

Abstract
Nuclear factor I (NFI) genes are expressed in multiple organs throughout development (Chaudhry et al., 1997; for review, seeGronostajski, 2000). All four NFI genes are expressed in embryonic mouse brain, with Nfia, Nfib, andNfix being expressed highly in developing cortex (Chaudhry et al., 1997). Disruption of the Nfia gene causes agenesis of the corpus callosum (ACC), hydrocephalus, and reduced GFAP expression (das Neves et al., 1999). Three midline structures, the glial wedge, glia within the indusium griseum, and the glial sling are involved in development of the corpus callosum (Silver et al., 1982; Silver and Ogawa, 1983; Shu and Richards, 2001). BecauseNfia−/−mice show glial abnormalities and ACC, we asked whether defects in midline glial structures occur inNfia−/−mice. NFI-A protein is expressed in all three midline populations. InNfia−/−, mice sling cells are generated but migrate abnormally into the septum and do not form a sling. Glia within the indusium griseum and the glial wedge are greatly reduced or absent and consequently Slit2 expression is also reduced. Although callosal axons approach the midline, they fail to cross and extend aberrantly into the septum. The hippocampal commissure is absent or reduced, whereas the ipsilaterally projecting perforating axons (Hankin and Silver, 1988; Shu et al., 2001) appear relatively normal. These results support an essential role for midline glia in callosum development and a role for Nfia in the formation of midline glial structures.
Keywords: glial wedge, glial sling, glial tunnel, corpus callosum, anterior commissure, hippocampal commissure, Slit2
Introduction
Midline glia are associated with the development of commissural projections in the mammalian brain (Katz et al., 1983; Guillery and Walsh, 1987; Godement and Mason, 1993; Silver, 1993; Silver et al., 1993; Reese et al., 1994; Marcus et al., 1995;Cummings et al., 1997; Pires-Neto et al., 1998). At the optic chiasm, specialized midline cells and glia regulate which axons project ipsilaterally and contralaterally by differentially guiding axons at the midline (Marcus et al., 1995; Wang et al., 1995). In the development of the corpus callosum, several midline populations, the glial sling, the glial wedge, and glia within the indusium griseum, have been implicated in callosal axon guidance (Silver et al., 1982;Silver and Ogawa, 1983; Shu and Richards, 2001).
In both lesioning and grafting experiments, Silver and colleagues (Silver et al., 1982; Silver and Ogawa, 1983) showed that the glial sling was important for the formation of the corpus callosum. Two other midline populations, the glial wedge and glia within the indusium griseum, may also be important for callosal formation (Shu and Richards, 2001). In collagen gels, glial wedge explants secrete a short-range diffusible repellent/growth suppressive molecule, and in organotypic slices, reorientation of the glial wedge and glia within the indusium griseum causes callosal axons to turn away from the midline (Shu and Richards, 2001). These results indicate a role for these midline populations in the development of the corpus callosum.
A large number of single gene mutations in mice result in ACC, yet in many cases it is not known how these genes function to prevent the formation of this commissure. In an effort to understand some of the critical developmental events required for callosal formation in vivo, we have investigated whether the development of midline structures may be disrupted in the Nfia mutant, which was reported previously to exhibit both agenesis of the corpus callosum and a reduction in glial fibrillary acidic protein (GFAP) expression (das Neves et al., 1999). Nfia belongs to the nuclear factor I (Nfi) family of transcription factors that function both in the regulation of adenoviral DNA replication and in viral and cellular gene expression, including the control of olfactory-specific genes (Nagata et al., 1982, 1983; Gronostajski et al., 1985; Hennighausen et al., 1985; Leegwater et al., 1985; Nowock et al., 1985; Baumeister et al., 1999; Behrens et al., 2000).
Given the role of Nfia in GFAP expression and the acallosal phenotype of Nfia knock-out animals, we hypothesized that midline glia known to be involved in callosal development may be disrupted in Nfia mutant mice. We found that Nfiais expressed in midline glial structures and that the development of these structures is severely impaired inNfia−/−mice. These data indicate that Nfia may regulate callosal development by regulating the development of midline glia.
Materials and Methods
Animals. Animals used for NFI-A immunohistochemistry were wild-type C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) bred on site at The University of Maryland, Baltimore animal facility under the care and approval of the accredited University of Maryland, Baltimore Institutional Animal Care and Use Committee.Nfia−/−animals used in all other experiments were generated and bred at The Lerner Research Institute (Cleveland, OH).Nfia−/−mice were generated using E14–1 embryonic stem cells (derived from strain 129P2) (Hooper et al., 1987; Festing et al., 1999) and backcrossed onto a random-bred Swiss background (strain Tac:N:NIHS-BC) as described previously (das Neves et al., 1999). TheNfia−/−allele was then backcrossed onto the C57BL/6NTac strain (Taconic, Germantown, NY) for >10 generations, and all embryonic and postnatal mice used in these experiments were derived from these mice. Unlike a number of 129 strains (Livy and Wahlsten, 1991, 1997), C57BL/6 mice do not display ACC. Furthermore, no defects in commissural tract formation were detected in wild-type or heterozygous Nfia animals on a C57BL/6 background; therefore, the phenotype described here is independent of any preexisting strain affect. To obtain timed-pregnant females, male and female mice were placed together overnight. Females were weighed and inspected the following morning for the presence of a vaginal plug. If present, this day was designated embryonic day 0. HeterozygousNfia+/− mice were bred to obtain litters of (+/+), (+/−), and (−/−) mice that were in a roughly Mendelian ratio. Embryos were genotyped by PCR according to the protocol described previously (das Neves et al., 1999). On the required gestational day, embryos were either immersion-fixed or perfused with 4% paraformaldehyde [for 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) labeling]. The heads were then removed and stored in 4% paraformaldehyde until use. Fixed heads were transported from Cleveland to Baltimore for processing.
