

Abstract
BH3 (Bcl-2 homology 3)-only proteins of the Bcl-2 family activate Bax or Bak during apoptosis to promote the release of pro-death factors sequestered in the mitochondrial intermembrane space. Previous results demonstrated that a synthetic BH3 peptide mimics the ability of the BH3-only protein Bid to promote Bax insertion and cytochrome c (cyt c) release from neural cell mitochondria. However, the BH3 peptide was deficient in promoting cyt c release from mitochondria without associated Bax, such as adult rat brain mitochondria. This study tested the hypothesis that the amphiphilic membrane-active cationic drugs dibucaine and propranolol block BH3 peptide-initiated cytc efflux by preventing the integration of Bax into the mitochondrial outer membrane. BH3 peptide-initiated release of cytc from GT1-7 neural cell mitochondria was inhibited by dibucaine and propranolol at concentrations of 100–300 μm. Recombinant Bax (100 nm) alone did not release cyt c from adult rat brain mitochondria; however, when BH3 peptide or caspase-8 cleaved Bid (cBid) was added, robust cyt c release was achieved that was inhibited completely by 200 μm dibucaine or propranolol. These drugs at similar concentrations also inhibited release of entrapped 10 kDa dextrans from protein-free liposomes treated with Bax and cBid. Contrary to the hypothesis that dibucaine and propranolol act by inhibiting the insertion of Bax into the mitochondrial outer membrane, membrane insertion of Bax was not inhibited in mitochondria or liposomes, indicating a mechanism of drug action downstream from this event. These results suggest that dibucaine and propranolol inhibit Bax-induced permeability changes through a direct interaction with the lipid membrane and present a novel target for the development of neuroprotective, antiapoptotic therapeutics.
Keywords: Bax, BH3, Bid, brain mitochondria, cytochromec, dibucaine, propranolol, apoptosis, permeability transition
Introduction
Bcl-2 family proteins are critical regulators of mammalian cell death and survival (Huang and Strasser, 2000; Lutz, 2000). The Bcl-2 protein family can be divided into subclasses on the basis of function and the inclusion of Bcl-2 homology (BH) domains. “BH3 domain-only” proteins, such as Bid and Bim, may act by either binding to and antagonizing the function of multidomain prosurvival proteins such as Bcl-2 and Bcl-XL(Degterev et al., 2001) or activating the multidomain pro-death proteins Bax or Bak (Desagher et al., 1999; Wei et al., 2000,2001).
It is known that BH3-only proteins and Bax play an essential role in programmed cell death execution in the nervous system during development (Deckwerth et al., 1996; Harris and Johnson, 2001; Vila et al., 2001). Increasing evidence also suggests that pathological apoptosis contributes to cell death in wide-ranging neurological disorders, including Parkinson's disease (Vila et al., 2001), amyotrophic lateral sclerosis (Martin, 1999; Vukosavic et al., 1999;Guegan et al., 2001), Alzheimer's disease (MacGibbon et al., 1997; Su et al., 1997), and ischemic (Krajewski et al., 1995) and traumatic (Lu et al., 2000) brain injury. Death-promoting Bcl-2 family members appear to participate primarily in the induction phase of apoptosis by releasing an assortment of mitochondrially compartmentalized proteins that are toxic to the cell, including the caspase activators cytochromec (cyt c) and Smac/DIABLO (second mitochondria-derived activator of caspase/direct IAP binding protein with low pI) (Green, 2000). Although organic compounds that mimic BH3 domain binding properties and induce apoptosis have been characterized recently (Degterev et al., 2001), pharmacological compounds that interfere with the BH3-mediated release of mitochondrial proteins have yet to be identified. The identification of such compounds is highly desirable, because it should lead to a new class of therapeutic candidates for a variety of neurological disorders.
We modeled the activity of BH3-only proteins previously by treating isolated neural cell mitochondria and permeabilized neural cells with a synthetic BH3 peptide and monitoring cyt c release. Results indicated that, like the insertion of Bax into the mitochondrial outer membrane that is mediated by the BH3-only protein Bid, BH3 peptide-induced cyt c release was associated with the integral membrane insertion of Bax (Polster et al., 2001). Although there seems to be considerable redundancy in upstream activators and downstream effectors of the apoptotic pathway, Bax and/or Bak is required uniquely in many tissues, including a sole dependence on Bax in at least some neurons (Deckwerth et al., 1996). The process of Bax mitochondrial insertion therefore presents an attractive target for drug intervention.
The amphiphilic cations propranolol and dibucaine are known to inhibit mitochondrial membrane activities, such as protein import and mitochondrial permeability transition, and we showed previously that these compounds also block cyt c release initiated by mitochondrial precursor targeting peptides (Kushnareva et al., 2001). The present study tested the hypothesis that dibucaine and propranolol inhibit BH3 peptide-induced cyt c efflux and that the inhibition is mediated by interference with the membrane insertion of Bax.
Materials and Methods
Materials. Rat forebrain mitochondria were isolated from adult (>6-week-old) Sprague Dawley rats according to the procedure of Rosenthal et al. (1987), yielding a combination of both nonsynaptosomal and synaptosomal purified mitochondria. In these studies, the nonspecific protease mixture nagarse was excluded from the tissue homogenization buffer because it was found to degrade mitochondrial outer membrane proteins. GT1-7 mitochondria were isolated according to the method of Moreadith and Fiskum (1984), with slight modifications. The BH3 peptide spanned amino acids 53–86 of Bax (53DASTKKLSECLKRIGDELDSNMELQRMIAAVDTD86) and was synthesized by the Wadsworth Center Biochemistry and Peptide Synthesis Core using an Applied Biosystems (Foster City, CA) 431A automated peptide synthesizer as described previously (Lohret and Kinnally, 1995). Peptide was prepared as dilute (15 μm to 15 mm) stocks in distilled water. Full-length untagged recombinant monomeric Bax was isolated as described previously (Suzuki et al., 2000). Bax was stored as a 0.1 or 0.2 mg/ml stock in 100 mm NaCl and 20 mmTris-HCl, pH 8.0, at 4°C. Caspase-8 cleaved human Bid (cBid) was obtained from R & D Systems (Minneapolis, MN) and stored in aliquots at −20°C. Cyclosporin A was obtained from Alexis Biochemicals (San Diego, CA). Polyclonal anti-Bax rabbit IgG was purchased from Upstate Biotechnology (Charlottesville, VA). Monoclonal anti-cyt c mouse IgG was from PharMingen (San Diego, CA). Monoclonal anti-porin mouse IgG was from Calbiochem (San Diego, CA). Dioleoylphosphatidylcholine (DOPC), dioleoylphosphatidylglycerol, and cardiolipin (CL) were purchased from Avanti Polar Lipids (Alabaster, AL). Other chemicals were from Sigma (St. Louis, MO), and all reagents were of the highest grade available.
