

Abstract
Mice deficient in the peripheral myelin component P0 mimic severe forms of inherited peripheral neuropathies in humans, with defective myelin formation and consequent axonal loss. We cross-bred these mice with the spontaneous mutant C57BL/Wldstypically showing protection from Wallerian degeneration because of fusion of the ubiquitination factor E4B (Ube4b) and nicotinamide mononucleotide adenylyltransferase (Nmnat) genes. We found that in the double mutants, the robust myelin-related axonal loss is reduced at 6 weeks and 3 months of age. Moreover, retrograde labeling from plantar nerves revealed an increased survival of motor axons. These motor axons appeared functionally active because both the amplitude of compound muscle action potentials and muscle strength were less reduced in the double mutants. At 6 months of age, reduction of axonal loss was no longer detectable in the double mutants when compared with littermates carrying the P0 null mutation only, although the Wlds gene was not reduced in its expression at this age. We conclude that myelin-related axonal loss is a process having some features in common with Wallerian degeneration. Introducing the Wldsgene would be a promising approach to delaying detrimental axonal loss in myelin disorders.
Keywords: protein zero, inherited neuropathies, Wallerian degeneration, myelin mutant, peripheral nervous system, Schwann cell
Introduction
One of the major and still unresolved problems of myelin-related disorders, such as demyelinating neuropathies and multiple sclerosis, is the progressive degeneration of axons that leads to irreversible symptoms and permanent disability (Trapp et al., 1998; Kornek et al., 2000; Martini, 2001). It is presently not known how abnormal myelin or demyelination leads to axonal destruction. In the case of inflammatory disorders, such as multiple sclerosis and immune-mediated peripheral neuropathies, the naked axons are particularly vulnerable because they are directly exposed to immune cells and their secreted cytotoxic products, such as proinflammatory cytokines, proteolytic enzymes, nitric oxide, and others (Toyka and Hartung, 1996; Redford et al., 1997; Trapp et al., 1999; Kornek et al., 2000; Smith et al., 2001). Moreover, axonal loss can be caused by mutant glial cells in the absence of overt inflammation. For instance, in proteolipid protein (PLP)-deficient mutants and in mice overexpressing PLP, axons degenerate in the CNS (Anderson et al., 1998;Griffiths et al., 1998). In the peripheral nervous system, MAG-deficient mutants and mice deficient in the myelin component P0, an established model for severe forms of inherited neuropathies, show substantial axonal degeneration (Carenini et al., 1997; Yin et al., 1998; Frei et al., 1999). The molecular mechanisms underlying axonal damage in myelin mutants are not yet understood. In the present study, we tested the possibility that the robust myelin-related axonal loss in P0-deficient mutants (Frei et al., 1999) follows a similar mechanism to that of Wallerian degeneration. For this purpose, we cross-bred the myelin mutants with the spontaneous mouse mutant C57BL/Wlds showing protection from Wallerian degeneration for at least 14 d after injury because of fusion of the genes for the ubiquitination factor E4B (Ube4b) and nicotinamide mononucleotide adenylyltransferase (Nmnat) (Conforti et al., 2000; Mack et al., 2001). Apart from mechanical injury, C57BL/Wlds mice have been shown recently to be resistant to paclitaxel (Taxol)-induced sensory neuropathy (Wang et al., 2002). We found that in distal nerve portions of 3-month-old but not of 6-month-old dysmyelinating C57BL/Wlds mutants, myelin-related axonal degeneration is significantly reduced. Moreover, in the younger double mutants the rescued motor axons appeared functionally intact, as reflected by elevated muscle response amplitudes and increased muscle strength. Thus, introducing the Wldsmutation into a myelin mutant significantly delays dysmyelination-induced axonal degeneration.
Materials and Methods
Animals and genotyping. All experiments have been approved by the local governmental authorities of the State of Bavaria. Homozygous C57BL/Wlds mutants were obtained from Harlan-Winkelmann (Borchen, Germany) and cross-bred with heterozygous P0 mutants taken from our own breeding colony. Cross-breeding of heterozygous P0 mice with other mouse mutants has been described previously (Schmid et al., 2000; Carenini et al., 2001). According to Mendelian law, the individuals of the F1 generation were expected to consist exclusively of mice heterozygous for theWlds mutation. All of these individuals were genotyped for the P0 mutation by conventional PCR as described previously (Schmid et al., 2000), and individuals heterozygously deficient for P0 were intercrossed, leading to progenies with homozygous, heterozygous, and no P0 deficiency. Pulsed field gel electrophoresis was used to genotype for the 170 kbWlds insertion, because this method reliably distinguishes homozygotes from heterozygotes (Mi et al., 2002). Briefly, unfixed spleen tissue was dissociated in PBS, filtered to make a single-cell suspension, and embedded in 0.5% (final) low-melting point agarose in a plug mold (Bio-Rad, Munich, Germany). After cell lysis in 0.5m EDTA, 1% sodium-N-laurylsarcosine, and 25 μg/ml proteinase K (48°C, 3 d), plugs containing high-molecular-weight DNA were washed extensively in Tris–HCl, pH 7.5, and 1 mm EDTA. NotI digestion produced restriction fragments of 220 kb (wild type), 390 kb (Wlds) or both, which were resolved on a Bio-Rad Chef-DR electrophoresis cell and detected by hybridization with a single copy probe on the basis of a sequence within the triplication (GenBank accession number AF260927).
Counting of axons and histological analysis. Mice were transcardially perfused with sodium cacodylate buffer containing 4% paraformaldehyde (PFA) and glutaraldehyde (2% each), as described previously (Carenini et al., 2001). Plantar nerves and median nerves were carefully removed under a dissecting microscope at the level of the ankle joint and wrist, respectively, and postfixed in the perfusion fixative. Tissue preparation for electron microscopy was performed as described previously (Carenini et al., 2001).
