

Abstract
Tumor necrosis factor-α (TNF) is implicated in the initiation of neuropathic pain. In vitro, TNF activates p38 mitogen-activated kinase. Accordingly, we investigated whether TNF activates the p38 cascade in vivo to trigger pain behavior after spinal nerve ligation (SNL). Treatment starting 2 d before SNL with the TNF antagonist etanercept (1 mg, i.p., every third day) attenuated mechanical allodynia. Treatment starting 1 or 7 d after SNL was ineffective. Similarly, intrathecal infusion of a p38 inhibitor (SB203580, 4 mg/d) was effective only if it was started before but not 7 d after SNL. For both treatments, the cessation of therapy resulted in increased allodynia. In separate experiments using Western blots and immunohistochemistry, ipsilateral lumbar spinal cord and L5 and L6 DRG were analyzed for total and phosphorylated p38 after SNL alone or SNL combined with etanercept pretreatment. In DRG, activated p38 was transiently elevated 5 hr after SNL and returned to baseline by 1 d after SNL. Phosphorylated p38 was localized in small TNF-positive DRG neurons. In spinal cord, p38 was activated between 5 hr and 3 d after SNL and returned to baseline within 5 d. In DRG, but not spinal cord, etanercept pretreatment blocked p38 activation. These data indicate that after SNL treatment, phosphorylated p38 levels in spinal cord and DRG are transiently elevated. In DRG, p38 activation is blocked by systemic TNF inhibition. Parallel inhibition of p38 activation and allodynia may represent a clinically relevant therapeutic window. These data suggest a sequential role for TNF and p38 in the induction of neuropathic pain.
Keywords: cytokines, tumor necrosis factor-α, dorsal root ganglion, spinal nerve ligation, p38 MAPK, neuropathic pain
Introduction
Peripheral nerve injury often results in neuropathic pain associated with hyperalgesia and allodynia (Zimmermann, 2001). Several lines of evidence suggest that proinflammatory cytokines such as tumor necrosis factor-α (TNF), which are released into the local milieu after injury, may initiate the cascade that underlies this facilitated processing (Wagner and Myers, 1996; Sommer and Schäfers, 1998; Sommer et al., 1998; Sorkin and Doom, 2000; Schäfers et al., 2002). Thus, after several types of nerve injury, TNF expression increases in both dorsal root ganglia (DRG) (Schäfers et al., 2002, 2003) and spinal cord (Hashizume et al., 2000). The inhibition of TNF reduces the hyperalgesia associated with two models of neuropathic pain: chronic constriction injury and partial nerve transection (Sommer et al., 2001a,b). The mechanisms by which TNF elicits pain behavior are still unclear. In cultured DRG neurons, exogenous TNF activates p38 mitogen-activated kinase (p38) (Pollock et al., 2002). Recently, p38 activation was shown to play a major role in the maintenance of pain arising from tissue or nerve inflammation (Ji et al., 2002; Milligan et al., 2003). Therefore, we hypothesized that activation of the p38 cascade may represent a route by which TNF could trigger changes in transcription as well as post-translational modifications of target proteins in the DRG and spinal cord that are correlated with the development of pain after nerve injury.
In the present study, we investigated whether the inhibition of TNF or p38 could block the development of or reverse already established allodynia after spinal nerve ligation (SNL). In addition, we looked at the time course of p38 activation in DRG and spinal cord after SNL. Finally, in addition to examining covariance among the susceptibility of SNL-associated allodynia to TNF antagonism, p38 antagonism, and p38 activation, we determined whether inhibition of TNF blocked the activation of p38, thus for the first time demonstrating a causal link between nerve injury, TNF, and p38.
Materials and Methods
Animals. Male Sprague Dawley rats weighing 120–150 gm were used in procedures approved by the Animal Care and Use Committee of the University of California San Diego. Experimental groups included: (1) rats with anti-TNF treatment and SNL, (2) rats with p38 inhibition and SNL, and (3) rats with SNL only.