Cell culture. GT1-7 cells stably transfected with a control puromycin-resistance vector (GT1-7 puro) were obtained from Dr. Dale Bredesen (Buck Research Institute, Novato, CA) and maintained as described previously (Murphy et al., 1996). PC12S cells, a morphological variant of rat pheochromocytoma PC12 cells that retain the ability to grow in tissue culture without poly-l-lysine treatment, were maintained as described previously (Fukuyama et al., 1993). SH-SY5Y human neuroblastoma cells were cultured in 10% fetal bovine serum and 1% penicillin and streptomycin and passaged every 3 d. Primary cortical neurons were isolated and cultured as described previously (McKenna et al., 2000).
Determination of cellular Bax protein concentration. GT1-7puro, SH-SY5Y, and PC12S cells and cortical neurons (6 din vitro) were harvested by trypsinization and counted with a hemacytometer. Cells were then pelleted at 210 × gfor 5 min and resuspended in cell lysis buffer (1% Triton X-100, 150 mm NaCl, 10 mmTris, 1 mm EDTA, and 0.5% Nonidet P-40) containing 36 μl/ml protease inhibitor cocktail (Sigma) to a concentration of 108 cells/ml. Cellular protein content was determined with the Biuret protein assay. Total cellular protein (30 μg) was separated by gel electrophoresis as described previously, and Bax was detected by immunoblot (Polster et al., 2001). Band intensities were analyzed densitometrically by use of the GelExpert system (NucleoTech, San Mateo, CA), and recombinant Bax protein standards (2, 4, and 6 ng) on the same blot were used to generate a calibration curve with a linear fit (R2 = 0.99).
Determination of cyt c release. Isolated GT1-7puro or adult rat brain mitochondria (0.25 mg/ml) were incubated in 0.25 ml of KCl assay medium consisting of 125 mm KCl, 2 mmKH2PO4, and 20 mm HEPES-KOH, pH 7.0 (KCl medium) that was supplemented with 4 mmMgCl2, 3 mm ATP, 0.8 mm ADP, 0.25 mm EGTA, 5 mm succinate, and 2 μmrotenone. For brain mitochondria, Bax (100 nm) or vehicle control (5 μl of 100 mm NaCl and 20 mm Tris-HCl, pH 8.0) was also included. BH3 peptide, vehicle control (water), or alamethicin was added after 2 min of incubation. At 6 min (GT1-7 mitochondria) or 16 min (brain mitochondria) after the addition of BH3 peptide, vehicle control, or alamethicin, mitochondria were pelleted by centrifugation at 13,400 × g for 5 min, and the supernatant and pellet were assayed for the presence of cyt c by immunoblot as described previously (Kushnareva et al., 2001). For quantitative comparisons, cyt c release was also determined with an ELISA kit (R & D Systems) according to the instructions of the manufacturer. Alamethicin treatment was used as a positive control representing maximum release of cyt c (Andreyev and Fiskum, 1999).
Measurement of mitochondrial respiration. Oxygen consumption was monitored at 30°C with a Clark-type oxygen electrode (Hansatech, Haverhill, MA) as described previously (Polster et al., 2001). Briefly, mitochondrial respiratory energy coupling was determined as the acceptor control ratio (ACR) in KCl medium in the presence of 0.5 mg/ml mitochondrial protein, 5 mm malate, 5 mm glutamate, 1 mm MgCl2, and 0.25 mm EGTA. The ACR was calculated as the ratio of the rate of ADP (0.8 mm)-stimulated respiration (state 3) to the resting rate (state 4) determined in the presence of the ATP synthase inhibitor oligomycin (2.5 μg/ml). The ACR values ranged from 5 to 10. Rates of oxygen consumption were calculated in nanomoles of O2 per milligram of mitochondrial protein per minute on the basis of a KCl medium oxygen content of 195 nmol/ml O2 at 30°C.
Measurement of mitochondrial membrane potential. Mitochondrial membrane potential was monitored with a Fluoro IV fluorescence spectrometer (Gilford, Oberlin, OH) by measurement of fluorescent changes caused by the extent of mitochondrial sequestration of the fluorescent cationic dye safranine-O [5 μm; excitation wavelength (λex) at 485 nm, emission wavelength (λem) at 586 nm] (Fiskum et al., 2000;Kowaltowski et al., 2000; Polster et al., 2001). Isolated rat brain mitochondria (0.25 mg/ml) were incubated in 0.5 ml of KCl medium including 5 mm succinate, 2 μm rotenone, 3 mm ATP, 4 mm MgCl2, 250 μm EGTA, and 5 μmsafranine-O. Bax (100 nm) or Bax vehicle was added before the addition of mitochondria, whereas BH3 peptide (60 μm) or vehicle was added after 2 min of incubation of mitochondria with the fluorescent dye. When present, 10 μm horse-heart cyt c(Sigma) was added to the assay medium before the mitochondria and did not affect the monitoring of safranine-O fluorescence.
Alkali extraction and detection of Bax, Bcl-XL, and voltage-dependent anion channels. The localization of mitochondrial Bax protein to sodium carbonate-extracted soluble fraction versus membrane fraction was determined essentially as described previously (Eskes et al., 2000). Proteins were separated by SDS-PAGE, and Bax was immunostained with primary rabbit polyclonal anti-Bax antibody (1:500 dilution; Upstate Biotechnology) plus secondary anti-rabbit IgG conjugated to horseradish peroxidase (1:10,000 dilution; Amersham Biosciences, Piscataway, NJ). Voltage-dependent anion channels (VDACs) were detected with a primary mouse monoclonal anti-porin antibody (1:4000 dilution ; Calbiochem) plus secondary anti-mouse IgG conjugated to horseradish peroxidase (1:10,000 dilution; Amersham Biosciences). Peroxidase activity was detected with the Enhanced Chemiluminescence detection kit (Amersham Biosciences) and x-ray film.
Preparation of liposomes and determination of fluorescent dextran release. Large unilamellar vesicles (LUVs) were formed by the freeze–thawing and extrusion method of Mayer et al. (1986). Briefly, DOPC/CL (1:1) was mixed in CHCl3/methanol (2:1), and the organic solvents were removed with an N2stream, followed by a 2 hr incubation under vacuum. Dry lipid films were resuspended in aqueous solutions containing 100 mg/ml fluorescein isothiocyanate-labeled dextrans of ∼10 kDa (FD-10), 100 mm KCl, 0.1 mm EDTA, and 10 mm HEPES, pH 7.0. Then, lipid samples were subjected to 15–20 freeze–thaw cycles, followed by 10 extrusions through two filters of 200 μm pore size (Nucleopore, Pleasanton, CA), with additional freeze–thaw cycles between the extrusions. Nonencapsulated FD-10 was removed using a Sephacryl S-400-HR column (Pierce, Rockford, IL) eluted with 100 mm KCl, 0.1 mm EDTA, and 10 mm HEPES, pH 7.0. Percentage of FD-10 release (fractional dequenching) was estimated according to the following equation: % Release = (F −F0/F100− F0) × 100, whereF is the measured fluorescence intensity after protein addition, F0 the initial fluorescence of the intact vesicle suspension, andF100 the fluorescence value after complete disruption of vesicle integrity by addition of Triton X-100 (final concentration of Triton X-100 in the cuvette, 0.3% w/v). λex was 490 nm, and λem was 520 nm (slits, 4 nm). Fluorometric measurements were conducted in an 8100 SIM-Aminco instrument with a thermostatted 1 cm path-length cuvette with constant stirring at 37°C.