The number of myelinated axons was counted on ultrathin sections, because demyelinated axons cannot be reliably quantified by light microscopy. We used a BioVision slow scan camera attached to aZeiss (Thornwood, NY) EM 10B and the corresponding software analySIS 3.0 Doku. To exclude bias, in all experiments axon counting was performed without knowledge of the C57BL/Wlds genotype. Statistical analysis was performed using a two-tailed t test, and pvalues of <0.05 were considered significant.
Spinal motoneuron labeling and counting. Mice were anesthetized with of a mixture of Ketanest (8 mg/ml) and Rompun (0.08%) in 0.9% NaCl (10 μl/gm body weight) and were placed on an operation hot plate. The plantar nerve of one side was exposed at the level of ankle joint, transected, and immediately introduced to 3% (in 0.1 m PBS) hydroxystilbamidine methanesulfonate (Fluorogold; Molecular Probes, Eugene, OR) in a 6-mm-long silicone tube for 1 hr. The tube was then removed, and the skin was sutured. The animals recovered for 5 d after Fluorogold exposure and then were perfused transcardially with a series of solutions: 0.9% NaCl (2–3 min), followed by 4% PFA in 0.1 mNa-acetate buffer, pH 6.5 (15 min), followed by 4% PFA in 0.1m Na-borate buffer, pH 9.5 (15 min), followed by 10% sucrose in 0.1 m Sörensen's buffer (15 min). The spinal cord (L2–S1) was dissected and frozen in Tissue-Tek freezing medium. Thirty-micrometer-thick cryosections were mounted and analyzed under a fluorescent microscope. Motoneurons labeled with Fluorogold were easily distinguishable in the ventral horn and were counted. Typically, the motor neurons supplying the plantar nerve occupied a column that was covering ∼120 serial sections (30 μm thick). To be sure that all of the corresponding motor neuron fragments had been considered, at least 80–100 sections rostral and caudal to the last labeled motoneuron fragments were investigated, so that a total number of ∼300 sections was scored for each animal. There was no contralateral motoneuron labeling, nor any labeling of terminals or neurons in the dorsal horn. To correct for multiple counting of the cells, the total number of motoneurons was recalculated using the method of Abercrombie (1946). Statistical analysis was performed using a two-tailed t test, and p values of <0.05 were considered significant.
Immunohistochemistry and Western blot analysis immunohistochemistry. Mice were transcardially perfused with 4% PFA in cacodylate buffer. Lumbar spinal cord was dissected and processed in 25% sucrose in 0.1 m PBS overnight. Ten micrometer cryosections of the spinal cord were treated with rabbit polyclonal anti-Wld antibody (1:250 dilution), raised against 18 aa at the junction between the Nmnat and Ube4b domains. The other primary antibody was mouse anti-tubulin III (Tuj1) monoclonal antibody (Research Diagnostics, Flanders, NJ) at 1:2500 dilution overnight.
Secondary antibodies were goat anti-rabbit Cy3 (1:200 dilution) and goat anti-mouse Cy2 (1:100 dilution) conjugated antibodies from Dianova (Hamburg, Germany).
Western blotting. Segments of lumbar spinal cord and combined dorsal root ganglia (L4–L5) were placed on ice in 20 vol of 60 mm Tris–HCl, pH 6.8, 8.0m urea, 2% (w/v) SDS, and 2% (v/v) 2-mercaptoethanol for 4 hr and then mechanically homogenized. They were mixed with an equal volume of 2× SDS-PAGE sample buffer and subjected to standard SDS-PAGE (10% polyacrylamide) and semidry blotting onto nitrocellulose. Wlds antigen was detected using anti-Wld antibody (see above), followed by horseradish peroxidase-coupled goat anti-rabbit secondary antibody (Dianova) and enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL). β-Tub 2.1 monoclonal antibody to β-tubulin (Sigma, St. Louis, MO) was used as a loading control.
Electrophysiology. Nerve conduction properties of sciatic nerves from 3-month-old mice were determined by established electrophysiological methods as described in detail previously (Zielasek et al., 1996). In all experiments, the investigator was not aware of the C57BL/Wlds genotype of the mice. Statistical analysis was performed using a one-tailedt test for grouped data.
Determination of muscle strength. Forelimb grip strength was determined using a custom-made automated grip strength meter as described previously (Masu et al., 1993). Animals were placed on a platform and allowed to grasp a ring. Mice were pulled away until they released the ring, and the strength was determined in Newtons by the electronic pull strain gauge. The ratio of male to females was 1:1 in each experimental group. In all experiments, the investigator was not aware of the C57BL/Wlds genotype of the mice. Statistical analysis was performed using an unpaired, two-tailedt test.
Results
Wlds reduces axonal degeneration in peripheral nerves of younger P0 mutants
The median and plantar nerves were chosen for the determination of axonal loss in the P0-deficient knock-out mutants (P0−/−). In 4-week-old P0−/− mice, numbers of axons were only slightly reduced, by ∼7% (p < 0.05) (Fig.1A) compared with age-matched wild-type mice, although features indicative of a severe dysmyelinating neuropathy were already evident at this age. At 6 weeks of age, axonal loss was more clearly detectable, with a reduction of ∼24% of fibers in plantar nerves (Fig. 1A,B). At 3 months of age in P0−/− mice these nerves showed a profound axonal loss when compared with age-matched wild-type mice (surviving axons numbered 60% of normal in the median and 59% of normal in the plantar nerve) (Fig. 1A,C,D), corroborating previous findings in the nerves of the toes (Frei et al., 1999).
Fig. 1.

