Specifically, blocking the electrical activity of hippocampal neurons using tetrodotoxin (TTX) does not prevent the formation of GABA receptor clusters (Craig et al., 1994). However, TTX does not block spontaneous synaptic release. Therefore, it is still possible that small amounts of GABA released at presynaptic sites may be sufficient to trigger receptor clustering. In a different experimental paradigm using TTX on cultured cortical neurons, blocking activity resulted in a ∼50% decrease in the number of synaptic sites with detectable GABAA receptors (Kilman et al., 2002). To evaluate the role of GABA in the formation of GABAergic synapses in vivo, we analyzed the formation of GABAergic neuromuscular junctions in the nematode Caenorhabditis elegans.
The GABA nervous system of C. elegans is composed of 26 neurons, 23 of which are inhibitory motor neurons that innervate body wall muscles (McIntire et al., 1993b). Muscle cells in C. elegans are mononucleate and send dendrite-like extensions, called muscle arms, to the axons of the motor neurons in the ventral and dorsal nerve cords (White et al., 1984). At the nerve cord, muscle arms elongate and form en passant synapses with motor axons. Muscle cells receive innervation from both cholinergic motor neurons that cause muscle contraction and GABAergic neurons that cause muscle relaxation (for review, see Jorgensen and Nonet, 1995). Thus, the muscles of the worm represent a simple system to study multi-innervation and receptor sorting.
Jin et al. (1999) demonstrated that presynaptic differentiation of GABAergic neuromuscular junctions is normal in the absence of GABA. In fact, GABAergic transmission is dispensable in C. elegans, and null mutations in unc-25, the single gene that encodes the glutamic acid decarboxylase, the biosynthetic enzyme for GABA, cause the animals to become uncoordinated, although they are fully viable (Jin et al., 1999). These data suggested that the number of synapses was not regulated by activity-dependent feedback from the muscle. However, these experiments could not evaluate whether presynaptic contacts or GABA secretion were involved in clustering receptors on the muscle. In this study, we analyze the differentiation of the postsynaptic domain of GABAergic neuromuscular junctions by visualizing GABA receptors in muscle. The subunits that comprise the GABAA-type receptor expressed in the body muscles are generated by alternative splicing from a single locus calledunc-49 (Bamber et al., 1999). Our results demonstrate that innervation from motor neurons is necessary to induce and maintain UNC-49 clusters. Moreover, differentiation of GABA receptor clusters requires transport of vesicular components to the synapse. However, GABA neurotransmission per se is dispensable for the formation of GABA receptor clusters.
Materials and Methods
C. elegans handling. Worms were cultured using standard methods (Brenner, 1974). L1 and L2 worms were obtained by moving gravid adults every 60 min to a fresh plate at 25°C. Embryos and larvae were cultured at 25°C. Cohorts of synchronized worms were obtained by harvesting the larvae at defined time points (12–22 hr after egg laying to isolate L1 larvae; 23–28 hr for L2 larvae).
The wild strain N2 and the following mutant alleles were used:unc-49(e407), lin-6(e1466),unc-25(e156), unc-5(e152), unc-5(e53),unc-104(e1265), unc-31(e928),unc-13(e51), and unc-18(e81).
UNC-49 antibodies. A DNA fragment encoding UNC-49 amino acids 23–67 was inserted into pGEX-3X (Amersham Biosciences, Uppsala, Sweden). The glutathione-S-transferase (GST)–UNC-49 fusion protein was expressed in Escherichia coli and purified. Rabbits were injected with 100 μg of fusion protein and boosted three times with 100 μg each. Antibodies were then purified as described previously (Miller et al., 2000) using the fusion protein GST–UNC-49 blotted on nitrocellulose.
Immunofluorescence staining. Worms were permeabilized using freeze-fracture (Duerr et al., 1999) and then fixed in −20°C methanol for 5 min and in −20°C acetone for 5 min. Samples were blocked for 1 hr at room temperature with 0.2% bovine gelatin. Purified UNC-49 antibodies were diluted 1:300 and incubated overnight at 4°C, and the secondary antibody Cy3-labeled goat anti-rabbit IgG (heavy and light) (Jackson ImmunoResearch, West Grove, PA) was diluted 1:900 and incubated 2.5–3 hr at room temperature. Worms were mounted in Vectashield medium (Biovalley, Marne-la-Vallee, France) on slides and imaged using fluorescence microscopy.
Molecular biology. In UNC-49B–GFP (green fluorescent protein), GFP is inserted in frame in the M3–M4 cytoplasmic loop of the UNC-49B subunit in a genomic fragment that contains all three GABA receptor subunit open reading frames plus 400 nt of theunc-49 promoter (Bamber et al., 1999). In this construct, we replaced GFP with YFP (yellow fluorescent protein) to generate pJL41. To construct pJL37, a 1.1 kb promoter fragment ofunc-47 (nucleotides −1195 to −3 relative to the translation start site) was inserted upstream to the translational fusion SNB-1 (synaptobrevin-1)–GFP derived from pSB120.65 (gift from M. Nonet, St. Louis, MO) (Nonet, 1999). The fragment encoding amino acids 58–225 in the GFP was replaced by the homologous CFP (cyan fluorescent protein) coding sequence PCR-amplified from the cameleon 2 cDNA (Miyawaki et al., 1997).
C. elegans transformation. Transformation was performed by microinjection of plasmid DNA into the gonad (Mello et al., 1991).unc-49(e407);lin-15(n765ts) worms were injected with a DNA mixture containing pJL37 (Punc-47:: snb-1–CFP) (20 ng/μl), a gel-purified AflII–MluI fragment of pJL41 (unc-49B–YFP missing the 5′ end of the gene) (20 ng/μl) and a 110/38 unc-49 PCR fragment (10 ng/μl) containing a 4 kb promoter fragment plus 2 kb of the 5′ coding sequence, as described by Bamber et al. (1999). pEKL15, a plasmid that contains the wild-type lin-15 gene (Clark et al., 1994), was coinjected at 40 ng/μl as a cotransformation marker. Progeny of injected animals were raised at the restrictive temperature forlin-15(n765ts). A rescued (non-Lin non-Unc) strain (oxEx171) was generated. This array was integrated by UV-4,5′,8-trimethylpsoralen exposure (Yandell et al., 1994) to generate the stable transgenic marker krIs1.
Microscopy. Animals were anesthetized with M9 buffer containing (in mm): 3.8 tricaine, 0.42 tetramisole, and 20 sodium azide; mounted on 1× M9, 2% agarose pads; and examined using a Leica (Nussloch, Germany) SP2 confocal microscope equipped for sequential scan of the emission lines. CFP was detected by using the 458 nm line, and YFP was excited by the 514 nm line. Confocal image reconstruction and merges were obtained with Metamorph image analysis system. Quantitative analysis of immunofluorescence was performed on confocal image reconstructions using Metamorph. For each image, background fluorescence level was based on fluorescence intensities outside of the nerve cord and used as a threshold to measure GABA receptor cluster size. Average fluorescence density per cluster was quantified by centering over each cluster a circle of 0.62 μm diameter in size and integrating the total fluorescence in this area.