Detection of Bax insertion in liposomes. To measure the amount of membrane-inserted protein, a method based on the fact that lipid-associated protein but not free protein floats in D2O buffer was used (Ostolaza and Goni, 1995). Briefly, liposomes prepared in D2O-based 100 mm KCl, 0.1 mm EDTA, and 10 mm HEPES, pH 7.0, were incubated with protein for 30 min in the same buffer, followed by a second incubation at pH 11.5 for 30 min and ultracentrifugation of the mixture (2 hr at 100,000 × g). The protein contents in lipid-associated and lipid-free fractions were determined on the basis of their fluorescence intensities at λex of 280 nm and λem of 345 nm after addition of the detergent dodecyl octaethyleneglycol mono ether. After incubation at an alkaline pH, the fraction of protein inserted into the membrane hydrophobic matrix remained associated with LUV, whereas the fraction of protein associated only peripherally with the membrane was detached from the vesicles.
Statistical analysis. A two-way ANOVA was used to determine drug and concentration differences for inhibition of cyt crelease by dibucaine and propranolol. Data expressed as percentage of inhibition of cyt c release were transformed by taking the square root before analysis, which tended to produce a more Gaussian distribution. No evidence of an interaction between the two factors was detected. A value of p < 0.05 was considered significant.
Results
Dibucaine and propranolol inhibit BH3 peptide-induced cytc release from GT1-7 puromitochondria
Pretreatment of isolated GT1-7 puro mitochondria with the amphiphilic cations dibucaine or propranolol resulted in a dose-dependent inhibition of cyt c efflux induced by BH3 peptide in the presence of the Ca2+chelator EGTA (Fig. 1A) (two-way ANOVA; p < 0.001). Propranolol was significantly more effective at suppressing cyt c release than dibucaine (p < 0.05), and essentially complete inhibition (95 ± 3.6%) was attained at 300 μm. Dibucaine is a local anesthetic, and propranolol has local anesthetic properties. However, the local anesthetics lidocaine (Fig. 1B), procaine, bupivicaine, etidicaine, and ropivicaine (data not shown) did not display any ability to inhibit cyt c release at concentrations up to 500 μm. For reference, the structures of several of these compounds are provided in Figure2. Because dibucaine and propranolol have the ability to inhibit phospholipase A2, we tested the ability of other phospholipase A2inhibitors to influence cyt c release by BH3 peptide. Chlorpromazine displayed a partial inhibition of cyt crelease at 500 μm (Fig. 1B), whereas the Ca2+-independent phospholipase A2 inhibitor bromoenol lactone was without effect (Fig. 1B).
Fig. 1.

Inhibition of BH3 peptide-induced cytochromec release from GT1-7 mitochondria by selective amphipathic cations. A, Mitochondria (0.25 mg/ml) isolated from neural GT1-7 puro cells were incubated at 30°C in KCl medium with 5 mm succinate, 2 μm rotenone, 4 mm MgCl2, 3 mm ATP, and 0.25 mm EGTA for 2 min, at which time BH3 peptide (0.5 μm), vehicle control, or alamethicin (80 μg/ml) was added. When present, dibucaine or propranolol was added 1 min before the addition of BH3 peptide or vehicle control. Cyt c was detected in supernatant and pellet fractions by ELISA after centrifugation of the mitochondrial suspension after 8 min total incubation. Cyt c release is expressed as the percentage of total cyt c that was present in the supernatant compared with the supernatant plus pellet. Percentage inhibition was calculated by subtracting background cytc release from cyt c release with treatments and expressing the value as the percentage inhibition of cytc release obtained with 0.5 μm BH3 peptide alone. B, Mitochondria (0.25 mg/ml) were incubated as inA, and vehicle control, BH3 peptide (1.5 μm), or alamethicin (Alm) (80 μg/ml) was added at 2 min. Dibucaine (500 μm), propranolol (500 μm), lidocaine (500 μm), chlorpromazine (500 μm), or bromoenol lactone (BEL) (100 μm), when present, were added 1 min before the addition of BH3 peptide. sup, Supernatant.
Fig. 2.

Structures of active (A) and inactive (B) compounds tested for the ability to inhibit the Bax-mediated release of cytochrome c.
Dibucaine and propranolol inhibit cyt c release from rat brain mitochondria induced by BH3 peptide and full-length recombinant Bax
We showed previously that BH3 peptide was incapable of inducing substantial cyt c release from isolated adult brain mitochondria devoid of detectable endogenous Bax (Polster et al., 2001). Full-length recombinant monomeric Bax at a concentration of 100 nm (based on a molecular weight of 21 kDa) also was unable to promote the exodus of cyt c (Fig.3). However, when BH3 peptide was added in the presence of recombinant Bax, robust cyt c release (∼40% as measured by cyt c ELISA) from adult brain mitochondria was observed (Fig. 3). Dibucaine and propranolol effectively blocked the release evoked by Bax plus BH3 peptide, with nearly complete inhibition occurring at 200 μm(Fig. 3). The background level of cyt c release in these experiments was <3% as measured by ELISA.
Fig. 3.

Dibucaine and propranolol display dose-dependent inhibition of cytochrome c release from adult brain mitochondria by BH3 peptide and Bax. Isolated adult rat brain mitochondria (0.25 mg/ml) were incubated under the conditions described in Figure 1 in the presence of 100 nm Bax or vehicle control, and drug or vehicle was added after 1 min. Vehicle control, BH3 peptide (60 μm), or alamethicin (Alm) (80 μg/ml) was added at 2 min to stimulate cyt crelease, and mitochondrial suspension was centrifuged after 18 min of incubation. Cyt c content in supernatant (sup) and pellet (pel) fractions was detected by immunoblot.
Dibucaine and propranolol can uncouple mitochondrial electron transport from ATP synthesis at concentrations only slightly greater than those used in our experiments (Pavlov and Glaser, 1998). The acceptor control ratio (state 3, ADP-stimulated respiration rate, to state 4, resting respiration rate) for brain mitochondria in the presence and absence of 200 μm dibucaine or propranolol was determined to examine the effect of the drugs on mitochondrial energy coupling. Exposure of brain mitochondria to dibucaine or propranolol treatment resulted in no substantial alteration in ADP-stimulated respiration or mitochondrial respiratory control (Fig. 4). Similar results were obtained if respiration was measured in the presence of succinate and rotenone instead of malate and glutamate (data not shown). These agents are therefore capable of inhibiting BH3 peptide–Bax-mediated cyt c release at a concentration that does not interfere with oxidative phosphorylation.