A, Schematic representation of numbers of axons in plantar nerves. Plantar nerves of P0−/− mice show a progressive axonal degeneration. At 1 month, ∼7% of axons are lost compared with age-matched wild types. At 3 months, degeneration occurs with a higher rate leading to 40% axonal loss. Axonal degeneration progresses with age, with a lower rate leading to ∼60% axonal loss in 1-year-old P0−/− mice. B, Schematic representation of axon numbers in plantar nerves of 1.5-month-old single and double mutants. P0−/−/C57BL/Wldsdouble mutants show a reduced axonal degeneration compared with age-matched P0−/−/wild type (WT) (p < 0.05).C, D, Schematic representation of numbers of axons in plantar (C) and median (D) nerves of 3-month-old P0+/+ and P0−/− mice with and without theWlds mutation. Note that theWlds mutation has no influence on axon numbers in P0+/+ genotypes. However, in P0−/− mice, loss of axons is significantly reduced in mice carrying the Wlds mutation. InC, values from P0−/− mice heterozygous for the Wlds mutation are also given, showing an intermediate axonal loss because of a dosage-dependent rescue effect of theWlds mutation. Error bars are SDs. ***p < 0.0001; **p < 0.002; *p < 0.05. n, Number of individuals of the respective genotype investigated.hom, Homozygous; het, heterozygous.
We then cross-bred the P0 knock-out mice with C57BL/Wlds mice. In this mutant, Wallerian degeneration after nerve injury is greatly delayed (Lunn et al., 1989). We aimed to investigate whether the Wldsmutation could rescue the myelin-related loss of axons seen in P0−/− mice at the age of 6 weeks and 3 months. The median and plantar nerves of C57BL/Wlds mice were morphologically indistinguishable from nerves of wild-type mice (data not shown) and contained a similar number of axons (Fig. 1C,D). P0−/− mutants with the homozygousWlds mutation showed the same dysmyelinating neuropathy as single mutant P0−/− mice (Fig.2). However, at 6 weeks of age, in plantar nerves, there was a significant reduction in the degree of axonal degeneration in the double mutants, in that only 15% degenerated instead of 25% (Fig. 1B). Even more striking was the rescue effect of the Wldsmutation at 3 months, when axonal loss in P0 single mutants had substantially increased compared with 6-week-old P0 mutants (Fig.1C). At 3 months of age, ∼40% degenerated in the P0−/− single mutants, whereas only 27% degenerated in the homozygous double mutants. In other words, ∼30% of those axons that degenerated in the P0−/− single mutants were rescued (Fig.1C). A similar rescue effect by theWlds mutation was detected in the median nerve (Fig. 1D). Here, again, 40% degenerated in the P0−/− single mutants, whereas ∼30% degenerated in the homozygous double mutants. Because protection from Wallerian degeneration by the Wlds gene is dosage dependent (Mack et al., 2001), we investigated whether P0−/− mice with a heterozygousWlds mutation showed a rescue of plantar axons. Although the relative increase in axon numbers was less conspicuous but still significant, the intermediate value observed in P0−/−/C57BL/Wldsheterozygotes further supports a protective role for theWlds mutation (Fig. 1C).
Fig. 2.
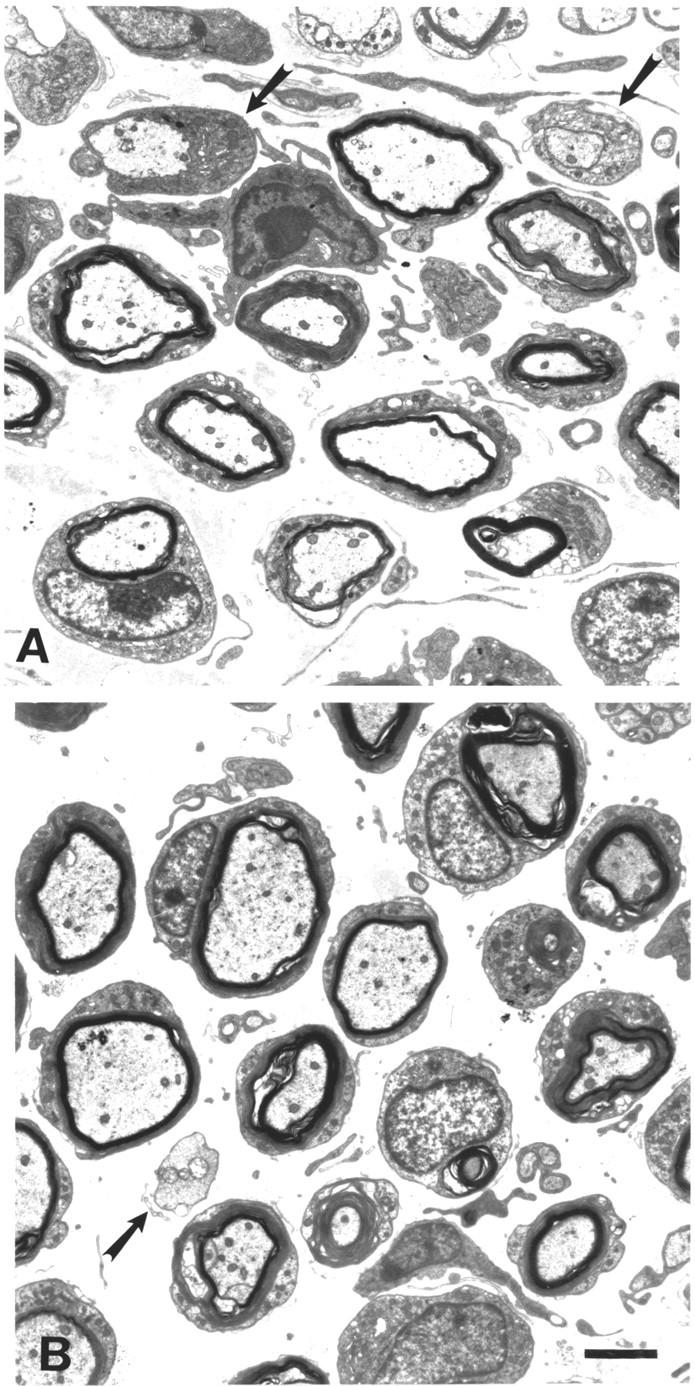
Electron micrographs of plantar nerves from P0−/− single mutants (A) and P0−/− mice with the homozygousWlds mutation (B). Note that P0−/− mutants with the homozygous Wlds mutation (B) show the same dysmyelinating neuropathy as single mutant P0−/− mice (A). Arrows point to myelin-competent axons devoid of myelin. Scale bar in B(for A and B), 2 μm.