Surgery. All surgical procedures used were performed under deep halothane anesthesia (5% for induction and 2% for maintenance) in 50% O2. The SNL was performed as described previously (Kim and Chung, 1992). Briefly, a midline incision above the lumbar spine exposed the left L6 transverse process. The process was then removed, the left L5 and L6 spinal nerves were isolated, and both nerves were tightly ligated with 6-0 silk.
Drug delivery. Recombinant TNF receptor (p75)–Fc fusion protein etanercept (Enbrel; Immunex, Seattle, WA) was used as a TNF antagonist (Mohler et al., 1993). Etanercept (1 mg/1 ml) or 0.9% saline was given every third day by intraperitoneal injection starting 2 d before or 1 or 7 d after SNL.
In a separate set of animals, 6.3-mm-long polyethylene (PE5) catheters were implanted into the intrathecal space (Yaksh and Rudy, 1976). Catheters were connected to primed osmotic pumps (Alzet, Cupertino, CA) filled with the p38 inhibitor SB203580 (Calbiochem, La Jolla, CA) or 0.9% saline. Pumps were placed subcutaneously on the upper back; animals received fluid at a rate of 0.5 μl/hr, resulting in drug delivery of 4 μg/d over a mean of 14 d starting 2 d before or 7 d after SNL.
Behavior. Mechanical sensitivity was assessed using von Frey hairs and the up–down method (Dixon, 1965), following the procedure described previously (Chaplan et al., 1994). The 50% probability withdrawal threshold was determined. Tests were performed on rats before drug treatment or surgery, before a second manipulation (surgery or drug), and then for several additional days on both ipsilateral and contralateral paws. Testing continued after the cessation of intraperitoneal treatment or after pumps ran dry to determine whether pain behavior changed in the absence of the inhibitor. All behavioral testing of animals was done by observers who were unaware of the treatment.
Western blots. At 5 hr or 1, 3, or 5 d after SNL, animals were quickly anesthetized with halothane; after decapitation, the ipsilateral lumbar enlargement and L5 and L6 DRG were removed. Tissue was immediately homogenized in 50 mm Tris buffer (pH 8.0, 0.5% Triton X-100, 150 mm NaCl, 1 mm EDTA, and phosphatase inhibitors) by sonication. Tissue extracts were denatured using NuPAGE 4–12% Bis-Tris gel electrophoresis and then electrophoretically transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). After blocking nonspecific binding sites with 5% low-fat milk in PBS containing 0.1% Tween 20 for 1 hr at room temperature, membranes were probed with antibodies overnight at 4°C. After washing, the antibody–protein complexes were probed with secondary antibodies labeled with horseradish peroxidase for 1 hr at room temperature and detected with chemiluminescent reagents. The nitrocellulose membranes were stripped with the Re-Blot Western blot recycling kit (Chemicon, Temecula, CA) and reblotted with different antibodies. The antibodies used detect phosphorylated p38 MAPK (mitogen-activated protein kinase; 1:1000), total p38 MAPK (1:500; Cell Signaling Technology, Beverly, MA), and β-actin (1:5000; Sigma, St. Louis, MO). Immunopositive bands were detected by ECL, and the bands were quantitatively analyzed using ImageQuant software (Molecular Dynamics, Sunnyvale, CA; Amersham Biosciences, Piscataway, NJ). Data for phospho-p38 are given in relation to the individual β-actin loading control and normalized to naive controls within the same blot.
Because rats exhibited increased phospho-p38 in both DRG and spinal cord 5 hr after SNL, an additional group was pretreated with etanercept or vehicle as in the behavioral experiments and then killed 5 hr after SNL to determine whether phosphorylation of p38 occurred secondary to increased TNF activity.