Fig. 4.
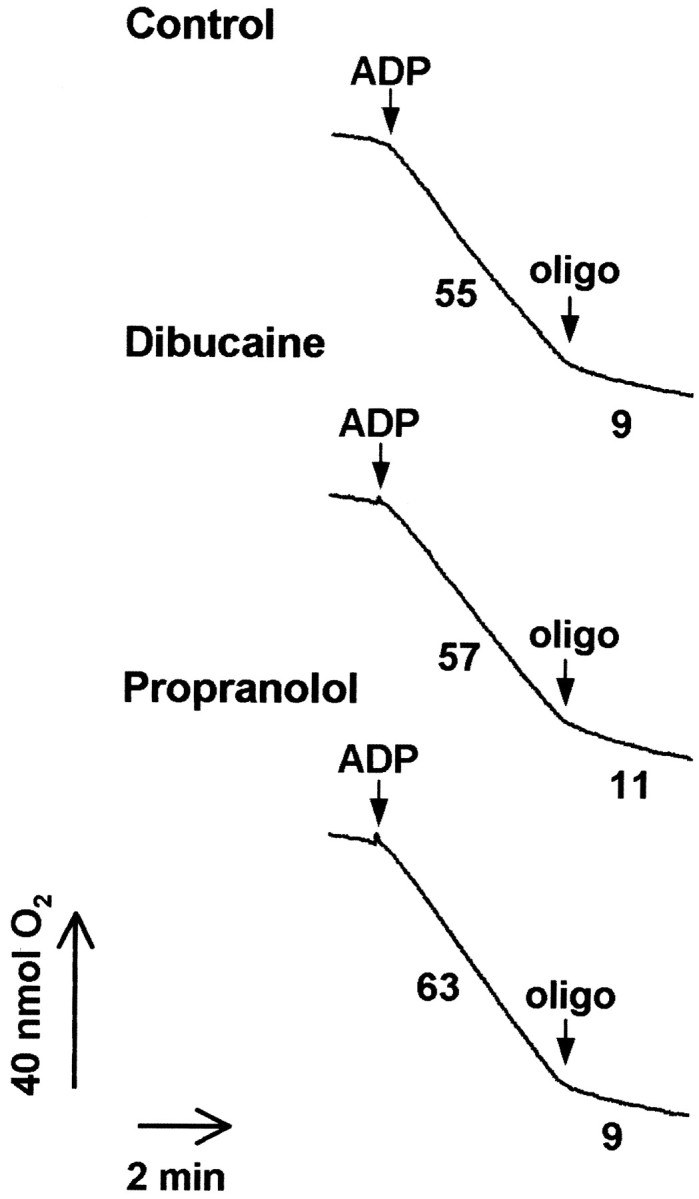
Dibucaine and propranolol do not impair mitochondrial respiration at concentrations that prevent cytochromec release. Isolated adult rat brain mitochondria (0.25 mg/ml) were incubated in KCl-based medium at 30°C with 5 mm malate, 5 mm glutamate, 1 mmMgCl2, and 0.25 mm EGTA. State 3 (phosphorylating) respiration was stimulated by the addition of 0.8 mm ADP. Oligomycin (oligo) (2.5 μg/ml) was added to produce state 4 (resting respiration). Numbersrepresent rates of oxygen consumption in nanomoles of O2per milligram of protein per minute. Dibucaine or propranolol (200 μm) was present in the incubation medium when indicated. The ACR (state 3/state 4) is an indicator of mitochondrial functional integrity. ACR values were 6.1 (control), 5.2 (dibucaine), and 7.0 (propranolol).
Estimation of cellular Bax concentrations
Although Bax alone did not release cyt c from isolated brain mitochondria at a concentration of 100 nm, other investigators have used Bax in the micromolar range to elicit cytc release (Jurgensmeier et al., 1998; Narita et al., 1998;Cao et al., 2001). To estimate the physiological level of Bax in healthy neural cells, the amount of total Bax protein in cell lysates of cortical neurons and neural GT1-7, SH-SY5Y, and PC12S cells was determined by densitometric quantification of immunoblots that included known amounts of recombinant Bax protein (Fig.5). Bax levels were quantified using only optical density values that were within the range in which a linear relationship with recombinant Bax was established (Fig. 5A). Cellular Bax concentrations were calculated on the basis of measurements of protein concentrations of the cellular lysates and were based on an assumption of an average cellular volume for most cell types of 200 pl per cell. Bax concentration in these cells ranged from 2.2 to 3.7 nm.
Fig. 5.

Determination of Bax levels from cell lysates. A, Cell lysates (30 μg) from cortical neurons and GT1-7 puro, SH-SY5Y, and PC12S cells were analyzed for Bax content by immunoblot together with known amounts of Bax (2, 4, and 6 ng). A very minor Bax-immunoreactive band probably corresponding to a dimer was present in recombinant Bax standards and included in optical density measurements because it was detected with multiple Bax antibodies. The band was not detected in cell lysates.B, A linear fit was applied to the densitometric quantification of Bax standards (R2 = 0.99), and the resulting equation was used to estimate Bax protein concentration.
Because Bax seems to release cyt c only after it binds and becomes integrated into the mitochondrial outer membrane, we also determined total cell Bax content relative to the cell mitochondrial content on the basis of our measurements of cell protein and an estimate of the mitochondrial fraction of cellular protein. These calculated values ranged from 0.7 to 1.1 μg of Bax per milligram of mitochondrial protein, assuming that mitochondria represent 10% of total cellular protein. The amount of Bax used in our experiments was 8 μg/mg of mitochondrial protein, which is even closer to the estimated normal cellular Bax/mitochondrial protein ratio than the in vitro Bax concentration is to the estimated total cellular Bax concentration.
BH3 peptide–Bax-induced cyt c release from adult brain mitochondria is not a result of mitochondrial permeability transition
BH3 peptide-initiated cyt c redistribution from neural cell mitochondria is independent of the mitochondrial permeability transition and loss of membrane potential but is associated with Bax integral membrane insertion (Polster et al., 2001). Cyt crelease induced from adult rat brain mitochondria by a combination of BH3 peptide and exogenous Bax (100 nm) was accompanied similarly by only partial loss of mitochondrial membrane potential (ΔΨ), as measured by the release of the cationic fluorescent dye safranine-O (Fig.6A). The addition of exogenous cyt c (10 μm) inhibited the partial drop in transmembrane potential, suggesting that the reduction in mitochondrial membrane potential is primarily because of the release of cyt c and associated respiratory inhibition (Fig. 6A). In contrast, a complete or nearly complete decline in ΔΨ that cannot be prevented by the addition of exogenous cyt c is normally associated with opening of the mitochondrial permeability transition pore. As in BH3 peptide-mediated cyt c efflux from neural cell mitochondria, the permeability transition pore inhibitor cyclosporin A was unable to block cytc release (Fig. 6B).