Wlds leads to improved conduction properties and reduced motor axon loss in P0 mutants
Next, we investigated whether the axons that had been rescued by the Wlds mutation and looked morphologically normal are functionally intact and active in the 3-month-old double mutants. In an initial series of experiments, we investigated the nerve-conduction properties with particular emphasis on the amplitude of compound muscle action potentials of the small foot muscles as an indicator of functional integrity of axons and synapses. In 3-month-old P0 wild-type and homozygous C57BL/Wlds mice, a biphasic muscle response with a normal amplitude of ∼12 mV was recorded on electrical stimulation of the distal sciatic nerve. In P0−/− single mutants of the same age, we found a polyphasic response with a strikingly reduced amplitude of the main component (∼1.5 mV), whereas the partially protected double homozygous (P0−/−/C57BL/Wlds-homozygous) mice primarily showed a biphasic response. Although there was still a clear decline in the amplitude compared with the wild-type mice, the double homozygous mice showed more than twice the amplitude found in the myelin single mutants (Fig.3A,B), and this difference was statistically significant (p < 0.05). The amplitudes of P0−/− mice with a heterozygous Wlds mutation still showed a trend toward higher values compared with P0−/− single mutants (Fig.3A).
Fig. 3.
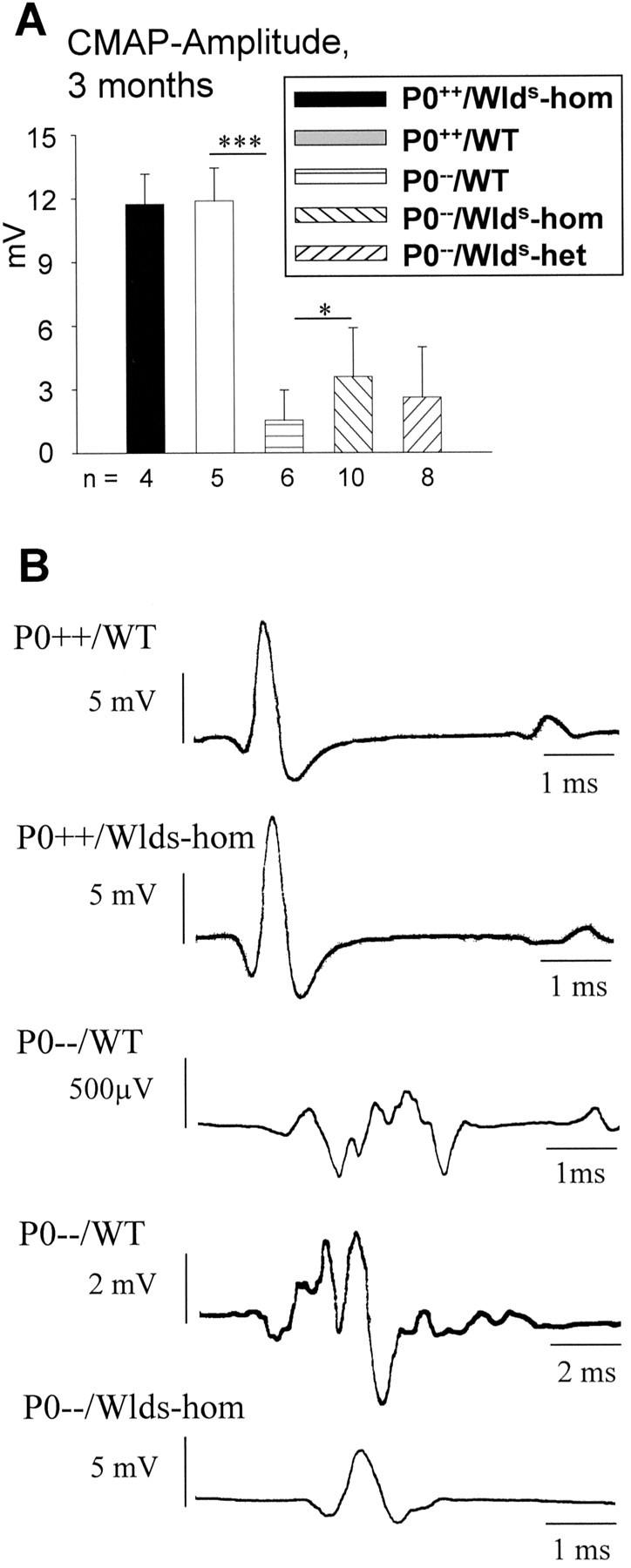
A, Schematic representation of amplitudes of compound action potentials from small foot muscles of 3-month-old P0+/+ and P0−/−mice with and without the Wldsmutation on distal stimulation of sciatic nerves. Note that theWlds mutation has no influence on amplitudes in P0+/+ genotypes. Amplitudes are markedly reduced in the absence of P0 (P0−/−), but reduction is less pronounced in the myelin mutants carrying the homozygousWlds mutation. Amplitudes from P0−/− mice heterozygous for theWlds mutation show a slight trend toward higher values compared with P0−/− single mutants. ***p < 0.0001 and *p< 0.05. n, Number of individuals of the respective genotype investigated. B, Original recordings of compound action potentials from small foot muscles of P0+/+ and P0−/− mice with and without the Wlds mutation on distal stimulation of sciatic nerves. Note the compact shape of muscle response in P0+/+ mice independent of theWlds mutation. P0−/− mice without theWlds mutation show predominantly dispersed responses, as shown here for two examples with relatively low and high amplitudes. Responses from P0−/−/C57BL/Wldsmice are usually more compact, as presented here for a typical case (lower recording). Note the different scales of time and amplitudes.CMAP, Compound muscle action potential;WT, wild type; hom, homozygous;het, heterozygous.
