

Abstract
Senile plaques found in the Alzheimer's disease brain are foci of local inflammatory reactions mediated by plaque-associated microglia. The interaction of microglia with compacted deposits of β-amyloid (Aβ) fibrils results in the stimulation of intracellular Tyr kinase-based signaling cascades and cellular activation, leading to the secretion of proinflammatory molecules. This study identifies a cell surface receptor complex that mediates the binding of microglia to Aβ fibrils and the subsequent activation of intracellular signaling pathways leading to a proinflammatory response. The receptor complex includes the B-class scavenger receptor CD36, the integrin-associated protein/CD47, and the α6β1-integrin. Antagonists of scavenger receptors, CD36, CD47, and α6β1 inhibited the adhesion of THP-1 monocytes to Aβ fibrils. In addition, peptide competitors of Aβ fibril interactions with CD36, scavenger receptors, CD47, and the α6β1-integrin inhibited Aβ stimulation of Tyr kinase-based signaling cascades in both THP-1 monocytes and murine microglia as well as interleukin 1β production. A scavenger receptor antagonist and antibodies specific for CD36 and the β1-integrin subunit also inhibited the Aβ-stimulated generation of reactive oxygen species. Importantly, the principal components of this receptor complex are shared with those for other fibrillar proteins and thus represent general elements through which myeloid lineage cells recognize complex fibrillar proteins. Identification of the cell surface molecules that interact with Aβ fibrils and mediate their activation of intracellular signaling cascades represents a potential intervention point in the treatment of Alzheimer's disease.
Keywords: Alzheimer's disease, β-amyloid, α6β1-integrin, CD36, CD47, microglia, THP-1 monocytes, scavenger receptor, signal transduction, receptors, Tyr kinase
Introduction
The contribution of an inflammatory component to the pathogenesis of Alzheimer's disease (AD) has been well documented (Akiyama et al., 2000). Microglia are the principal immune effector cells in the brain and undergo a conversion into a reactive proinflammatory phenotype after their association with fibrillar β-amyloid (Aβ) deposits comprising the senile plaques in the AD brain. Epidemiological studies have demonstrated the decreased incidence and severity of AD in patient populations treated with nonsteroidal anti-inflammatory drugs (Rich et al., 1995; Breitner, 1996; Stewart et al., 1997; in't Veld et al., 2001), which were correlated with a 65% reduction in plaque-associated reactive microglia (Mackenzie and Munoz, 1998). These data support a role for microglia in the pathophysiology of AD and as a target of anti-inflammatory therapy.
The interaction of microglia with fibrillar forms of Aβ peptides initiates cellular activation, as evidenced by the enhanced expression of immune cell markers by these cells, including CD45, major histocompatibility complex class I and II antigens, Fcγ receptors, and complement receptors 3 and 4 (Akiyama et al., 2000). The plaque-associated microglia also exhibit increased levels of intracellular phospho-Tyr staining (Wood and Zinsmeister, 1991), which is a consequence of the activation of Tyr kinase-based signaling pathways on exposure of the cells to Aβ fibrils (McDonald et al., 1997, 1998; Combs et al., 1999). Significantly, these Aβ-stimulated microglial signaling events lead to the production and secretion of a broad range of proinflammatory molecules and neurotoxic factors that contribute to the sustained inflammatory response and neuronal loss observed in AD (Combs et al., 2000, 2001; Yates et al., 2000).
There has been considerable interest in the identity of the cell surface receptor(s) that mediates microglial activation on interaction with Aβ fibrils. A number of receptors have been reported to bind Aβ, namely scavenger receptor class A (SR-A; El Khoury et al., 1996;Paresce et al., 1996), SR-B1 (Husemann et al., 2001), the neuronal α7 nicotinic acetylcholine receptor (Dineley et al., 2001; Pettit et al., 2001), the receptor for advanced glycation end products (RAGE; Yan et al., 1996), the serpin enzyme complex (Boland et al., 1996), the formyl peptide chemotactic receptor (FPR; Lorton, 1997; Le et al., 2001), heparan sulfate proteoglycans (Giulian et al., 1998; Scharnagl et al., 1999), and the α5β1-integrin (Matter et al., 1998). However, only SR-A and RAGE have been shown to interact with fibrillar forms of Aβ, and neither of these receptors is linked to activation of intracellular signaling cascades leading to a proinflammatory response. Recently, while this work was in progress, the B class scavenger receptor CD36 was also shown to act as a receptor for fibrillar Aβ (Coraci et al., 2002; Moore et al., 2002).
Our approach to the identification of putative Aβ receptors arose from the recognition that myeloid lineage cells use multiple cell surface receptors to bind fibrillar proteins, and the assembly of ensembles of receptors is necessary for cellular activation (Ishibashi et al., 1994; Bornstein, 1995; Wong et al., 1996). We evaluated the involvement of a number of such receptors by monitoring their ability to induce intracellular signaling events and generation of proinflammatory molecules such as interleukin 1β (IL-1β) and reactive oxygen species (ROS). We report the identification of a multireceptor complex comprising the B-class scavenger receptor CD36, α6β1-integrin, and the integrin-associated protein CD47. This complex mediates activation of microglia and other myeloid cells (e.g., THP-1 monocytes) on interaction with fibrillar Aβ peptides.
Materials and Methods
Materials. The anti-β1-integrin antibody Lia1/2 used for intracellular signaling assays was from Immunotech (Marseille, France). The β1-integrin antibody used for adhesion assays and all α subunit integrin antibodies were from Chemicon (Temecula, CA). Anti-β2-integrin antibody was from Roche Molecular Biochemicals (Indianapolis, IN). The anti-Fyn JD3 antibody was from Dr. S. Brady-Kalnay (Case Western Reserve University). The glutathione S-transferase (GST)-CD36-(93–120) peptide was a gift from Dr. S. Frieda Pearce (Cornell University, Ithaca, NY). The anti-CD36 monoclonal antibody OKM5 was from Ortho-Clinical Diagnostics (Raritan, NJ). Mouse IgG was from Sigma (St. Louis, MO). The 4N1K and RHD peptides were purchased from Bachem (Philadelphia, PA) and reconstituted in sterile distilled water. Fucoidan, pertussis toxin, and N-formyl-Met-Leu-Phe (fMLP) were purchased from Sigma and reconstituted in sterile distilled water. Aβ peptides corresponding to human Aβ amino acids 25–35 and 1–42 were purchased from American Peptide Co. (Sunnyvale, CA), and that corresponding to amino acids 1–40 was from California Peptide Research, Inc. (Napa, CA). The peptides were resuspended in sterile distilled water followed by incubation at 37°C for 1 week to allow fibrillarization. The method used to fibrillarize Aβ25–35, 1–40, and 1–42 has been well characterized (Burdick et al., 1992;Lorenzo and Yankner, 1994; Terzi et al., 1994); however, the composition of the fibrillar solutions was not tested and may have included Aβ oligomers as well as fibrils. Nonfibrillar Aβ1–40 was prepared by resuspending the peptide in sterile distilled water and used immediately. Soluble RAGE (sRAGE) was a gift from Dr. Mark Kindy (University of Kentucky). The anti-phospho-Tyr antibody 4G10 was obtained from Upstate Biotechnology (Lake Placid, NY). Anti-phospho-extracellular-regulated kinase (ERK) antibody was obtained from New England Biolabs (Beverly, MA). Anti-ERK2 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-IL-1β was obtained from the National Cancer Institute (Frederick, MD). Affinity-purified horseradish peroxidase-conjugated sheep anti-mouse and donkey anti-rabbit antibodies were from Amersham Biosciences (Piscataway, NJ).
Tissue culture. Human THP-1 monocytes (American Type Culture Collection, Manassas, VA) were grown in RPMI 1640 medium (Whittaker Bioproducts, Walkersville, MD) containing 10% heat-inactivated fetal bovine serum (FBS), 5 × 10−5m2-mercaptoethanol, 5 mm HEPES, and 15 μg/ml gentamycin in 5% CO2. Microglia were derived from postnatal day 1–2 mouse brains (C57BL/6J) as described previously (McDonald et al., 1997).