Fig. 6.

BH3 peptide-induced cytochrome crelease from brain mitochondria in the presence of recombinant Bax is not caused by mitochondrial inner membrane permeability transition.A, Isolated adult rat brain mitochondria (mito) (0.25 mg/ml) were incubated under the conditions described in Figure 1 with the fluorescent cationic dye safranine-O (5 μm), which exhibits membrane potential-dependent accumulation and quenching. Treatment with alamethicin (Alm) (80 μg/ml) plus the mitochondrial uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (100 nm) corresponds to complete mitochondrial membrane potential depolarization. BH3 peptide (60 μm), Bax (100 nm), or BH3 peptide plus Bax was added at 2 min, and membrane potential was recorded for 10 min. When indicated, exogenous cyt c (10 μm) was present. Arrows signify timing of additions, andP denotes peptide. B, Mitochondria (0.25 mg/ml) were incubated as in A. Bax (100 nm) and cyclosporin A (CsA) (1 μm) were present when indicated. BH3 peptide (60 μm) was added at 2 min to stimulate cyt c release, and mitochondrial suspension was centrifuged after 18 min of incubation. Cytc content in the supernatant (sup) fraction was detected by immunoblot.
Dibucaine and propranolol do not prevent Bax membrane insertion in adult brain mitochondria
We demonstrated previously that the BH3 peptide was able to promote the insertion of endogenous Bax in neural GT1-7 cell mitochondria (Polster et al., 2001). Sodium carbonate alkali extraction and membrane fractionation were performed on mitochondrial pellets after treatment of brain mitochondria in suspension with exogenous Bax in the presence and absence of BH3 peptide to test whether BH3 peptide could also promote the membrane insertion of exogenous Bax in brain mitochondria. Little Bax was detected in either the alkali-extracted fraction or the nonextracted membrane fraction when mitochondria were treated with Bax alone, indicating that Bax did not interact strongly with mitochondria in the absence of BH3 peptide (Fig.7). When brain mitochondria were treated with both BH3 peptide and Bax, substantial Bax was found by immunoblot in the alkali-unextractable membrane fraction. Dibucaine and propranolol were unable to block this membrane integration of Bax (Fig.7), despite their strong inhibition of the release of cyt c(Fig. 3B). VDACs were present only in the membrane fraction, and levels did not vary among samples, demonstrating adequate fractionation and even gel loading (Fig. 7).
Fig. 7.

Dibucaine and propranolol do not prevent membrane insertion of exogenous recombinant Bax in brain mitochondria. Isolated adult rat brain mitochondria (0.25 mg/ml) were incubated in the presence of 100 nm Bax or vehicle control under the conditions described in Figure 1, and drug or vehicle was added after 1 min. BH3 peptide (60 μm) was added after 2 min when indicated to stimulate cyt c release, and mitochondrial suspension was centrifuged after 18 min of incubation. The mitochondrial pellet was resuspended to a concentration of 3 mg/ml in sodium carbonate, pH 11.5, and incubated for 20 min at 4°C. Sodium carbonate-extracted (extracted) and nonextracted (membrane) fractions were separated by ultracentrifugation. Both fractions were solubilized with 2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate detergent and analyzed by Bax and VDAC immunoblots.
Dibucaine and propranolol prevent cytochromec release from adult brain mitochondria initiated by Bax and caspase-8 cleaved Bid
Because cleavage of the BH3-only protein Bid and release of cyt c are implicated in cell death after ischemic brain injury (Plesnila et al., 2001; Yin et al., 2002), we tested whether dibucaine and propranolol could inhibit cyt c release by cleaved Bid and Bax from adult brain mitochondria. As was the case for cyt c release mediated by BH3 peptide and Bax, dibucaine and propranolol (300 μm) effectively inhibited cytc release initiated by 30 nm cBid and 100 nm Bax (Fig.8A). Release was not blocked by lidocaine or bupivicaine. Thus, dibucaine and propranolol were capable of inhibiting Bax-induced cyt c release triggered by either a synthetic or physiological BH3 peptide.
Fig. 8.

Dibucaine and propranolol inhibit the release of cytochrome c from brain mitochondria and entrapped 10 kDa dextrans from liposomes induced by cBid plus Bax. A, Isolated adult rat brain mitochondria (0.25 mg/ml) were incubated as in Figure 3, except that 30 nm cBid instead of BH3 peptide was incubated with 100 nm Bax to trigger cyt crelease. Cyt c content in the supernatant (sup) fraction was detected by immunoblot.B, Representative kinetics of cBid- and Bax-induced FD-10 release from liposomes in the absence (Control) and presence of indicated drugs. In all cases, drugs (200 μm) and liposomes (DOPC/CL at 1:1; 30 μm) were incubated together for 1 min, followed by the addition of 100 nm monomeric Bax and 10 nmcBid. C, Dose dependence of drug-induced inhibition of cBid- and Bax-mediated liposome leakage. A 0% inhibition corresponds to that obtained in the absence of any drug. Mean and SEM values are shown for four to seven independent measurements. D, Percentage of membrane-inserted Bax in the LUVs used in the experiment described in C. Bax insertion was determined fluorometrically as described in Materials and Methods.
Dibucaine and propranolol inhibit release of 10 kDa dextran from protein-free liposomes by Bax and cBid
Although phospholipase A2 inhibitors that interact directly with the enzyme (e.g., bromoenol lactone) (Hazen et al., 1991) rather than the lipid substrate were incapable of inhibiting cyt c release by BH3 peptide plus Bax, the possibility remained that the inhibition of cyt c release by dibucaine and propranolol could be the result of an effect on a different phospholipase enzyme or other mitochondrial protein(s). Therefore, we examined the ability of these drugs to influence Bax-induced permeability changes in artificial liposomes that lack mitochondrial proteins. As was the case for whole mitochondria, the addition of cBid (10 nm) and monomeric Bax (100 nm) but not Bax alone was able to permeabilize liposomes, as measured by the release of a 10 kDa entrapped fluorescent dextran (Fig. 8B). Lidocaine, procaine, and bupivicaine were without effect. The dose dependence of inhibition by dibucaine and propranolol was similar to that obtained using GT1-7 or brain mitochondria (Fig. 8C), suggesting that the drugs are probably acting by the same mechanism. Bax was inserted into the liposome membrane in an alkali-resistant manner to the same degree in the presence or absence of dibucaine and propranolol, demonstrating that, like the observations in isolated mitochondria, the drugs inhibit Bax-induced permeability downstream of Bax membrane insertion (Fig.8D).