Immunohistochemistry. Wild-type brains stained for NFI-A were perfused with 0.9% saline followed by a mixture of 4% paraformaldehyde and then postfixed in 4% paraformaldehyde at 4°C until sectioning. Heads from Nfia litters were immersion-fixed in 4% paraformaldehyde at 4°C until the brains were removed for sectioning. Before sectioning, the brains were blocked in 3% noble agar and then sectioned at 50 μm on a Vibratome (Leica, Deerfield, IL). Immunohistochemistry was performed in a manner similar to that described previously (Shu et al., 2000). Sections of acrolein-fixed brains were first incubated in 1% sodium borohydride (w/v) (Sigma, St. Louis, MO) PBS, pH 7.4, for 10 min to remove the acrolein and then processed in an identical manner to the brains fixed only in 4% paraformaldehyde. Sections were washed three times in PBS, blocked in 2% normal goat serum (v/v) (Jackson ImmunoResearch Laboratory, West Grove, PA) and 0.2% Triton X-100 (v/v) (Sigma) in PBS for 2 hr, and then incubated in primary antibody [rabbit anti-NFI-A, at either 1:1000 or 1:75,000 (affinity-purified antibody, catalog #16111460; Geneka Biotechnology, Montreal, Canada); rabbit anti-GFAP, 1:30,000 (Dako, Glostrup, Denmark); rabbit anti-neurofilament M C-terminal, 1:10,000 (Chemicon, Temecula, CA); mouse anti-neuronal nuclei (NeuN), 1:3000 (Chemicon)] overnight. On day 2, sections were washed three times in PBS and then incubated in biotinylated goat anti-rabbit secondary antibody (1:600; Jackson ImmunoResearch Laboratory) for 1 hr. After three washes in PBS, sections were incubated in avidin–biotin solution (1:500, Vector Laboratories, Burlingame, CA) for 1 hr, followed by three washes in PBS. Sections were then immersed in a nickel/3,3′ diaminobenzidine (DAB) chromogen solution (2.5% nickel sulfate and 0.02% DAB in 0.175m sodium acetate) activated with 0.01% (v/v) H2O2 until a dark purple/black precipitate formed. Sections were washed in PBS, mounted, and coverslipped in DPX mounting medium (Electron Microscopy Services). For brain lipid binding protein (BLBP) (Feng et al., 1994; Kurtz et al., 1994) immunohistochemistry, brains were perfused in 4% paraformaldehyde and vibratome sectioned and blocked as described above. BLBP primary antibody (polyclonal; a gift from Dr. N. Heinz, Rockefeller University, New York, NY) was used at 1:8000 (diluted in blocking solution) overnight, washed three times, and then incubated with a donkey anti-rabbit Cy3-conjugated antibody (1:400; Jackson ImmunoResearch Laboratories). For double immunohistochemical staining of NFI-A or BLBP and GFAP, sections were stained as described above for the first primary antibody (anti-NFI-A or anti-BLBP) and then incubated in donkey anti-rabbit Cy2- or Cy3-conjugated antibody (1:400; Jackson ImmunoResearch Laboratories). Sections were then reblocked in PBS with 2% normal donkey serum and 0.2% Triton X-100 (Sigma) and then incubated in the second primary antibody (1:1000, rabbit-anti-GFAP; Dako) overnight. Sections were washed as described for single labeling and then incubated in a donkey anti-rabbit Cy2- or Cy3-conjugated secondary antibody (1:400; Jackson ImmunoResearch Laboratories) for 2 hr. The sections were then washed, mounted, and coverslipped with polyvinyl alcohol (PVA)/1,4-diazabicyclo(2.2.2)octane (DABCO) mounting medium [consisting of 9.6 gm PVA (Sigma)/DABCO (Sigma) in a solution containing 24 gm glycerol, 48 ml 0.2 m Tris-HCl, pH 8–8.5, and 24 ml H2O] for confocal microscopy.