Because the increased amplitudes of compound muscle action potentials might be indicative of improved motor axon survival, we aimed to confirm the putative preservation of motor axons by an independent technique. Retrograde transport of Fluorogold and counting of the corresponding spinal motoneurons is an established technique for scoring intact motor axons in a peripheral nerve (Brushart, 1993). Using this technique, a significantly lower number of retrogradely labeled motoneurons was counted in P0−/−mice (136 ± 21) than in age-matched wild-type mice (177 ± 26) (Fig. 4). Most interestingly, the loss of ∼20% of retrogradely labeled motoneurons as seen in the single mutants could no longer be detected in the double mutants (171 ± 18), demonstrating again the protective role of theWlds gene (Fig. 4).
Fig. 4.

Schematic representation of retrogradely labeled spinal motoneurons of 3-month-old mice using Fluorogold. P0−/−/WT mice show a significantly lower number of back-labeled spinal (lumbosacral) motoneurons than P0+/+/WT mice (*p < 0.05). In 3-month-old P0−/−/C57BL/Wldsdouble mutants, however, the number of labeled motoneurons is as high as in P0+/+ mice, indicating a significant preservation of P0−/− motor axons onWlds expression. n, Number of individuals of the respective genotype investigated.WT, Wild type; hom, homozygous;het, heterozygous.
Wlds leads to improved muscle strength in P0 mutants
Next, we addressed the question of whether, as a consequence of improved motor axon survival, the Wldsmutation would also lead to improved muscle strength in P0−/− mutants by performing quantitative grip strength analysis. Wild-type mice at 3 months of age showed a grip strength of ∼1.3 N, a very similar value to that obtained in age-matched homozygous C57BL/Wlds mice (Fig. 5). As expected, P0−/− mice were weaker, with only 70% of the strength of mice with normal myelination. Three-month-old P0−/− double mutants with the homozygousWlds mutation displayed significantly more strength than the single mutant P0−/−mice (Fig. 5), reflecting the milder axonal loss in median nerves of the double mutants (Fig. 1D). P0−/− mice with a heterozygousWlds mutation still showed a mild yet nonsignificant trend toward higher values compared with P0−/− single mutants (Fig. 5). Thus, cross-breeding of C57BL/Wlds mice with P0−/− mice not only reduced axonal loss but also improved electrophysiological properties and physical strength compared with P0−/− single mutants.
Fig. 5.

Schematic representation of muscle strength (grip test of forelimb) of P0+/+ and P0−/− mice (3 months of age) with and without theWlds mutation. TheWlds mutation has no influence on muscle strength in P0+/+ genotypes. Muscle strength is strongly reduced in P0−/−/WT mice, but reduction is less pronounced in P0−/−/Wlds-homozygous mutants. Muscle strength of P0−/− mice carrying a heterozygous Wlds mutation show a small, nonsignificant trend toward higher values compared with P0−/− single mutants. ***p < 0.0005; *p <0.05. n, Number of individuals of the respective genotype investigated. WT, Wild type; hom, homozygous; het, heterozygous.
Wlds does not lead to reduced axon loss in 6-month-old P0−/− mutants