Discussion
Pharmacological attempts at inhibiting apoptosis after CNS injury have focused primarily on caspase inhibition (Eldadah and Faden, 2000). Although there has been success with these agents in animal models of ischemia and trauma, there are concerns about their ability to cross the blood–brain barrier and their effectiveness in different models of neural cell death (Deshmukh et al., 2000). Another important consideration is that caspase activation often occurs after mitochondrial alterations (e.g., release of cyt c) that can compromise the ability of mitochondria to maintain normal cellular energy (ATP) levels and can promote mitochondrial reactive oxygen species generation (Starkov et al., 2002). Therefore, a more effective approach to inhibiting cell death and subcellular injury might entail pharmacological protection against events that precede caspase activation, including the release of mitochondrial proapoptotic proteins mediated by Bax and BH3 domain-only proteins.
In this study, we identified two drugs belonging to a class of molecules known as amphiphilic cations that inhibit cyt crelease mediated by BH3 peptide–Bax interaction. Dibucaine and propranolol displayed dose-dependent inhibition of cyt crelease at concentrations from 50 to 300 μm, with propranolol exhibiting significantly greater maximum inhibition than dibucaine (i.e., 95 vs 60% in Fig. 1A). Although these agents can interfere with mitochondrial respiration and energy coupling (Pavlov and Glaser, 1998), these effects were not observed at concentrations that provided maximal inhibition of cytc release (Fig. 4). Both of these compounds exhibit local anesthetic properties that can influence the fluidity of biological membranes (Papahadjopoulos et al., 1975; Weitman et al., 1989; Tanji et al., 1992); however, other local anesthetics did not share the ability to prevent cyt c release in response to BH3 peptide–Bax interaction (Fig. 1B). Because dibucaine and propranolol inhibit phospholipase A2 activity with an IC50 similar to the concentration that exhibited half-maximal inhibition of cyt c release from GT1-7 neural cell mitochondria (Hostetler and Matsuzawa, 1981), we tested the possibility that phospholipase A2activity is required for the release of cyt c by BH3 peptide–Bax interaction. Chlorpromazine was partially effective at inhibiting cyt c release and, like propranolol, inhibits phospholipase A through its interaction with phospholipid substrates (Kubo and Hostetler, 1985). However, the Ca2+-independent phospholipase A2 inhibitor bromoenol lactone, which interacts directly with the enzyme (Hazen et al., 1991), did not protect against cyt c release by BH3 peptide. Ca2+-dependent phospholipase A2 isoforms are unlikely to be involved because of the presence of the Ca2+-chelator EGTA in the incubation medium.
Although previous observations indicated that the mechanism of BH3 peptide-induced cyt c release in neural cell mitochondria was dependent on Bax and independent of the inner membrane permeability transition (Polster et al., 2001), it was necessary to confirm these observations directly using adult brain mitochondria that possess only barely detectable endogenous Bax. In addition to our observation that BH3 peptide does not release cyt c from adult brain mitochondria in the absence of Bax (Polster et al., 2001), the results described in Figure 3 indicate that exogenous Bax at concentrations up to 100 nm does not release cyt c in the absence of the BH3 peptide. In the presence of both Bax and the BH3 peptide, mitochondrial cyt c release was accompanied by a mild reduction in the electrical potential existing across the mitochondrial inner membrane (Fig. 6A). Because this partial depolarization was inhibited by the inclusion of exogenous cytc in the mitochondrial suspension, the small drop in membrane potential was most likely a result of the release of cytc and subsequent respiratory inhibition (Polster et al., 2001). Protection against depolarization by exogenous cyt c, together with the lack of protection by cyclosporin A against cytc release (Fig. 6A,B), provides strong evidence that the inner membrane permeability transition is not involved in Bax-mediated cyt c release, at least at the concentrations of Bax and the BH3 peptide used in this and our previous study (Polster et al., 2001). Therefore, the ability of dibucaine and propranolol to inhibit the release of cyt c by Bax and BH3 peptide cannot be explained by their reported ability to inhibit the mitochondrial permeability transition (Broekemeier et al., 1985;Sokolove and Kinnally, 1996; Hoyt et al., 1997; Kowaltowski and Castilho, 1997).
The lack of cyt c release with monomeric Bax alone is contrary to some published reports that demonstrated Bax-induced release of cyt c (Jurgensmeier et al., 1998; Narita et al., 1998; Cao et al., 2001) but consistent with others (Antonsson et al., 2000; Gogvadze et al., 2001). To examine whether the concentrations of Bax used in our experiments were near the physiological level of Bax expression, we estimated the cellular Bax protein concentration in lysates from four different types of neural cells, including primary cortical neurons. The concentration of Bax in these cells ranged from 2.2 to 3.7 nm, assuming an average cellular volume for most cell types of 200 pl. Additional calculations of cellular Bax relative to mitochondrial content revealed a range of 0.7–1.1 μg of Bax per milligram of mitochondrial protein. Because Bax is upregulated in many acute and chronic neurodegenerative conditions (Sadoul, 1998), our use of Bax in vitro at 8 μg/mg mitochondrial protein is probably within the range of concentrations that exists within cells under pathological conditions. However, the micromolar concentrations of Bax often used in studies with isolated mitochondria are highly unlikely to exist in vivo and represent Bax/mitochondrial ratios in excess of 100 μg/mg mitochondrial protein.
BH3 peptide promotes the insertion of endogenous Bax in GT1-7 mitochondria, as indicated by the conversion of Bax from alkali extractable to alkali inextractable (Polster et al., 2001). To determine whether propranolol and dibucaine inhibit Bax-induced cytc release by preventing the integration of Bax into the mitochondrial outer membrane in response to BH3 peptide, we tested the effect of these drugs on the alkali extractability of exogenous Bax incubated with isolated brain mitochondria. As anticipated from findings of endogenous Bax insertion in GT1-7 mitochondria treated with BH3 peptide, BH3 peptide was necessary to promote the membrane insertion of exogenous Bax in adult brain mitochondria. However, dibucaine and propranolol were not able to inhibit Bax membrane insertion in brain mitochondria (Fig. 7), even at concentrations that completely prevented cyt c release (Fig. 3). This finding suggests that dibucaine and propranolol inhibit cyt crelease at an uncharacterized step downstream of Bax insertion. Bax mitochondrial membrane insertion and dimerization or complex formation can occur in healthy nonapoptotic cells (Makin et al., 2001), in agreement with our observation that Bax insertion is not sufficient for cyt c efflux.
