

Abstract
The dopamine (DA) innervation of medial prefrontal cortex (mPFC) regulates cognitive activity in a complex manner. Alterations of DA function, particularly via the DA D1 receptor class (D1R), are implicated in both schizophrenia and drug addiction, yet the precise roles of DA in modulating mPFC excitability remain unclear. We focused on DA modulation of voltage-gated K+ current (VGKC) in acutely dissociated rat mPFC pyramidal neurons. We defined three components of the whole-cell VGKC according to biophysical and pharmacological properties. The A-type current (IA), with rapid activation and inactivation kinetics, was completely inactivated by prolonged holding of the membrane potential at −40 mV and was sensitive to the K+ channel blocker 4-aminopyridine (4-AP) but not tetraethylammonium (TEA) or dendrotoxin (DTX). The slowly inactivating K+ current (ID), with rapid activation but relatively slow inactivation, was the major contributor to VGKC and was completely inactivated at −40 mV and sensitive to TEA and DTX but less so to 4-AP. The very slowly inactivating K+ current (IK) was elicited by command steps to more depolarized potentials from a prolonged holding potential of −40 mV and was sensitive to all three blockers. Stimulation of DA D2 receptors failed to alter any component of whole-cell VGKC. Stimulation of DA D1Rs selectively suppressedID, an effect mimicked by the adenylyl cyclase activator forskolin, the active cAMP analog Sp-cAMP, and the protein phosphatase inhibitor okadaic acid. Inhibition of protein kinase A (PKA) with either PKI or Rp-cAMP abolished D1R modulation. Thus, the DA D1R/cAMP/PKA signaling pathway mediates modulation of ID by DA in rat mPFC pyramidal neurons.
Keywords: dopamine, prefrontal cortex, potassium current, dopamine D1 receptors, drug addiction, schizophrenia
Introduction
The prefrontal cortex (PFC) is a major association area connected to all areas of neocortex and to various allocortical, limbic, and other regions. This provides the PFC the ability to prioritize and reference stimuli to internal representations, direct attention, and monitor temporal event sequencing (Fuster, 2001; Miller and Cohen, 2001). Dopamine (DA) is essential for proper information processing in the PFC (Goldman-Rakic et al., 2000). In particular, an optimal level of DA D1-class receptor (D1R) stimulation is required. When PFC D1R stimulation is either significantly reduced or augmented, disruption of working memory results (Sawaguchi and Goldman-Rakic, 1991; Williams and Goldman-Rakic, 1995; Zahrt et al., 1997; Seamans et al., 1998).
The mesocortical DA projection arises primarily from the ventral tegmental area (VTA) and terminates mainly on pyramidal neurons in deep layers V and VI of medial PFC (mPFC) (Bjorklund et al., 1978; Emson and Koob, 1978; Berger et al., 1991; Carr et al., 1999). Many of these neurons project to the VTA and nucleus accumbens, providing the means by which the PFC can modulate the mesoaccumbens DA system (Sesack and Pickel, 1992; Carr and Sesack, 2000). Alterations of DA function within these systems have been implicated in various neuronal disorders, particularly schizophrenia (for review, see Lewis, 1995; Knable and Weinberger, 1997) and drug addiction (for review, see Vanderschuren and Kalivas, 2000; Tzschentke, 2001).
Despite clear evidence that DA modulates behavioral output from the PFC, specific knowledge of the mechanisms by which DA alters the activity of PFC pyramidal neurons remains limited. Indeed, the electrophysiological actions of DA within the PFC have long been an enigma (for review, see Yang et al., 1999). Recent current- and voltage-clamp recordings have begun to provide clues about the targets of DA modulation. DA was reported to decrease persistent Na+ current in mPFC pyramidal neurons (Geijo-Barrientos and Pastore, 1995), although others observed the opposite effect (Yang and Seamans, 1996; Gorelova and Yang, 1997) or no modulation (Maurice et al., 2001). DA D1R stimulation was recently shown to suppress the transient Na+current in acutely dissociated rat mPFC neurons (Maurice et al., 2001) and to remove outward rectification in PFC neurons recorded in brain slices (Yang and Seamans, 1996; Gorelova and Yang, 1997). A decrease in excitability of PFC neurons has recently been attributed to DA D2-class receptor (D2R) stimulation (Gulledge and Jaffe, 1998).
Here we focused on DA receptor modulation of voltage-gated K+ currents (VGKCs) in acutely dissociated rat mPFC pyramidal neurons. VGKCs are responsible for setting the resting potential, repolarizing and hyperpolarizing the cell, and shaping voltage trajectories in the subthreshold voltage range for action potentials (Hille, 2001). As such, they may provide a specific target by which DA may regulate cognitive function.
Materials and Methods
Acute dissociation. Pyramidal neurons from mPFC layers V and VI were acutely dissociated from brain slices obtained from 4- to 5-week-old rats. Rats were anesthetized with methoxyflurane (Mallinckrodt Veterinary Incorporated, Mundelein, IL) and decapitated. Brains were quickly removed, blocked, and sliced on a DSK microslicer (Campden Instrument) in a 1–2°C sucrose solution containing (in mm): 234 sucrose, 2.5 KCl, 1 Na2HPO4, 11 glucose, 4 MgSO4, 0.1 CaCl2, and 15 HEPES, pH 7.3, 300 mOsm/l. Coronal slices (400 μm) were incubated for 1–4 hr at room temperature in a sodium bicarbonate-buffered Earle's balanced salt solution bubbled with 95%O2/5% CO2 and containing (in mm): 1 kynurenic acid, 1 pyruvic acid, 0.1 N-nitroarginine, and 0.005 glutathione, pH 7.4, 300 mOsm/l. Individual slices were placed in a Ca2+-free buffer containing (in mm): 140 Na-isethionate, 2 KCl, 4 MgCl2, 23 glucose, and 15 HEPES, pH 7.4, 300 mOsm/l, and the mPFC was isolated under a dissecting microscope. The mPFC tissue was then placed into an oxygenated, HEPES-buffered HBSS containing 1.5 mg/ml protease (type XIV) at 35°C for 30 min. The enzyme chamber also contained (in mm): 1 kynurenic acid, 1 pyruvic acid, 0.1 N-nitroarginine, and 0.005 glutathione, pH 7.4, 300 mOsm/l.
After enzymatic treatment, the tissue was rinsed several times in Ca2+-free buffer and triturated with a graded series of fire-polished Pasteur pipettes. The cell suspension was placed in a 35 mm Lux Petri dish (Nunc, Naperville, IL), which was mounted on an inverted microscope. Cells were then given several minutes to settle before recording.
Whole-cell recordings. Whole-cell recordings were performed using standard techniques. Electrodes were pulled from Corning (Corning, NY) 7052 glass (Flaming/Brown P-97 puller; Sutter Instrument Co., Novato, CA) and fire-polished (MF-83 microforge; Narishige, Hempstead, NY) just before use. The intracellular recording solution for recording outward K+ currents contained (in mm): 60 K2SO4, 60N-methyl-glucamine, 10 HEPES, 5 BAPTA, 12 phosphocreatine, 3 MgATP, 0.2 Na3GTP, 2 MgCl2, and 0.5 CaCl2, pH 7.2, 275 mOsm/l. The normal extracellular recording solution contained (in mm): 140 Na-isethionate, 10 HEPES, 12 glucose, 17.5 sucrose, 1–4 KCl, 4 MgCl2, 0.01 TTX, pH 7.35, 300 mOsm/l. All reagents were obtained from Sigma (St. Louis, MO) except ATP and GTP (Boehringer Mannheim, Indianapolis, IN); BAPTA, Sp-8-Brom-cAMP, Rp-cAMP, H8, active and inactive forskolin, and okadaic acid (OA) were from Calbiochem (La Jolla, CA). Extracellular recording solutions were applied via one of a series of four glass capillaries (∼250 μm inner diameter) in which gravity-fed flow was regulated by electronic valves (Bio-logic). Recordings were obtained with an Axon Instruments (Foster City, CA) 200A patch-clamp amplifier and controlled and monitored with a Pentium PC running pCLAMP (version 8.0) with a 125 kHz interface (Axon Instruments). Electrode resistances were ∼1–4 MΩ in bath. After formation of the gigaohm seal and subsequent cell rupture, series resistance was compensated (70–80%) and periodically monitored. Recordings were restricted to neurons with pyramidal soma and small remnants of the apical dendrites. The neuron was not included in analysis unless its whole-cell capacitance was between 8 and 14 pF and its series resistance was below 8 MΩ and steady (<10% oscillation). Recordings were performed at room temperature (22–24°C).