Immunohistochemistry. At different time points after SNL (0 d, 5 hr, 1 d, 4 d, 8 d), the ipsilateral L5 DRG was removed, mounted in Tissue-Tek ornithine transcarbamylase embedding compound, quickly frozen, and stored at −80°C. DRG were sectioned on a cryostat at a thickness of 10 μm. Sections were thaw-mounted onto Superfrost Plus Slides and air-dried for 30 min. Sections were postfixed for 10 min in acetone at −20°C and blocked with 10% BSA/PBS. Serial sections were used to determine colocalization of phospho-p38 with other proteins. The following antibodies (Abs) were used for immunofluorescence: polyclonal phospho-p38 MAPK Ab (1:100; Cell Signaling Technology), total p38 MAPK Ab (1:100; Cell Signaling Technology), polyclonal rabbit anti-protein gene product 9.5 (PGP 9.5) Ab (1:500; Ultraclone, Wellow, UK), polyclonal rabbit anti-TNF Ab (1:100; Serotec, Oxford, UK) and polyclonal rabbit anti-glial fibrillary acid protein Ab (GFAP, 1:100; ICN Pharmaceutical, Frankfurt, Germany). Sections were incubated at 4°C for 24 hr and washed three times for 10 min each in PBS. An ABC system (Vector Laboratories, Burlingame, CA) and DAB or Cy3-conjugated or Cy2-conjugated secondary Ab (1:100, 2 hr; Amersham Biosciences, Freiburg, Germany) were used for detection. Isolectin B4 (IB4)-binding histochemistry was performed by adding IB4 (1:100; Sigma, Taufkirchen, Germany) to a final concentration of 0.1 mm with CaCl2, MgCl2, and Triton X-100. IB4 binding was detected by incubating in ExtrAvidin-conjugated fluorescein isothiocyanate (1:200; Sigma). The omission of all primary antibodies phospho-p38, total p38, GFAP, PGP 9.5, and TNF resulted in negative staining.
Statistical analysis. To determine differences from baseline at individual time points or doses, data were analyzed by repeated-measures two-way ANOVA, followed by Tukey post hoctests (GraphPad Instat, version 3.0; GraphPad Software, San Diego, CA). To determine differences between treatment groups, behavioral data were analyzed by one-way ANOVA with Tukey post hoc tests. Percentage differences in allodynia were calculated by comparing areas under the curve for drug- and vehicle-treated groups. Unpaired t tests were used for Western blot data. References associated with p values of <0.05 were considered to be statistically significant. All data are given as means ± SE.
Results
Inhibition of TNF or p38 attenuates SNL-induced mechanical allodynia
Etanercept treatment that started 2 d before SNL attenuated mechanical allodynia by 41% (Fig.1A). The etanercept-related attenuation dissipated when drug treatment was stopped (Fig. 1A). Etanercept treatment starting only 1 d after SNL had no effect on the allodynia even when the dosage was doubled during days 12–20 after SNL (Fig.1B). Etanercept treatment started 7 d after SNL also had no effect on mechanical withdrawal thresholds (Fig.1C). Because TNF signaling is linked to the activation of p38 in many cells (including DRG), we next investigated the effects of p38 inhibition in rats with SNL. Animals treated with intrathecal SB203580 displayed an almost identical pattern to those with systemic TNF antagonism. Pretreatment with the phospho-p38 antagonist reduced the SNL-induced allodynia by 43%, whereas post-treatment had no effect (Fig. 1D). Cessation of drug treatment resulted in a loss of the anti-allodynic effect.
Fig. 1.

Mechanical allodynia induced by SNL is attenuated by the systemic inhibition of TNF and the intrathecal inhibition of p38. A, Treatment with the TNF antagonist etanercept (▴) starting 2 d before SNL (d-2) attenuates mechanical allodynia induced by SNL.B, Treatment with etanercept started 1 d after SNL (▴) has no effect on mechanical thresholds, even when the etanercept dose is doubled (dark shaded bar).C, Treatment with etanercept started 7 d after SNL has no effect. D, Treatment with the p38 inhibitor SB203580 (d-2) but not treatment starting 7 d after SNL attenuates mechanical allodynia induced by SNL. *p < 0.05, **p < 0.005, ***p < 0.001 versus vehicle-treated rats;n = 6–12 per treatment group.