In a subsequent step, we investigated whether the rescue effect of the Wlds mutation is still detectable in P0−/− mice of >3 months of age. By investigating axon numbers in plantar nerves of 6-month-old P0−/−/C57BL/Wldshomozygous animals, P0−/−/C57BL/Wldsheterozygotes, and P0−/− mice, we no longer observed a decrease in axon loss in the presence of theWlds mutation (Fig.6A). Correspondingly, neither the amplitude of compound muscle action potentials nor the grip strength was significantly elevated in P0−/− mice carrying the homozygous or heterozygous Wlds mutation (Fig.6B,C). These results indicate that theWlds mutation delays, but does not completely abolish, the myelin-related axonal loss in P0−/− mice.
Fig. 6.
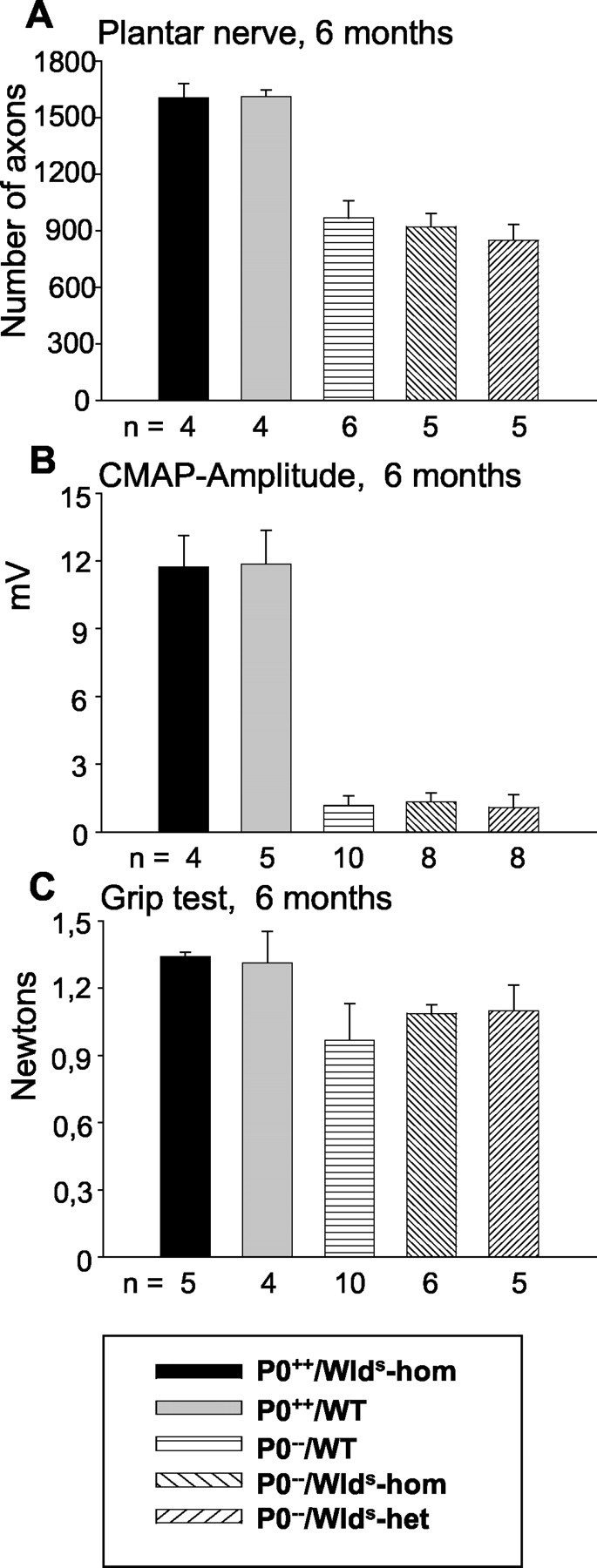
Schematic presentation of numbers of axons in plantar nerve (A), amplitudes of compound muscle action potentials (B), and muscle strength (C) of 6-month-old P0+/+, P0−/−, and P0−/−/C57BL/Wldsmice. A, In 6-month-old P0−/−/C57BL/Wldsdouble mutants, axonal loss is no longer reduced compared with P0−/−/WT mice. B, The amplitudes of compound muscle action potentials of small foot muscles of P0−/−/C57BL/Wldsdouble mutants are not significantly different from those of P0−/−/WT mutants. C, The muscle strength (grip test) of 6-month-old P0−/−/C57BL/Wldsdouble mutants is not significantly higher than that of P0−/−/WT mutants. n, Number of individuals of the respective genotype investigated.CMAP, Compound muscle action potential;WT, wild type; hom, homozygous;het, heterozygous.
Wlds protein expression is not decreasing with age
Next, we examined whether loss of axonal protection in older double mutants is caused by any decrease inWlds expression. Wlds protein expression has been reported to be unaltered in brain homogenates with aging (Gillingwater et al., 2002), but it has not been studied in motor or sensory neurons, nor has it been studied in the context of the P0−/− mutation. Using a highly specific polyclonal antibody against amino acids 71–88 of the Wlds protein (T. G. A. Mack and M. P. Coleman, unpublished observations), we showed the nuclear localization of the Wlds protein in the spinal motoneurons of 6-week-old and 6-month-old Wlds mutants (Fig.7E,F), but no signal was observed in the absence of the Wldsmutation (Fig. 7A,B). Furthermore, we observed no overt difference in the intensity of Wldsimmunoreaction in spinal motoneurons from young versus aged Wlds mice (Fig. 7C,D). In contrast, there was a clear difference in Wlds protein expression between homozygous and heterozygous Wlds mutants, proving that with the immunohistochemical method applied we can detect a decrease in protein expression of ∼50% (Fig.7D,F). Similarly, we could find no evidence of a decrease in Wlds protein expression in spinal cord or dorsal root ganglia between 6-week-old and 6-month-old double mutants (P0−/−/C57BL/Wlds-homozygous) by Western blot analysis (Fig. 8).
Fig. 7.
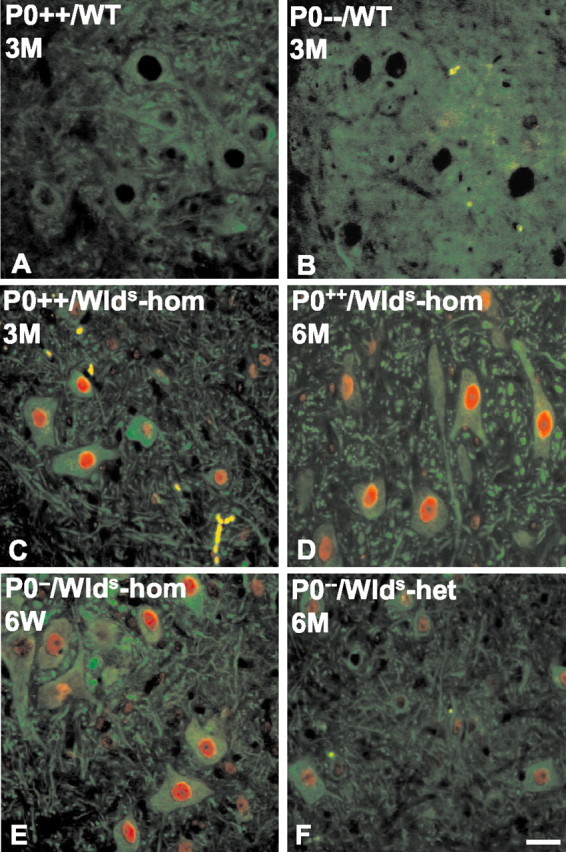
Wlds protein immunoreactivity is not decreased in the spinal motoneurons of young versus old C57BL/Wlds single or P0−/−/C57BL/Wldsdouble mutants. A primary antibody against Wldsstains nuclei of the spinal motoneurons of C57BL/Wlds mice (C,red). β-Tubulin III outlines the cell body and processes (green). The nuclear Wlds signal is missing in P0+/+or P0−/− mice that do not express this chimeric gene (A, B). There is no detectable decrease in Wlds immunoreactivity in young versus old C57BL/Wlds single mutant mice (C, D). Reduced expression of Wlds protein is detectable inWlds-heterozygous versusWlds-homozygous mutants (D, F). Scale bar, 20 μm.WT, Wild type; hom, homozygous;het, heterozygous.
Fig. 8.