In situ hybridization. Digoxigenin was used to label Tbr-1 probes (a gift from Dr. J. L. Rubenstein, University of California, San Francisco, San Francisco, CA). Nonradioactive in situ hybridization was performed on 45 μm floating Vibratome sections. Sections were washed in PBS, pH 7.4, and acetylated for 10 min with 0.25% acetic anhydride in 1% triethanolamine solution. Two washes in PBS for 5 min were followed by an equilibration in 2× standard saline citrate (SSC) for 5 min. Sections were prehybridized overnight at room temperature in 50% formamide, 5× Denhardt's solution, 5× SSC, and 250 μg/ml baker's yeast tRNA. The hybridization mixture was the same as the prehybridization mixture but also contained 3 ng/ml DIG-labeled riboprobe. Hybridization was performed at 60°C overnight. Sections were washed at 60°C for 5 min in 5× SSC, 1 min in 2× SSC, 30 min in 50% formamide containing 0.2× SSC, and 5 min in 0.2× SSC. Sections were then rinsed in Tris-buffered saline (TBS; 100 mm Tris-HCl, pH 7.5, and 150 mm NaCl) for 5 min and incubated for 1 hr in 1% blocking reagent (Roche, Hertforshire, UK) in TBS. For immunodetection, sections were incubated for 1 hr with an anti-DIG Fab fragment conjugated to alkaline phosphatase (anti-DIG-AP; Roche) at a dilution of 1:5000. Sections were then rinsed twice for 15 min in TBS and once in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, and 5 mm MgCl2. The color reaction was performed with nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate (Roche). Sections were rinsed in 10 mm Tris-HCl, pH 8.0, 5 mm EDTA (TE), mounted on glass slides, and coverslipped with Hydromount (National Diagnostics, Atlanta, GA). Controls consisted of sections subjected to the complete in situ hybridization procedure, but with no probe or hybridized with a Tbr-1 sense probe. These exhibited no specific hybridization signal.
For Slit2, Robo1, and Robo2 in situ hybridization, probes were made from 1 μg DNA (plasmids were gifts from Dr. M. Tessier-Lavigne, Stanford, CA), radiolabeled with [S35]UTP (40 mCi/ml). Probes were purified by DNase digestion and precipitated in 0.6m NH4OAC and 100% ethanol. Slit2 and Robo1 probes were hydrolyzed, but the Robo2 probe did not need to be hydrolyzed. For hydrolyzing the probes, 20 μl of 40 mm sodium bicarbonate and 30 μl of 60 mm sodium carbonate were added to the probes and then incubated at 65°C for 30 min. Probes were again precipitated in NH4OAC and ethanol and then counted in a scintillation counter. A concentration of 400,000–600,000 cpm/μl was generally obtained from the above procedures. Cryostat sections (10 μm) were pretreated with 4% paraformaldehyde for 20 min and 2× SSC at 65°C for 30 min. This was followed by a 10 min treatment with Proteinase K (20 μg/ml in TE) and fresh acetylation mix with 3.25 ml triethanolamine and 0.7 ml acetic anhydride in 250 ml DEPC water. Sections were dehydrated through a 70–100% ethanol. Probes (50,000 cpm/μl) were diluted in the hybridization buffer with 50% deionized formamide, 10% dextran sulfate, 1× Denhardt's solution, 0.3m NaCl, 10 mm Tris, pH 7.5, 10 mm sodium phosphate, pH 6.8, 5 mm EDTA, 25 mm DTT, and 50 mm β-mercaptoethanol. Hybridization was performed at 55°C overnight. Sections were washed in 5× SSC and 20 mm β-mercaptoethanol at 55°C for 30 min, and then a solution of 2× SSC, 50% formamide, and 20 mm β-mercaptoethanol at 65°C for 30 min at room temperature for an additional 1 hr. Sections were further washed in TE, RNase A, and SSC before being dehydrated through ethanol series with NH4OAC. Slides were dipped in NTB-2 emulsion (Kodak) and stored at room temperature for 4 weeks before being developed in D-19 and fixer (Kodak).
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate and 4-(4-(dihexadecylamino)styryl)-N-methyl-pyridinium iodide labeling of axons. Tract tracing experiments were performed using a 3% solution of 4-(4-(dihexadecylamino)styryl)-N-methyl-pyridinium iodide (DiA) (Molecular Probes, Eugene, OR) and/or a 10% solution of 1,1′-dioctadecyl-3,3,3′,3′ tetramethylindocarbocyanine perchlorate (DiI) (Molecular Probes) in dimethylformamide (Sigma). Brains from embryonic day (E) 15, E17, E18, and postnatal day 0 (P0)Nfia litters were perfused with 0.9% saline followed by 4% paraformaldehyde and then postfixed in 4% paraformaldehyde until injection. Embryos were genotyped by PCR as described previously (das Neves et al., 1999). Dye injections were performed using a fine-tipped (pulled) glass micropipette attached to a Picospritzer (Parker Instrumentation, Fairfield, NJ). DiI injections were made in the cortex just lateral to the midline from rostral to caudal of one hemisphere, or at the midline of a hemisected brain (see Fig. 1H) to label callosal axons. In some brains, a coronal cut was made caudally through the hippocampus to expose the structure. DiA was then injected into the hippocampus to label the fornix and the hippocampal commissural axons. Brains were stored in the dark at room temperature for at least 4 weeks to allow DiI and DiA transportation. Brains were blocked in 3% noble agar and cut at 50 μm on a Vibratome. The sections were then mounted and coverslipped with PVA/DABCO mounting medium for confocal microscopy as described above. Some sections (see Fig. 1H) were counterstained with 10 nm Sytox green (Molecular Probes) in DMSO for 1–2 hr and then washed three times in PBS before mounting and analysis.