Cell stimulation. THP-1 cells and microglia were collected and resuspended in 37°C HBSS for 30 min at 37°C in the absence or presence of sRAGE, pertussis toxin, fucoidan, GST-CD36-(93–120), 4N1K, or the RHD peptide or at 4°C in the presence of anti-α1, 2, 4, 5,6, v or anti-β1 antibody for intracellular signaling analysis. Cells were then stimulated by the addition of 60 μm Aβ peptides for 3 min at 37°C, collected by centrifugation, and lysed at 4°C in Triton buffer (1% Triton X-100, 20 mm Tris, pH 7.5, 100 mm NaCl, 40 mm NaF, 1 mm EDTA, 1 mm EGTA, and 1 mmNa3VO4). All experiments were performed a minimum of three times.
IL-1β assay. THP-1 cells were plated 48 hr before stimulation in 2% FBS. The cells (3 × 106) were collected and resuspended in 37°C 0.5% FBS with 0.5 μg/ml lipopolysaccharide for 4 hr in the presence of fucoidan, 4N1K, and RHD peptide and stimulated with 60 μm Aβ25–35. Cells were collected by centrifugation and lysed at 4°C in radioimmunoprecipitation assay (RIPA) buffer (1% Triton X-100, 20 mm Tris, pH 7.5, 100 mm NaCl, 40 mmNaF, 0.2% SDS, 0.5% deoxycholate, 1 mm EDTA, 1 mm EGTA, and 1 mmNa3VO4).
Immunoprecipitation and Western blotting. After cell lysis, insoluble material was removed by centrifugation at 10,000 ×g at 4°C for 10 min. Protein concentrations were determined by the method of Bradford (1976) using bovine serum albumin as a standard. Lysates were added to 30 μl of protein A-agarose with the primary antibody (2 μg of primary antibody/mg of lysate) and incubated with rocking for 2 hr at 4°C. Immune complexes were washed three times in Triton X-100 buffer. Lysates and immune complexes were resolved by 7.5 or 12% SDS-PAGE and Western blotted with primary antibody (4G10, 1:1500), anti-phospho-ERK (1:1000), anti-ERK2 (1:2000), anti-Fyn (1:1000), or anti-IL-1β (1:1000) overnight at 4°C. Antibody binding was detected by enhanced chemiluminescence (Pierce, Rockford, IL). IL-1β, 4G10, and phospho-ERK blots were stripped by incubation in stripping buffer (62.5 mm Tris, pH 6.8, 100 mmβ-mercaptoethanol, and 2% SDS) for 30 min at 50°C and then reprobed with anti-Fyn or anti-ERK2 antibodies. Quantitation of the Aβ-stimulated Tyr phosphorylation was obtained by imaging the ECL signal using a Bio-Rad (Hercules, CA) VersaDoc, and the integrated optical density of the individual lanes was obtained using Quantity One software.
Respiratory burst. Intracellular superoxide radical generation was assayed by measuring nitroblue tetrazolium (NBT) reduction (Pick, 1986; McDonald et al., 1997). THP-1 cells (2 × 106) were resuspended at 37°C for 30 min in the presence or absence of fucoidan, anti-CD36 antibody, anti-β1 antibody, or an isotype-specific mouse IgG and with 1 mg/ml NBT. Cells were collected by centrifugation and sonicated in 4°C RIPA buffer. NBT reduction was measured by the change in absorbance at 550 nm. The assays were performed in triplicate.
Cell adhesion assay. THP-1 monocytes were preincubated with antibody or peptide antagonist for 30 min in 4°C serum-free RPMI 1640 medium. The fibrillar Aβ peptides (2 μg) were applied directly to a glass slide and allowed to dry. The adherent Aβ fibrils could be visualized by eye. Although the percentage of peptide that bound was not determined, it was constant between individual spots in each experiment. Approximately 50,000 cells were applied to either the glass slide or the Aβ fibrils and allowed to adhere for 5 min. Cells were washed at 37°C, and the number of adherent cells were counted microscopically. The data are reported as cell number bound per square millimeter. The assays were performed in triplicate.
Immunochemistry. Microglia were fixed in ice-cold acetone for 15 min, washed, blocked with PBS containing 20% goat serum, incubated with 20 μg/ml mouse anti-murine CD36 IgA or control IgA for 60 min, washed, incubated with 1 μg/ml biotinylated goat anti-mouse IgA for 30 min at room temperature, washed, and incubated with 2 mg/ml avidin-conjugated Alexa 488 (Molecular Probes, Eugene, OR) in PBS at room temperature. All antibodies were diluted in PBS containing 3% goat serum.
Statistical analysis. Experiments were done in triplicate. Mean values ± SEM for each experiment were determined, and values statistically different from controls were calculated using one-way ANOVA. The Tukey–Kramer multiple-comparisons post test was used to determine P values.
Results
Stimulation of protein-Tyr phosphorylation in THP-1 monocytes by fibrillar Aβ peptides
We have previously demonstrated that fibrillar Aβ peptides, Aβ1–40, Aβ1–42, and Aβ25–35, stimulated protein-Tyr phosphorylation in THP-1 monocytes and microglia (McDonald et al., 1997, 1998; Combs et al., 1999). Quantitative analysis of the response revealed that the three peptides elicited similar increases in protein-Tyr phosphorylation in THP-1 cells, with the Aβ25–35 peptide exhibiting a modestly greater response than the longer peptides (Fig.1). These data indicate that fibrillar Aβ peptides containing the terminal 10 amino acids that form a β-pleated sheet are sufficient to stimulate intracellular signaling cascades (Terzi et al., 1994). The response to the various fibrillar Aβ peptides was qualitatively similar, indicating that they act through common mechanisms to initiate intracellular signaling events.
Fig. 1.

Fibrillar Aβ25–35, 1–40, and 1–42 peptides stimulate comparable increases in protein-Tyr phosphorylation in THP-1 monocytes. THP-1 monocytes were stimulated with fibrillar Aβ25–35, 1–40, and 1–42 peptides for 3 min. Cell lysates were analyzed by Western blot analysis using the anti-phospho-Tyr antibody 4G10. The integrated optical density (IOD) ratio of the protein-Tyr phosphorylation signal stimulated by the different Aβ ligands was quantitated.
Scavenger receptors mediate interactions of Aβ fibrils with THP-1 monocytes and microglia
Senile plaque-associated microglia are activated after the interaction of Aβ fibrils with microglia (Akiyama et al., 2000). Previous reports have provided evidence for a role for both SR-A and SR-B1 in the binding of fibrillar Aβ peptides to neonatal microglia, resulting in the subsequent generation of ROS (El Khoury et al., 1996;Paresce et al., 1996; Husemann et al., 2001). Scavenger receptors constitute a structurally diverse family of cell surface receptors that exhibit broad substrate specificity and participate in both cellular adhesion and uptake of ligands (Krieger and Herz, 1994). Sulfated polysaccharides such as fucoidan act to inhibit SR–ligand interactions in an SR class-independent manner. We have verified that incubation of THP-1 monocytes with fucoidan inhibited the ability of these cells to bind to surfaces coated with fibrillar Aβ peptides (Fig.2A). Because THP-1 monocytes do not normally express high levels of SR-A or SR-B1 unless differentiated into macrophages (Hsu et al., 1996), fucoidan-mediated inhibition of cell binding to Aβ fibrils provoked us to examine the involvement of additional SR class members.
Fig. 2.

Scavenger receptors are expressed on microglia and are involved in cellular adhesion to fibrillar Aβ. THP-1 monocytes were preincubated in serum-free RPMI 1640 medium with 300 μg/ml fucoidan (A) or 100 nmGST-CD36-(93–120) (B), and the cell suspension was applied to the fibrillar Aβ (2 μg) bound to a glass slide. Dose-dependent inhibition of adhesion of THP-1 monocytes to Aβ25–35 fibrils in the presence of 20–100 nm GST-CD36-(93–120) (C) and 50 nm GST or GST-CD36 (D) was evaluated. Cells were treated as described in (A, B). The number of adherent cells per square millimeter was recorded. The data shown represent the mean ± SEM of triplicate determinations. (*p < 0.001)E, Primary murine microglia were stained with an anti-CD36 antibody (left panel) or control Ig (middle panel) as described in Materials and Methods or visualized by phase microscopy (right panel).