Data analysis. Decay time constants were determined by fitting current recording with single, double, or triple exponential functions of the form I =A0 +A1exp(−t/τ1) +A2exp(−t/τ2) +A3exp(−t/τ3).A0 toA3 are amplitude coefficients. τ1 to τ3 refer to time constants. Dose–response data were fit with a Langmuir isotherm of the form C/(C + IC50), whereC is the concentration of blockers. A two-site model of pharmacological blockade was fit withI/Imax = A/[1 + EXP(LOG(C)/B)] + (1 −A)/[1 + EXP(C)/D], whereC is the concentration of blockers, and B andD are two IC50 values. Recovery of VGKC was fit with I/Imax = A × [1 − EXP(−t/τ1)] + (1 − A)/[1 − EXP(−t/τ2)], in which Iis the current, t is time, and τ1and τ2 are the time constants of recovery from inactivation. Statistica (Statsoft, Tulsa, OK) was used for statistical analysis. Origin (Microcal Northampton, MA) was used to plot the current traces and graphs. Box–whisker was used to plot most of the graph presentation because of the small sample size. The box plot presented the distribution as a box, with the median as a central line and the hinges as the edge of the box, which divided the top and bottom halves of the distribution in two. The inner fence, started from the edge of the box, runs to the limits of the distribution, excluding outliers, which are defined as points that are two times the interquartile range beyond the inner fence. The outliers are shown as open circles. The averaged data are typically shown as mean ± SE.
Results
Three components of VGKC in mPFC pyramidal neurons
Whole-cell VGKC was elicited in mPFC neurons using multiple protocols. In a protocol with short depolarization duration, the membrane potential was held at −70 mV. A hyperpolarization prepulse (2 sec) to −100 mV was used to deinactivate inactivated K+ channels that could not be recovered by holding at −70 mV. The following 100 msec depolarization steps (from −100 to + 30 mV with 10 mV increments) elicited the whole-cell VGKC (Fig. 1A). This protocol elicited similar traces in most of the recorded neurons (n = 16 of 20) in which whole-cell current showed a rapid activation and slow inactivation. A fast inactivating component was observed in 20% of the recorded neurons (n = 4 of 20) (Fig. 2A), as discussed later. Depolarization steps inactivated whole-cell currents as shown in Figure 1B (n = 6). The membrane potential was held at −70 mV. The current that was elicited by a test step to +30 mV showed a voltage-dependent inactivation after the 600 msec prepulse (stepped from −100 to +30 mV with 10 mV increments). We chose this short prepulse to generate a pseudo-steady-state inactivation instead of true steady-state inactivation for technical convenience. The peak current–voltage (I–V) curve is summarized in Figure1C. Whole-cell VGKC in this neuron began to activate at −60 mV, suggesting that VGKC may be activated at physiological resting membrane potentials (−70 to −50 mV). A different set of protocols (Fig. 1D,E) was used to isolate individual components of VGKC by taking advantage of their inactivating kinetics. The membrane potential was held at −70 mV. The first protocol started with a hyperpolarization prepulse (4 sec) to −100 mV, which deinactivated most inactivated K+channels. The following 4 sec test steps from −70 to +30 mV (at 10 mV increments) elicited the whole-cell current (n = 5) (Fig. 1D). The second protocol started with a depolarization prepulse (10–20 sec, −40 mV), which inactivated components of the current with relatively short inactivation kinetics (several seconds). The following test steps (from −70 to +30 mV at 10 mV increments) elicited relatively small currents with little inactivation (n = 5) (Fig. 1E). We operationally defined these noninactivating traces asIK. This current did not change when extracellular Cl− was replaced by gluconate (n = 4; data not shown), excluding involvement of Cl− channels. The slowly inactivating current was obtained (Fig. 1F) after subtraction of IK from the whole-cell currents shown in Figure 1D. This current activated rapidly and inactivated slowly and was operationally termedID. The I–V relationship ofIK and ID are plotted in Figure 1G. Both currents begin to activate at approximately −40 mV (n = 5 for each group).
Fig. 1.
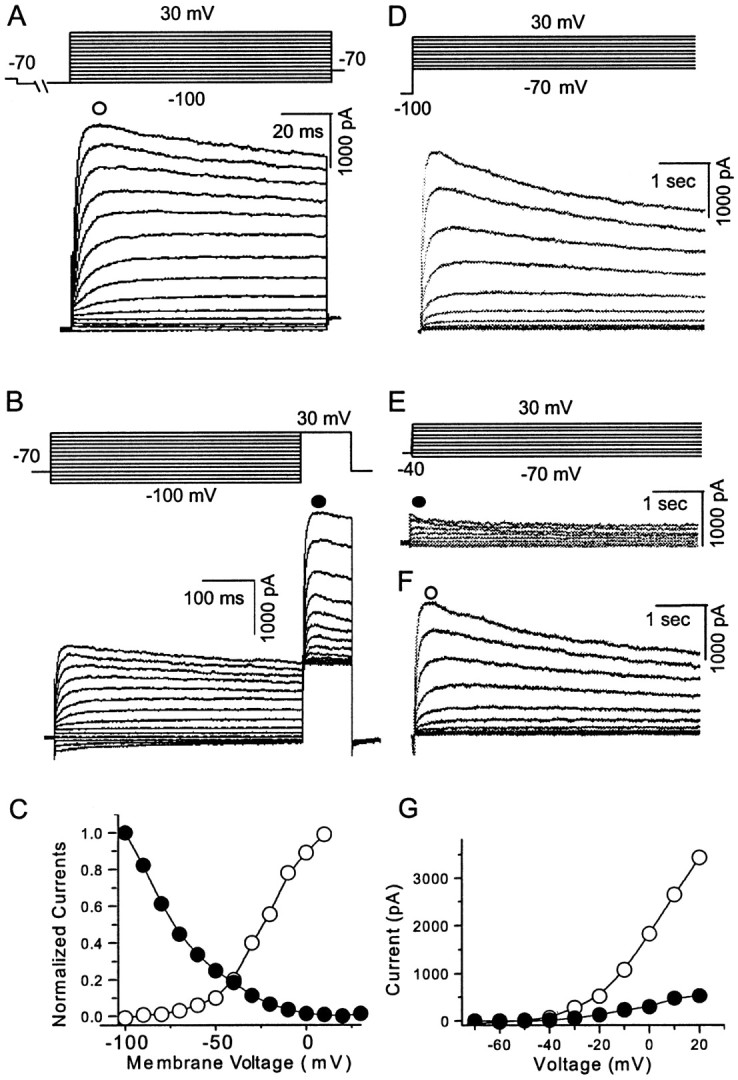
Whole-cell VGKCs in acutely dissociated mPFC pyramidal neurons. A, After holding the membrane potential at −70 mV and administering a 2 sec prepulse to −100 mV, test protocols from −100 to 30 mV with 10 mV increments elicited the whole-cell currents (n = 16 of 20).B, Whole-cell VGKCs could be partially inactivated by a depolarizing prepulse. After a holding potential at −70 mV and a 600 msec prepulse from −100 to 30 mV (10 mV increments), 100 msec test steps to 30 mV elicited currents showing a (pseudo) steady-state inactivation (n = 6). C, Current was measured at the time point indicated as an open orfilled circle in A and B. The activation and inactivation observed in A andB were plotted as I–V curves.D, Whole-cell VGKC was elicited by a similar activation protocol but with longer time course (4 sec) (n = 5). E, After the holding potential at −40 mV, the same test steps as in D elicited currents showing little inactivation. This current is operationally termedIK (n = 5).F, A slowly inactivating current was obtained by subtraction of traces in E from D. This slowly inactivating component is operationally termedID. G,I–V curves of IK andID indicate that both currents begin to activate at approximately −40 mV (n = 5). Currents were measured at the time points indicated byopen or filled circles inE and F.
Fig. 2.

A-type K+ component of whole-cell VGKC. A, A rapid inactivating component could be observed in a small population of neurons (n = 4 of 20) using the protocol described in Figure 1A. The rapidly inactivating component became more obvious at more depolarized steps. Because both activation and inactivation of this component are rapid and consistent with A-type current, it is operationally called IA.B, Activation curves for the early and later components are plotted with the values measured at the time points indicated byfilled and open circles, respectively.C1 and C2 show the activation of the same neuron before and after TEA perfusion, respectively. In most neurons, whole-cell VGKC activation protocols alone failed to show obviousIA. However, after perfusion with 10 mm TEA, IA was revealed (n = 4). D, Recovery protocol shows that IA deinactivates more rapidly than ID. After the holding potential at −70 mV, a prepulse to −40 mV inactivated most of the VGKC. A second hyperpolarizing prepulse to −100 mV recovered the K+ channels from the inactive state. The time course for this hyperpolarization prepulse varied from 10 msec to 1 sec with a nonlinear increment. IA could be recovered with a brief hyperpolarization step but contributes <25% of the whole-cell current (n = 4). E, The recovery time and the current amplitudes (measured at the time points indicated with open and filled circles in D) were plotted. The relationship of the early component (IA+ ID) could be fit with two exponentials (τ1 = 18 msec; τ2 = 890 msec). The relationship of the late component could be fit with one exponential (τ = 900 msec) (n = 5). From F1 toF4, IA was isolated biophysically (n = 4). F1, A neuron with obvious IA component was selected. By running a regular protocol, the whole-cell VGKC was elicited. This current containedIA, ID, and IK. F2, The same testing step as in F1 after a 200 msec depolarization prepulse elicited a K+trace. Because the prepulse had inactivatedIA, the following trace contained onlyID andIK. F3, TheIA component was isolated after subtraction of the trace in F2 from F2. The trace was shown in F4 in a higher magnification.