Fig. 1.
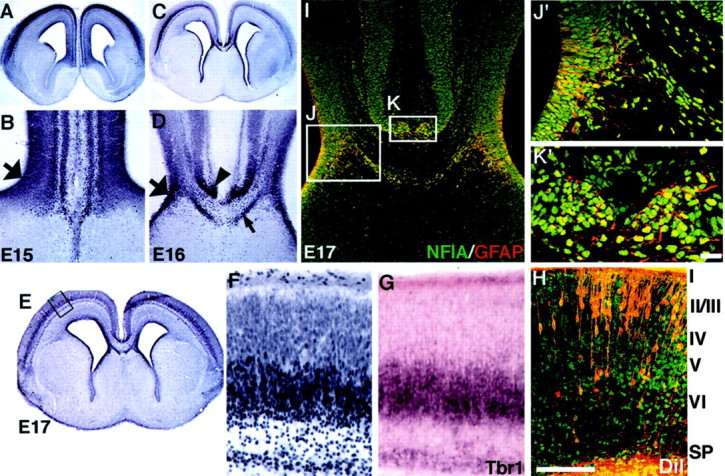
NFI-A expression in the developing mouse cortex. Sections of wild-type (C57BL/6J) brains were processed for NFI-A immunohistochemistry at E15 (A, B), E16 (C, D), and E17 (E,F, H–K). At E15, NFI-A is expressed in the dorsal telencephalon, including the neocortical plate and the cingulate cortex, but stops at the corticoseptal boundary (A, B). Cells within the region of the glial wedge express NFI-A (B,large arrow). At E16 and E17, expression became more confined to the deeper layers of the cortex (C,E) and is evident at the midline in the indusium griseum (D, arrowhead), the glial sling (D, small arrow), and the glial wedge (D, large arrow). At higher power, NFI-A labeling in the subplate and layer 6 (F) overlaps with Tbr-1 labeling in an adjacent section (G). Retrograde labeling of callosal neurons at E17 with DiI (counterstained with Sytox green) in an area similar to F andG shows that most of the callosally projecting neurons are not in an area of high NFI-A labeling (H). At E17, NFI-A is expressed in midline glial populations (I, J′,K′, green nuclei) double-labeled with GFAP immunohistochemistry (I, J′, andK′, red labeling in the cytoplasm). Cells within the glial wedge (boxed region labeledJ) and the indusium griseum (boxed region labeled K) express both NFI-A and GFAP as seen in higher-power views of these regions (J′ and K′, respectively). The glial sling is NFI-A positive (D, small arrow) but GFAP negative (I). Scale bar: (in K′)A, C, 600 μm; B,D, 150 μm; I, 50 μm;J′, K′, 20 μm; (inH) E, 1000 μm;F–H, 100 μm.
Preparation of figures. Bright-field images were scanned directly into Photoshop (Adobe) on an upright microscope (Leica) equipped with a digital scanning camera (Phase One). All fluorescent images were collected on a scanning laser confocal microscope (OlympusFluoview) and then transferred to Photoshop (Adobe) for processing. Collages were made of the images, and the appropriate labels were added. No image processing was performed other than enhancing the brightness and contrast and cropping the image. Figures were made on a Macintosh G4 computer.
Results
Expression of NFI-A at the developing cortical midline
To characterize the expression of NFI-A protein, we performed immunohistochemistry on developing mouse brain sections using a polyclonal NFI-A antibody (Geneka Biotechnology, Montreal, Canada). This anti-peptide antisera is directed against a peptide epitope present only on NFI-A proteins and not present in NFI-B, -C, or -X proteins. At E15, NFI-A was expressed within the ventricular zone and the emerging cortical plate (Fig.1A,B). At E16 and E17, NFI-A expression was confined to the subplate and the deeper layers of the neocortex (Fig. 1C,E). A higher power view of the cortical plate at E17 shows that NFI-A labeling is mostly confined to the subplate, layer 6, and possibly the most ventral part of layer 5 (Fig. 1F). Very low levels of expression were seen in the upper layers of the cortical plate (Fig. 1F). To examine this expression further, we performed in situ hybridization for Tbr-1, which labels the subplate and layer 6 (Hevner et al., 2001), on an adjacent section (Fig. 1G). NFI-A and Tbr-1 overlapped in their expression patterns, indicating that by this criterion at least, NFI-A labeled both the subplate and layer 6. Most of the callosal axons arise from neurons in layers 2/3 and 5 (Wise and Jones, 1976). To determine whether NFI-A was expressed in callosal neurons, we retrogradely labeled callosal neurons at E17 with DiI from an injection into the corpus callosum at the midline (Fig. 1H). By comparing the DiI labeling with NFI-A labeling, we found that most DiI-filled cells were in regions that did not express NFI-A (Fig. 1, compare F, H). This indicated that most of the callosally projecting axons probably do not express NFI-A.