Results
GABA receptors form postsynaptic clusters at neuromuscular junctions
The distribution of GABA neuromuscular junctions was initially described in C. elegans using electron microscopy (White et al., 1976, 1984). Synapses were identified on the basis of the accumulation of synaptic vesicles and the presence of an electron-dense presynaptic specialization at the membrane juxtaposed to the muscle. However, it was not possible to observe electron-dense postsynaptic densities. To test whether GABA receptors accumulated in specialized domains or were distributed more loosely in the muscle membranes, we analyzed the distribution of the UNC-49 GABA receptor by immunofluorescence. An antibody was raised against the N-terminal region of the UNC-49 receptor. This fragment is present in all predicted UNC-49 subunits that are generated by alternative splicing from the unc-49 gene (Bamber et al., 1999). Antibody staining of wild-type animals generated a punctate pattern along the ventral and dorsal nerve cords and two large spots in the nerve ring where the muscle cells in the head are innervated (Fig.1A). These puncta were absent in unc-49(e407) null mutants; however, some nonspecific staining was seen in the pharynx and above the preanal ganglia in the null mutant (data not shown). At high magnification, GABA receptor clusters could be visualized as individual spots measuring ∼0.7 μm in diameter (Fig. 1B). Between clusters, weak diffuse staining was detected along the cord, and this staining was absent in unc-49(e407) mutants. Because muscle cells were permeabilized during the staining procedure, we do not know whether these receptors were inserted in the plasma membrane or were part of an intracellular pool. No UNC-49 labeling could be detected in body wall muscle cell somas or in muscle arms. Therefore, UNC-49 antibody staining demonstrated that muscle GABA receptors are clustered in discrete domains along the nerve cords.
Fig. 1.
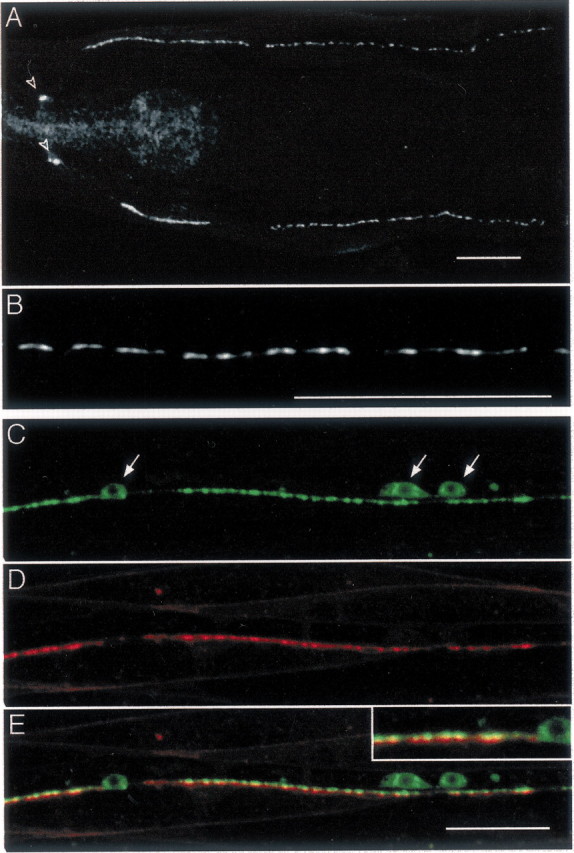
UNC-49 GABA receptors form postsynaptic clusters at neuromuscular junctions. A, Immunostaining of UNC-49. Young wild-type adults were immunolabeled with rabbit anti-UNC-49 antibody. UNC-49 receptors are present in the nerve ring (open arrowheads) and along ventral and dorsal nerve cords. In thispanel, as well as in all following figures, anterior is to the left and ventral is down.B, High magnification of the ventral nerve cord reveals that UNC-49 clusters into individual puncta. C, Visualization of GABAergic varicosities in the ventral nerve cord of a transgenic worm expressing in GABAergic neurons the synaptic vesicle protein synaptobrevin SNB-1 fused to CFP. Note that some SNB-1–CFP accumulates in motor neuron soma (arrows).D, In the same transgenic animal, muscle GABA receptors are visualized by expressing UNC-49 fused to YFP. UNC-49–YFP fluorescence is detected in muscle cells in a punctate pattern along the nerve cord. Note that some weak UNC-49–YFP signal outlines muscle cell membranes. E, Merging SNB-1–CFP and UNC-49–YFP signals shows that UNC-49 clusters are localized opposite GABA release sites (inset, detail of ventral cord labeling). Scale bars, 20 μm (for all images).
To evaluate whether GABA receptor clusters were juxtaposed to GABA synapses, we visualized presynaptic and postsynaptic domains in the same living animal. Presynaptic terminals were visualized using the synaptic vesicle protein synaptobrevin, also called VAMP (vesicle-associated membrane protein), tagged with the GFP variant CFP, and expressed in GABA neurons using the promoter of the vesicular GABA transporter gene, unc-47 (McIntire et al., 1997) (Fig. 1C). Postsynaptic GABA receptor fields were visualized by tagging the UNC-49B subunit with the GFP variant YFP. Expression of the UNC-49B–YFP subunit from a modifiedunc-49 genomic fragment (see Materials and Methods) was able to rescue the uncoordinated phenotype of the unc-49(e407)mutant. YFP fluorescence was detected in the nerve ring and along the ventral and dorsal cords with a punctate pattern (Fig.1D) very similar to what was observed with anti-UNC-49 antibody staining. Transgenic worms expressing both SNB-1–CFP and UNC-49–YFP were analyzed by confocal microscopy using sequential excitation to avoid bleed-through between CFP and YFP signals. After merging CFP and YFP images, we observed a one-to-one association between presynaptic and postsynaptic clusters in the cords and in the nerve ring. (Fig. 1E). These data indicate that GABA receptors form high-density clusters opposite presynaptic GABA release sites at neuromuscular junctions.
GABAergic innervation precedes GABA receptor clustering
Does innervation have a role in GABA receptor clustering? In the simplest model, motor neuron innervation causes the GABA receptors to cluster. Alternatively, clusters could be prepatterned on the surface of the muscle. In this case, the muscle expresses positional cues that drive the formation of synapses at specific positions. Such a mechanism is used in certain neuromuscular junctions in vertebrate muscles where acetylcholine receptors are distributed along a predetermined pattern (Yang et al., 2000, 2001; Lin et al., 2001). To address this question, we analyzed the temporal relationship of presynaptic and postsynaptic components during development.
In L1 larvae, the GABA DD motor neurons innervate the ventral body wall muscles, but the dorsal muscles do not receive GABAergic innervation. At the end of the L1 larval stage, the DD motor neurons rewire and innervate dorsal muscles (White et al., 1978; Hallam and Jin, 1998). Concurrently, the postembryonic GABA motor neurons, the VDs, are generated and innervate the ventral muscles. Clusters of GABA receptors were detected in the ventral muscles of L1 larvae by antibody staining of the wild type (Fig.2A,arrowheads) or by fluorescence visualization of the YFP-tagged receptor (Fig. 2B). These synapses were formed in a highly reproducible pattern (n = 100 animals) with a gap containing no synapses in the segment of the axon just anterior to each cell body. No GABA receptors were detected in the dorsal muscles of L1 larvae (Fig. 2B,C). Early in the L2 larval stage (23–24 hr after egg laying at 25°C), synaptic vesicles were detected in the dorsal cord in 100% of the individuals; however, UNC-49–YFP was detected in only 39% of the animals (n = 23) (Fig. 2D). At 24–25 hr after egg laying, 75% of the animals contained dorsal UNC-49–YFP aggregates (n = 20) that aligned with synaptic vesicle clusters. Finally, at 26 hr after egg laying, 100% of the L2 larvae (n = 23) had an adult staining pattern with colocalized SNB-1–CFP and UNC-49–YFP clusters along the entire dorsal cord (Fig.2E). UNC-49–YFP was never observed as a diffuse signal before innervation. At the onset of detection, YFP-tagged GABA receptors formed small puncta that became brighter and bigger over time. Thus, the appearance of synaptic vesicles at presynaptic sites precedes the formation of postsynaptic receptor clusters in the dorsal nerve cord.
Fig. 2.