In 20% of the recorded neurons (n = 4 of 20), the protocol shown in Figure 1A elicited a whole-cell current with a rapid (several milliseconds) inactivating component (Fig. 2A). Because the kinetics of this rapidly inactivating component are similar to that of A-type currents identified previously in cortical pyramidal neurons (Foehring and Surmeier, 1993), we operationally defined it as the A-current orIA. When IAis present, it contributes only to the initiation of whole-cell VGKC because it inactivates completely within the first several 10 msec. The I–V curve of both early and late components measured from the same neuron is plotted in Figure2B. Here, both components of the current begin to activate at −40 mV. In general, the whole-cell VGKC in mPFC neurons begins to activate at −42 ± 3 mV (n = 15). A lack of obvious IA in most recorded neurons could be explained if IA is too small and is totally obscured by ID. To test this hypothesis, we applied 1 mm TEA, which is an effective blocker of non-A-type K+current, to neurons that showed no obviousIA in whole-cell VGKC (Figs.1A, 2C1). The hiddenIA was revealed (Fig. 2C2) in this neuron after ID was inhibited by TEA perfusion. A similar effect was observed in three other neurons. Obviously the limits of pharmacological isolation of K+ currents must be noted. Low concentrations of TEA (1 mm) blocked only ∼50% of ID, whereas higher concentrations (50 mm) of TEA also blockedIA (see Fig. 4A).
Fig. 4.

Pharmacological analysis also revealed multiple components of the whole-cell VGKC. A modified step to 10 mV elicited whole-cell VGKC when the holding potential was −100 mV but elicitedIK when the holding potential was −40 mV. ID was obtained with subtraction of IK from whole-cell VGKC.A1 and A2 show thatID andIK were blocked by different concentrations of TEA. A3, A4, The dose–response curves for TEA blockade of bothIK (A3) andID (A4) are best fit with two Langmuir isotherms, suggesting two TEA-sensitive components of these currents. B1 and B2show that ID andIK are blocked by different concentrations of 4-AP. B3, B4, The dose–response curve for 4-AP block ofIK (B3) is best fit by two Langmuir isotherms, suggesting at least two 4-AP-sensitive components of this current, whereas ID(B4) appears to possess only one 4-AP-sensitive component. C1 and C2 show thatID andIK are blocked by different concentrations of DTX. C3, C4, The dose–response curves for DTX blockade ofIK (C3) andID are best fit by two Langmuir isotherms, suggesting at least two DTX-sensitive components of these currents. For all graphs, three to six cells are represented at each data point.
Biophysical isolation of IA andID
To characterize further IA in mPFC neurons, biophysical methods were used to isolateIA and ID. Deinactivation by hyperpolarization was shown by a recovery protocol in which the membrane potential was held at −70 mV. A 4 sec depolarization step to −40 mV inactivated bothIA and ID. The membrane potential was then stepped to the recovery voltage (−100 mV) for varying durations (5, 10, 20, 50, 100, 200, 500, 1000 msec). A test step to +30 mV elicited the whole-cell VGKC (Fig.2D). IA was recovered from inactivation with relatively short hyperpolarization steps and contributed only slightly to the whole-cell VGKC. Recovery ofID required relatively long hyperpolarization steps. After full recovery,ID totally obscuredIA. The currents were measured from 10 msec after onset of the current, at whichIA reached its peak, as well as from 400 msec after onset current, at which IA was completely inactivated. The relationships between these two currents and their recovery time are plotted in Figure 2E. The early current from this cell was well fit with two exponentials with time constants of 18 and 890 msec, respectively. The late current was well fit with one exponential with a time constant of 900 msec, consistent with the 890 msec time constant during the early current. The averaged time constants are 39 ± 10 msec (n = 5) and 925 ± 207 msec (n = 5). Clearly,IA and IDcould be distinguished by different recovery kinetics.
Another protocol was also used to isolateIA from whole-cell VGKC by taking advantage of the rapid inactivation of IA(Fig. 2E). In the first part of this protocol, the test step to +30 mV after prolonged holding at −100 mV elicited whole-cell VGKC, which includes IA,ID, and IK(Fig. 2F1). In the second part of this protocol, a 200 msec prepulse to 30 mV fully inactivatedIA but was short enough to maintain most of ID and IKbecause of their slow inactivation. So the following test step from −70 to +30 mV elicited a current including onlyID and IK(Fig. 2F2). Subtraction of whole-cell K+ current composed ofID and IK(Fig. 2F2) from the whole-cell K+ current composed ofIA, ID, andIK (Fig. 2F1) resulted in pure IA (n = 4) (Figs.2F3, 4).
We have described three components of the whole-cell VGKC,IA, ID, andIK, in mPFC neurons, similar to that described in sensorimotor cortex pyramidal neurons (Foehring and Surmeier, 1993). Another way to distinguish different components in a combined current is to analyze current decay (Hoshi et al., 1990). An outward current trace was elicited by a 200 msec step to +30 mV from a holding potential of −100 mV (Fig.3A, inset). The point at which VGKC starts to decay is defined at 10 msec after the onset of peak current. The digitized current was taken from the 10 msec point to total decay. Using this protocol, the decay could be fit by two exponentials with time constants (τ) of 15 and 250 msec. The two components became more obvious when the current was converted to logarithmic values (Fig. 3A). The late current was extrapolated and subtracted from the early current. The VGKC was peeled into two linear exponentials in the logarithmic coordinates (Fig.3A). Another protocol with similar activation steps but longer duration (4 sec) was used to generate the decay current (Fig.3B, inset). The decay was analyzed with a similar peeling process, and two decay time constants were obtained (200 msec and 2.9 sec). No faster τ could be obtained because the step duration is too long to catch short inactivating (τ = 15 msec) components. The fast τ (200 msec) in Figure 3B is consistent with the slow τ (250 msec) in Figure 3A, which suggests that they are the same component. The less than perfect consistency may be attributable to the heterogeneous expression of VGKC but is more likely the result of different time courses used to truncate the current. In Figure 3B, the decay of the 250 msec component occupies the entire time course of the analysis and should be more accurate.
Fig. 3.

Multiple components of whole-cell VGKC revealed by inactivation kinetics. A, A cell with obviousIA was selected for this example. Whole-cell VGKC was elicited during a 200 msec step to 30 mV from −100 mV, as shown in the inset. The current amplitude was digitized and put in a LOG coordinate. The late part, but not the early part, of the current displayed a linear relationship with a time constant of 250 msec. This linear relationship was then extrapolated forward and subtracted from the early part of the digitized current, which resulted in another linear relationship with a time constant of 15 msec. This exponential peeling of decay within the time course demonstrated two components (n = 7).B, Whole-cell VGKC was elicited during a longer (4 sec) step to 30 mV from −100 mV, as shown in the inset. Exponential peeling of the components of decay of this trace revealed two time constants of 200 msec and 2.9 sec (n = 6).C, Whole-cell VGKC was elicited during a 40 sec step to 30 mV from −100 mV, as shown in the inset. Exponential peeling of the components of decay of this trace revealed two time constants of 3.4 and 25 sec (n = 3).D, A protocol as shown in the inset was used to elicit the K+ tail current. Exponential fitting revealed two components in the tail current (using the 40 mV step as the example) with time constants of 8 and 43 msec (n = 4). Taken together, these data support four distinct inactivation time constants, one consistent withIA, one consistent withIK, and two as distinct components ofID.
Another similar protocol but with a much longer step (40 sec) generated a VGKC trace that was well fit with two exponentials (τ = 3.4 and 25 sec, respectively). Again the 3.4 sec τ in Figure3C is consistent with the 2.9 sec τ in Figure3B. Taken together, there are four components according to the time constants of current decay: 12 ± 3 msec (n = 7), 220 ± 26 msec (n = 6), 3.4 ± 0.5 sec (n = 6), and 33 ± 7 sec (n = 3). The strategy to isolate these four time constants is to progressively increase the voltage step duration. When the step was much longer than a time constant, the component to this τ decayed too quickly to be taken into account in such a long step. On the contrary, the decay of the component with matched τ that predominated in this analysis can be calculated accurately. The rapidly inactivating (12 ± 3 msec) component is consistent withIA. The very slowly inactivating (33 ± 7 sec) component is consistent with IK. We propose that there are two components inID, with decay τ of 220 ± 26 msec and 3.4 ± 0.5 sec, respectively. The pseudo-steady-state inactivation data (see Fig. 5D) indicate the likelihood that there are at least two components in ID, consistent with our hypothesis.