At the cortical midline, NFI-A expression was observed within the indusium griseum (Fig. 1D, arrowhead,I,K,K′), the glial sling (Fig. 1D, small arrow), and the glial wedge cell bodies (Fig. 1B,D,large arrows,I,J,J′). Double staining of NFI-A in the nucleus (green) and GFAP in the cytoplasm (red) in the same section revealed many cells labeled for both NFI-A and GFAP in the glial wedge and the indusium griseum glia (Fig.1I,J′,K′).
Abnormal development of midline glia in the Nfiaknock-out mouse
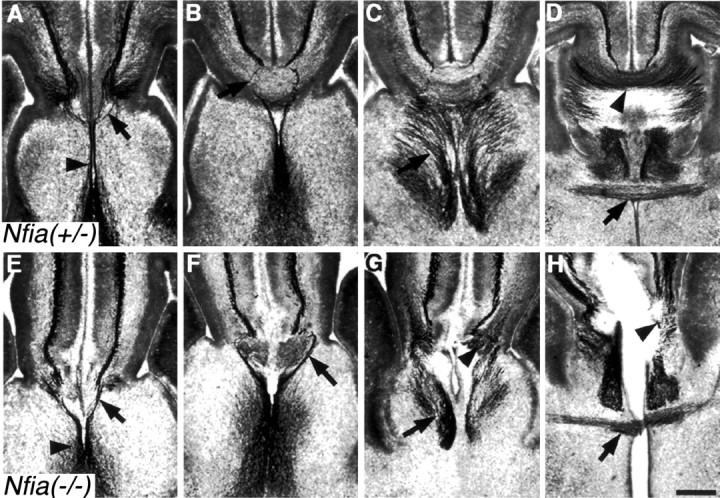
We used GFAP immunohistochemistry to examine the development of the glial wedge and glia within the indusium griseum because this is a reliable marker of these glial populations. Although it had been shown previously that GFAP expression was greatly reduced in theNfia−/−adult mice (das Neves et al., 1999), we found that GFAP was still expressed at embryonic stages and therefore could be used to identify these structures in the knock-out. In heterozygousNfia+/− mice the glial wedge formed normally at E15 (Fig.2A) but was not present inNfia−/−littermates (Fig. 2B). At E17 the glial wedge was clearly evident on either side of the midline and underlying the developing corpus callosum in heterozygote mice (Fig. 2C,white arrow); however, only a few GFAP-positive processes within the wedge were present in theNfia−/−mice (Fig. 2D, white arrow), and the corticoseptal boundary was not well defined. Glia within the indusium griseum also formed normally by E17 in the heterozygotes (Fig.2C, white arrowhead), but in theNfia−/−mice some glia were present at the midline (Fig. 2D,white arrowhead); however, it was not clear whether these were displaced/malformed indusium griseum glia or, alternatively, midline zipper glia (Silver et al., 1982, 1993). Glia within the hippocampus, particularly the dentate gyrus and the fimbria, were present in theNfia+/− mice (Fig. 2E, white arrow) but were also reduced in theNfia−/−mice (Fig. 2F, white arrow). Finally, we also examined the formation of the glial tunnel that surrounds the anterior commissure (Katz et al., 1983; Silver et al., 1993; Cummings et al., 1997; Pires-Neto et al., 1998). This structure is best seen in sagittal sections. In Nfia+/− mice the glial tunnel formed normally with fibers surrounding the anterior commissure (Fig. 2G, arrow). InNfia−/−mice, however, the