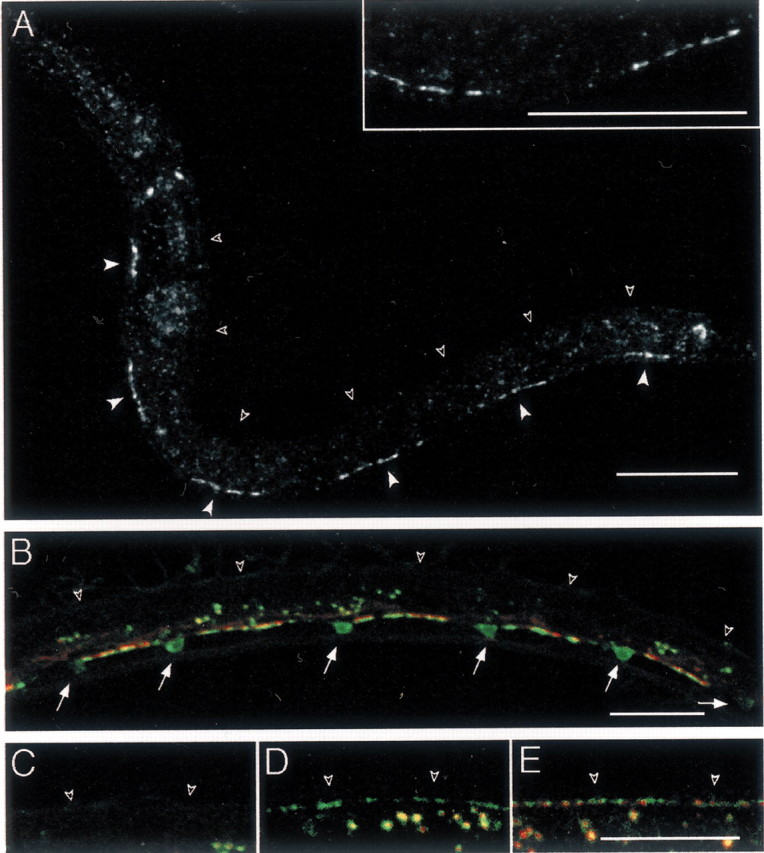
GABA receptor clusters are present only at sites of innervation in L1 larvae. A, Immunostaining of UNC-49 in wild-type L1 larvae. L1 larvae were immunolabeled 4 hr after hatching at 25°C. At this stage, UNC-49 was found ventrally as six segments (closed arrowheads) interrupted by gaps; however, no staining could be observed in the dorsal nerve cord (open arrowheads). Inset shows a higher magnification of the ventral nerve cord revealing that UNC-49 receptors form clusters. B, SNB-1–CFP (in green) expressed in GABAergic neurons stains synaptic vesicle clusters present along the ventral nerve cord and the soma of the six GABAergic DD motor neurons present in young L1 larvae (arrows). Clusters of UNC-49–YFP (red) are detected only along the ventral nerve cord. Open arrowheads indicate dorsal cord position. The fluorescence in the middle of the animal corresponds to autofluorescent gut granules. C–E, Dorsal cord labeling (open arrowheads) in representative larvae grown at 25°C after egg laying for 16 hr (L1 larval stage) (C), 24 hr (early L2 stage) (D), and 26 hr (L2 larva) (E). Presynaptic vesicles (labeled with SNB-1–CFP in green) are detected before UNC-49–YFP receptor clusters (red). Scale bars, 20 μm (for all images).
However, the time required to fold CFP and YFP may affect the interpretation of our results. Specifically, SNB-1–CFP is already folded in synaptic vesicles of the ventral nerve cord. The vesicles simply need to be transported to the dorsal nerve cord. In contrast, UNC-49–YFP needs to be folded de novo, because no staining is apparent before the appearance of clusters. Therefore, we analyzed the appearance of UNC-49 clusters by immunostaining. These data confirm the results obtained using YFP-tagged receptors. During the early L2 larval stage (23–24 hr after egg laying at 25°C), UNC-49 immunoreactivity was detected only at the anterior end of the dorsal nerve cord. At 25–26 hr after egg laying, labeling of the receptor was intense along the entire dorsal cord (data not shown). These results suggest that GABA receptor aggregates are not assembled before innervation but appear only after the motor neurons have differentiated presynaptic specializations.
Innervation is required for the maintenance of GABA receptor clusters
Time course analysis of UNC-49 clusters suggests that presynaptic innervation triggers the differentiation of postsynaptic domains. To address whether innervation is required for maintenance of receptor clusters, we analyzed the distribution of UNC-49 in alin-6(e1466) background. In lin-6 mutants, most of the postembryonic cell divisions are aborted (Sulston and Horvitz, 1981). DD neurons are generated normally during embryonic development in these mutants, and they rewire properly at the L1-L2 transition (White et al., 1978). However, VD motor neurons do not differentiate in L1 larvae. As a result, ventral muscles are innervated in the L1 but lose all innervation in the L2 stage.
In L1 lin-6 mutant larvae, presynaptic and postsynaptic staining was present in the ventral cord and was indistinguishable from wild-type L1 larvae (data not shown). In lin-6(e1466) L4 larvae, SNB-1–CFP was detected only in the dorsal cord, confirming that DDs rewired properly, and, as expected, no GABA innervation was observed ventrally (Fig. 3A). UNC-49–YFP clusters colocalized with presynaptic varicosities along the dorsal cord (Fig. 3A,B). However, in ventral muscle cells, UNC-49–YFP clusters were absent, and only scattered YFP fluorescence was detected in some cells (Fig. 3B). These results suggest that innervation is required not only for the formation but also for the maintenance of GABA receptor clusters.
Fig. 3.

Innervation is required for the maintenance of UNC-49 clusters. A, SNB-1–CFP expressed in the GABA nervous system of lin-6(e1466) L4 larvae. In this mutant, the GABAergic ventral motor neurons are absent, and no synaptic varicosities are observed along the ventral cord (closed arrowheads). The fluorescence in the middle of the animal corresponds to autofluorescent gut granules. B, Localization of UNC-49–YFP in the same individual, showing a disorganization of UNC-49–YFP expression pattern ventrally (closed arrowheads) while dorsally presynaptic and postsynaptic markers appear wild type (open arrowheads). Scale bar, 20 μm.
Presynaptic innervation is sufficient to specify the site of GABA receptor clustering
Innervation is required for the clustering of GABA receptors; however, it is not clear whether it is sufficient. For example, commissures from the GABA motor neurons first contact the muscles on the lateral edges and grow underneath them (Knobel et al., 1999). Despite this extensive region of contact between the motor neuron and its target, synapses and GABA receptor clusters do not form along these segments. It is possible that a permissive environment for receptor clustering is found only along the dorsal midline, perhaps because of signals from the nerve cord or hypodermal ridge. To determine whether synaptic innervation alone is sufficient to trigger GABA receptor clustering, we analyzed the distribution of GABA receptors inunc-5 mutants.
unc-5 encodes a netrin receptor required for the dorsal migration of the GABA motor neuron axons (Hedgecock et al., 1990). Inunc-5 loss-of-function mutants, dorsal motor neuron axons do not reach the dorsal nerve cord but frequently extend along the lateral side of the animal. Dorsal muscle cells do not extend muscle arms to the dorsal midline but sometimes extend muscle arms ventrally to contact ectopic axons. In the same way, some ventral muscles extend arms dorsally to contact nearby sublateral axons. Electron microscopy identified presynaptic specializations at contacts between ectopic axons and muscle arms (Hedgecock et al., 1990). To assess whether clustering of GABA receptors could occur outside of the ventral and dorsal midlines, we visualized tagged GABA receptors inunc-5 mutant animals. In unc-5(e152) mutants, synaptic vesicle clusters and GABA receptor clusters were punctate along the ventral cord as in wild-type animals (Fig.4). Only a few synaptic vesicle clusters were detected along the dorsal midline, because most motor neurons fail to extend to the dorsal cord; however, synaptic vesicle clusters were found along the lateral side of these animals. The space between clusters was highly variable, as was the shape and size of individual clusters. Ectopic muscle arms could be faintly detected extending ventrally from dorsal muscles toward lateral axons (Fig.4B, inset). At the tip of these arms, UNC-49 receptors formed clusters. Merging of SNB-1–CFP and UNC-49–YFP images revealed that UNC-49 aggregates at ectopic presynaptic varicosities (Fig. 4C). These results indicated that interactions between motor neuron axons and muscles are sufficient to cause GABA receptor clustering, although the size and spacing of GABA neuromuscular junctions might be influenced by additional regulation at the nerve cord.