Fig. 5.
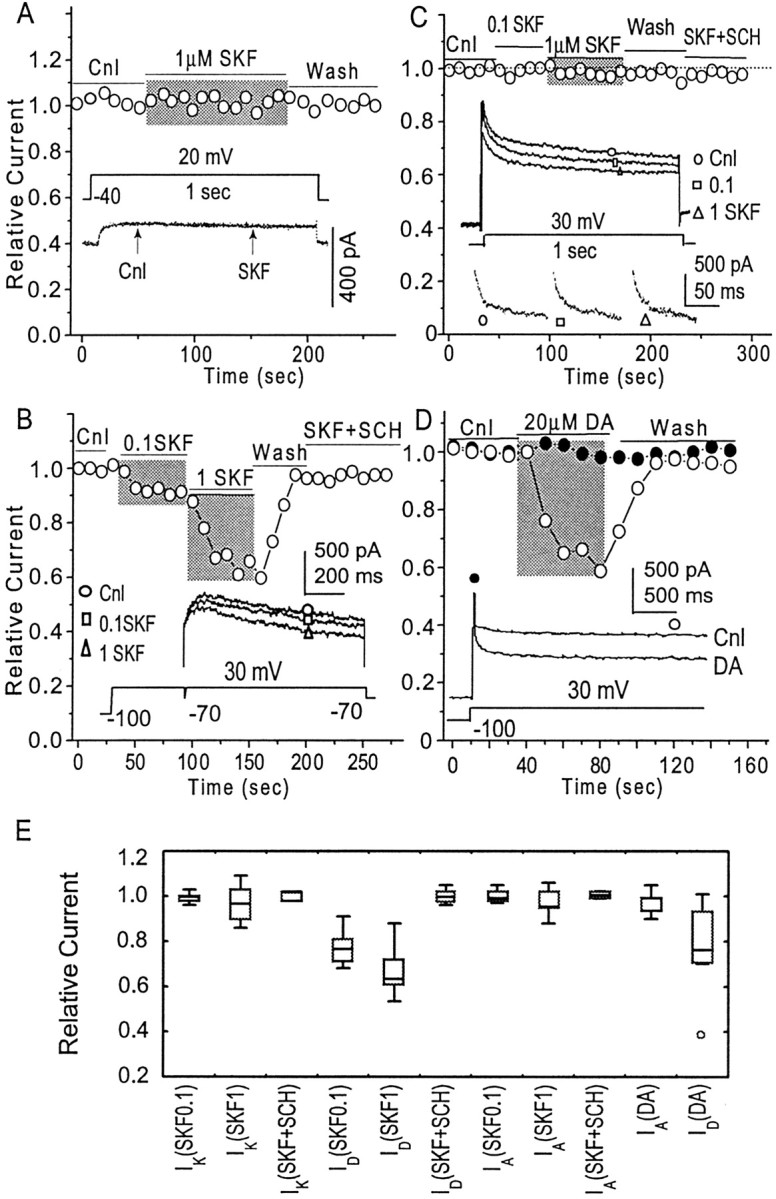
Dopamine D1 receptor-mediated inhibition of VGKC.A, Activation of D1 receptors by the selective agonist SKF 81297 (0.1 and 1 μm) failed to alterIK (n = 5). Theinset shows two traces ofIK in the presence and absence of SKF 81297 (0.1 μm), clearly indicating the lack of effect of D1 receptor stimulation on this current. B, Activation of D1 receptors with SKF 81297 produced a dose-dependent suppression of ID(n = 25), which was prevented by the D1 receptor antagonist SCH 23390 (n = 5). Inset traces clearly show the dose-dependent modulation.C, Activation of D1 receptors by SKF 81297 (1 μm) failed to alterIA (n = 5).Inset traces show the overall effect of SKF 81297 and the effect on the isolated IA. D, The late component of VGKC (mostlyID) was measured at the open circle in inset; peak VGKC (mostlyIA) was measured at the beginning of the current, as indicated by the filled circle ininset. Perfusion of DA (20 μm) failed to alter peak VGKC (mostly IA) (n = 5; filled circles) but significantly inhibited the late component of VGKC (mostlyID) (n = 6;open circles). E, The effects of DA and SKF 81297 on the various components of VGKC are summarized in a box–whisker plot. Concentrations of 0.1 μm SKF 81297 (n = 5), 1 μm SKF 81297 (n = 5), or 0.1 μm SKF 81297 with 1 μm SCH 23390 (n = 3) failed to alter the amplitude of IK. Perfusion of 0.1 μm SKF 81297 produces a 19 ± 3% inhibition of ID(n = 15). Perfusion of 1 μm SKF 81297 produces a 34 ± 17% inhibition ofID (n = 10). Perfusion of 0.1 μm SKF 81297 together with 1 μm SCH 23390 does not alter the amplitude ofID(IRelative = 0.96 ± 0.03;n = 5). Perfusion of 0.1 (n = 3) or 1 μm SKF 81297 (n = 5), or SKF 81297 (0.1 μm) with SCH 23390 (1 μm) (n = 3), does not alter the amplitude of IA. Perfusion of DA does not alter IA(IRelative = 0.93 ± 0.09;n = 5) but inhibitsID(IRelative = 0.78 ± 0.06;n = 6.).
We next examined current decay with a tail current protocol (Fig.3D). This experiment was performed in 40 mm external K+ and Cl− was replaced with methanesulfonate. The membrane potential was held at −100 mV. The tail currents were elicited by stepping to −70 mV after 10 msec voltage commands to steps from −60 to +30 mV with 10 mV increments. In cells withIA, the tail current was well fit by two exponentials with time constants of 8 and 43 msec (Fig. 3D). Similar 2-τ deactivation was also observed in three other neurons (4 and 19 msec; 9 and 28 msec; 7 and 33 msec), suggesting that there are at least two components involved in the tail current. Note that it is difficult to correlate the deactivation τ values with inactivation τ values.
Pharmacological isolation of K+ currents
The biophysical studies above suggest that mPFC pyramidal neurons express at least four components of the whole-cell VGKC. Are they attributable to different channels or to similar channels with different gating properties? The following pharmacological studies attempted to address this question.
Three pharmacological agents, TEA, 4-AP, and DTX, were applied toID and IKcurrents. IK was elicited by a 100 msec step to +10 mV from a holding potential of −40 mV (Fig.4A2,B2,C2). A whole-cell VGKC was elicited by a similar protocol except that the prepulse was −100 mV. ID was generated by subtraction of IK from this whole-cell current (Fig. 4A1,B1,C1).
IA was TEA insensitive (Fig.2C2), but IK andID were reversibly blocked by TEA across a broad concentration range (Fig.4A3,A4). The relationship between relative current (I/Imax) ofIK and LOG[TEA] was well fit with a two-site model (Langmuir isotherm) with IC50values of 8 μm and 4 mm(Fig. 4A3). The dose–response curve forID was also well fit with a two-site model with IC50 values of 25 μmand 10 mm (Fig. 4A4).IA in mPFC pyramidal neurons was sensitive to 4-AP and blocked by 60% at low (1–5 mm)concentrations (data not shown). Both IKand ID currents were also sensitive to 4-AP in our study (Fig. 4B3,B4). The relationship between relative current and LOG [4-AP] forIK was well fit with a two-site model with IC50 values of 60 μm and 6 mm (Fig. 4B3). The effects of 4-AP on ID currents are relatively limited. The maximal blockade was ∼35% when 10 mm 4-AP was applied. The dose–response curve was well fit with a single-site model with an IC50 of 30 μm (Fig. 4B4).IA was insensitive to low (1–10 nm) concentrations of DTX, but bothID and IKwere blocked by DTX across a broad concentration range. The dose–response curve for IK was best fit with a two-site model with IC50 values of 800 pm and 52 nm (Fig.4C3). The does–response curve forID was well fit with a two-site model with IC50 values of 200 pm and 40 nm (Fig. 4C4). The two IC50 values usually suggest two binding sites in the channels or two kinds of channels. The similar pharmacology of VGKC (multiple binding sites or multiple channels) was reported previously in sensorimotor cortical neurons (Foehring and Surmeier, 1993).
DA modulation of VGKC
The whole-cell VGKC is composed of three components,IA, ID, andIK, each of which display different biophysical and pharmacological properties. In this section of the study, we examined DA receptor modulation of each VGKC component. In this set of simplified protocols, IK was elicited when the membrane potential was stepped to +30 mV (2 sec) from prolonged (>10 sec) holding at −40 mV (Fig.5A). The current amplitude was measured at the time point 200 msec after current initiation. To elicitID, the membrane potential was held at −100 mV. The prepulse (100 msec) to +30 mV was used to inactivate available IA. After a brief hyperpolarization (1 msec) to −70 mV, the membrane potential was again stepped to +30 mV, which elicited the combined current ofID and IK(Fig. 5B). Because IK is so small compared with ID, and becauseIK is insensitive to D1R stimulation (see below), the combined current is regarded asID in the following functional studies. The current amplitude was measured at the time point 100 msec after onset. The detailed isolation of IA was described in Figure 2F. PeakIA was measured and was regarded asIA amplitude. The relative current, orIRelative, is defined as current amplitude during perfusion of drug divided by current amplitude in control (IPerfusion/IControl).