p38 phosphorylation in DRG and spinal cord after SNL
Five hours after SNL, Western blots indicated that phospho-p38 in the ganglia of the ligated nerves had increased substantially. This increase was short-lived; by 1 d after injury, phospho-p38 had returned to basal levels (Fig.2A). This transient increase in phospho-p38 corresponded with the therapeutic window seen for both TNF and phospho-p38 inhibitors. Total p38 in the DRG did not change after SNL, indicating that the increase in phospho-p38 after SNL is caused by increased phosphorylation rather than by elevated substrate. In the spinal cords of these animals, the early onset of p38 activation was similar to that seen in DRG; however, phospho-p38 remained elevated in the spinal cord through day 1 after SNL (Fig.2B). As in DRG, total p38 did not change.
Fig. 2.

SNL induces transient p38 activation in DRG neurons and spinal cord. A, Time course of p38 phosphorylation in DRG neurons after SNL. Phospho-p38 (p-p38) is transiently increased 5 hr after SNL.B, Time course of p38 phosphorylation in lumbar spinal cord after SNL; phospho-p38 is increased between 5 hr and 3 d after SNL. *p < 0.05, **p < 0.005 versus control; n = 6 per time point.
Phospho-p38 is predominantly expressed in small sensory DRG neurons
Immunohistochemistry confirmed that there were low levels of phospho-p38 in DRG under basal conditions; more importantly, 5 hr after ligation there was a clear increase in both staining density and the number of cells positive for activated p38 that returned to control levels before day 4 (Fig.3A–C). Labeling with phospho-p38 and PGP 9.5 on serial sections confirmed the predominantly neuronal localization of activated p38 after SNL (Fig. 3D). There was no staining of phospho-p38 in activated satellite cells that stained positively for GFAP, although these cells frequently encircled the phospho-p38-stained neurons (Fig. 3E). Phospho-p38 colocalized with IB4, indicating that p38 activation occurs in small, presumably nociceptive, neurons (Fig. 3F).
Fig. 3.
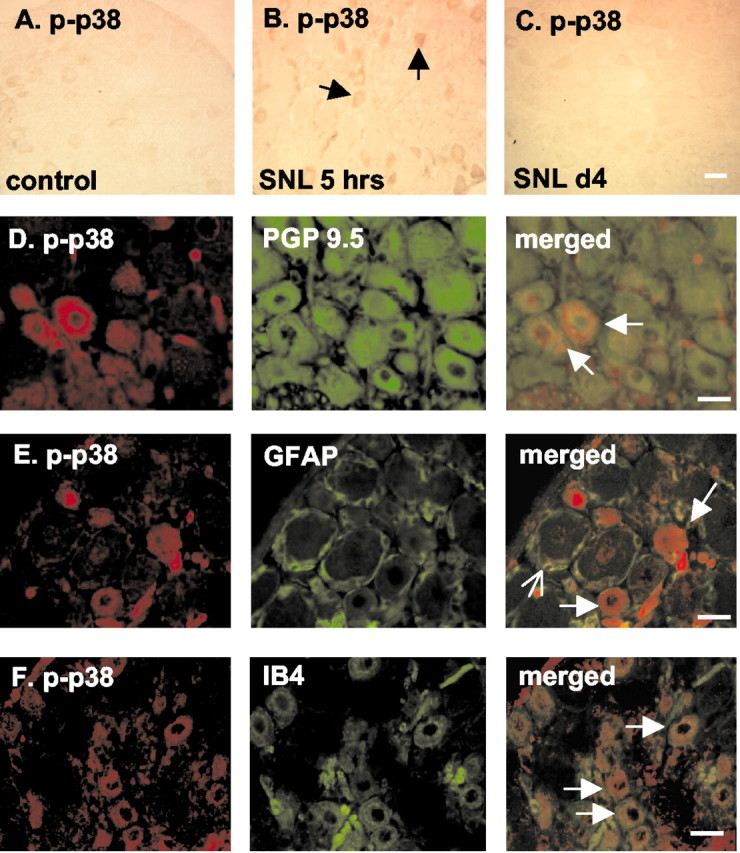
Phospho-p38 (p-p38) is activated in small, presumably nociceptive DRG neurons and not in activated satellite cells. A–C, p38 phosphorylation is increased in DRG neurons (arrows) 5 hr after SNL.D, Serial staining of phospho-p38 (red) with PGP 9.5 (green), a marker for neuronal cells, in DRG neurons indicates that p38 is predominantly activated in neuronal cells (arrows). E, Serial staining of phospho-p38 (red) with GFAP (green, open arrow), a marker for activated satellite cells, indicates that p38 (arrows) is not phosphorylated in activated satellite cells 5 hr after SNL.F, Double staining demonstrates colocalization (arrows) of phospho-p38 (red) with IB4 (green) in DRG neurons after SNL, suggesting p38 activation in small, presumably nociceptive DRG neurons. Scale bars, 30 μm. n = 4 per time point.