Fig. 4.
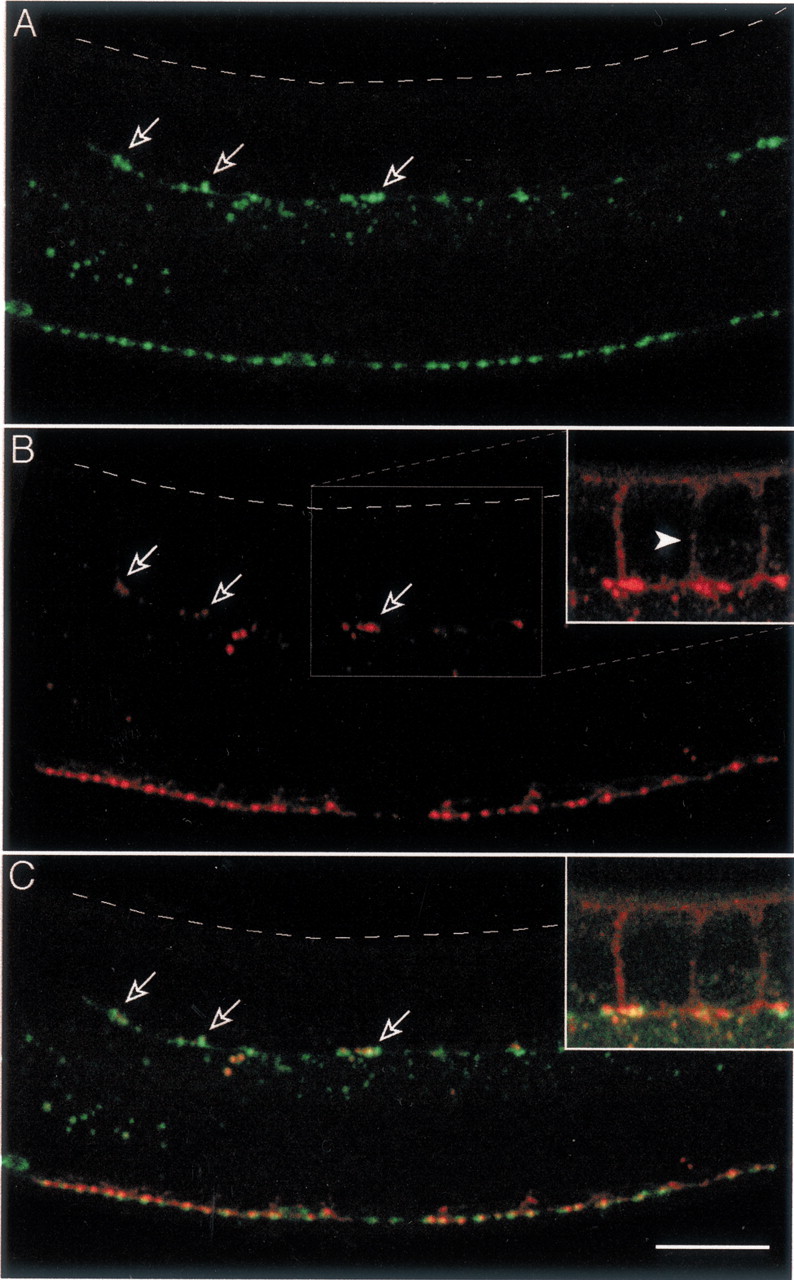
GABA receptors cluster at ectopic GABA release sites in unc-5 mutants. A, Localization of SNB-1–CFP expressed in the GABAergic system ofunc-5(e152) adult mutant. The migration of motor neuron axons to the dorsal cord is disrupted. Presynaptic varicosities are observed in ectopic axons located along the lateral side of the body (open arrows). The dashed line indicates the dorsal side of the worm. B, Localization of UNC-49–YFP expressed in the same individual. UNC-49–YFP is found along the ventral cord and at ectopic lateral positions (open arrows). Inset shows a high amplification of the UNC-49–YFP signal, which enables the visualization of ectopic muscle arms extending ventrally from dorsal muscles (arrowhead). C, Merging SNB-1–CFP and UNC-49–YFP signals demonstrates that lateral clusters of UNC-49–YFP are colocalized with ectopic presynaptic varicosities (inset as in B). Scale bar, 20 μm.
Receptor clustering is triggered by a factor transported by the neuronal kinesin UNC-104
Our results demonstrate that formation of UNC-49 clusters requires innervation. These data suggest that a presynaptic factor is instructing the differentiation of postsynaptic domains in muscle. One possibility is that this factor is released from synaptic vesicles. To test this hypothesis, we analyzed GABA receptor clusters inunc-104 mutants. unc-104 encodes a neuronal kinesin required for synaptic vesicle transport (Hall and Hedgecock, 1991; Zhou et al., 2001). In partial loss-of-function mutants such asunc-104(e1265), axonal outgrowth is normal but most synaptic vesicles remain trapped in neuron cell bodies, resulting in a complete absence of synaptic vesicles in the dorsal nerve cord as determined by electron microscopy (Hall and Hedgecock, 1991). Consistent with these data, we could not detect any SNB-1–CFP in the dorsal nerve cord ofunc-104(e1265) mutants (Fig.5A). Instead, fluorescence appeared concentrated in motor neuron cell bodies, and only some weak vesicular staining was observed along the ventral neurites.
Fig. 5.