IK was insensitive to D1R stimulation. Activation of D1Rs with the selective agonist SKF 81297 (0.1 and 1 μm) failed to induce any change in current amplitude of IK (n = 5 for each concentration) (Fig. 5A,E). However, in the same neuron, SKF 81297 induced a significant inhibition of ID amplitude (Fig. 5B). The D1R-mediated inhibition of ID was dose dependent. Perfusion of neurons with 0.1 μm SKF 81297 induced 19 ± 3% inhibition ofID amplitude (n = 15;p < 0.05; paired t test between control and perfusion of SKF 81297,) whereas perfusion with 1 μm SKF 81297 induced 34 ± 5% inhibition of ID amplitude (n = 10;p < 0.05; paired t test between control and perfusion of SKF 81297) (Fig.5B,E). The D1R-mediated inhibition (by SKF 81297, 0.1 μm) was reversible after wash and was antagonized by the DA D1R-selective antagonist SCH 23390 (1 μm;n = 5;IRelative = 0.98 ± 0.02) (Fig.5B,E). In most neurons (6 of 7),ID was also inhibited by the partial DA D1R agonist SKF38393 (0.1 μm; data not shown). In another neuron, whole-cell VGKC was elicited by a voltage step (1 sec) to +30 mV from a holding potential of −100 mV (Fig.5C, inset). An obvious inhibition in the late component (ID) of VGKC could be observed when the neuron was perfused with SKF 81297 (0.1 and 1 μm) (Fig. 5C, inset).IA from this neuron was isolated from each perfusion condition (Fig. 5C, inset), displaying no response to D1R stimulation (Fig. 5C). In most neurons, no alteration in IA amplitude was observed when D1Rs were stimulated by SKF 81297 at concentrations of 0.1 μm (IRelative= 1.03 ± 0.04; n = 5,) or 1 μm (IRelative = 0.95 ± 0.05; n = 5,) (Fig.5C,E).
No obvious alterations in whole-cell VGKC (or either component of VGKC) were observed when D2Rs were stimulated by the selective D2R agonist quinpirole (0.1 and 1 μm; n = 9; data not shown). We next examined how DA, the endogenous agonist, affects VGKC. Whole-cell VGKC was elicited when the membrane potential was stepped to +30 mV (4 sec) from a holding potential of −100 mV. In the neuron shown in Figure 5D, inset, the very rapid inactivating component (IA) was observed. The amplitude of the peak current (mostlyIA) was measured at the time point 10 msec, as indicated by the filled circle. The amplitude of the late component (mostly ID) was measured at the time point 3 sec, as indicated by the open circle in Figure5D, inset. Perfusion of neurons with DA (20 μm) did not alter peak VGKC (mostlyIA) (IRelative = 0.93 ± 0.05;n = 5), but induced a washable inhibition of the late component (mostly ID) (IRelative = 0.78 ± 0.06;n = 6) (Fig. 5D,E). A summary of DA-induced inhibition of VGKC (Fig. 5E) indicates that (1) activation of D1R does not affectIK and IA but reliably inhibits ID, and (2) DA inhibits VGKC via its action on D1Rs.
We next examined the effects of DA on the I–V curve of whole-cell VGKC, which could be obtained by the protocols described in Figure 1C. DA-induced (30 sec perfusion) inhibition of current amplitude could be observed in both activation traces and pseudo-inactivation traces (Fig.6A). DA did not alter activation but shifted inactivation in the hyperpolarizing direction (Fig. 6B) [half-inactivation voltage (Vh) of VGKC shifted 5.4 ± 1.2 mV in the hyperpolarizing direction by DA; n = 3]. We argued above that DA modulates VGKC via its action on D1Rs, so it was not surprising that stimulation of D1Rs by SKF 81297 induced a similar left-shift of inactivation in VGKC, but activation remained intact (Fig. 6C). It could also be observed that two components (two Boltzmann equations) were involved in the inactivation, which was interpreted as two components in the earlier section of this study. It is worth mentioning that the variability ofVh among neurons is high in our preparations, which may be attributable to the diverse expression of K+ channels/currents, the inadequate voltage clamp, the incomplete compensation of junction potential, or other factors. However, in all recorded neurons theVh consistently exhibited a left-shift (ΔVh = 5.3 ± 1.1 mV;n = 6) in responding to D1R activation, suggesting that D1R suppression of VGKC might be mediated by the enhanced inactivation of K+ channels.
Fig. 6.
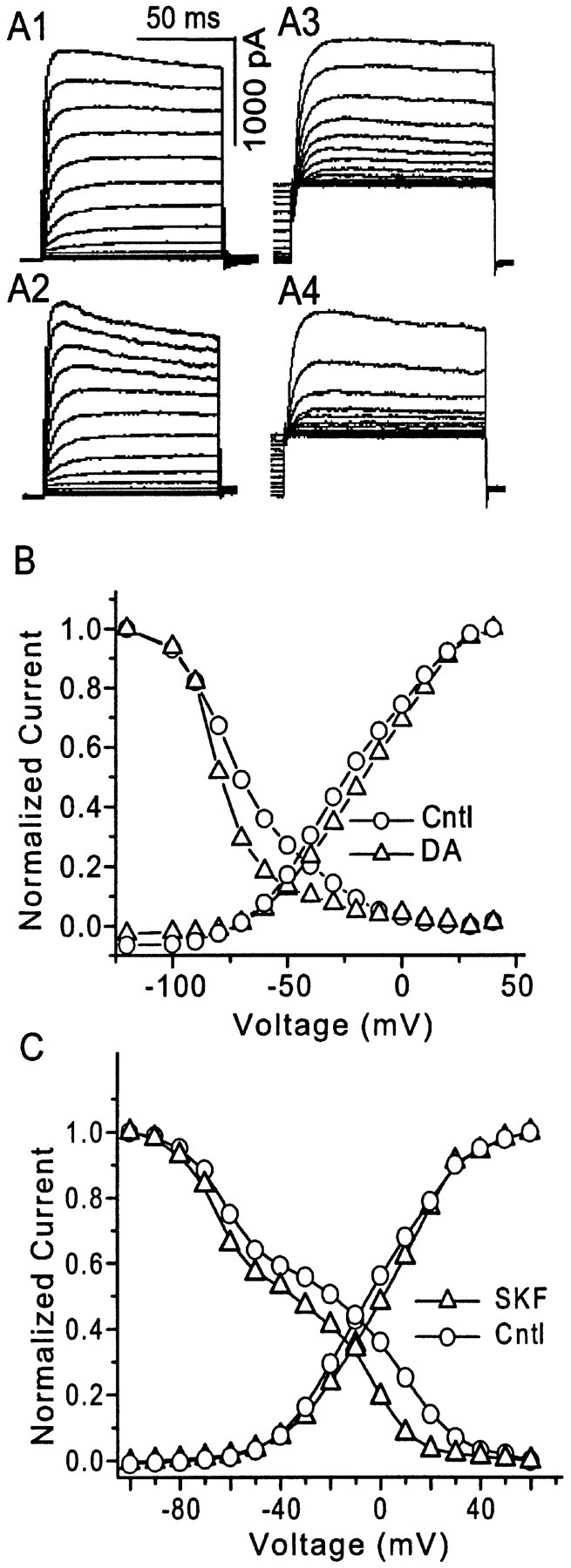
The effects of dopamine (DA) and SKF 81297 on the VGKC I–V relationship.A1, A2, The activation protocol (as in Fig. 1A) elicited VGKC before and during perfusion of DA (20 μm). A3,A4, The inactivation protocol (as in Fig.1B) elicited VGKC in the same pyramidal mPFC neuron before and during perfusion of 20 μm DA.B, The I–V curve for whole-cell VGKC from the neuron shown in A is plotted. DA clearly produces a hyperpolarizing shift in the inactivation curve of VGKC in mPFC neurons (n = 3 of 3). C, A similar shift of the inactivation curve was observed during perfusion of the D1 receptor agonist SKF 81297 (0.1 μm). The inactivation of these neurons could be best fit with two Boltzmann equations, suggesting that two components with distinct inactivation kinetics are involved in the whole-cell VGKC.
Mechanisms responsible for D1R modulation ofID
D1Rs are positively coupled to the adenylyl cyclase/cAMP/protein kinase A (PKA) signal transduction pathway (Stoof and Kebabian, 1984). By activation of this pathway, D1Rs have been shown to suppress whole-cell Na+ current in ventral and dorsal striatal medium spiny neurons (Surmeier et al., 1992; Zhang et al., 1998), PFC pyramidal neurons (Maurice et al., 2001), and hippocampal pyramidal neurons (Cantrell et al., 1997, 1999). To determine whether this signal transduction pathway is also responsible for the D1R modulation of VGKC, we stimulated or inhibited each component of this signal cascade and examined the role of this pathway in D1R-mediated modulation of VGKC.