SNL-induced p38 activation is mediated by TNF
p38 was predominantly activated in DRG neurons that were also TNF-positive (Fig.4A). To test whether TNF plays a role in p38 activation after SNL, the TNF antagonist etanercept (1 mg) was given 2 d before SNL. The spinal lumbar enlargement and L5 and L6 DRG were harvested 5 hr after SNL and analyzed for p38 phosphorylation using Western blots. As seen in Figure2, SNL induced an increase in phospho-p38 in both spinal cord and DRG. Systemic pretreatment with etanercept markedly inhibited the SNL-induced activation of p38 in DRG (Fig. 4B), but not in spinal cord (Fig. 4C). These data suggest that p38 activation in DRG after nerve injury is mediated by TNF.
Fig. 4.

TNF induces p38 activation in DRG after SNL.A, Phospho-p38 (p-p38,red) is colocalized with TNF (green) in small DRG neurons after SNL (arrows). Scale bar, 30 μm. B, Systemic anti-TNF treatment with etanercept (Eta, 1 mg/rat, i.p.) suppresses the SNL-induced increase in phospho-p38 in DRG 5 hr after SNL. C, Systemic etanercept does not suppress the increase of phospho-p38 in lumbar spinal cord after SNL. *p < 0.05, **p < 0.005 versus control; n = 6 per group.
Discussion
After a variety of nerve injuries, including transection of the L5 spinal nerve, increased TNF in both DRG and spinal cord is associated with neuropathic pain (DeLeo et al., 1997; Wagner et al., 1998;Schäfers et al., 2002). In parallel with these findings, antagonism of TNF reduces pain behavior (Wagner et al., 1998; Sommer et al., 2001a,b). The present data obtained using the SNL model support the hypothesis that TNF plays a causal role in neuropathic pain, but importantly, they also introduce several novel points. Pretreatment with a TNF antagonist must be started before (or perhaps at the time of) injury rather than 1 d after to be effective in reducing the mechanical allodynia that arises from SNL. This is in contrast to the ability of the same or similar TNF antagonists to reduce already established pain behavior 6 d after chronic constriction injury in the mouse (Sommer et al., 2001a) and the ability of combined high-dose antagonism of TNF and interleukin-1 receptor to cause a reduction in mechanical allodynia several days after spinal-nerve transection in the rat (Sweitzer et al., 2001). The brevity of the therapeutic window after this acute nerve injury may be partially a function of the proximity of the injury to the DRG and the resultant shorter time necessary for transport of injury factors to the neuronal cell bodies (Shubayev and Myers, 2001, 2002; Schäfers et al., 2002).