Western blot analysis of young versus old P0−/−/C57BL/Wldsdouble mutants. Lumbar spinal cord (SPC) and lumbar (L4–L5) dorsal root ganglia (DRG) homogenates of 6-week-old versus 6-month-old P0−/−/C57BL/Wldsdouble mutants show a similar Wlds expression (Wlds). Transgenic (Tg Wlds) C57BL/Wlds and wild-typelanes show that the observed band is specific for Wlds mice. β-Tubulin (β-Tub) expression in spinal cord and dorsal root ganglia homogenates are shown as controls. hom, Homozygous.
Discussion
Here, we demonstrate that, in 6-week-old and 3-month-old P0−/− mice, a unique neuroprotective gene, Wlds, can partially rescue axons prone to undergoing myelin-related degeneration and in doing so alleviate the symptoms of mice suffering from inherited dysmyelination. We also present the first evidence that theWlds mutation can protect axons in vivo from the effects of chronic neurological disease, in addition to its well documented delay in axonal degeneration on acute injury (Gillingwater and Ribchester, 2001). The effect of theWlds mutation on the disorder is robust, because it rescues ∼30% of those axons that would degenerate in the 3-month-old P0−/− single mutants. It is of note that motor axons have been rescued by Wlds, as reflected by an increase in muscle strength, by elevated amplitudes of the muscle responses, and by an increased number of retrogradely labeled spinal motoneurons. Additionally, a reduced temporal dispersion of the muscle responses was found, possibly reflecting improved conduction velocities of some slowly conducting fibers in the double mutants. It is tempting to speculate that in the P0−/− single mutants, these particularly slowly conducting fibers comprise those prone to or being in the stage of degeneration, thus leading to reduced myelin function because of impaired axon–glia signaling.
We were able to show that in the double mutants especially motor axons are well preserved, because in 3-month-old P0−/−/Wldsmice, the number of back-labeled motoneuron cell bodies was as high as in P0+/+ mice. Comparing this robust rescue effect on motor axons with the compound muscle action potential amplitudes and grip test strength of the double mutants, the physiological and functional rescue effect has to be considered lower than one would expect from the high number of preserved motor axons. The most probable explanation for this discrepancy is the recent finding that presynaptic motor nerve terminals are less well preserved by theWlds mutation than the motor axon (Gillingwater et al., 2002).
Our findings imply that myelin-related degeneration of axons has some features in common with injury-induced Wallerian degeneration. So far, the principle mechanisms that underlie Wallerian degeneration are only incompletely understood. It has been shown previously that Ca2+ influx and activation of the Ca2+-dependent protease calpain are involved (George et al., 1995; Wang et al., 2000). Wallerian degeneration does not seem to follow caspase-3-mediated apoptosis (Finn et al., 2000), although it would seem likely that an active mechanism of some kind is involved, because it appears to be a regulated process. The recent characterization of the mutation underlying delayed Wallerian degeneration in C57BL/Wlds mice revealed an 85 kb triplication on chromosome 4 (Coleman et al., 1998) creating the chimeric gene Ube4b/Nmnat (Conforti et al., 2000) that, when transmitted to transgenic mice, confers protection from Wallerian degeneration (Mack et al., 2001). Thus, characterization of the mechanism by which this gene protects the axon should give valuable insights into mechanisms underlying both injury-induced and dysmyelination-related degeneration of axons.
The rescue effect of the Wlds mutation in P0−/− mice is transient. Whereas in 3-month-old mice ∼30% of axons prone to degeneration could be rescued, at 6 months the rescue effect was no longer detectable. This might be related to the fact that in C57BL/Wlds mice the rescue of motor nerve terminals and the ability of peripheral nerves to transmit compound action potentials is reduced considerably between the 2 and 4 months of age (Perry et al., 1992; Gillingwater et al., 2002). However, this age dependency on axon rescue does not correlate with preservation of axon structure (Crawford et al., 1995; Gillingwater et al., 2002). We also demonstrate that the loss of axons in 6-month-old P0−/−/Wlds double homozygotes cannot be explained by any decrease in Wlds protein expression with age. Thus, the most likely explanation is that Wldsprotected axons in these experiments for only a limited period. It is now important to find ways to extend this period of protection, perhaps, for example, by expressing Wldsprotein at still higher levels.
Because the Wlds mutation significantly prohibits myelin-related axonal degeneration, our study may be relevant for the clinical aspects of axon degeneration in inherited dysmyelinating peripheral neuropathies of the Charcot-Marie-Tooth type in humans. The major medical problem of patients suffering from these relatively frequent disorders (Skre, 1974) is progressive and disabling weakness and muscle atrophy on the basis of distal motor axonal loss (Berciano et al., 2000; Martini, 2001). Similarly, in other relatively frequent myelin disorders, such as multiple sclerosis, axonal loss seems to be the major reason for the clinical impact of the disease (Trapp et al., 1998; Kornek et al., 2000; Kuhlmann et al., 2002). Thus, additional studies will be necessary to investigate the reasons for the functional decline of the Wlds protein on the myelin-related axonopathy in adult animal models of incurable human nerve disorders.
Footnotes
This work was supported by grants from the German Research Council (SFB-581, to R.M. and K.V.T.), Gemeinnützige Hertie-Stiftung (GHS-191/00/01, to R.M.), Federal Ministry of Education and Research (FKZ: 01-KS-9502, to M.P.C.), and Center for Molecular Medicine Cologne, University of Cologne (ZMMK, to M.P.C. and W.M.); and local research funds from the University of Würzburg. We are grateful to Heinrich Blazyca and Carolin Kiesel for excellent technical assistance, Dr. Robert Adalbert for genotyping one group of double mutants, Helga Brünner for animal care, and Drs. Jochen Ulzheimer, Martin Berghoff, and in particular Igor Kobsar for help with data processing and statistics.
Correspondence should be addressed to Dr. Rudolf Martini, Department of Neurology, Developmental Neurobiology, University of Würzburg, Josef-Schneider-Strasse 11, D-97080 Würzburg, Germany. E-mail:rudolf.martini@mail.uni-wuerzburg.de.