Perfusion of 20 μm 8-Br-Sp-cAMP, a membrane-permeable cAMP analog with strong PKA activating effect, induced a significant inhibition of the late component of whole-cell VGKC (mostlyID) but did not suppress the fast-inactivating component (mostly IA) (Fig. 7A). Whole-cell VGKC amplitude (measured at the end of the traces) was decreased by 26 ± 6% (n = 5; p < 0.05; pairedt test between control and perfusion of cAMP) (Fig. 7A,F). Perfusion of 10 μm forskolin, a membrane-permeable activator of AC, inhibited VGKC by 22 ± 4% (n = 6), whereas incubation with H8 (10 μm), the nonspecific PKA inhibitor, enhanced VGKC by 21 ± 5% (n = 5) (Fig. 7B,F). Basal VGKC could also be enhanced by PKI, a more specific PKA inhibitor (n = 2; data not shown). OA was used to examine whether the K+ channel is the direct substrate of PKA. OA is a strong inhibitor of multiple phosphatases, inhibition of which blocks dephosphorylation of VGKC channels, resulting in more phosphorylated channels. Perfusion of 100 nm OA suppressed VGKC (17 ± 4%;n = 4) (Fig. 7C,F). The OA-induced inhibition of VGKC could not be washed out because of its irreversible binding to phosphatases. The above experiments suggest that (1) sequential activation of adenylyl cyclase, cAMP, and PKA is sufficient for VGKC inhibition, (2) constitutive activity of PKA tonically inhibits VGKC, and (3) K+channels underlying VGKC exist in a dynamic balance between phosphorylation and dephosphorylation states and can be modulated in either direction.
Fig. 7.
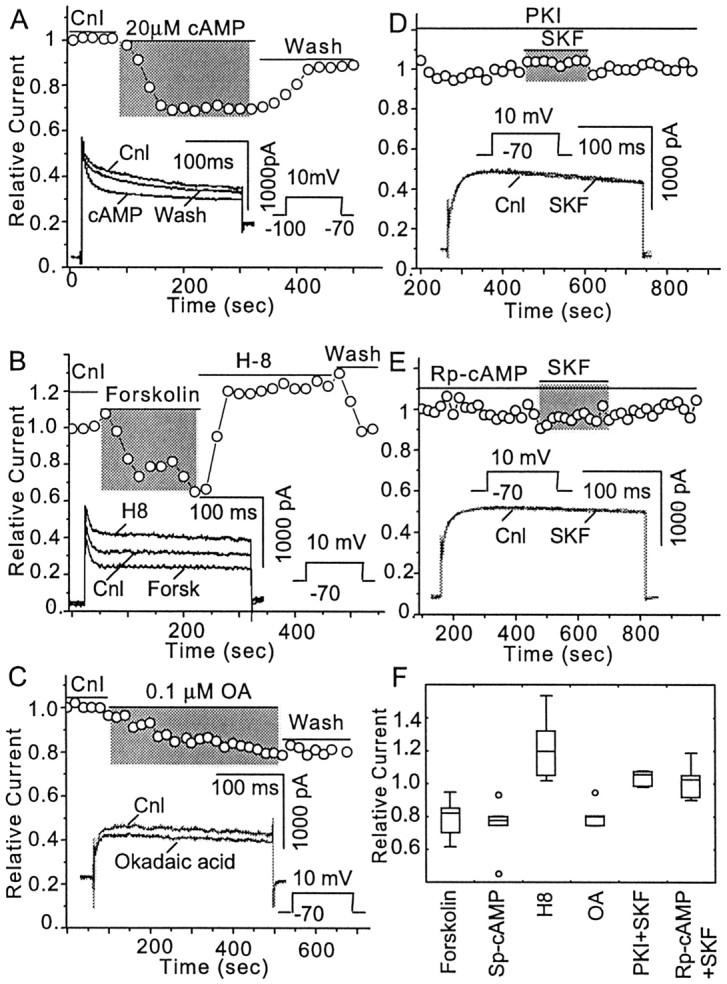
The adenylyl cyclase/cAMP/protein kinase A (PKA) signaling system mediates DA D1 receptor inhibition ofID. A, Sp-cAMP (20 μm), a membrane-permeable cAMP analog that activates protein kinase A directly, inhibitedID (n = 6) but notIA. B, Forskolin (10 μm), a membrane-permeable stimulator of adenylyl cyclase, also inhibited ID(n = 6). Perfusion of the same neuron with H8 (10 μm), a membrane-permeable PKA inhibitor, increasedID (n = 5).C, The membrane-permeable protein phosphatase inhibitor okadaic acid (100 nm) inhibitedID (n = 4).D, The PKI subunit (1 U/ml) was included in the pipette solution and diffused into the cell when the whole-cell configuration was formed. Once the cAMP/PKA pathway was fully blocked by PKI, indicated as the VGKC reached steady state, the inhibitory effect of SKF 81297 (1 μm) was also blocked (n = 4). E, Perfusion of Rp-cAMP (50 μm), a membrane-permeable cAMP analog with strong inhibitory effects on PKA (n = 5), also blocked the inhibition by SKF 81297 (1 μm). F, This plot summarizes the contribution of each component of the AC/cAMP/PKA pathway to D1R-mediated modulation of VGKC. Perfusion of Sp-Br-cAMP suppresses VGKC by 26 ± 6% (n = 5). Perfusion of forskolin suppresses VGKC by 22 ± 4% (n = 6), whereas perfusion of H8 enhances VGKC by 21 ± 5%. Perfusion of okadaic acid suppresses VGKC by 17 ± 4% (n = 4). VGKCs become resistant to the stimulation of D1R when the cell is treated with either PKI (IRelative = 1.04 ± 0.05;n = 4) or Rp-cAMP (IRelative = 1.02 ± 0.05;n = 5).
Other signal transduction cascades are also reported to participate in D1R signaling (Undie et al., 2000). Is the AC/cAMP/PKA pathway also necessary in mediating D1R effects on VGKC? The next two experiments addressed this question. The pipette solution contained 1 U/ml PKI subunit, a PKA inhibitor. After cell rupture, PKI diffused into the neuron, blocking PKA-mediated phosphorylation. After full blockade of PKI was obtained (monitored as the amplitude of VGKC became stable), perfusion with SKF 81297 no longer reduced VGKC (IRelative = 1.04 ± 0.05;n = 4) (Fig.7D,F). Similar experiments were performed with another PKA inhibitor, Rp-cAMP, the nonfunctional cAMP analog that neutralizes intracellular cAMP activity so that PKA activity is blocked. Perfusion of Rp-cAMP (50 μm) initially induced an enhancement of VGKC (data not shown). After the PKA is fully inactivated, which was indicated as VGKC became stable, SFK 81297 failed to suppress VGKC (IRelative = 1.02 ± 0.05;n = 5) (Fig.7E,F). In sum, the above results suggest that the AC/cAMP/PKA pathway is essential to D1R modulation of VGKC.
Discussion
The effects of DA on mPFC pyramidal neurons have remained an enigma for >25 years (for review, see Yang et al., 1999). Although recent studies have begun to elucidate the means by which DA modulates neuronal excitability in subcortical areas (for review, see Nicola et al., 2000), such clarification has been slower to emerge in the mPFC, perhaps because previous in vivo and in vitrocharacterizations have been even more controversial than long-standing disputes regarding striatal neurons. Our whole-cell voltage-clamp studies provide the first direct demonstration of DA receptor modulation of isolated VGKCs in pyramidal neurons of the mPFC. Although this technical approach has the disadvantages of a lack of neuronal processes as a result of the dissociation process and a potential disruption of cytosolic messenger systems, it provides excellent voltage control and easy access to pharmacological agents, thereby allowing isolation of whole-cell VGKC and the ability to examine the effects of DA on individual components of VGKC. Our results clearly show that DA D1Rs, but not D2Rs, selectively decrease a slowly inactivating K+ conductance via the classical D1R/AC/cAMP/PKA signaling pathway.
Biophysical and pharmacological dissection of VGKCs
We identified at least three major contributors to whole-cell K+ current in mPFC pyramidal neurons,IA, ID, andIK, as indicated by both biophysical and pharmacological dissection. These findings are consistent with previously described K+ currents in sensorimotor cortical neurons (Foehring and Surmeier, 1993). Although four time constants were obtained from studies of current decay, it is difficult to know whether three or more types of K+ channels are responsible for these components as revealed by different biophysical properties. Multiple K+ channels can express homogeneous biophysics (Coetzee et al., 1999), and identical channels can express heterogeneous biophysics, as a result of post-translational modification and interactions with auxiliary subunits (Heinemann et al., 1996; Holmqvist et al., 2001). Here we focused on current type as opposed to distinct channel proteins.