A combination of cleaved Bid and monomeric Bax or oligomeric Bax alone can release large dextrans (10 and 2000 kDa) from liposomes in the absence of other proteins and without permanent disruption of vesicle morphology (Kuwana et al., 2002). These results were used to support the hypothesis that Bax-induced changes in lipid morphology (e.g., the formation of either lipidic pores or inverted micelles) are responsible for Bax-induced cyt c release (Hardwick and Polster, 2002). In agreement with the view that Bax forms lipidic pores, another study found that Bax-induced liposome permeabilization depends on membrane monolayer curvature, with positive monolayer curvature promoting and negative monolayer curvature inhibiting Bax-induced vesicle permeabilization (Basañez et al., 2002). The observation that dibucaine and propranolol interact strongly with the lipidic part of the membrane and the finding that dibucaine promotes changes in the polymorphic behavior of cardiolipin associated with increased negative monolayer curvature (Cullis et al., 1978) suggests that these drugs may inhibit cyt c release by blocking Bax-induced changes in lipid structure. Consistent with this hypothesis, dibucaine and propranolol were able to inhibit the release of vesicle-encapsulated 10 kDa dextrans downstream of Bax membrane insertion and in the absence of other proteins when liposomes were treated with cBid and Bax (Fig. 8). Similar results were obtained when dextran release was triggered by adding oligomeric Bax alone (our unpublished data), demonstrating that the drugs act at a step downstream of Bax oligomerization and membrane insertion. Studies are currently under way to investigate the relationship between the effect of dibucaine and propranolol on lipid phase transitions and their inhibition of the Bax-induced increase in membrane permeability.
Continued study of the mechanism of action of these and similar drugs should lead to a greater understanding of the mechanism of mitochondrial outer membrane permeabilization by BH3 death domain protein–Bax interaction and possibly lead to the development of neuroprotective, antiapoptotic therapeutics.
Footnotes
This work was supported by National Institutes of Health Grant NS-34152 to G.F. G.B. was supported by the Ministerio de Ciencia y Tecnología and by the Universidad del País Vasco. We thank A. Carey and C. Shifflett for expert technical assistance and Dr. A. Starkov for helpful discussions.
Correspondence should be addressed to Dr. Gary Fiskum, Department of Anesthesiology, University of Maryland, Baltimore, Medical Sciences Teaching Facility 5.34, 685 West Baltimore Street, Baltimore, MD 21201. E-mail: gfisk001@umaryland.edu.
References
- 1.Andreyev A, Fiskum G. Calcium induced release of mitochondrial cytochrome c by different mechanisms selective for brain versus liver. Cell Death Differ. 1999;6:825–832. doi: 10.1038/sj.cdd.4400565. [DOI] [PubMed] [Google Scholar]
- 2.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- 3.Basañez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM, Zimmerberg J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem. 2002;277:49360–49365. doi: 10.1074/jbc.M206069200. [DOI] [PubMed] [Google Scholar]
- 4.Broekemeier KM, Schmid PC, Schmid HH, Pfeiffer DR. Effects of phospholipase A2 inhibitors on ruthenium red-induced Ca2+ release from mitochondria. J Biol Chem. 1985;260:105–113. [PubMed] [Google Scholar]
- 5.Cao G, Minami M, Pei W, Yan C, Chen D, O'Horo C, Graham SH, Chen J. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Cullis PR, Verkleij AJ, Ververgaert PH. Polymorphic phase behaviour of cardiolipin as detected by 31P NMR and freeze-fracture techniques: effects of calcium, dibucaine and chlorpromazine. Biochim Biophys Acta. 1978;513:11–20. doi: 10.1016/0005-2736(78)90107-4. [DOI] [PubMed] [Google Scholar]
- 7.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 8.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 9.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deshmukh M, Kuida K, Johnson EM., Jr Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma. 2000;17:811–829. doi: 10.1089/neu.2000.17.811. [DOI] [PubMed] [Google Scholar]
- 12.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiskum G, Kowaltowski AJ, Andreyev A, Kushnareva YE, Starkov AA. Apoptosis-related activities measured with isolated mitochondria and digitonin-permeabilized cells. Methods Enzymol. 2000;322:222–234. doi: 10.1016/s0076-6879(00)22023-5. [DOI] [PubMed] [Google Scholar]
- 14.Fukuyama R, Chandrasekaran K, Rapoport SI. Nerve growth factor-induced neuronal differentiation is accompanied by differential induction and localization of the amyloid precursor protein (APP) in PC12 cells and variant PC12S cells. Brain Res Mol Brain Res. 1993;17:17–22. doi: 10.1016/0169-328x(93)90067-y. [DOI] [PubMed] [Google Scholar]
- 15.Gogvadze V, Robertson JD, Zhivotovsky B, Orrenius S. Cytochrome c release occurs via Ca2+-dependent and Ca2+-independent mechanisms that are regulated by Bax. J Biol Chem. 2001;276:19066–19071. doi: 10.1074/jbc.M100614200. [DOI] [PubMed] [Google Scholar]
- 16.Green DR. Apoptotic pathways: paper wraps stone blunts scissors. Cell. 2000;102:1–4. doi: 10.1016/s0092-8674(00)00003-9. [DOI] [PubMed] [Google Scholar]
- 17.Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci. 2001;21:6569–6576. doi: 10.1523/JNEUROSCI.21-17-06569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardwick JM, Polster BM. Bax, along with lipid conspirators, allows cytochrome c to escape mitochondria. Mol Cell. 2002;10:963–965. doi: 10.1016/s1097-2765(02)00751-7. [DOI] [PubMed] [Google Scholar]
- 19.Harris CA, Johnson EM., Jr BH3-only bcl-2 family members are coordinately regulated by the jnk pathway and require bax to induce apoptosis in neurons. J Biol Chem. 2001;276:37754–37760. doi: 10.1074/jbc.M104073200. [DOI] [PubMed] [Google Scholar]
- 20.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2: mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 21.Hostetler KY, Matsuzawa Y. Studies on the mechanism of drug-induced lipidosis: cationic amphiphilic drug inhibition of lysosomal phospholipases A and C. Biochem Pharmacol. 1981;30:1121–1126. doi: 10.1016/0006-2952(81)90451-2. [DOI] [PubMed] [Google Scholar]
- 22.Hoyt KR, Sharma TA, Reynolds IJ. Trifluoperazine and dibucaine-induced inhibition of glutamate-induced mitochondrial depolarization in rat cultured forebrain neurones. Br J Pharmacol. 1997;122:803–808. doi: 10.1038/sj.bjp.0701442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang DC, Strasser A. BH3-only proteins: essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 24.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kowaltowski AJ, Castilho RF. Ca2+ acting at the external side of the inner mitochondrial membrane can stimulate mitochondrial permeability transition induced by phenylarsine oxide. Biochim Biophys Acta. 1997;1322:221–229. doi: 10.1016/s0005-2728(97)00078-9. [DOI] [PubMed] [Google Scholar]