The narrow therapeutic window that we observed for etanercept corresponded to the activation of phospho-p38 in the DRG and initiation of p38 activation in the spinal cord. Intrathecal administration of SB203580, a p38 antagonist, was also effective only when started before the injury; thus, both TNF and p38 activation seem to be necessary for the initiation of mechanical allodynia. A perplexing fact is that despite the return of phospho-p38 to basal levels in the DRG and spinal cord within the first few days, interruption of either antagonist after ≥10 d of therapy results in a complete loss of the anti-allodynic effect. After SNL, a peripheral generator likely develops that drives central sensitization and pain behavior through a TNF–p38 linkage. Cessation of therapy could unblock this pathway and shift the p38 activation normally seen at 5 hr to begin at a later time point. Future experiments will examine this possibility.
At present, it is unknown whether the essential inhibitory action of etanercept or SB203580 is in the spinal cord, DRG, or both, although the systemic administration of etanercept might be expected to have a greater action on DRG, in which the blood–nerve barrier is more open (Devor, 1999). Low CNS penetration may also explain the lack of direct etanercept blockade of spinal p38 activation seen in our study. Blockade of p38 activation in DRG still allows some persistent nociceptive input to reach the spinal cord (Ji et al., 2002), which may induce spinal activation and subsequent spinal release of proinflammatory cytokines (Milligan et al., 2003). Despite this, the present data suggest that blockade of p38 activation at the level of DRG is critical for the attenuation of SNL-induced pain behavior.
Activation of p38 as it relates to pain behavior is relatively unexplored. After peripheral inflammation, p38 is activated in a subset of DRG neurons consisting of predominantly capsaicin-sensitive (containing the heat and capsaicin receptor vanilloid receptor 1) small cells (Ji et al., 2002). We noted colocalization of phospho-p38 with a different population of small nociceptive neurons, those staining for IB4.
In the spinal cord, p38 was activated between 5 hr and 1 d after SNL. One and 3 weeks after sciatic-nerve injury, spinal p38 phosphorylation is not seen in any spinal cord cell type, whereas two other MAP kinases, ERK (extracellular signal-regulated kinase) and JNK (c-Jun N-terminal protein kinase), are phosphorylated in astrocytes of the L5 spinal cord and nucleus gracilis (Ma and Quirion, 2002). This may be because of the timing, because phospho-p38 was no longer seen 7 d after SNL. Unlike nerve injury, peripheral inflammation induced by Freund's adjuvant resulted in no change in phospho-p38 in the spinal cord (Ji et al., 2002). However, direct spinal activation (intrathecal substance P) phosphorylates p38 located in OX-42-positive microglia (Svensson et al., 2002). Differences among the various models seem to cause substantial differences in spinal p38 activation.
After peripheral inflammation, intrathecal SB203580 was effective both before and after treatment in partially antagonizing heat hyperalgesia; it was totally ineffective, regardless of timing, in ameliorating the accompanying mechanical allodynia (Ji et al., 2002). Mechanical allodynia generated by sciatic-nerve inflammation was totally and immediately reversed by a nonspecific p38 antagonist (Milligan et al., 2003). Thus, inactivation of p38 appears to block the sensitization of different modalities in inflammatory and neuropathic pain models.
TNF and p38 activation in DRG could be linked at two points. TNF could trigger p38 activation, or phosphorylated p38 could induce the upregulation of TNF protein. Both mechanisms have been shown in a variety of cell types (Raingeaud et al., 1995; Barbin et al., 2001;Bhat et al., 2002; Jeohn et al., 2002; Park et al., 2002; Pollock et al., 2002). Alternatively, both mechanisms could be involved with TNF and/or other proinflammatory cytokines produced subsequent to the MAPK activation acting in a positive feedback loop to trigger increased p38 phosphorylation. Because TNF and phospho-p38 are colocalized in a substantial number of DRG neurons, including presumptive nociceptive neurons, any of these alternatives is a likely possibility. However, colocalization of TNF receptors and phospho-p38 has not yet been demonstrated. In cultured DRG neurons, exogenous TNF phosphorylates p38 within minutes (Pollock et al., 2002). Longer exposure of DRG neurons to TNF resulted in increased phospho-p38 (Park et al., 2002). In neural tissue other than DRG, TNF-induced activation of p38 occurs in neurons as well as within glia (Raingeaud et al., 1995; Barbin et al., 2001;Bhat et al., 2002). Phosphorylation of p38 in glia leads to increased TNF gene expression as well as TNF protein (Bhat et al., 2002; Jeohn et al., 2002). The present data indicate that TNF antagonism blocks activation of p38 in DRG after SNL, and that this results in an attenuation of pain behavior.