The fast decaying component of the whole-cell VGKC (τ = 12 ± 3 msec) is kinetically consistent with the fast inactivating component IA, whereas the very slow decay component (τ = 33 ± 7 sec) is kinetically consistent with IK. The other two components (τ = 220 ± 26 msec and 3.4 ± 0.5 sec) are consistent with the slowly inactivating componentID. Although the two components ofID could not be isolated with activation protocols (Fig. 1), they were dissociable during inactivation in some neurons (Fig. 5D). This conclusion is consistent with recent findings in pyramidal neurons from several cortical regions, for example, in young neocortical neurons (Korngreen and Sakmann, 2000; Bekkers and Delaney, 2001), visual cortical neurons (Locke and Nerbonne, 1997), neocortex layer I neurons (Zhou and Hablitz, 1996), and sensorimotor cortical neurons (Foehring and Surmeier, 1993).
In dissociated mPFC neurons, IA was often too small to be detected. Similar results were observed in acutely dissociated sensorimotor cortical pyramidal neurons (Foehring and Surmeier, 1993), cultured hippocampal pyramidal neurons (Murakoshi and Trimmer, 1999), or dissociated striatal spiny neurons (Surmeier et al., 1991), but in dissociated rat hippocampal neurons, A-current was shown to prevail, and it contributed 61% of total K+ currents (Martina et al., 1998). The difference in expression of A-currents could be interpreted as cellular diversity between hippocampal and mPFC pyramidal neurons. An alternative interpretation is developmental regulation. Sensorimotor, striatal, and mPFC neurons that exhibited small A-current were dissociated from mature animals (4 weeks or older), whereas hippocampal neurons that displayed predominant A-current were obtained from young rats (11–16 d) (Martina et al., 1998). Both cultured and premature neonatal striatal neurons exhibit large A-currents, whereas in mature striatal neurons, the A-current is much less prominent (Surmeier et al., 1991). Thus, different developmental ages could be responsible for expression of different channel proteins.
Pharmacological studies confirm our biophysical findings of multiple currents involved in the whole-cell VGKC.IK displayed two IC50 values for all three blockers (4-AP, TEA, and DTX) that we used. ID also displayed two IC50 values for TEA and DTX. This suggests that both ID andIK contain at least two types of currents.ID displayed only one IC50 for 4-AP, but the maximum blockade by 4-AP is only ∼50%, indicating that there is at least one component ofID that is 4-AP insensitive. In principle, the two types of currents, identified by the two IC50 values, likely result from two distinct types of channels, but this prediction only works well in homogenous expression systems and may not hold in electrophysiological studies of native neurons, in which two different IC50 values are not sufficient to conclude that there are two different channel types (Kirsch and Drewe, 1993). The inactivation analysis shows that there are two components (two τ values) in ID, which is consistent with the pharmacological results (two IC50 values). There is no direct biophysical evidence indicating the composition (two components) of IK, but the pharmacological data show two components (two IC50 values). This could indicate that IK contains two components that share similar biophysical properties but differ in binding to pharmacological agents. However, because the decay time constant of the slow component in ID is relatively large (3.4 sec), in the protocol that we used to isolateIK, prolonged holding (10–20 sec) at −40 mV may not totally inactivate ID, resulting in contamination of IK. Therefore, the alternative interpretation is that the uninactivated slow component of ID may contribute to one of the IC50 values ofIK. Both biophysical (two τ values) and pharmacological (two IC50 values) analyses argue that there are two components in ID, so in theory the correlation of the τ values with the IC50 values could be obtained by totally blocking the component with small IC50 (with low concentration) and calculating the τ of the rest of the current. However, technical limitations prevented us from resolving this question. In a small number of our experiments, low concentrations of the blockers never “totally” blocked the one component, and when we increased concentrations, the other component was also considerably affected.
Dopamine modulation of VGKC
The mesocortical DA input arises from the VTA and predominately terminates on pyramidal neurons in deep layers of mPFC (Bjorklund et al., 1978; Emson and Koob, 1978; Berger et al., 1991; Carr et al., 1999). Both D1Rs and D2Rs are expressed in pyramidal neurons, although the density of D2R appears to be considerably lower (Gaspar et al., 1995; Vincent et al., 1995). It is widely accepted that D1Rs positively couple to the AC/cAMP/PKA signal transduction pathway, whereas D2Rs inhibit this transduction system (Stoof and Kebabian, 1984; Sibley and Monsma, 1992). Thus, we expected VGKC to be modulated in opposite ways by D1Rs and D2Rs. Surprisingly, most recorded neurons did not respond to D2R agonists. Does this indicate a lack of D2R expression in mPFC pyramidal neurons? Apparently not, because our parallel studies demonstrate that when inwardly rectifying K+ current (IRKC) and VGKC were sequentially elicited in the same mPFC neuron, D2R agonists modulate only IRKC but do not affect VGKC (Dong and White, 2001), suggesting selective targeting of specific K+channels for D2R activation.
D1R activation suppresses only ID in mPFC neurons, suggesting that the channel(s) conductingID is selectively targeted by D1Rs as opposed to channels conductingIA andIK. A similar inhibition of VGKC induced by PKA activation was observed in the soma of striatal medium spiny neurons (Surmeier and Kitai, 1993) as well as in dendrites of hippocampal pyramidal neurons (Hoffman and Johnston, 1999). Delayed rectifier K+ current (IK), as well as the late component of ID, is primarily responsible for action potential duration, whereas transient K+ currents,IA and the early component ofID, preferentially control subthreshold responses to excitatory inputs and firing frequency (Debanne et al., 1997; Hille, 2001). Consistently, intracellular recordings from mPFC slices show that perfusion of either D1R-selective agonists or DA itself, but not D2R-selective agonists, reduces first spike latency, lowers firing threshold, and increases firing frequency, but does not alter the shape of the action potential (Yang and Seamans, 1996).
Mechanism of DA D1R modulation
Our studies indicate that D1Rs modulateID by activation of the AC/cAMP/PKA signal transduction pathway. Low concentrations (0.1 μm) of D1R agonists induced similar inhibition, which was abolished by the D1R-selective antagonist SCH 23390 (1 μm). Increasing intracellular cAMP levels with either forskolin or Sp-cAMP mimicked D1R-mediated inhibition ofID, suggesting that stimulation of the AC/cAMP/PKA pathway is sufficient to modulateID. Neutralization of either cAMP or PKA, with Rp-cAMP and PKI, respectively, abolished D1R-induced inhibition of ID, suggesting that the AC/cAMP/PKA pathway is essential in D1R-mediated modulation. Phosphorylation is a common mechanism by which membrane channel proteins are subject to modification. Indeed, phosphorylation-induced inhibition of several types of K+ channels has been reported in many other systems (for review, see Levitan, 1988).
The actions of DA on mPFC pyramidal neurons have long been controversial. Why does DA increase excitability in some situations while decreasing it in others? Our study suggests that DA can influence the activity of mPFC pyramidal neurons by modulation of VGKCs. Activation of DA D1Rs can increase neuronal excitability by inhibition of VGKC, whereas both D1R and D2R stimulation can suppress IRKC (Dong and White, 2001), but DA may also decrease whole-cell sodium current to decrease excitability (Maurice et al., 2001). Clearly, the actions of DA on mPFC pyramidal neurons depend on the timing and strength of synaptic inputs as well as on the membrane potential range at which mPFC neurons are operating (Yang et al., 1999).
Footnotes
This work was supported by United States Public Health Service Grant DA 12618 and Senior Scientist Award DA 00456 from the National Institute on Drug Abuse (F.J.W.). We thank Lori Baker and Kerstin Ford for excellent technical assistance, Dr. Robert Foehring for his critical comments on this manuscript, and Drs. Donald C. Cooper, D. James Surmeier, and Xiu-Ti Hu for expert advice.
Correspondence should be addressed to Dr. Francis J. White, Department of Cellular and Molecular Pharmacology, Finch University of Health Sciences/The Chicago Medical School, 3333 Green Bay Road, North Chicago, IL 60064. E-mail: francis.white@finchcms.edu.