- 26.Kowaltowski AJ, Smaili SS, Russell JT, Fiskum G. Elevation of the resting mitochondrial membrane potential of neural cells by cyclosporin A, BAPTA-AM, and Bcl-2. Am J Physiol Cell Physiol. 2000;279:C852–C859. doi: 10.1152/ajpcell.2000.279.3.C852. [DOI] [PubMed] [Google Scholar]
- 27.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubo M, Hostetler KY. Mechanism of cationic amphiphilic drug inhibition of purified lysosomal phospholipase A1. Biochemistry. 1985;24:6515–6520. doi: 10.1021/bi00344a031. [DOI] [PubMed] [Google Scholar]
- 29.Kushnareva YE, Polster BM, Sokolove PM, Kinnally KW, Fiskum G. Mitochondrial precursor signal peptide induces a unique permeability transition and release of cytochrome c from liver and brain mitochondria. Arch Biochem Biophys. 2001;386:251–260. doi: 10.1006/abbi.2000.2201. [DOI] [PubMed] [Google Scholar]
- 30.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 31.Lohret TA, Kinnally KW. Targeting peptides transiently block a mitochondrial channel. J Biol Chem. 1995;270:15950–15953. doi: 10.1074/jbc.270.27.15950. [DOI] [PubMed] [Google Scholar]
- 32.Lu J, Moochhala S, Kaur C, Ling E. Changes in apoptosis-related protein (p53, Bax, Bcl-2 and Fos) expression with DNA fragmentation in the central nervous system in rats after closed head injury. Neurosci Lett. 2000;290:89–92. doi: 10.1016/s0304-3940(00)01307-0. [DOI] [PubMed] [Google Scholar]
- 33.Lutz RJ. Role of the BH3 (Bcl-2 homology 3) domain in the regulation of apoptosis and Bcl-2-related proteins. Biochem Soc Trans. 2000;28:51–56. doi: 10.1042/bst0280051. [DOI] [PubMed] [Google Scholar]
- 34.MacGibbon GA, Lawlor PA, Sirimanne ES, Walton MR, Connor B, Young D, Williams C, Gluckman P, Faull RL, Hughes P, Dragunow M. Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimer's disease hippocampus. Brain Res. 1997;750:223–234. doi: 10.1016/s0006-8993(96)01351-0. [DOI] [PubMed] [Google Scholar]
- 35.Makin GW, Corfe BM, Griffiths GJ, Thistlethwaite A, Hickman JA, Dive C. Damage-induced Bax N-terminal change, translocation to mitochondria and formation of Bax dimers/complexes occur regardless of cell fate. EMBO J. 2001;20:6306–6315. doi: 10.1093/emboj/20.22.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Mayer LD, Hope MJ, Cullis PR. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim Biophys Acta. 1986;858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- 38.McKenna MC, Stevenson JH, Huang X, Tildon JT, Zielke CL, Hopkins IB. Mitochondrial malic enzyme activity is much higher in mitochondria from cortical synaptic terminals compared with mitochondria from primary cultures of cortical neurons or cerebellar granule cells. Neurochem Int. 2000;36:451–459. doi: 10.1016/s0197-0186(99)00148-5. [DOI] [PubMed] [Google Scholar]
- 39.Moreadith RW, Fiskum G. Isolation of mitochondria from ascites tumor cells permeabilized with digitonin. Anal Biochem. 1984;137:360–367. doi: 10.1016/0003-2697(84)90098-8. [DOI] [PubMed] [Google Scholar]
- 40.Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci USA. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostolaza H, Goni FM. Interaction of the bacterial protein toxin alpha-haemolysin with model membranes: protein binding does not always lead to lytic activity. FEBS Lett. 1995;371:303–306. doi: 10.1016/0014-5793(95)00927-2. [DOI] [PubMed] [Google Scholar]
- 43.Papahadjopoulos D, Jacobson K, Poste G, Shepherd G. Effects of local anesthetics on membrane properties. I. Changes in the fluidity of phospholipid bilayers. Biochim Biophys Acta. 1975;394:504–519. doi: 10.1016/0005-2736(75)90137-6. [DOI] [PubMed] [Google Scholar]
- 44.Pavlov PF, Glaser E. Inhibition of protein import into mitochondria by amphiphilic cations: potential targets and mechanism of action. Biochem Biophys Res Commun. 1998;252:84–91. doi: 10.1006/bbrc.1998.9590. [DOI] [PubMed] [Google Scholar]
- 45.Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, Moskowitz MA. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Polster BM, Kinnally KW, Fiskum G. BH3 death domain peptide induces cell type-selective mitochondrial outer membrane permeability. J Biol Chem. 2001;276:37887–37894. doi: 10.1074/jbc.M104552200. [DOI] [PubMed] [Google Scholar]
- 47.Rosenthal RE, Hamud F, Fiskum G, Varghese PJ, Sharpe S. Cerebral ischemia and reperfusion: prevention of brain mitochondrial injury by lidoflazine. J Cereb Blood Flow Metab. 1987;7:752–758. doi: 10.1038/jcbfm.1987.130. [DOI] [PubMed] [Google Scholar]
- 48.Sadoul R. Bcl-2 family members in the development and degenerative pathologies of the nervous system. Cell Death Differ. 1998;5:805–815. doi: 10.1038/sj.cdd.4400438. [DOI] [PubMed] [Google Scholar]
- 49.Sokolove PM, Kinnally KW. A mitochondrial signal peptide from Neurospora crassa increases the permeability of isolated rat liver mitochondria. Arch Biochem Biophys. 1996;336:69–76. doi: 10.1006/abbi.1996.0533. [DOI] [PubMed] [Google Scholar]
- 50.Starkov AA, Polster BM, Fiskum G. Regulation of hydrogen peroxide production by brain mitochondria by calcium and Bax. J Neurochem. 2002;83:220–228. doi: 10.1046/j.1471-4159.2002.01153.x. [DOI] [PubMed] [Google Scholar]
- 51.Su JH, Deng G, Cotman CW. Bax protein expression is increased in Alzheimer's brain: correlations with DNA damage, Bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol. 1997;56:86–93. doi: 10.1097/00005072-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 53.Tanji K, Ohta Y, Kawato S, Mizushima T, Natori S, Sekimizu K. Decrease by psychotropic drugs and local anaesthetics of membrane fluidity measured by fluorescence anisotropy in Escherichia coli. J Pharm Pharmacol. 1992;44:1036–1037. [PubMed] [Google Scholar]
- 54.Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vukosavic S, Dubois-Dauphin M, Romero N, Przedborski S. Bax and Bcl-2 interaction in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;73:2460–2468. doi: 10.1046/j.1471-4159.1999.0732460.x. [DOI] [PubMed] [Google Scholar]
- 56.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 57.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weitman SD, Phelan AM, Lech JJ, Lange DG. Propranolol-induced alterations in rat erythrocyte membrane fluidity and apparent phase-transition temperatures: a depth-dependent process. Biochem Pharmacol. 1989;38:2949–2955. doi: 10.1016/0006-2952(89)90002-6. [DOI] [PubMed] [Google Scholar]
- 59.Yin XM, Luo Y, Cao G, Bai L, Pei W, Kuharsky DK, Chen J. Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia. J Biol Chem. 2002;277:42074–42081. doi: 10.1074/jbc.M204991200. [DOI] [PubMed] [Google Scholar]