References
- 1.Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- 2.Anderson TJ, Schneider A, Barrie BA, Klugmann M, McCulloch MC, Kirkham D, Kyriakides E, Nave K-A, Griffiths IR. Late-onset neurodegeneration in mice with increased dosage of the proteolipid protein gene. J Comp Neurol. 1998;394:506–519. doi: 10.1002/(sici)1096-9861(19980518)394:4<506::aid-cne8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 3.Berciano J, Garcia A, Calleja J, Combarros O. Clinico-electrophysiological correlation of extensor digitorum brevis muscle atrophy in children with Charcot-Marie-Tooth disease 1A duplication. Neuromuscul Disord. 2000;10:419–424. doi: 10.1016/s0960-8966(99)00114-5. [DOI] [PubMed] [Google Scholar]
- 4.Brushart TME. Motor axons preferentially reinnervate motor pathways. J Neurosci. 1993;13:2730–2738. doi: 10.1523/JNEUROSCI.13-06-02730.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carenini S, Montag D, Cremer H, Schachner M, Martini R. Absence of myelin-associated glycoprotein (MAG) and the neural cell adhesion molecule (N-CAM) interferes with the maintenance, but not with the formation of peripheral myelin. Cell Tissue Res. 1997;287:3–9. doi: 10.1007/s004410050727. [DOI] [PubMed] [Google Scholar]
- 6.Carenini S, Mäurer M, Werner A, Blazyca H, Toyka KV, Schmid CD, Raivich G, Martini R. The role of macrophages in demyelinating peripheral nervous system of mice heterozygously deficient in P0. J Cell Biol. 2001;152:301–308. doi: 10.1083/jcb.152.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci USA. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crawford TO, Hsieh ST, Schryer BL, Glass JD. Prolonged axonal survival in transected nerves of C57BL/Ola mice is independent of age. J Neurocytol. 1995;24:333–340. doi: 10.1007/BF01189060. [DOI] [PubMed] [Google Scholar]
- 10.Finn JT, Weil M, Archer F, Siman R, Srinivasan R, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frei R, Mötzing S, Kinkelin I, Schachner M, Koltzenburg M, Martini R. Loss of distal axons and sensory Merkel cells and features indicative of muscle denervation in hindlimbs of P0-deficient mice. J Neurosci. 1999;19:6058–6067. doi: 10.1523/JNEUROSCI.19-14-06058.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J Physiol (Lond) 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillingwater TH, Thomson D, Mack TG, Soffin EM, Mattison RJ, Coleman MP, Ribchester RR. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J Physiol (Lond) 2002;543:739–755. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- 16.Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H. Multiple sclerosis and chronic autoimmune encephalomyelitis. A comparative quantitative study of axonal injury in active, inactive and remyelinated lesions. Am J Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–2212. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- 18.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;11:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 19.Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlto A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;12:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 20.Martini R. The effect of myelinating Schwann cells on axons. Muscle Nerve. 2001;24:456–466. doi: 10.1002/mus.1027. [DOI] [PubMed] [Google Scholar]
- 21.Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H. Disruption of the CNTF gene results in motor neuron degeneration. Nature. 1993;365:27–32. doi: 10.1038/365027a0. [DOI] [PubMed] [Google Scholar]
- 22.Mi W, Conforti L, Coleman MP. Genotyping methods to detect a unique neuroprotective factor for axons (Wld(s)). J Neurosci Methods. 2002;113:215–218. doi: 10.1016/s0165-0270(01)00501-5. [DOI] [PubMed] [Google Scholar]
- 23.Perry HV, Brown MC, Tsao JW. The effectiveness of the gene which slows the rate of Wallerian degeneration in C57BL/Ola mice declines with age. Eur J Neurosci. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- 24.Redford EJ, Kapoor R, Smith KJ. Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain. 1997;120:2149–2157. doi: 10.1093/brain/120.12.2149. [DOI] [PubMed] [Google Scholar]
- 25.Schmid CD, Stienekemeier M, Oehen S, Bootz F, Zielasek J, Gold R, Toyka KV, Schachner M, Martini R. Immune deficiency in mouse models for inherited peripheral neuropathies leads to improved myelin maintenance. J Neurosci. 2000;20:729–735. doi: 10.1523/JNEUROSCI.20-02-00729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 27.Smith KJ, Kapoor R, Hall SM, Davies M. Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol. 2001;49:470–476. [PubMed] [Google Scholar]
- 28.Toyka KV, Hartung HP. Chronic inflammatory polyneuritis and related neuropathies. Curr Opin Neurol. 1996;9:240–250. doi: 10.1097/00019052-199606000-00016. [DOI] [PubMed] [Google Scholar]
- 29.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 30.Trapp BD, Ransohoff RM, Fisher E, Rudick RA. Neurodegeneration in multiple sclerosis: relationship to neurological disability. The Neuroscientist. 1999;5:48–57. [Google Scholar]
- 31.Wang MS, Wu Y, Culver DG, Glass JD. Pathogenesis of axonal degeneration: parallels between Wallerian degeneration and vincristine neuropathy. J Neuropathol Exp Neurol. 2000;59:599–606. doi: 10.1093/jnen/59.7.599. [DOI] [PubMed] [Google Scholar]
- 32.Wang MS, Davis AA, Culver DG, Glass JD. Wlds mice are resistant to paclitaxel (Taxol) neuropathy. Ann Neurol. 2002;52:442–447. doi: 10.1002/ana.10300. [DOI] [PubMed] [Google Scholar]
- 33.Yin X, Crawford TO, Griffin JW, Tu PH, Lee VMY, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zielasek J, Martini R, Toyka KV. Functional abnormalities in P0-deficient mice resemble human hereditary neuropathies linked to P0 gene mutations. Muscle Nerve. 1996;19:946–952. doi: 10.1002/(SICI)1097-4598(199608)19:8<946::AID-MUS2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]