Y. Dong's present address: Department of Psychiatry, Stanford University School of Medicine, Palo Alto, CA 94304-5485.
References
- 1.Bekkers JM, Delaney AJ. Modulation of excitability by α-dendrotoxin-sensitive potassium channels in neocortical pyramidal neurons. J Neurosci. 2001;21:6553–6560. doi: 10.1523/JNEUROSCI.21-17-06553.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger B, Gaspar P, Verney C. Dopaminergic innervation of the cerebral cortex: unexpected differences between rodents and primates. Trends Neurosci. 1991;14:21–27. doi: 10.1016/0166-2236(91)90179-x. [DOI] [PubMed] [Google Scholar]
- 3.Bjorklund A, Divac I, Lindvall O. Regional distribution of catecholamines in monkey cerebral cortex, evidence for a dopaminergic innervation of the primate prefrontal cortex. Neurosci Lett. 1978;7:115–119. doi: 10.1016/0304-3940(78)90153-2. [DOI] [PubMed] [Google Scholar]
- 4.Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel α subunit. J Neurosci. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantrell AR, Scheuer T, Catterall WA. Voltage-dependent neuromodulation of Na+ channels by D1-like dopamine receptors in rat hippocampal neurons. J Neurosci. 1999;19:5301–5310. doi: 10.1523/JNEUROSCI.19-13-05301.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr DB, Sesack SR. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci. 2000;20:3864–3873. doi: 10.1523/JNEUROSCI.20-10-03864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr DB, O'Donnell P, Card JP, Sesack SR. Dopamine terminals in the rat prefrontal cortex synapse on pyramidal cells that project to the nucleus accumbens. J Neurosci. 1999;19:11049–11060. doi: 10.1523/JNEUROSCI.19-24-11049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz DM, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- 9.Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Action-potential propagation gated by an axonal I(A)-like K+ conductance in hippocampus. Nature. 1997;389:286–289. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- 10.Dong Y, White FJ. Dopamine (DA) modulation of K+ currents in prefrontal cortex neurons: effects of chronic cocaine. Soc Neurosci Abstr. 2001;27:1578. [Google Scholar]
- 11.Emson PC, Koob GF. The origin and distribution of dopamine-containing afferents to the rat frontal cortex. Brain Res. 1978;142:249–267. doi: 10.1016/0006-8993(78)90634-0. [DOI] [PubMed] [Google Scholar]
- 12.Foehring RC, Surmeier DJ. Voltage-gated potassium currents in acutely dissociated rat cortical neurons. J Neurophysiol. 1993;70:51–63. doi: 10.1152/jn.1993.70.1.51. [DOI] [PubMed] [Google Scholar]
- 13.Fuster JM. The prefrontal cortex—an update: time is of the essence. Neuron. 2001;30:319–333. doi: 10.1016/s0896-6273(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 14.Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 15.Geijo-Barrientos E, Pastore C. The effects of dopamine on the subthreshold electrophysiological responses of rat prefrontal cortex neurons in vitro. Eur J Neurosci. 1995;7:358–366. doi: 10.1111/j.1460-9568.1995.tb00331.x. [DOI] [PubMed] [Google Scholar]
- 16.Goldman-Rakic PS, Muly EC, III, Williams GV. D1 receptors in prefrontal cells and circuits. Brain Res Rev. 2000;31:295–301. doi: 10.1016/s0165-0173(99)00045-4. [DOI] [PubMed] [Google Scholar]
- 17.Gorelova N, Yang CR. Dopamine D1 receptor stimulation modulates a slowly inactivating Na+ current in layer V–VI prefrontal cortex neurons. Soc Neurosci Abstr. 1997;23:1771. [Google Scholar]
- 18.Gulledge AT, Jaffe DB. Dopamine decreases the excitability of layer V pyramidal cells in the rat prefrontal cortex. J Neurosci. 1998;18:9139–9151. doi: 10.1523/JNEUROSCI.18-21-09139.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinemann SH, Rettig J, Graack HR, Pongs O. Functional characterization of Kv channel beta-subunits from rat brain. J Physiol (Lond) 1996;493:625–633. doi: 10.1113/jphysiol.1996.sp021409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hille B. Ionic channels of excitable membranes. Sinauer; Sunderland, MA: 2001. [Google Scholar]
- 21.Hoffman DA, Johnston D. Neuromodulation of dendritic action potentials. J Neurophysiol. 1999;81:408–411. doi: 10.1152/jn.1999.81.1.408. [DOI] [PubMed] [Google Scholar]
- 22.Holmqvist MH, Cao J, Knoppers MH, Jurman ME, DiStefano PS, Rhodes KJ, Xie Y, An WF. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J Neurosci. 2001;21:4154–4161. doi: 10.1523/JNEUROSCI.21-12-04154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 24.Kirsch GE, Drewe JA. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J Gen Physiol. 1993;102:797–816. doi: 10.1085/jgp.102.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knable MB, Weinberger DR. Dopamine, the prefrontal cortex and schizophrenia. J Psychopharmacol. 1997;11:123–131. doi: 10.1177/026988119701100205. [DOI] [PubMed] [Google Scholar]
- 26.Korngreen A, Sakmann B. Voltage-gated K+ channels in layer 5 neocortical pyramidal neurones from young rats: subtypes and gradients. J Physiol (Lond) 2000;525:621–639. doi: 10.1111/j.1469-7793.2000.00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levitan IB. Modulation of ion channels in neurons and other cells. Annu Rev Neurosci. 1988;11:119–136. doi: 10.1146/annurev.ne.11.030188.001003. [DOI] [PubMed] [Google Scholar]
- 28.Lewis DA. Neural circuitry of the prefrontal cortex in schizophrenia. Arch Gen Psychiatry. 1995;52:269–272. doi: 10.1001/archpsyc.1995.03950160019004. [DOI] [PubMed] [Google Scholar]
- 29.Locke RE, Nerbonne JM. Three kinetically distinct Ca2+-independent depolarization-activated K+ currents in callosal-projecting rat visual cortical neurons. J Neurophysiol. 1997;78:2309–2320. doi: 10.1152/jn.1997.78.5.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martina M, Schultz JH, Ehmke H, Monyer H, Jonas P. Functional and molecular differences between voltage-gated K+ channels of fast-spiking interneurons and pyramidal neurons of rat hippocampus. J Neurosci. 1998;18:8111–8125. doi: 10.1523/JNEUROSCI.18-20-08111.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller EK, Cohen JD. An integrated theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- 33.Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicola SM, Surmeier DJ, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 35.Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 1991;251:947–950. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- 36.Seamans JK, Floresco SB, Phillips AG. D1 receptor modulation of hippocampal-prefrontal cortical circuits integrating spatial memory with executive functions in the rat. J Neurosci. 1998;18:1613–1621. doi: 10.1523/JNEUROSCI.18-04-01613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sesack SR, Pickel VM. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol. 1992;320:145–160. doi: 10.1002/cne.903200202. [DOI] [PubMed] [Google Scholar]
- 38.Sibley DR, Monsma FJ. Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 39.Stoof JC, Kebabian JW. Two dopamine receptors: biochemistry, physiology and pharmacology. Life Sci. 1984;35:2281–2296. doi: 10.1016/0024-3205(84)90519-8. [DOI] [PubMed] [Google Scholar]
- 40.Surmeier DJ, Kitai ST. D1 and D2 dopamine receptor modulation of sodium and potassium currents in rat neostriatal neurons. In: Arbuthnott GW, Emson PC, editors. Chemical signaling in the basal ganglia. Elsevier; Amsterdam: 1993. pp. 309–324. [DOI] [PubMed] [Google Scholar]
- 41.Surmeier DJ, Stefani A, Foehring RC, Kitai ST. Developmental regulation of a slowly-inactivating potassium conductance in rat neostriatal neurons. Neurosci Lett. 1991;122:41–46. doi: 10.1016/0304-3940(91)90188-y. [DOI] [PubMed] [Google Scholar]
- 42.Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci USA. 1992;89:10178–10182. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol. 2001;63:241–320. doi: 10.1016/s0301-0082(00)00033-2. [DOI] [PubMed] [Google Scholar]
- 44.Undie AS, Berki AC, Beardsley K. Dopaminergic behaviors and signal transduction mediated through adenylate cyclase and phospholipase C pathways. Neuropharmacology. 2000;39:75–87. doi: 10.1016/s0028-3908(99)00106-9. [DOI] [PubMed] [Google Scholar]
- 45.Vanderschuren LJMJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology. 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- 46.Vincent SL, Khan Y, Benes FM. Cellular colocalization of dopamine D1 and D2 receptors in rat medial prefrontal cortex. Synapse. 1995;19:112–120. doi: 10.1002/syn.890190207. [DOI] [PubMed] [Google Scholar]
- 47.Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- 48.Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V–VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang CR, Seamans JK, Gorelova N. Developing a neuronal model for the pathophysiology of schizophrenia based on the nature of electrophysiological actions of dopamine in the prefrontal cortex. Neuropsychopharmacology. 1999;21:161–194. doi: 10.1016/S0893-133X(98)00112-2. [DOI] [PubMed] [Google Scholar]
- 50.Zahrt J, Taylor JR, Mathew RG, Arnsten AF. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci. 1997;17:8528–8535. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X-F, Hu X-T, White FJ. Whole-cell plasticity in cocaine withdrawal: reduced sodium current in nucleus accumbens neurons. J Neurosci. 1998;18:488–498. doi: 10.1523/JNEUROSCI.18-01-00488.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou FM, Hablitz JJ. Layer I neurons of the rat neocortex. II. Voltage-dependent outward currents. J Neurophysiol. 1996;76:668–682. doi: 10.1152/jn.1996.76.2.668. [DOI] [PubMed] [Google Scholar]