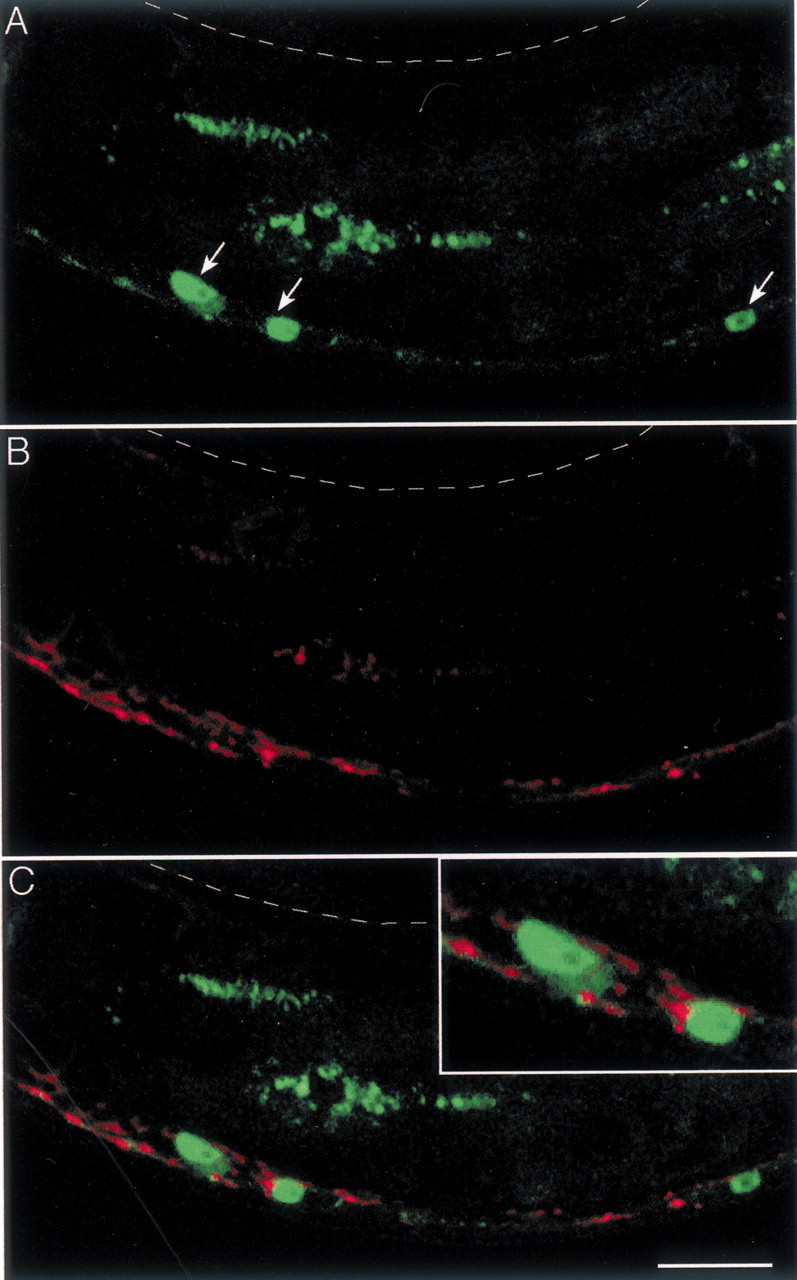
The neuronal kinesin UNC-104 is required for the differentiation of both presynaptic and postsynaptic domains.A, Expression of SNB-1–CFP in the GABA neurons of an adult unc-104(e1265) mutant. Synaptic vesicles are highly concentrated in the cell bodies of motor neurons (arrows); few synaptic vesicles are present along the ventral cord, but no SNB-1–CFP is detected in the dorsal cord (indicated by the dashed line). B, Localization of UNC-49–YFP in the same animal. C, The overlay of the CFP and YFP images shows that UNC-49–YFP receptors cluster in contact with the soma of the motor neurons (inset) and also form large aggregates in the ventral nerve cord, mostly at places where residual synaptic vesicles are detected. Scale bar, 20 μm.
In these animals, a faint and diffuse UNC-49–YFP signal was observed in dorsal muscle cells, but no clusters were present along the dorsal cord (Fig. 5B). Furthermore, along the ventral side of theunc-104(e1265) mutants, unusually large and irregular UNC-49–YFP aggregates were found along the ventral cord where residual numbers of synaptic vesicles accumulated (Fig. 5C). UNC-49–YFP clusters were also found directly in contact with the cell bodies of the GABA motor neurons (Fig. 5C,inset). These data suggest that, in a wild-type animal, a factor required for clustering of GABA receptors is transported from the cell bodies to synaptic sites by the neuronal kinesin UNC-104.
GABA is not necessary for the differentiation of postsynaptic domains
The most obvious candidate for a signaling molecule carried by synaptic vesicles is the neurotransmitter itself, GABA. GABA is synthesized by glutamic acid decarboxylase, which is encoded by a single gene, unc-25. Using tagged synaptic vesicle markers and electron microscopy, Jin et al. (1999) demonstrated previously that the distribution and density of presynaptic contacts to muscles were normal in unc-25 mutants. Similarly, we observed a normal distribution of the presynaptic marker SNB-1–CFP inunc-25(e156) mutants (Fig.6A). To test whether GABA is required to signal the differentiation of postsynaptic domains, we determined the distribution of GABA receptors in anunc-25-null mutant.
Fig. 6.
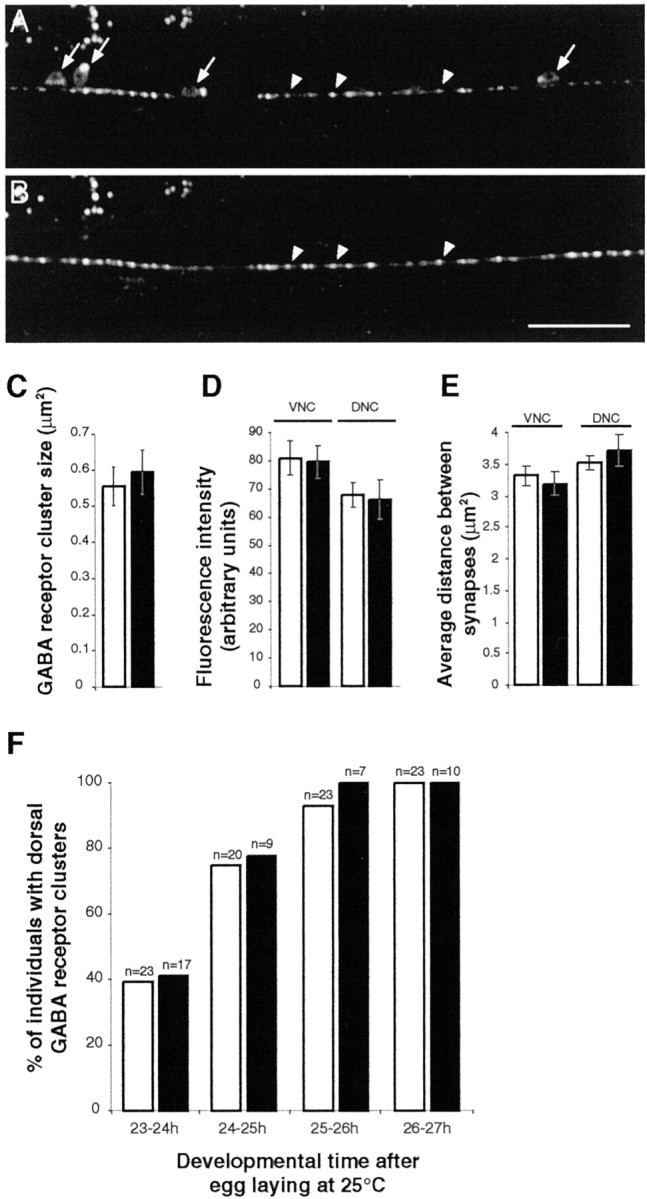
UNC-49 GABA receptor clustering is normal in the absence of GABA. A, SNB-1–CFP expression in GABAergic neurons of the ventral cord in an adult unc-25 mutant (arrows, motor neuron soma; arrowheads, synaptic varicosities). unc-25 encodes GAD, the biosynthetic enzyme of GABA, and in the mutant unc-25(e156), no GABA is detected (McIntire et al., 1993a). B, Localization of UNC-49–YFP in the ventral cord of the same animal. Presynaptic vesicle aggregates and GABA receptor clusters are colocalized in a pattern indistinguishable from what is observed in wild-type animals (arrowheads). Scale bar, 20 μm. C, UNC-49–YFP cluster size in the dorsal cord of wild-type (white) and unc-25(e156)(black) animals. Wild type, 0.56 ± 0.05 μm2 (average ± SEM; n = 3 worms; 95 clusters); unc-25, 0.59 ± 0.06 μm2 (n = 3 worms; 74 clusters). D, Fluorescence intensity of UNC-49–YFP clusters in wild type (white) andunc-25(e156) (black). Average ± SEM are, respectively, 80.96 ± 6.16 (n = 4 worms; 152 clusters) and 79.56 ± 5.71 (n = 8; 362) in the ventral nerve cord (VNC) and 67.85 ± 4.40 (n = 4; 100) and 66.17 ± 6.89 (n = 6; 182) in the dorsal nerve cord (DNC). E, Synapse densities in wild type (white) andunc-25(e156) (black). Average spacing ± SEM between clusters are, respectively, 3.19 ± 0.18 μm (n = 18 worms) and 3.31 ± 0.15 μm (n = 16) in ventral nerve cord (VNC) and 3.72 ± 0.26 μm (n = 15) and 3.52 ± 0.12 μm (n = 18) in dorsal nerve cord (DNC). F, Time course analysis of GABA receptor cluster formation in the dorsal cord of N2 (white) and unc-25(e156)(black) L2 larvae. As early as 23–24 hr after egg laying, synaptic varicosities are detected in 100% of L2 larvae, but the detection of GABA receptor clusters is delayed. UNC-49–YFP clusters are detected with a similar time course in wild-type (white) and unc-25(e156)(black) animals.