We examined whether the B class scavenger receptor CD36 participated in monocyte adhesion to fibrillar Aβ and found that the binding of THP-1 monocytes to Aβ25–35 and Aβ1–40 fibrils was inhibited by a peptide derived from the extracellular domain of CD36 (residues 93–120 fused to GST; Fig. 2B) with aKi of ∼50nm (Fig. 2C). The inhibition of cell adhesion was specific to GST-CD36-(93–120) fusion peptide, because GST alone did not have any effect on cell binding to Aβ fibrils (Fig. 2D). Amino acids 93–120 of CD36 represent the minimal region of the CD36 receptor required for the binding of the fibrillar angiogenesis peptide thrombospondin-1 (TSP-1; Pearce et al., 1995). Anti-CD36 antibodies directed to other epitopes on CD36 did not inhibit cell adhesion (data not shown; Coraci et al., 2002), indicating that the interaction of Aβ fibrils was restricted to the same domain as that required for TSP-1 binding. We wished to verify that CD36 was expressed on microglia. We demonstrated the expression of CD36 on neonatal murine microglia by fluorescent immunohistochemistry (Fig.2E). Microglia stained with control Ig showed no staining (Fig. 2E).
A primary goal of these studies was to establish the identity of cell surface receptors linked to activation of intracellular signaling pathways. CD36 is physically associated with members of the Src family of Tyr kinases (Huang et al., 1991; Bull et al., 1994), which transduce signals from this receptor (Jimenez et al., 2000). We have previously demonstrated that exposure of murine microglia and THP-1 monocytes to Aβ25–35, Aβ1–40, or Aβ1–42 fibrils resulted in the activation of Tyr kinase-based signaling pathways, including activation of the Src family kinase Lyn and Syk, leading to the activation of the ERK and p38 MAP kinase cascades (McDonald et al., 1997, 1998; Combs et al., 1999). We found that exposure of cells to CD36 antagonists, either fucoidan or the GST-CD36-(93–120) peptide, inhibited the Aβ-stimulated activation of intracellular protein-Tyr phosphorylation and subsequent ERK phosphorylation in THP-1 monocytes (Fig.3A,B) and microglia (Fig.3C). A control GST peptide did not have any effect on Aβ-stimulated intracellular signaling (data not shown). We were unable to demonstrate ligand-dependent changes in the association of Src family kinases with CD36 (data not shown).
Fig. 3.
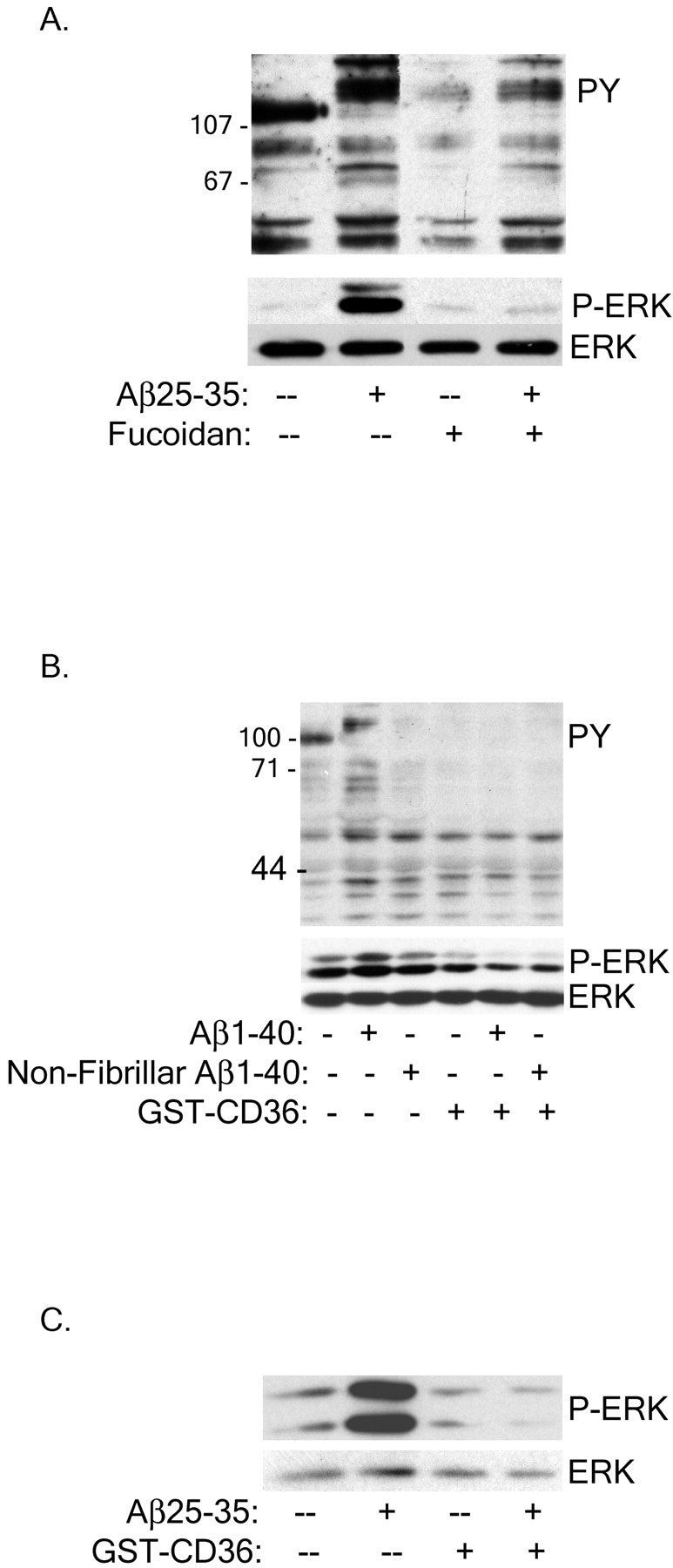
Scavenger receptors mediate Aβ stimulation of intracellular Tyr signaling cascades in THP-1 monocytes and microglia. THP-1 monocytes were preincubated with 300 μg/ml fucoidan (A) or 100 nm GST-CD36-(93–120) (B) for 30 min before stimulation with fibrillar or nonfibrillar Aβ. C, Primary murine microglia were treated with 100 nm GST-CD36-(93–120) before Aβ stimulation. Cell lysates were analyzed by Western blot using the anti-phospho-Tyr antibody 4G10 (PY) or an anti-phospho-ERK antibody (P-ERK). P-ERK blots were stripped and reprobed with an anti-ERK antibody (ERK) as a protein-loading control.
The α6β1-integrin and the integrin-associated protein CD47 mediate Aβ-stimulated phospho-Tyr signaling and adhesion in THP-1 monocytes and microglia
The integrins represent a family of molecules that activate intracellular signaling cascades and promote adhesion to other cells as well as matrix proteins. The identification of the signaling molecules transducing Aβ-stimulated responses suggested the participation of this superfamily of receptors in the binding of other fibrillar proteins (McDonald et al., 1997, 1998). Evidence for the involvement of the β1-integrin as an element of the Aβ receptor complex was obtained using a peptide containing the consensus β1-integrin-binding epitope RHD. This peptide comprises residues 1–11 of Aβ (DAEFRHDSGYE) and contains a sequence closely related to the canonical integrin recognition site peptide RGD. However, although RGD binds to most integrins, heterodimers containing the β1 subunit display an RHD binding preference (Ghiso et al., 1992). The RHD-containing peptide inhibited Aβ-mediated intracellular Tyr and ERK phosphorylation in THP-1 monocytes (Fig.4A) and microglia (Fig.4B). It is evident that the epitope present in the fibrillar Aβ species that interacts with the β1-integrin is distinct from the RHD sequence, because fibrils derived from Aβ25–35 are also antagonized by the anti-β1 antibody. To confirm that the β1-integrin subunit was specifically involved in mediating Aβ stimulation of protein-Tyr phosphorylation, THP-1 monocytes were incubated with an anti-β1antibody. Western blot analysis confirmed inhibition of Aβ-mediated intracellular phospho-Tyr signaling by the β1antibody (Fig. 4C). To identify the corresponding α-integrin subunit, we used a panel of antibodies and found that an antibody to α6 (Fig. 4D), but not other α-integrin subunits (Fig. 4E; α3-integrin data not shown), inhibited both intracellular phospho-Tyr signaling and ERK phosphorylation.
Fig. 4.