number of GFAP-expressing glial fibers was greatly reduced, and the region occupied by the anterior commissure was much thinner in the rostrocaudal axis (Fig. 2H,arrow), although it appeared normal in the dorsoventral axis (see Fig. 7H). These results suggest thatNfia may participate in the development of glia in the forebrain, perhaps by regulating GFAP expression.
Fig. 2.
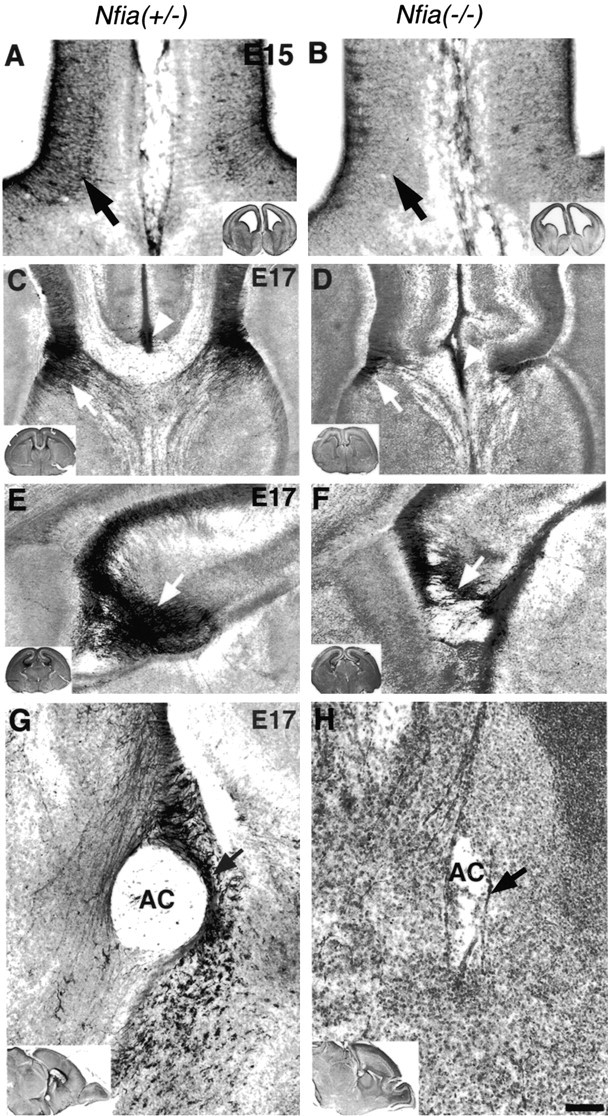
Development of medial glial populations inNfia−/−mice. At E15 the long radial glial-like processes of the glial wedge are evident by GFAP staining inNfia+/−mice (A, arrow), but are not evident in the knock-out (B), indicating a possible delay in glial wedge development. At E17 the glial wedge (C,arrow) and the indusium griseum glia (C,arrowhead) are clearly evident in the heterozygote. In the knock-out the glial wedge is greatly reduced (D,arrow) and additional glia arise at the fusion point between the two hemispheres (D,arrowhead), but it is not clear whether these are displaced indusium griseum glia or midline “zipper” glia, which are thought to participate in midline fusion. In the hippocampus, a large number of glial processes are evident, particularly in the dentate gyrus and the fimbria (E, arrow) but are greatly reduced in the knock-out (F,arrow). In sagittal sections (G,H), the glial tunnel is evident inNfia+/−mice but greatly reduced in the knock-out (H,arrow). Insets in each figure show a low-power view of each section. Scale bar: (in H)A, B, 100 μm; C,D, 150 μm; E, F, 70 μm; G, H, 10 μm.
Fig. 7.
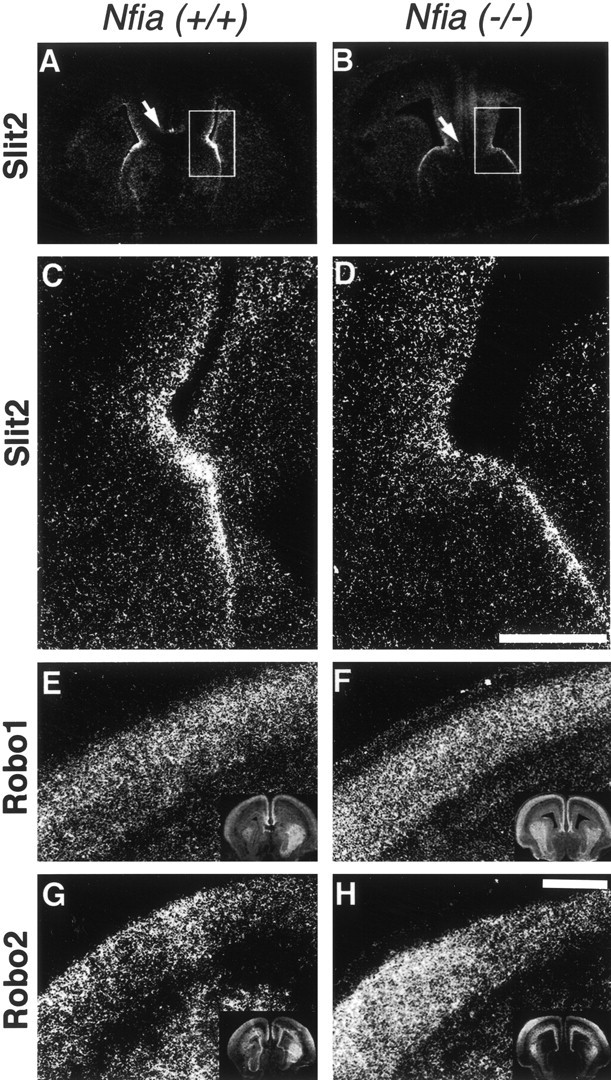
Slit and Robo expression in theNfia mutant. E17Nfia−/−brains (B, D, F,H) or brains from wild-type littermates (A, C, E, G) were processed for in situ hybridization using S35-labeled probes of Slit2 (A–D), Robo1 (E, F), and Robo2 (G, H). Slit 2 was still expressed in the mutant, but expression in the indusium griseum was absent (compare A, B, region marked by arrows). At higher power it was also evident that Slit2 expression in the glial wedge was decreased (compareC and D, which correspond to theboxed region in A and B, respectively). However, neither Robo1 nor Robo 2 expression in the cortical plate was decreased (compare E withF and G with H, respectively; low-power views of each section are shown in theinsets). Scale bars: (in D)A, B, 800 μm; C,D, 150 μm; (in H):E–H, 400 μm.