In adult unc-25(e156) animals, GABA receptors were clustered into puncta, and these puncta were aligned with presynaptic vesicle clusters (Fig. 6A,B). The size of the clusters and the average fluorescence intensity per cluster were similar inunc-25 mutants and in wild type (Fig. 6C,D). Furthermore, synapse densities along the ventral and dorsal nerve cords were identical in unc-25 and wild-type young adults (Fig.6E). Because GABA could play a role in the early formation of GABA receptor clusters, we performed a time course analysis of UNC-49–YFP expression in unc-25 mutants. No difference was observed between wild-type and unc-25 mutants during the formation of GABAergic synapses on dorsal body wall muscles (Fig. 6F). These results demonstrate that the neurotransmitter GABA is not required for the clustering of GABA receptors in C. elegans.
Discussion
In this study, we characterized the interactions between GABAergic inhibitory motor neurons and body muscle cells during synapse formation in C. elegans. We found that the formation of GABA receptor clusters followed the appearance of synaptic vesicles at presynaptic sites, suggesting that the formation of postsynaptic GABA receptor clusters is instructed by presynaptic inputs. Signaling from the presynaptic terminal is dependent on the neuronal kinesin UNC-104. However, the neurotransmitter GABA is not required for the formation of GABA synapses. These results suggest that synapse formation does not depend on neurotransmission during the initial wiring of the nervous system.
The differentiation of GABAergic neuromuscular junctions results from bidirectional interactions between nerve and muscle
Morphological analysis of inhibitory neuromuscular junctions indicates that neurotransmitter release sites and receptor fields are tightly aligned. Using fluorescently tagged proteins in living specimens, we observed a one-to-one codistribution between synaptic vesicle aggregates and GABA receptor clusters. At the ultrastructural level, 95% of the synaptic vesicles are contained within an area of 0.5 μm centered around the active zone (Crump et al., 2001). As revealed by UNC-49 antibody staining, GABA receptors are contained in clusters with an apparent diameter of 0.7 μm. Because the resolution of light microscopy overestimates the actual size of the clusters, these results suggest that receptor fields closely match neurotransmitter release sites.
How is the differentiation of two congruent subcellular domains in motor neurons and in body wall muscle cells achieved? Previous work demonstrated that presynaptic differentiation requires a signal from the muscle. When muscle cell precursors are laser ablated in theC. elegans embryo so that dorsal muscle cells are completely absent, neural processes still extend dorsally, but no presynaptic varicosity can be detected using anti-GABA immunostaining (Plunkett et al., 1996). Thus, the development of presynaptic release sites depends on inputs from the muscle. Our data indicate that the formation of the postsynaptic domain relies on a signal emanating from the motor neuron. First, presynaptic varicosities start to differentiate before the postsynaptic compartment. Synaptic vesicles tagged with SNB-1–CFP were detected before the appearance of UNC-49–YFP receptor clusters. Second, loss of innervation to the ventral muscles disrupts the maintenance of GABA receptor clusters, indicating that continuous input from the motor neurons is required to maintain receptor clusters. Third, displacing synaptic vesicle release sites to ectopic positions causes receptor clusters to form at these locations. Specifically, inunc-5 mutants, ectopic motor axons that run along the lateral hypoderm are intercepted by muscle arms, and receptor clusters form at these lateral positions. More dramatically, in mutants that lack the synaptic vesicle kinesin UNC-104, synaptic vesicles remain in the cell body of the motor neuron, and GABA receptors are found clustered against the motor neuron soma. This cannot be explained by a primary defect of GABA receptor trafficking in muscle ofunc-104, because unc-104 gene expression is detected only in neurons, and neuronal expression of an UNC-104–GFP fusion protein rescues the UNC-104 phenotype (Zhou et al., 2001). Interestingly, proteins of the presynaptic active zone are properly localized in unc-104 mutants. Specifically, liprin/SYD-2 (Zhen and Jin, 1999), Rab-3 interacting molecule/UNC-10 (Koushika et al., 2001), and RPM-1 (Schaefer et al., 2000; Zhen et al., 2000) are correctly localized in the dorsal cord; however, these incomplete synapses cannot direct the clustering of GABA receptors. Therefore, sites of presynaptic differentiation can be specified in the motor neuron independently from the formation of synaptic vesicle aggregates. However, GABA receptor clusters will differentiate only after synaptic vesicles have reached presynaptic sites.
The formation of GABAergic neuromuscular junctions does not depend on GABAergic neurotransmission
Because the density of receptors present on the postsynaptic membrane is a critical parameter of synaptic strength, it would be reasonable to predict that GABA release directly controls the formation of postsynaptic GABA receptor clusters. In one model, GABAergic neurotransmission would be required for the differentiation or the maintenance of the postsynaptic domains. Disrupting GABAergic transmission would therefore cause a loss of GABA receptor clusters on the muscle membrane. In another model, homeostatic control of receptor number could be achieved by neurotransmission. In this model, the loss of neurotransmission would cause an increase in postsynaptic GABA receptors. These hypotheses could be tested using mutants with impaired GABAergic neurotransmission. Our results indicate that the morphological differentiation of GABAergic synapses is independent of neurotransmission.
First, presynaptic varicosities and GABA receptor clusters differentiate normally in mutants in which evoked synaptic vesicle release is absent. UNC-49–GFP was shown previously to be properly clustered in unc-13 mutants (Richmond et al., 1999). Using double-transgenic worms expressing SNB-1–CFP and UNC-49–YFP, we confirmed that GABA receptor clusters were morphologically normal and were opposite GABA release sites in unc-13 mutants (data not shown). Interestingly, electrophysiological recording from muscle cell indicates that the response to exogenous application of GABA is normal in unc-13 mutants (Richmond et al., 1999), demonstrating that GABA receptors are inserted in the plasma membrane and functional. Similarly, we observed normal differentiation of presynaptic and postsynaptic compartments at GABAergic neuromuscular junctions ofunc-18 mutants that are also defective in synaptic release (data not shown). However, electrophysiological analysis ofunc-13 and unc-18 mutants indicates that spontaneous release of synaptic vesicles occurs at very low frequency (Richmond et al., 1999; J. Richmond, personal communication). Because it was possible that low amounts of neurotransmitter could be enough to initiate the differentiation of a postsynaptic domain, we used a mutant that lacked GABA altogether. unc-25 encodes the biosynthetic enzyme for GABA, glutamic acid decarboxylase. Presynaptic differentiation of GABAergic synapses is normal in the absence of GABA (Jin et al., 1999). Our data indicate that postsynaptic differentiation (i.e., the clustering of GABA receptors) is normal in the absence of GABA as well. Even in 4-d-oldunc-25 adult mutants, presynaptic and postsynaptic specializations could still be detected despite the absence of GABAergic neurotransmission (data not shown). Therefore, differentiation of GABAergic neuromuscular junctions occurs independently of GABA neurotransmission.
Synaptogenesis: a neurotransmission-independent process?