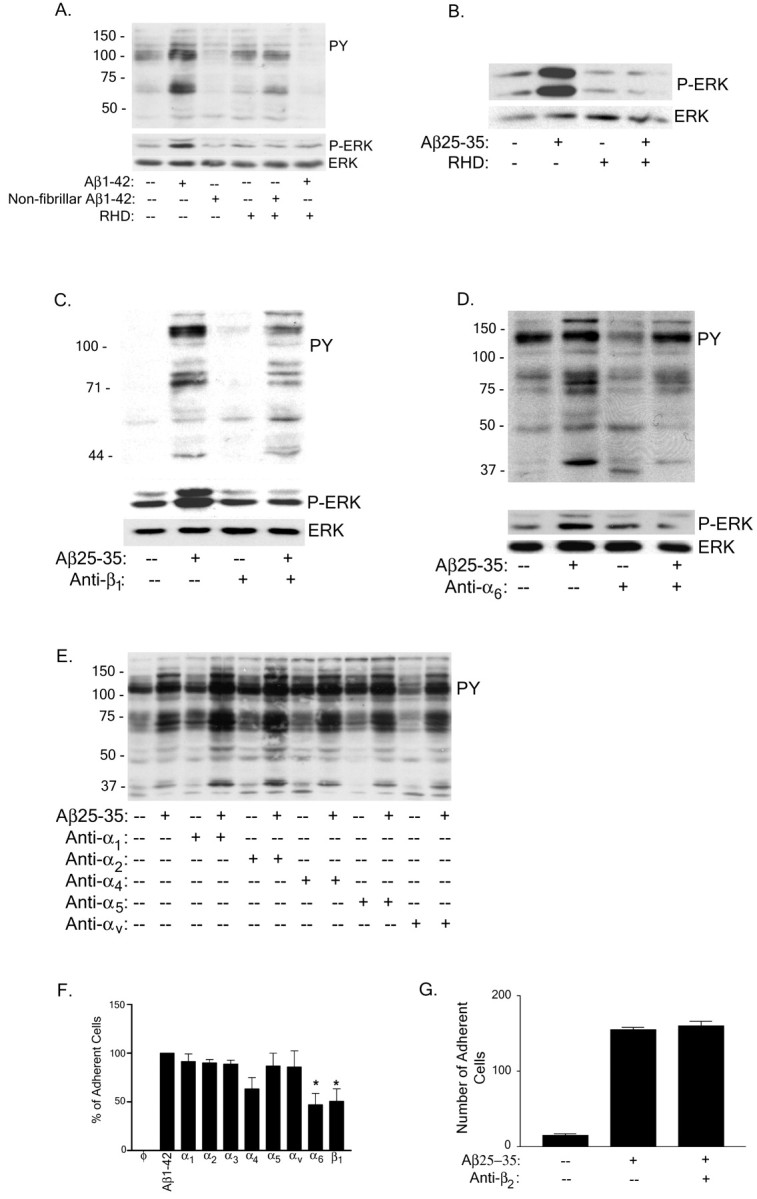
The α6β1-integrin is required for Aβ binding and stimulation of THP-1 monocytes.A, THP-1 monocytes were preincubated with 100 μg/ml RHD peptide for 30 min before fibrillar or nonfibrillar Aβ1–40 stimulation. Cell lysates were analyzed by Western blot using the anti-phospho-Tyr antibody 4G10 (PY) or an anti-phospho-ERK antibody (P-ERK).B, Primary murine microglia were pretreated with RHD peptide as described in A but stimulated with fibrillar Aβ25–35 and analyzed by P-ERK Western blot analysis of cell lysates. Blots were stripped and reprobed with anti-ERK antibody (ERK) as a protein-loading control.C–E, THP-1 monocytes were preincubated with antibodies (40 μg/ml) to the β1 (C), α6 (D), and α1,2,4,5,v (E) integrins as described in A and stimulated with fibrillar Aβ25–35. Cell lysates were evaluated by Western blot analysis, using the 4G10, P-ERK, and ERK antibodies. F, G, Adhesion of THP-1 cells to fibrillar Aβ evaluated by preincubation in serum-free RPMI 1640 medium with antibodies to the α1–6, v- and β1-integrin subunits (40 μg/ml;F) or anti-β2-integrin subunit (40 μg/ml; G) and added to 2 μg of fibrillar Aβ bound to a glass slide. The number of adherent cells per square millimeter was recorded. The data shown represent the mean ± SEM of triplicate determinations (*p < 0.05;F).
One of the primary functions of integrins is to mediate cellular adhesion, and we next evaluated which integrins were responsible for cellular binding to Aβ fibrils using a panel of antibodies to β1-integrin partners. We found that antibodies to the α6- and β1-integrin subunits decreased cell adhesion of THP-1 monocytes to Aβ1–42 fibrils. We were unable to implicate other α subunits (α1–5 and αv; Fig. 4F) or the β2 (Fig. 4G) and β3 (data not shown) integrin subunits in cellular adhesion. These data demonstrate that the α6β1-integrin is the principal integrin species responsible for the interaction of Aβ fibrils with the cells.
The integrin-associated protein CD47 is a transmembrane receptor expressed on many cell types and acts primarily to modulate integrin-dependent signaling through its physical and functional association with integrins (Porter and Hogg, 1998) as well as intracellular signaling molecules, including the Tyr kinases Lyn and Syk (Chung et al., 1997). We tested whether CD47 was involved in Aβ-stimulated intracellular signaling cascades and adhesion events. A peptide antagonist of CD47, 4N1K, is derived from the cell-binding domain of TSP-1 and specifically blocks the interaction of CD47 with TSP-1 (Chung et al., 1997). Incubation of THP-1 monocytes with 4N1K inhibited the ability of THP-1 monocytes to adhere to Aβ1–42 fibrils (Fig. 5A). In addition, blocking the association of CD47 with Aβ fibrils inhibited Aβ fibril stimulation of intracellular protein-Tyr and ERK phosphorylation in THP-1 monocytes (Fig. 5B).
Fig. 5.

Inhibition of CD47 interactions with Aβ fibrils blocks Aβ-mediated THP-1 monocyte activation. A, THP-1 monocytes were preincubated in serum-free RPMI 1640 medium with 100 μm CD47 antagonist peptide 4N1K and added to 2 μg of fibrillar Aβ bound to a glass slide. The number of adherent cells per square millimeter was recorded. The data shown represent the mean ± SEM of triplicate determinations (*p < 0.01).B, THP-1 monocytes were preincubated with 100 μm 4N1K for 30 min before fibrillar Aβ25–35 or Aβ1–42 stimulation. Cell lysates were analyzed by Western blot using the anti-phospho-Tyr antibody 4G10 (PY) or an anti-phospho-ERK antibody (P-ERK). The P-ERK blot was stripped and reprobed with an anti-ERK antibody (ERK) as a protein-loading control.
Aβ-stimulated signaling requires elements of the Aβ-binding receptor complex
It has been well documented that exposure of microglia and monocytes to fibrillar forms of Aβ stimulates NADPH oxidase activation and a respiratory burst, resulting in the generation and secretion of ROS (Meda et al., 1995; El Khoury et al., 1996; McDonald et al., 1997; Bianca et al., 1999; Van Muiswinkel et al., 1999). Preincubation of THP-1 monocytes with fucoidan, an anti-CD36 antibody, and an anti-β1-integrin subunit antibody inhibited the Aβ-stimulated respiratory burst (Fig.6A–C). Thus, these data support an essential role for scavenger receptors, specifically CD36, as well as the β1-integrin in mediating the Aβ-stimulated respiratory burst in microglia cells and monocytes.
Fig. 6.