Given that Nfia may regulate GFAP expression, it may be that there were glia present at the midline but not expressing GFAP. To investigate this, we labeled sections with BLBP, a second marker for these glial populations, using a polyclonal anti-BLBP antibody (a gift from Dr. N. Heinz) (Feng et al., 1994) (Fig.3). BLBP labels both structures in wild-type littermates at E17 (Fig.3B,H) as shown by double immunohistochemistry for both GFAP and BLBP [Fig.3C,I, yellow-labeled glial; labeled with arrows (glial wedge) and arrowheads(indusium griseum glia)]. Although some glial wedge processes were present inNfia−/−mice, there were fewer, and those that were present were shorter and did not coalesce into the typical wedge-shaped structure (Fig.3F, arrow). Labeling of the indusium griseum glia with BLBP showed that this population was also absent or greatly reduced inNfia−/−mice compared with wild-type littermates (Fig.3I,L, arrowheads). BLBP expression was unchanged in other regions of the forebrain, including the cortex and radial glial within the striatum (data not shown), indicating that the reduced expression of BLBP in the glial wedge and indusium griseum glia was unlikely to be caused by the regulation of BLBP by Nfia.
Fig. 3.

GFAP and BLBP labeling of the glial wedge and indusium griseum glia. To examine whether the reduction in GFAP expression observed in theNfia−/−mice reflected abnormalities in these glial structures, we double-labeled sections at E17 with both GFAP (A,D, G, J) and BLBP (B, E, H,K) and overlaid the images to examine labeling in the same cells (C, F, I,L). GFAP and BLBP labeled glial processes (not cell bodies) in both the glial wedge (A and B, respectively) and indusium griseum (G andH, respectively) in wild-type animals. However, both the glial wedge (C, F, I,L, arrows) and indusium griseum glia (C, I, L,arrowheads) were either greatly reduced or absent inNfia−/−mice. Glial wedge processes appeared shorter and failed to coalesce into a wedge shape (F, arrow). Scale bar: (in L) A–F, 150 μm;G–L, 500 μm.
Another structure associated with the development of the corpus callosum is the glial sling (Silver et al., 1982). In rodents the glial sling is not GFAP positive (Silver et al., 1993) but can be labeled with the neuronal marker NeuN (Mullen et al., 1992), which, along with morphological and anatomical criteria, is a reliable method for identifying the sling (Preston et al., 2000; T. Shu and L. J. Richards, unpublished observation). As described previously by Silver and colleagues (Silver et al., 1982), the sling is obvious morphologically as a group of cell bodies at the midline between the two cerebral hemispheres (Fig.4A, white arrow). InNfia−/−mice, at E17 the sling cells are generated but migrate into the septum and do not form the sling structure (Fig. 4B,white arrow). In more rostral regions where the lateral edges of the sling are present inNfia+/− mice (Fig. 4C, white arrow), the sling cells are generated inNfia−/−mice (Fig. 4D, white arrow) but also migrate into the septum at this level (Fig. 4D,black arrow).
Fig. 4.

Abnormal development of the glial sling in theNfia−/−mice. Expression of NeuN immunohistochemistry at E17 in eitherNfia+/−(A, C) orNfia−/−(B, D) mice is shown. In heterozygote mice, the glial sling forms a continuous band of cells across the midline that spans the two cerebral hemispheres (A,arrow). InNfia−/−mice, sling cells migrate aberrantly into the septum (B,arrow). In more rostral regions, the lateral edges of the sling are present in the heterozygote (C,arrow) and the knock-out (D, white arrow), but at this level sling cells also migrate aberrantly into the septum in the knock-out (D,black arrow). Scale bar: (in D)A–D, 150 μm.
Abnormal development of midline commissural projections: the corpus callosum and the hippocampal commissure
We examined the development of medial cortical projections in theNfia knock-out mice to investigate when the callosal abnormality is first evident. We used DiI labeling between E15 and P0 to view the trajectory of axons during the period in which the corpus callosum normally forms in mice. At E15 we observed no difference in the cortical projections among Nfia+/+,+/−, or−/−littermates (data not shown). This is just before the time when the first axons from the cingulate cortex cross the midline to pioneer the corpus callosum in mice (Rash and Richards, 2001). At E17, however, when a significant number of axons have crossed the midline in theNfia+/+ orNfia+/− mice (n = 7/7) (Fig.5A,C,E), fewer axons appeared to reach the midline in theNfia−/−mice (n = 6/6) (Fig. 5B), and those that had, failed to cross (Fig.5D,F). In both the genu (Fig. 5, compare C, D) and the body (Fig. 5, compare E, F) of the corpus callosum (or where it would normally form), cortical axons in theNfia−/−mice reached the midline and instead of turning to cross the midline, grew down into the septum, ignoring the corticoseptal boundary. Once in the septum, some axons began to turn laterally, forming the characteristic beginnings of Probst bundles (Fig. 5F,arrow). At E18 when significantly more axons have normally crossed the midline (Fig. 5G), swirls of axons characteristic of Probst bundles were present on either side of the midline in theNfia−/−mice (Fig. 5H, arrow).
Fig. 5.