Is synapse formation independent of neurotransmission in other systems and species? The roles of neurotransmission and electrical activity during synaptogenesis have been investigated extensively at the Drosophila neuromuscular junction. At these synapses, the formation of glutamate receptor fields is dependent on presynaptic innervation (Broadie and Bate, 1993; Saitoe et al., 1997). Moreover, postsynaptic activity regulates the release of the neurotransmitter via retrograde signaling (for review, see Featherstone and Broadie, 2000;Keshishian, 2002). Whether neurotransmission per se is required for the differentiation of postsynaptic domains remains controversial. It was initially reported that Drosophila mutantssyntaxin-1A and shibire, which lack spontaneous neurotransmitter release, also fail to cluster receptors at neuromuscular junctions (Saitoe et al., 2001). However, recent data suggest that, in syntaxin and shibire mutants, many muscles are not properly innervated despite the penetration of the nerve into the musculature. In these mutant flies, noninnervated muscles lack postsynaptic receptor clusters, but whenever a presynaptic contact is made, glutamate receptor fields are indistinguishable from wild-type individuals (Featherstone et al., 2002). These results would suggest that, at Drosophila neuromuscular junctions too, neurotransmission is not required for synaptogenesis.
Recent analysis of mouse knock-outs with impaired synaptic vesicle release suggests that neurotransmission is dispensable for the initial formation of the mammalian brain. synaptobrevin 2 (Schoch et al., 2001) or SNAP-25 (Washbourne et al., 2002) homozygous mutants exhibit a total block of evoked release of synaptic vesicles but retain spontaneous vesicle release. These mutants die at birth, but the overall structure of the fetal brain is normal and synapses differentiate properly. Deletion of Munc18–1 abolishes both evoked and spontaneous vesicular release. In these mutants, initial development of the brain is normal, but neurons undergo apoptosis after initial brain assembly is completed (Verhage et al., 2000). Apoptosis is probably not caused by the lack of neurotransmission, because Munc13–1/2 mutants also have a complete block of vesicular release but develop normally up to birth. In Munc13–1/2 mutant brain, normal synapses are observed by electron microscopy, and in primary cultures of hippocampal neurons, synapses differentiate and persist at normal density in culture (Varoqueaux et al., 2002). Specifically, GABA receptors are expressed and functional in these cells on the basis of electrophysiological experiments, suggesting that neurotransmission is dispensable for the initial steps of synapse formation.
This question has been best studied at the mouse neuromuscular junction using choline acetyltransferase knock-out mice, which do not synthesize the neurotransmitter ACh (Misgeld et al., 2002). Despite several abnormalities of the neuromuscular system, many of which can be related to muscle inactivity, the differentiation of the neuromuscular junction per se is preserved. Large ACh receptor clusters are present in front of well differentiated synaptic varicosities. Such structures are completely missing in agrin andMusk knock-out mice (DeChiara et al., 1996; Burgess et al., 1999) demonstrating that the neurotransmitter is not the key player of synapse formation at the vertebrate neuromuscular junction.
The role of GABA during brain development has been addressed using mice knock-outs that are deficient for the two GAD isoforms and do not contain detectable levels of GABA. Animals die at birth because of cleft palate, but histological analysis of GABAergic neuron populations does not reveal any gross morphological defect (Ji et al., 1999). Unfortunately, detailed analysis of the few existing GABA synapses present in newborn animals has not been reported to our knowledge.
What is the GABA receptor clustering factor?
Our data indicate that GABA receptor clustering activity is transported to and delivered at the presynaptic terminus. It is cotransported with synaptic vesicles but is not the neurotransmitter GABA. The GABA receptor clustering factor could be another intravesicular component such as a neuropeptide. unc-31encodes the homolog of CAPS (Ca2+-dependent activator protein for secretion) (Avery et al., 1993), which is necessary for dense core vesicle release in adrenal cells (Berwin et al., 1998) and inDrosophila neurons (Renden et al., 2001). We observed normal GABA receptor clusters in the mutant unc-31(e928) (data not shown). However, we cannot rule out that residual release of neuropeptide persists in this mutant. Alternatively, the GABA receptor aggregating factor could be a nonvesicular protein transported by the kinesin UNC-104. For example, it might be cotransported in the “synaptic packets” during development; these large complexes contain synaptic vesicles together with proteins of the presynaptic domains such as synapsin, amphyphisin, and the calcium channel subunit α1 (Ahmari et al., 2000). Distinguishing between these possibilities will require the identification of the GABA receptor clustering activity.
Footnotes
This work was funded by the Institut National de la Santé et de la Recherche Médicale, the Ministère de la Recherche (Action Concertée Incitative Jeunes Chercheurs 5016 CDR 5), and the Fondation pour la Recherche Médicale. C.G. was supported by a fellowship from the Ministère de la Recherche. We are greatly indebted to Erik Jorgensen; he contributed scientific insight, reagents, and unpublished data throughout the entire project and provided critical comments during the preparation of this manuscript. We thank Bruce Bamber for sharing the UNC-49B–GFP construct, Mike Nonet for the VAMP–GFP construct, Janet Richmond for sharing unpublished results, the Caenorhabditis Genetic Center for strains, and Antoine Triller and Robby Weimer for critical reading of this manuscript.
Correspondence should be addressed to Dr. Jean-Louis Bessereau, Laboratoire de Biologie Cellulaire de la Synapse Normale et Pathologique, Institut National de la Santé et de la Recherche Médicale, Unité 497, Ecole Normale Supérieure, 46 rue d'Ulm, 75005 Paris, France. E-mail: jlbesse@wotan.ens.fr.