In summary, pretreatment but not post-treatment with a TNF or a phospho-p38 antagonist attenuates mechanical allodynia induced by SNL. Antagonism of TNF not only reduces pain behavior but also blocks activation of p38. After SNL, there is a brief increase in phospho-p38 in DRG neurons, many of which are presumably nociceptive. Most of these cells also express TNF. Together, these data implicate the TNF–p38 signal transduction cascade in the DRG as a significant participant in the generation of mechanical allodynia after nerve injury.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant SCHA 924/1-1 (M.S.) and National Institutes of Health Grant NS35630 (L.S.S.). We thank L. Biko, B. Dekant, E. Higuera, and C. Geis for expert technical assistance.
Correspondence should be addressed to Dr. Maria Schäfers, Anesthesiology Research Laboratory, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093. E-mail: mschaefers@ucsd.edu.
References
- 1.Barbin G, Roisin MP, Zalc B. Tumor necrosis factor-α activates the phosphorylation of ERK, SAPK/JNK, and p38 kinase in primary cultures of neurons. Neurochem Res. 2001;26:107–112. doi: 10.1023/a:1011086426652. [DOI] [PubMed] [Google Scholar]
- 2.Bhat NR, Feinstein DL, Shen Q, Bhat AN. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells: roles of nuclear factors, nuclear factor κB, cAMP response element-binding protein, CCAAT/enhancer-binding protein-β, and activating transcription factor-2. J Biol Chem. 2002;277:29584–29592. doi: 10.1074/jbc.M204994200. [DOI] [PubMed] [Google Scholar]
- 3.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 4.DeLeo JA, Colburn RW, Rickman AJ. Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res. 1997;759:50–57. doi: 10.1016/s0006-8993(97)00209-6. [DOI] [PubMed] [Google Scholar]
- 5.Devor M. Unexplained peculiarities of the dorsal root ganglion. Pain [Suppl] 1999;6:S27–S35. doi: 10.1016/S0304-3959(99)00135-9. [DOI] [PubMed] [Google Scholar]
- 6.Dixon W. The up-and-down method for small samples. J Am Stat Assoc. 1965;60:967–978. [Google Scholar]
- 7.Hashizume H, DeLeo JA, Colburn RW, Weinstein JN. Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine. 2000;25:1206–1217. doi: 10.1097/00007632-200005150-00003. [DOI] [PubMed] [Google Scholar]
- 8.Jeohn GH, Cooper CL, Wilson B, Chang RC, Jang KJ, Kim HC, Liu B, Hong JS. p38 MAP kinase is involved in lipopolysaccharide-induced dopaminergic neuronal cell death in rat mesencephalic neuron-glia cultures. Ann NY Acad Sci. 2002;962:332–346. doi: 10.1111/j.1749-6632.2002.tb04078.x. [DOI] [PubMed] [Google Scholar]
- 9.Ji R, Samad T, Jin S, Schmoll R, Woolf C. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 10.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 11.Ma W, Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- 12.Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohler KM, Torrance DS, Smith CA, Goodwin RG, Stremler KE, Fung VP, Madani H, Widmer MB. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993;151:1548–1561. [PubMed] [Google Scholar]
- 14.Park JG, Yuk Y, Rhim H, Yi SY, Yoo YS. Role of p38 MAPK in the regulation of apoptosis signaling induced by TNF-α in differentiated PC12 cells. J Biochem Mol Biol. 2002;35:267–272. doi: 10.5483/bmbrep.2002.35.3.267. [DOI] [PubMed] [Google Scholar]