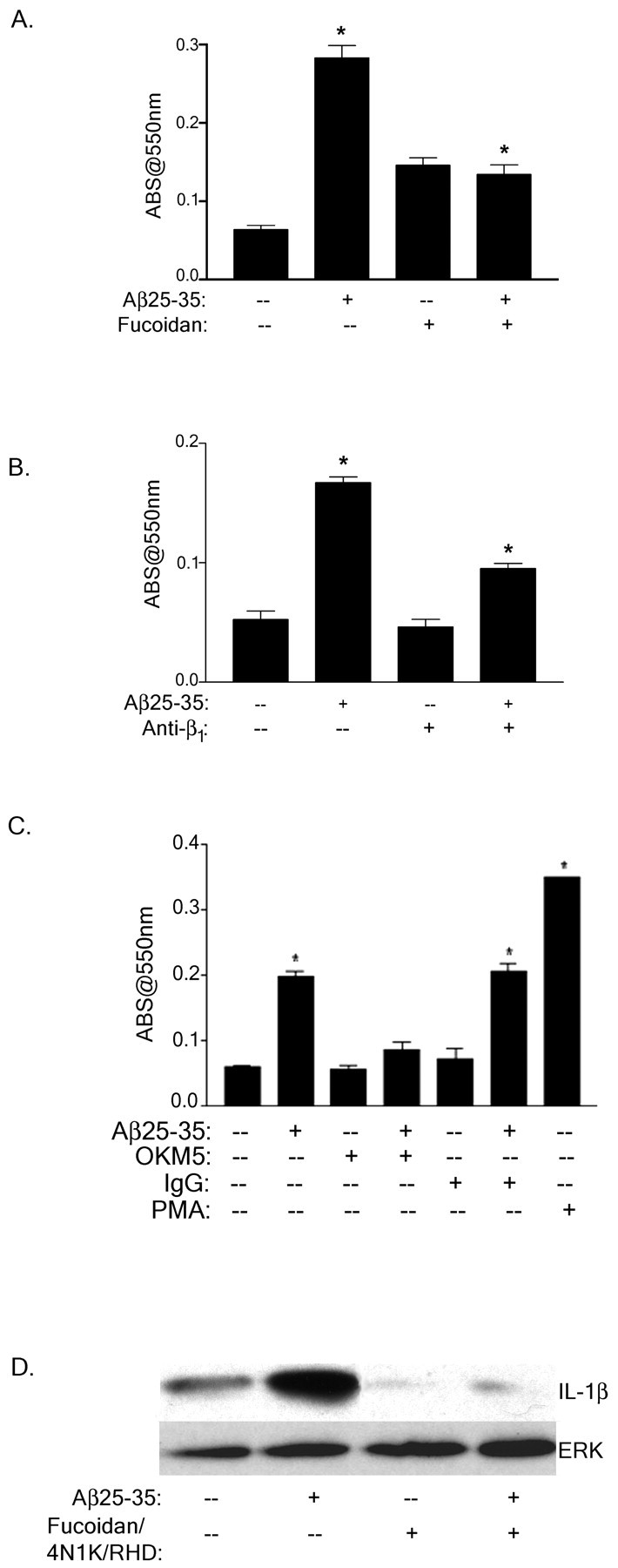
The Aβ receptor complex mediates the stimulation of ROS and IL-1β production. A–C, THP-1 monocytes were incubated with fucoidan (300 μg/ml; A), anti-β1 (40 μg/ml; B), isotype-specific IgG (C) phorbol 12-myristate 13-acetate (PMA, C) or anti-CD36 (OKM5, 40 μg/ml; C) in serum-free RPMI containing NBT and stimulated with fibrillar Aβ25–35 for 25 min at 37°C. Superoxide anion generation was measured by the change in absorbance at 550 nm. The data shown represent the mean ± SEM of triplicate determinations (*p < 0.001;A–C). D, Mature IL-1β production in Aβ-stimulated THP-1 monocytes was evaluated in the presence of 300 μg/ml fucoidan, 100 μm 4N1K, and 100 μg/ml RHD peptide. Cell lysates were resolved by 12% SDS-PAGE, transferred to polyvinylidene difluoride, and probed with the anti-IL-1β antibody. The blot was stripped and reprobed with an anti-ERK antibody (ERK) as a protein-loading control.
The proinflammatory cytokine IL-1β is elevated in AD and augments the inflammatory response of the disease through its autocrine and paracrine actions (Akiyama et al., 2000). It has previously been demonstrated that microglia secrete IL-1β as well as other cytokines on exposure to fibrillar Aβ and other immune stimuli (Araujo and Cotman, 1992; Meda et al., 1995, 1996, 1999; Lorton et al., 1996; Fiala et al., 1998; Murphy et al., 1998; Combs et al., 2000, 2001). We therefore assessed the role of the putative Aβ receptors in Aβ-stimulated IL-1β production. The 4 hr time course required to measure IL-1β levels in response to extracellular stimuli technically excluded our ability to assess the effects of the occupancy of the individual receptors on this response. However, blocking the association of Aβ fibrils with the Aβ receptor complex using antagonists of CD47, the β1-integrin, and scavenger receptors inhibited Aβ-mediated stimulation of IL-1β production (Fig. 6D). Therefore, the engagement of the Aβ-binding receptor complex is required for the ability of Aβ fibrils to mediate intracellular changes and subsequent production of neurotoxic and proinflammatory factors.
To formally test whether the concerted actions of the receptor elements were required for the activation of intracellular signaling cascades, we evaluated the effects of antagonists of the individual receptor elements on the activation of Src kinases. We report that Fyn, like its family member Lyn, is activated in response to Aβ stimulation. Importantly, Fyn phosphorylation is dependent on the interaction of each receptor with fibrillar Aβ (Fig.7). The activation of Fyn and its association with downstream signaling elements are inhibited after blockade of scavenger receptors, specifically CD36, CD47, and the β1-integrin (Fig. 7).
Fig. 7.

Members of the Aβ receptor complex are required for Aβ stimulation of Fyn phosphorylation. THP-1 monocytes were preincubated in serum free RPMI 1640 medium with 300 μg/ml fucoidan, 100 nm GST-CD36-(93–120), 100 μm 4N1K, or 100 μg/ml RHD for 30 min before stimulation with fibrillar Aβ. Cell lysates were immunoprecipitated with the anti-Fyn JD3 antibody. Cell lysates and immunoprecipitates were resolved by 7.5% SDS-PAGE and analyzed by Western blot using the anti-phospho-Tyr antibody 4G10 (PY). Blots were stripped and reprobed with an anti-Fyn JD3 antibody (FYN) as a protein-loading control.
A Gi-linked receptor is not involved in Aβ stimulation of Tyr kinase-based signaling pathways in THP-1 monocytes
The heterotrimeric small G-protein Gi is an effector of some CD47-linked receptors and the chemotactic FPR (Gao et al., 1996; Chung et al., 1997; Lorton, 1997; Frazier et al., 1999; Wang et al., 1999; Le et al., 2001). We tested the involvement of Gi-proteins in Aβ-mediated stimulation of intracellular signaling cascades. Inhibition of Gi action by pertussis toxin treatment of THP-1 monocytes did not affect Aβ stimulation of protein-Tyr phosphorylation or ERK phosphorylation (Fig.8). The specificity of pertussis toxin activity was monitored using the chemotactic peptide fMLP, which stimulates ERK phosphorylation after binding of FPR receptors in a pertussis toxin-sensitive manner. We conclude that a Gi-linked receptor is not involved in Aβ stimulation of intracellular signaling in THP-1 monocytes, and the participation of CD47 in this event is independent of its ability to functionally associate with Gi-proteins.
Fig. 8.

A Gi-linked receptor is not involved in Aβ-stimulated intracellular signaling. THP-1 monocytes were preincubated with 200 ng/ml pertussis toxin (PTX) for 30 min at 37°C and stimulated with Aβ25–35 or 1 μm fMLP for 3 min. Western blots of cellular lysates were probed with the anti-phospho-Tyr antibody 4G10 (PY) or with the anti-phospho-ERK antibody (P-ERK). Blots were stripped and reprobed with the anti-ERK antibody (ERK).
RAGE does not mediate Aβ stimulation of Tyr kinase-based signaling pathways in THP-1 monocytes
RAGE was previously reported to be an Aβ-binding protein, capable of interacting with both fibrillar and nonfibrillar forms of Aβ and provoking microglia activation (Yan et al., 1996; Lue et al., 2001). However, it is unclear whether there is a preference of RAGE for aggregated forms of Aβ (Yan et al., 1996). We (McDonald et al., 1997,1998; Combs et al., 1999) and others (Bianca et al., 1999) have failed to demonstrate that engagement of RAGE on microglia or THP-1 monocytes stimulates cellular effects similar to those induced by fibrillar Aβ. In the present study, we extended our analysis of RAGE as a candidate Aβ-binding receptor mediating stimulation of intracellular signaling. The addition of sRAGE at 2× (120 μm) or 20× (1.2 mm) the concentration of fibrillar Aβ did not alter Aβ-mediated stimulation of protein-Tyr phosphorylation, as demonstrated by Western blot analysis (Fig.9). We verified that sRAGE alone did not affect the ability of Aβ fibrils to stimulate intracellular signaling, nor did sRAGE alone stimulate any changes in intracellular Tyr phosphorylation. Although we cannot rule out that RAGE may participate in the adhesion of cells to Aβ and act in common with other cell surface proteins such as SR-A, we do not find evidence for the involvement of RAGE in Aβ-mediated stimulation of intracellular signaling cascades.