References
- 1.Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- 2.Avery L, Bargmann CI, Horvitz HR. The Caenorhabditis elegans unc-31 gene affects multiple nervous system-controlled functions. Genetics. 1993;134:455–464. doi: 10.1093/genetics/134.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bamber BA, Beg AA, Twyman RE, Jorgensen EM. The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci. 1999;19:5348–5359. doi: 10.1523/JNEUROSCI.19-13-05348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berwin B, Floor E, Martin TF. CAPS (mammalian UNC-31) protein localizes to membranes involved in dense-core vesicle exocytosis. Neuron. 1998;21:137–145. doi: 10.1016/s0896-6273(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 5.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broadie K, Bate M. Innervation directs receptor synthesis and localization in Drosophila embryo synaptogenesis. Nature. 1993;361:350–353. doi: 10.1038/361350a0. [DOI] [PubMed] [Google Scholar]
- 7.Burgess RW, Nguyen QT, Son YJ, Lichtman JW, Sanes JR. Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron. 1999;23:33–44. doi: 10.1016/s0896-6273(00)80751-5. [DOI] [PubMed] [Google Scholar]
- 8.Clark SG, Lu X, Horvitz HR. The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics. 1994;137:987–997. doi: 10.1093/genetics/137.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craig AM, Boudin H. Molecular heterogeneity of central synapses: afferent and target regulation. Nat Neurosci. 2001;4:569–578. doi: 10.1038/88388. [DOI] [PubMed] [Google Scholar]
- 10.Craig AM, Blackstone CD, Huganir RL, Banker G. Selective clustering of glutamate and gamma-aminobutyric acid receptors opposite terminals releasing the corresponding neurotransmitters. Proc Natl Acad Sci USA. 1994;91:12373–12377. doi: 10.1073/pnas.91.26.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crump JG, Zhen M, Jin Y, Bargmann CI. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron. 2001;29:115–129. doi: 10.1016/s0896-6273(01)00184-2. [DOI] [PubMed] [Google Scholar]
- 12.DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 13.Duerr JS, Frisby DL, Gaskin J, Duke A, Asermely K, Huddleston D, Eiden LE, Rand JB. The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J Neurosci. 1999;19:72–84. doi: 10.1523/JNEUROSCI.19-01-00072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Featherstone DE, Broadie K. Surprises from Drosophila: genetic mechanisms of synaptic development and plasticity. Brain Res Bull. 2000;53:501–511. doi: 10.1016/s0361-9230(00)00383-x. [DOI] [PubMed] [Google Scholar]
- 15.Featherstone DE, Rushton E, Broadie K. Developmental regulation of glutamate receptor field size by nonvesicular glutamate release. Nat Neurosci. 2002;5:141–146. doi: 10.1038/nn789. [DOI] [PubMed] [Google Scholar]
- 16.Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- 17.Hallam SJ, Jin Y. lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature. 1998;395:78–82. doi: 10.1038/25757. [DOI] [PubMed] [Google Scholar]
- 18.Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. 1990;4:61–85. doi: 10.1016/0896-6273(90)90444-k. [DOI] [PubMed] [Google Scholar]
- 19.Ji F, Kanbara N, Obata K. GABA and histogenesis in fetal and neonatal mouse brain lacking both the isoforms of glutamic acid decarboxylase. Neurosci Res. 1999;33:187–194. doi: 10.1016/s0168-0102(99)00011-5. [DOI] [PubMed] [Google Scholar]
- 20.Jin Y, Jorgensen E, Hartwieg E, Horvitz HR. The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J Neurosci. 1999;19:539–548. doi: 10.1523/JNEUROSCI.19-02-00539.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jorgensen E, Nonet M. Neuromuscular junctions in the nematode C. elegans. Semin Dev Biol. 1995;6:207–220. [Google Scholar]
- 22.Keshishian H. Is synaptic homeostasis just wishful thinking? Neuron. 2002;33:491–492. doi: 10.1016/s0896-6273(02)00597-4. [DOI] [PubMed] [Google Scholar]
- 23.Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J Neurosci. 2002;22:1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirsch J, Betz H. Glycine-receptor activation is required for receptor clustering in spinal neurons. Nature. 1998;392:717–720. doi: 10.1038/33694. [DOI] [PubMed] [Google Scholar]
- 25.Knobel KM, Jorgensen EM, Bastiani MJ. Growth cones stall and collapse during axon outgrowth in Caenorhabditis elegans. Development. 1999;126:4489–4498. doi: 10.1242/dev.126.20.4489. [DOI] [PubMed] [Google Scholar]
- 26.Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post-docking role for active zone protein Rim. Nat Neurosci. 2001;4:997–1005. doi: 10.1038/nn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levi S, Vannier C, Triller A. Strychnine-sensitive stabilization of postsynaptic glycine receptor clusters. J Cell Sci. 1998;111:335–345. doi: 10.1242/jcs.111.3.335. [DOI] [PubMed] [Google Scholar]
- 28.Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- 29.McIntire SL, Jorgensen E, Horvitz HR. Genes required for GABA function in Caenorhabditis elegans. Nature. 1993a;364:334–337. doi: 10.1038/364334a0. [DOI] [PubMed] [Google Scholar]
- 30.McIntire SL, Jorgensen E, Kaplan J, Horvitz HR. The GABAergic nervous system of Caenorhabditis elegans. Nature. 1993b;364:337–341. doi: 10.1038/364337a0. [DOI] [PubMed] [Google Scholar]
- 31.McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- 32.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller KG, Emerson MD, McManus JR, Rand JB. RIC-8 (Synembryn): a novel conserved protein that is required for Gqα signaling in the C. elegans nervous system. Neuron. 2000;27:289–299. doi: 10.1016/s0896-6273(00)00037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–648. doi: 10.1016/s0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- 35.Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 36.Nonet ML. Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J Neurosci Methods. 1999;89:33–40. doi: 10.1016/s0165-0270(99)00031-x. [DOI] [PubMed] [Google Scholar]
- 37.Plunkett JA, Simmons RB, Walthall WW. Dynamic interactions between nerve and muscle in Caenorhabditis elegans. Dev Biol. 1996;175:154–165. doi: 10.1006/dbio.1996.0103. [DOI] [PubMed] [Google Scholar]
- 38.Rasmussen H, Rasmussen T, Triller A, Vannier C. Strychnine-blocked glycine receptor is removed from synapses by a shift in insertion/degradation equilibrium. Mol Cell Neurosci. 2002;19:201–215. doi: 10.1006/mcne.2001.1074. [DOI] [PubMed] [Google Scholar]
- 39.Renden R, Berwin B, Davis W, Ann K, Chin CT, Kreber R, Ganetzky B, Martin TF, Broadie K. Drosophila CAPS is an essential gene that regulates dense-core vesicle release and synaptic vesicle fusion. Neuron. 2001;31:421–437. doi: 10.1016/s0896-6273(01)00382-8. [DOI] [PubMed] [Google Scholar]
- 40.Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruegg MA, Bixby JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–27. doi: 10.1016/s0166-2236(97)01154-5. [DOI] [PubMed] [Google Scholar]
- 42.Saitoe M, Tanaka S, Takata K, Kidokoro Y. Neural activity affects distribution of glutamate receptors during neuromuscular junction formation in Drosophila embryos. Dev Biol. 1997;184:48–60. doi: 10.1006/dbio.1996.8480. [DOI] [PubMed] [Google Scholar]
- 43.Saitoe M, Schwarz TL, Umbach JA, Gundersen CB, Kidokoro Y. Absence of junctional glutamate receptor clusters in Drosophila mutants lacking spontaneous transmitter release. Science. 2001;293:514–517. doi: 10.1126/science.1061270. [DOI] [PubMed] [Google Scholar]
- 44.Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 45.Schaefer AM, Hadwiger GD, Nonet ML. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26:345–356. doi: 10.1016/s0896-6273(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 46.Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- 47.Sulston JE, Horvitz HR. Abnormal cell lineages in mutants of the nematode Caenorhabditis elegans. Dev Biol. 1981;82:41–55. doi: 10.1016/0012-1606(81)90427-9. [DOI] [PubMed] [Google Scholar]
- 48.Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci USA. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 50.Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- 51.White JG, Southgate E, Thomson JN, Brenner S. The structure of the ventral nerve cord of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1976;275:327–348. doi: 10.1098/rstb.1976.0086. [DOI] [PubMed] [Google Scholar]
- 52.White JG, Albertson DG, Anness MA. Connectivity changes in a class of motoneurone during the development of a nematode. Nature. 1978;271:764–766. doi: 10.1038/271764a0. [DOI] [PubMed] [Google Scholar]
- 53.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1984;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 54.Yandell MD, Edgar LG, Wood WB. Trimethylpsoralen induces small deletion mutations in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1994;91:1381–1385. doi: 10.1073/pnas.91.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X, Li W, Prescott ED, Burden SJ, Wang JC. DNA topoisomerase IIβ and neural development. Science. 2000;287:131–134. doi: 10.1126/science.287.5450.131. [DOI] [PubMed] [Google Scholar]
- 56.Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001;30:399–410. doi: 10.1016/s0896-6273(01)00287-2. [DOI] [PubMed] [Google Scholar]
- 57.Young SH, Poo MM. Spontaneous release of transmitter from growth cones of embryonic neurones. Nature. 1983;305:634–637. doi: 10.1038/305634a0. [DOI] [PubMed] [Google Scholar]
- 58.Zhen M, Jin Y. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature. 1999;401:371–375. doi: 10.1038/43886. [DOI] [PubMed] [Google Scholar]
- 59.Zhen M, Huang X, Bamber B, Jin Y. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 2000;26:331–343. doi: 10.1016/s0896-6273(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 60.Zhou HM, Brust-Mascher I, Scholey JM. Direct visualization of the movement of the monomeric axonal transport motor UNC-104 along neuronal processes in living Caenorhabditis elegans. J Neurosci. 2001;21:3749–3755. doi: 10.1523/JNEUROSCI.21-11-03749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]