- 15.Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ, Scott RH. TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology. 2002;42:93–106. doi: 10.1016/s0028-3908(01)00163-0. [DOI] [PubMed] [Google Scholar]
- 16.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 17.Schäfers M, Geis C, Brors D, Yaksh TL, Sommer C. Anterograde transport of tumor necrosis factor-α in the intact and injured rat sciatic nerve. J Neurosci. 2002;22:536–545. doi: 10.1523/JNEUROSCI.22-02-00536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schäfers M, Geis C, Svensson CI, Luo ZD, Sommer C. Selective increase of tumor necrosis factor-α in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci. 2003;17:791–804. doi: 10.1046/j.1460-9568.2003.02504.x. [DOI] [PubMed] [Google Scholar]
- 19.Shubayev VI, Myers RR. Axonal transport of TNF-α in painful neuropathy: distribution of ligand tracer and TNF receptors. J Neuroimmunol. 2001;114:48–56. doi: 10.1016/s0165-5728(00)00453-7. [DOI] [PubMed] [Google Scholar]
- 20.Shubayev VI, Myers RR. Anterograde TNF-α transport from rat dorsal root ganglion to spinal cord and injured sciatic nerve. Neurosci Lett. 2002;320:99–101. doi: 10.1016/s0304-3940(02)00010-1. [DOI] [PubMed] [Google Scholar]
- 21.Sommer C, Schäfers M. Painful mononeuropathy in C57BL/Wld mice with delayed Wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res. 1998;784:154–162. doi: 10.1016/s0006-8993(97)01327-9. [DOI] [PubMed] [Google Scholar]
- 22.Sommer C, Marziniak M, Myers RR. The effect of thalidomide treatment on vascular pathology and hyperalgesia caused by chronic constriction injury of rat nerve. Pain. 1998;74:83–91. doi: 10.1016/S0304-3959(97)00154-1. [DOI] [PubMed] [Google Scholar]
- 23.Sommer C, Schäfers M, Marziniak M, Toyka KV. Etanercept reduces hyperalgesia in experimental painful neuropathy. J Peripher Nerv Syst. 2001a;6:67–72. doi: 10.1046/j.1529-8027.2001.01010.x. [DOI] [PubMed] [Google Scholar]
- 24.Sommer C, Lindenlaub T, Teuteberg P, Schäfers M, Hartung T, Toyka KV. Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res. 2001b;913:86–89. doi: 10.1016/s0006-8993(01)02743-3. [DOI] [PubMed] [Google Scholar]
- 25.Sorkin LS, Doom CM. Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripher Nerv Syst. 2000;5:96–100. doi: 10.1046/j.1529-8027.2000.00012.x. [DOI] [PubMed] [Google Scholar]
- 26.Svensson CI, Marsala M, Yaksh TL. Intrathecal substance P induced hyperalgesia, blocked by p38 MAPK inhibitor, caused activation of p38 MAPK in spinal microglia selectively. Soc Neurosci Abstr. 2002;28:652.6. [Google Scholar]
- 27.Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- 28.Wagner R, Myers RR. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. NeuroReport. 1996;7:2897–2901. doi: 10.1097/00001756-199611250-00018. [DOI] [PubMed] [Google Scholar]
- 29.Wagner R, Myers RR, O'Brien JS. Prosaptide prevents hyperalgesia and reduces peripheral TNFR1 expression following TNF-α nerve injection. NeuroReport. 1998;9:2827–2831. doi: 10.1097/00001756-199808240-00026. [DOI] [PubMed] [Google Scholar]
- 30.Yaksh TL, Rudy TA. Analgesia mediated by a direct spinal action of narcotics. Science. 1976;192:1357–1358. doi: 10.1126/science.1273597. [DOI] [PubMed] [Google Scholar]
- 31.Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]