Fig. 9.

RAGE is not involved in Aβ-mediated intracellular signaling. THP-1 monocytes were incubated for 30 min at 37°C and stimulated with Aβ25–35, 2 or 20× sRAGE, or both for 3 min. Western blots of cellular lysates were probed with the anti-phospho-Tyr antibody 4G10 (PY).
Discussion
There has been substantial interest in the identity of cell surface molecules that bind Aβ, because this represents the first potential point of intervention in the events leading to the pathophysiology of AD. Our approach to the identification of microglial Aβ receptors derived from consideration of the mechanisms through which macrophages interact with foreign elements, including protein fibrils. These cells use ensembles of receptors whose individual receptor elements exhibit relatively broad substrate specificity. The resulting receptor complex can mediate adhesion, migration, a respiratory burst, or proinflammatory responses. For example, the fibrillar proteins Bordetella pertussis filamentous hemagglutinin (FHA) and TSP-1 provoke cellular activation, a response similar to that observed in Aβ-stimulated microglia. TSP-1 interacts with CD36, CD47, and β1- and β3-integrins and several other cell surface proteins (Bornstein, 1995), whereas FHA binds to monocytes via CD87 (urokinase receptor), αmβ2- and αvβ3-integrins, and CD47 (Ishibashi et al., 1994; Wong et al., 1996). Thus, we reasoned that Aβ fibrils, which consist of repetitive units linked through C-terminal β-pleated sheet domains, may use some of the same receptors to interact with microglia and monocytes. Importantly, we evaluated the participation of candidate receptor elements by monitoring the ability of these molecules to stimulate Aβ-activated intracellular signaling pathways whose activation is functionally linked to the production and secretion of proinflammatory and neurotoxic factors (McDonald et al., 1997; Bianca et al., 1999; Combs et al., 1999; Meda et al., 1999).
We report the identification of a multicomponent Aβ receptor complex consisting of CD36, α6β1-integrin, and CD47. This complex mediates the adhesion of Aβ fibrils to microglia and subsequent activation of intracellular Tyr kinase-based signal transduction cascades, leading to the stimulation of a respiratory burst and IL-1β cytokine production (Fig.10). It is significant that this receptor does not interact with nonfibrillar forms of Aβ, whereas association with all fibrillar forms of Aβ was shown to elicit cellular responses (Pike et al., 1993; Lorton et al., 1996; McDonald et al., 1997, 1998; Combs et al., 1999).
Fig. 10.

Model of Aβ fibril interaction with a microglial cell surface receptor complex resulting in activation of intracellular signal transduction cascades.
Scavenger receptors are expressed on cells of monocytic lineage and interact with a structurally diverse range of ligands. The A class of SRs has been shown to participate in binding of Aβ by microglia and has been implicated in chemotaxis, ROS generation, and Aβ peptide uptake (Hartung et al., 1986; Klegeris et al., 1994; Meda et al., 1995;El Khoury et al., 1996; Paresce et al., 1996). An exclusive role for SR-A in these events appears unlikely in view of recent reports that microglia from SR-A null animals are only modestly impaired in their ability to bind Aβ peptides (Husemann et al., 2001). Moreover, transgenic animals overexpressing the amyloid precursor protein but lacking SR-A expression exhibited levels of amyloid deposition similar to those of SR-A-expressing control animals (Huang et al., 1999). The compensatory action of B-class SRs may explain these findings.
We have provided evidence that the B-class SR CD36 plays an essential role in both the binding of Aβ fibrils to the cells and the initiation of intracellular signaling events. This view is supported by the report that macrophage binding to oxidized low-density lipoprotein is mediated by SR-A, whereas subsequent intracellular production of H2O2 is dependent on CD36 (Maxeiner et al., 1998). Moreover, despite considerable work, it has not been possible to conclusively show that SR-A acts independently to elicit activation of intracellular signaling cascades. CD36 is physically associated with Src family protein kinases, and it has recently been demonstrated that TSP-1 binding results in the activation of these Tyr kinases (Jimenez et al., 2000). Importantly, TSP-1 and Aβ peptides interact with a common domain on CD36, and we have demonstrated that binding of Aβ fibrils to microglia and monocytes elicits the activation of the Src family kinases Lyn and Fyn (McDonald et al., 1997; Combs et al., 1999). These data support our hypothesis that CD36 (and possibly SR-BI) is a critical component of the Aβ receptor complex linking fibril binding to stimulation of signal transduction pathways and that SR-A acts primarily to tether Aβ fibrils to the cell surface of microglia and is not critical for Aβ stimulation of intracellular signaling events (Maxeiner et al., 1998;McDonald et al., 1998; Husemann et al., 2001).
While this manuscript was under review, Coraci et al. (2002) and Moore et al. (2002) published compelling evidence that CD36 acts as a receptor for Aβ fibrils and participates in both cellular adhesion and initiation of intracellular signaling events. Their observations are consistent with those reported here and provide further support for critical involvement of this scavenger receptor in Aβ-stimulated activation of microglia.
Integrins are likely candidate receptors for Aβ on microglia cells, because they promote adhesion to other cells and matrix proteins. We have shown that the α6β1-integrin is a central element of the receptor complex and is essential for eliciting a cellular response to Aβ and adhesion of Aβ fibrils. Ligation of the β1-integrin subunit has been shown to increase the protein-Tyr phosphorylation of paxillin, focal adhesion kinase, Syk, and Fyn, leading to the production of proinflammatory cytokines such as IL-1β (Lin et al., 1995; Chen et al., 2000). Many of the Tyr kinases that are stimulated by Aβ interaction with microglia and monocytic lineage cells are also components of integrin signaling pathways, including those linked to ERK phosphorylation (McDonald et al., 1997, 1998; Combs et al., 1999). It was previously reported that the α5β1-integrin recognizes nonfibrillar Aβ (Matter et al., 1998). However, we did not detect any effect of a blocking antibody directed against α5. Thus, α5β1 is unlikely to be involved in microglial activation by fibrillar Aβ in AD.
We have also implicated CD47 in mediating Aβ stimulation of THP-1 monocytes and microglia. CD47 is an integral membrane protein that functionally interacts with both β1- and β3-integrins, where it serves to modulate integrin signaling functions and cellular adhesion. Importantly, CD47 physically interacts with both integrins and intracellular signaling elements and may act to assemble signaling complexes and functionally integrate signals generated through ligand binding at the cell surface (Porter and Hogg, 1998). In addition, CD47 can act independently to promote cell–cell adhesion through its interactions with fibrillar proteins such as TSP-1 and cell surface proteins on adjacent cells (Jiang et al., 1999; Babic et al., 2000) and has been demonstrated to have a role in integrin-independent signaling events (Brown and Frazier, 2001). We have demonstrated a critical requirement for CD47 in cellular binding of fibrillar Aβ and the subsequent activation of intracellular protein-Tyr kinase cascades.
We provide evidence for a receptor complex that acts both to mediate the adhesion of cells to Aβ fibrils and to activate intracellular signaling cascades. There is now extensive literature documenting the influence of cellular contacts with the extracellular matrix and other cells on the organization and coupling to intracellular signaling systems (Woods and Shimizu, 2001). These data have clearly demonstrated that the process of adhesion itself activates specific signaling pathways and also influences the efficiency of other receptors in stimulating their downstream effectors. We cannot distinguish the relative contribution of the individual receptor elements to activation of intracellular signaling pathways as distinct from those responsible for adhesion, but it seems probable that these events are intimately linked.
Microglial recognition of Aβ fibrils uses a cell surface receptor complex that capitalizes on the broad substrate specificity of the individual receptor elements. We postulate that the engagement of Aβ fibrils with the core CD36, α6β1, and CD47 receptors serves to bind the fibril to the cell surface, leading to immobilization and the focal aggregation of the receptors into a complex. The cooperative action of the receptor core elements confers a unique binding capability to the receptor complex that is a composite of the actions of the individual receptors. It is important to note that CD36, CD47, and α6β1 are physically associated with Tyr kinases as well as other intracellular signaling molecules. Moreover, it has been reported that the α6β1-integrin exists in a complex with CD36 (Thorne et al., 2000; Miao et al., 2001), and β1-containing integrins have been shown to physically interact with CD47 (Wang and Frazier, 1998; Chung et al., 1999). We demonstrate a direct role of these receptors in transducing signals generated from microglial interactions with Aβ fibrils, resulting in the stimulation of proinflammatory pathways. The assembly of the receptor complex results in activation of Tyr kinases and downstream signaling pathways in a manner directly analogous to that of other immune receptor complexes. Importantly, disrupting the interaction of Aβ fibrils with any component of the receptor complex inhibits Aβ stimulation of intracellular signaling cascades. The peptides interacting with the binding sites of the individual receptors do not exhibit agonist activity, suggesting that the fibrillar peptide serves to focally assemble a signaling complex. It is possible that additional cell surface receptors are involved in Aβ stimulation of microglial activation, either in the binding of Aβ fibrils to the cell surface or the activation of intracellular signaling cascades. It is noteworthy that both CD36 and α6β1 expression is induced on activation of myeloid-derived cells (Tontonoz et al., 1998;Kloss et al., 2001). Thus, the identification of an Aβ-binding receptor complex provides a therapeutic intervention point, targeting the specific cell surface event resulting in the proinflammatory activation of microglia associated with Aβ deposits in the AD brain.
Footnotes
This work was supported by National Institutes of Health Grants AG 16740 and AG 08012 and Training Grant GM 08056-18 (M.E.B.). J.H. was was supported by Columbia University Alzheimer's Disease Research Center Pilot Grant Award AG 08702 and the Alzheimer's Association. We thank the Blanchett Hooker Rockefeller Foundation and the Coins for Alzheimer's Research Trust Fund of Rotary International for generous support of our work. We thank Dr. Colin Combs for assistance and helpful comments on this manuscript and Dr. Frieda Pearce and Dr. Mark Kindy for providing us with reagents.
Correspondence should be addressed to Dr. Gary Landreth, Alzheimer Research Laboratory, E504, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail:gel2@po.cwru.edu.
References
- 1.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Araujo DM, Cotman CW. Beta-amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer's disease. Brain Res. 1992;569:141–145. doi: 10.1016/0006-8993(92)90380-r. [DOI] [PubMed] [Google Scholar]
- 3.Babic I, Schallhorn A, Lindberg FP, Jirik FR. SHPS-1 induces aggregation of Ba/F3 pro-B cells via an interaction with CD47. J Immunol. 2000;164:3652–3658. doi: 10.4049/jimmunol.164.7.3652. [DOI] [PubMed] [Google Scholar]
- 4.Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils: a possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 5.Boland K, Behrens M, Choi D, Manias K, Perlmutter DH. The serpin-enzyme complex receptor recognizes soluble, nontoxic amyloid-β peptide but not aggregated, cytotoxic amyloid-β peptide. J Biol Chem. 1996;271:18032–18044. doi: 10.1074/jbc.271.30.18032. [DOI] [PubMed] [Google Scholar]
- 6.Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol. 1995;130:503–506. doi: 10.1083/jcb.130.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 8.Breitner JC. The role of anti-inflammatory drugs in the prevention and treatment of Alzheimer's disease. Annu Rev Med. 1996;47:401–411. doi: 10.1146/annurev.med.47.1.401. [DOI] [PubMed] [Google Scholar]
- 9.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 10.Bull HA, Brickell PM, Dowd PM. Src-related protein tyrosine kinases are physically associated with the surface antigen CD36 in human dermal microvascular endothelial cells. FEBS Lett. 1994;351:41–44. doi: 10.1016/0014-5793(94)00814-0. [DOI] [PubMed] [Google Scholar]
- 11.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 12.Chen LM, Bailey D, Fernandez-Valle C. Association of beta 1 integrin with focal adhesion kinase and paxillin in differentiating Schwann cells. J Neurosci. 2000;20:3776–3784. doi: 10.1523/JNEUROSCI.20-10-03776.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung J, Gao AG, Frazier WA. Thrombospondin acts via integrin-associated protein to activate the platelet integrin alphaIIbbeta3. J Biol Chem. 1997;272:14740–14746. doi: 10.1074/jbc.272.23.14740. [DOI] [PubMed] [Google Scholar]
- 14.Chung J, Wang XQ, Lindberg FP, Frazier WA. Thrombospondin-1 acts via IAP/CD47 to synergize with collagen in alpha2beta1-mediated platelet activation. Blood. 1999;94:642–648. [PubMed] [Google Scholar]
- 15.Combs CK, Johnson DJ, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of β-amyloid and prion proteins. J Neurosci. 1999;19:928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Combs CK, Karlo JC, Kao SC, Landreth GE. β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella G, Luster AD, Silverstein SC, El Khoury J. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 21.Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med. 1998;4:480–489. [PMC free article] [PubMed] [Google Scholar]
- 22.Frazier WA, Gao AG, Dimitry J, Chung J, Brown EJ, Lindberg FP, Linder ME. The thrombospondin receptor integrin-associated protein (CD47) functionally couples to heterotrimeric Gi. J Biol Chem. 1999;274:8554–8560. doi: 10.1074/jbc.274.13.8554. [DOI] [PubMed] [Google Scholar]
- 23.Gao AG, Lindberg FP, Finn MB, Blystone SD, Brown EJ, Frazier WA. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J Biol Chem. 1996;271:21–24. doi: 10.1074/jbc.271.1.21. [DOI] [PubMed] [Google Scholar]
- 24.Ghiso J, Rostagno A, Gardella JE, Liem L, Gorevic PD, Frangione B. A 109-amino-acid C-terminal fragment of Alzheimer's-disease amyloid precursor protein contains a sequence, -RHDS-, that promotes cell adhesion. Biochem J. 1992;288:1053–1059. doi: 10.1042/bj2881053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giulian D, Haverkamp LJ, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher AE. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer's disease. J Biol Chem. 1998;273:29719–29726. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 26.Hartung HP, Kladetzky RG, Melnik B, Hennerici M. Stimulation of the scavenger receptor on monocytes-macrophages evokes release of arachidonic acid metabolites and reduced oxygen species. Lab Invest. 1986;55:209–216. [PubMed] [Google Scholar]
- 27.Hsu HY, Nicholson AC, Hajjar DP. Inhibition of macrophage scavenger receptor activity by tumor necrosis factor-α is transcriptionally and post-transcriptionally regulated. J Biol Chem. 1996;271:7767–7773. doi: 10.1074/jbc.271.13.7767. [DOI] [PubMed] [Google Scholar]
- 28.Huang F, Buttini M, Wyss-Coray T, McConlogue L, Kodama T, Pitas RE, Mucke L. Elimination of the class A scavenger receptor does not affect amyloid plaque formation or neurodegeneration in transgenic mice expressing human amyloid protein precursors. Am J Pathol. 1999;155:1741–1747. doi: 10.1016/S0002-9440(10)65489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang MM, Bolen JB, Barnwell JW, Shattil SJ, Brugge JS. Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc Natl Acad Sci USA. 1991;88:7844–7848. doi: 10.1073/pnas.88.17.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Husemann J, Loike JD, Kodama T, Silverstein SC. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J Neuroimmunol. 2001;114:142–150. doi: 10.1016/s0165-5728(01)00239-9. [DOI] [PubMed] [Google Scholar]
- 31.in't Veld BA, Ruitenberg A, Hofman A, Stricker BH, Breteler MM. Antihypertensive drugs and incidence of dementia: the Rotterdam Study. Neurobiol Aging. 2001;22:407–412. doi: 10.1016/s0197-4580(00)00241-4. [DOI] [PubMed] [Google Scholar]
- 32.Ishibashi Y, Claus S, Relman DA. Bordetella pertussis filamentous hemagglutinin interacts with a leukocyte signal transduction complex and stimulates bacterial adherence to monocyte CR3 (CD11b/CD18). J Exp Med. 1994;180:1225–1233. doi: 10.1084/jem.180.4.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang P, Lagenaur CF, Narayanan V. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J Biol Chem. 1999;274:559–562. doi: 10.1074/jbc.274.2.559. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 35.Klegeris A, Walker DG, McGeer PL. Activation of macrophages by Alzheimer beta amyloid peptide. Biochem Biophys Res Commun. 1994;199:984–991. doi: 10.1006/bbrc.1994.1326. [DOI] [PubMed] [Google Scholar]
- 36.Kloss CU, Bohatschek M, Kreutzberg GW, Raivich G. Effect of lipopolysaccharide on the morphology and integrin immunoreactivity of ramified microglia in the mouse brain and in cell culture. Exp Neurol. 2001;168:32–46. doi: 10.1006/exnr.2000.7575. [DOI] [PubMed] [Google Scholar]
- 37.Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP). Annu Rev Biochem. 1994;63:601–637. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 38. Le Y, Gong W, Tiffany HL, Tumanov A, Nedospasov S, Shen W, Dunlop NM, Gao JL, Murphy PM, Oppenheim JJ, Wang JM. Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci 21 2001. RC123(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin TH, Rosales C, Mondal K, Bolen JB, Haskill S, Juliano RL. Integrin-mediated tyrosine phosphorylation and cytokine message induction in monocytic cells: a possible signaling role for the Syk-tyrosine kinase. J Biol Chem. 1995;270:16189–16197. doi: 10.1074/jbc.270.27.16189. [DOI] [PubMed] [Google Scholar]
- 40.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorton D. Beta-amyloid-induced IL-1 beta release from an activated human monocyte cell line is calcium- and G-protein-dependent. Mech Aging Dev. 1997;94:199–211. doi: 10.1016/s0047-6374(96)01847-7. [DOI] [PubMed] [Google Scholar]
- 42.Lorton D, Kocsis JM, King L, Madden K, Brunden KR. Beta-amyloid induces increased release of interleukin-1 beta from lipopolysaccharide-activated human monocytes. J Neuroimmunol. 1996;67:21–29. doi: 10.1016/0165-5728(96)00030-6. [DOI] [PubMed] [Google Scholar]
- 43.Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation end products (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 44.Mackenzie IR, Munoz DG. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology. 1998;50:986–990. doi: 10.1212/wnl.50.4.986. [DOI] [PubMed] [Google Scholar]
- 45.Matter ML, Zhang Z, Nordstedt C, Ruoslahti E. The alpha5beta1 integrin mediates elimination of amyloid-beta peptide and protects against apoptosis. J Cell Biol. 1998;141:1019–1030. doi: 10.1083/jcb.141.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maxeiner H, Husemann J, Thomas CA, Loike JD, El Khoury J, Silverstein SC. Complementary roles for scavenger receptor A and CD36 of human monocyte-derived macrophages in adhesion to surfaces coated with oxidized low-density lipoproteins and in secretion of H2O2. J Exp Med. 1998;188:2257–2265. doi: 10.1084/jem.188.12.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald D, Bamberger M, Combs C, Landreth G. β-Amyloid fibril activate parallel mitogen-activated protein kinase pathways in microglia and THP-1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997;17:2284–2294. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meda L, Cassatella MA, Szendrei GI, Otvos L, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 50.Meda L, Bernasconi S, Bonaiuto C, Sozzani S, Zhou D, Otvos L, Jr, Mantovani A, Rossi F, Cassatella MA. Beta-amyloid (25–35) peptide and IFN-gamma synergistically induce the production of the chemotactic cytokine MCP-1/JE in monocytes and microglial cells. J Immunol. 1996;157:1213–1218. [PubMed] [Google Scholar]
- 51.Meda L, Baron P, Prat E, Scarpini E, Scarlato G, Cassatella MA, Rossi F. Proinflammatory profile of cytokine production by human monocytes and murine microglia stimulated with beta-amyloid[25–35]. J Neuroimmunol. 1999;93:45–52. doi: 10.1016/s0165-5728(98)00188-x. [DOI] [PubMed] [Google Scholar]
- 52.Miao WM, Vasile E, Lane WS, Lawler J. CD36 associates with CD9 and integrins on human blood platelets. Blood. 2001;97:1689–1696. doi: 10.1182/blood.v97.6.1689. [DOI] [PubMed] [Google Scholar]
- 53.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:49982–49988. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 54.Murphy GM, Jr, Yang L, Cordell B. Macrophage colony-stimulating factor augments beta-amyloid-induced interleukin-1, interleukin-6, and nitric oxide production by microglial cells. J Biol Chem. 1998;273:20967–20971. doi: 10.1074/jbc.273.33.20967. [DOI] [PubMed] [Google Scholar]
- 55.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 56.Pearce SFA, Wu J, Silverstein RL. Recombinant GST/CD36 fusion proteins define a thrombospondin binding domain: evidence for a single calcium-dependent binding site on CD36. J Biol Chem. 1995;270:2981–2986. doi: 10.1074/jbc.270.7.2981. [DOI] [PubMed] [Google Scholar]
- 57. Pettit DL, Shao Z, Yakel JL. β-Amyloid(1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci 21 2001. RC120(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pick E. Microassays for superoxide and hydrogen peroxide production and nitroblue tetrazolium reduction using an enzyme immunoassay microplate reader. Methods Enzymol. 1986;132:407–421. doi: 10.1016/s0076-6879(86)32026-3. [DOI] [PubMed] [Google Scholar]
- 59.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porter JC, Hogg N. Integrins take partners: cross-talk between integrins and other membrane receptors. Trends Cell Biol. 1998;8:390–396. doi: 10.1016/s0962-8924(98)01344-0. [DOI] [PubMed] [Google Scholar]
- 61.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 62.Scharnagl H, Tisljar U, Winkler K, Huttinger M, Nauck MA, Gross W, Wieland H, Ohm TG, Marz W. The betaA4 amyloid peptide complexes to and enhances the uptake of beta-very low density lipoproteins by the low density lipoprotein receptor-related protein and heparan sulfate proteoglycans pathway. Lab Invest. 1999;79:1271–1286. [PubMed] [Google Scholar]
- 63.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 64.Terzi E, Holzemann G, Seelig J. Alzheimer beta-amyloid peptide 25–35: electrostatic interactions with phospholipid membranes. Biochemistry. 1994;33:7434–7441. doi: 10.1021/bi00189a051. [DOI] [PubMed] [Google Scholar]
- 65.Thorne RF, Marshall JF, Shafren DR, Gibson PG, Hart IR, Burns GF. The integrins alpha3beta1 and alpha6beta1 physically and functionally associate with CD36 in human melanoma cells: requirement for the extracellular domain OF CD36. J Biol Chem. 2000;275:35264–35275. doi: 10.1074/jbc.M003969200. [DOI] [PubMed] [Google Scholar]
- 66.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 67.Van Muiswinkel FL, Raupp SF, de Vos NM, Smits HA, Verhoef J, Eikelenboom P, Nottet HS. The amino-terminus of the amyloid-beta protein is critical for the cellular binding and consequent activation of the respiratory burst of human macrophages. J Neuroimmunol. 1999;96:121–130. doi: 10.1016/s0165-5728(99)00019-3. [DOI] [PubMed] [Google Scholar]
- 68.Wang XQ, Frazier WA. The thrombospondin receptor CD47 (IAP) modulates and associates with alpha2 beta1 integrin in vascular smooth muscle cells. Mol Biol Cell. 1998;9:865–874. doi: 10.1091/mbc.9.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang XQ, Lindberg FP, Frazier WA. Integrin-associated protein stimulates alpha2beta1-dependent chemotaxis via Gi-mediated inhibition of adenylate cyclase and extracellular-regulated kinases. J Cell Biol. 1999;147:389–400. doi: 10.1083/jcb.147.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wong WS, Simon DI, Rosoff PM, Rao NK, Chapman HA. Mechanisms of pertussis toxin-induced myelomonocytic cell adhesion: role of Mac-1(CD11b/CD18) and urokinase receptor (CD87). Immunology. 1996;88:90–97. doi: 10.1046/j.1365-2567.1996.d01-646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wood JG, Zinsmeister P. Tyrosine phosphorylation systems in Alzheimer's disease pathology. Neurosci Lett. 1991;121:12–16. doi: 10.1016/0304-3940(91)90637-9. [DOI] [PubMed] [Google Scholar]
- 72.Woods ML, Shimizu Y. Signaling networks regulating beta1 integrin-mediated adhesion of T lymphocytes to extracellular matrix. J Leukoc Biol. 2001;69:874–880. [PubMed] [Google Scholar]
- 73.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 74.Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J Neurochem. 2000;74:1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x. [DOI] [PubMed] [Google Scholar]