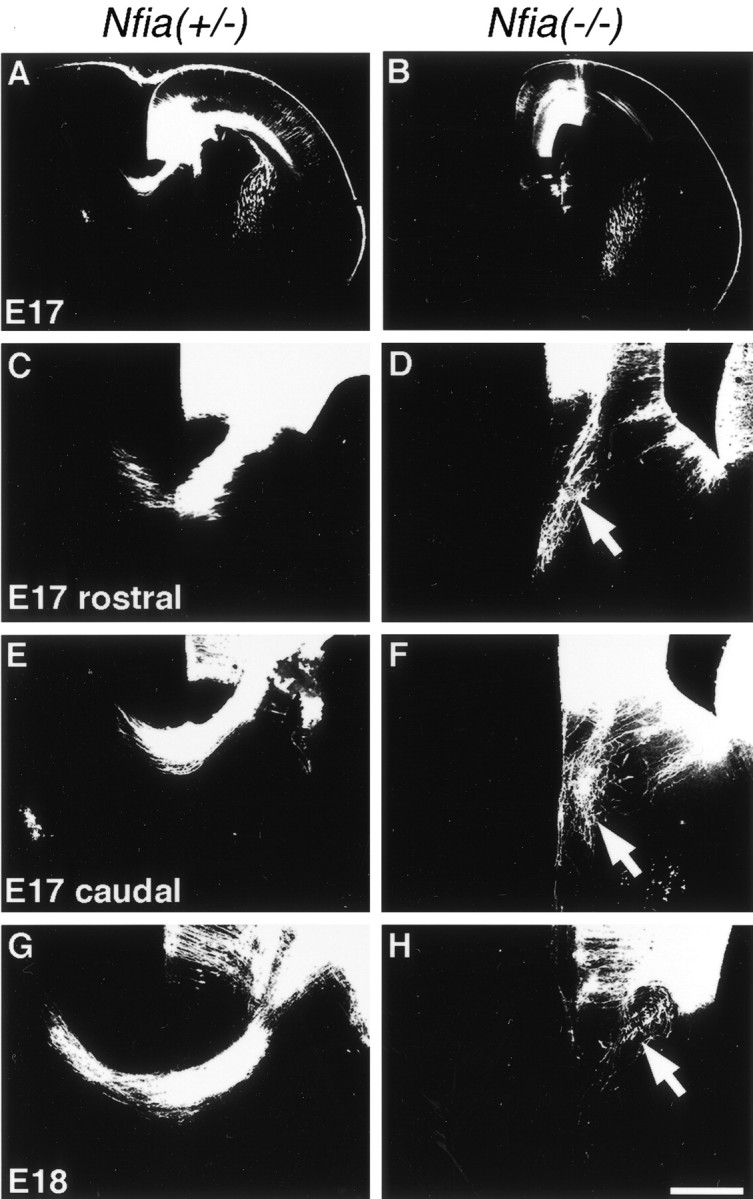
Development of callosal projections inNfia−/−mice. Fixed brains at either E17 (A–F) or E18 (G, H) were injected with DiI to label callosal projections. In heterozygote mice, callosal axons crossed the midline at E17 and E18 (A, C, E, G) but failed to cross the midline in the knock-out (B, D, F, H). Instead, these axons projected aberrantly into the septum and began to curl back on themselves, characteristic of the formation of Probst bundles (D, F, H, arrows). Scale bar: (inH) A, B, 600 μm;C, E, 200 μm; D,F, 100 μm; G, H, 150 μm.
In the most caudal regions of the corpus callosum (the splenium), callosal axons (Fig. 6, red axons) cross the midline dorsal to the hippocampal commissure (Fig. 6, green axons). The hippocampal commissure forms over an extended period of development from E14.8 to birth (Wahlsten, 1981;Valentino and Jones 1982; Super and Soriano, 1994; Livy and Wahlsten, 1997). By E17, many axons of the hippocampal commissure have crossed the midline in Nfia+/+ orNfia+/− mice (n = 3/3) (Fig. 6C). In theNfia−/−mutant, we did not observe any axons crossing the midline to form the hippocampal commissure at E17 (n = 3/3) (Fig.6B,D). Furthermore, the two commissures normally remain distinct at this age, with the callosal axons crossing dorsal to the hippocampal commissure (n= 3/3) (Fig. 6A,C). In theNfia−/−mice, as in the body and the genu, callosal axons of the splenium did not cross the midline at E17. Within the rostral (Fig.6B) and caudal (Fig. 6D) regions of the splenium at E17, callosal axons (red) and axons of the fornix (Fig. 6B, green) or the hippocampal commissure (Fig. 6D, green) overlapped at the border of the two commissures and did not remain in distinct axonal bundles (n = 2/2) (Fig.6B,D, arrows). At P0 we also observed aberrations in both the hippocampal commissure and the corpus callosum. As shown in the Nfia+/+or Nfia+/−controls, the callosal axons (red) cross the midline in a distinct bundle dorsal to the fornix (Fig. 6E,green) and the hippocampal commissural axons (Fig.6G, green) (n = 5/5); however, in theNfia−/−mice, we again observed overlap of the axons in the corpus callosum and fornix (Fig. 6F, arrow) and the corpus callosum and the hippocampal commissure (n = 4/4) (Fig.6H, arrow). In addition, at P0 we did observe some cases (n = 2/4) in which a few axons from the hippocampus and occasionally a small number of callosal axons that appeared to leave the Probst bundle crossed the midline together (Fig.6F, arrowhead). This occurred only in the splenium but not in the body and genu regions of the callosum at P0 (n = 4/4).
Fig. 6.
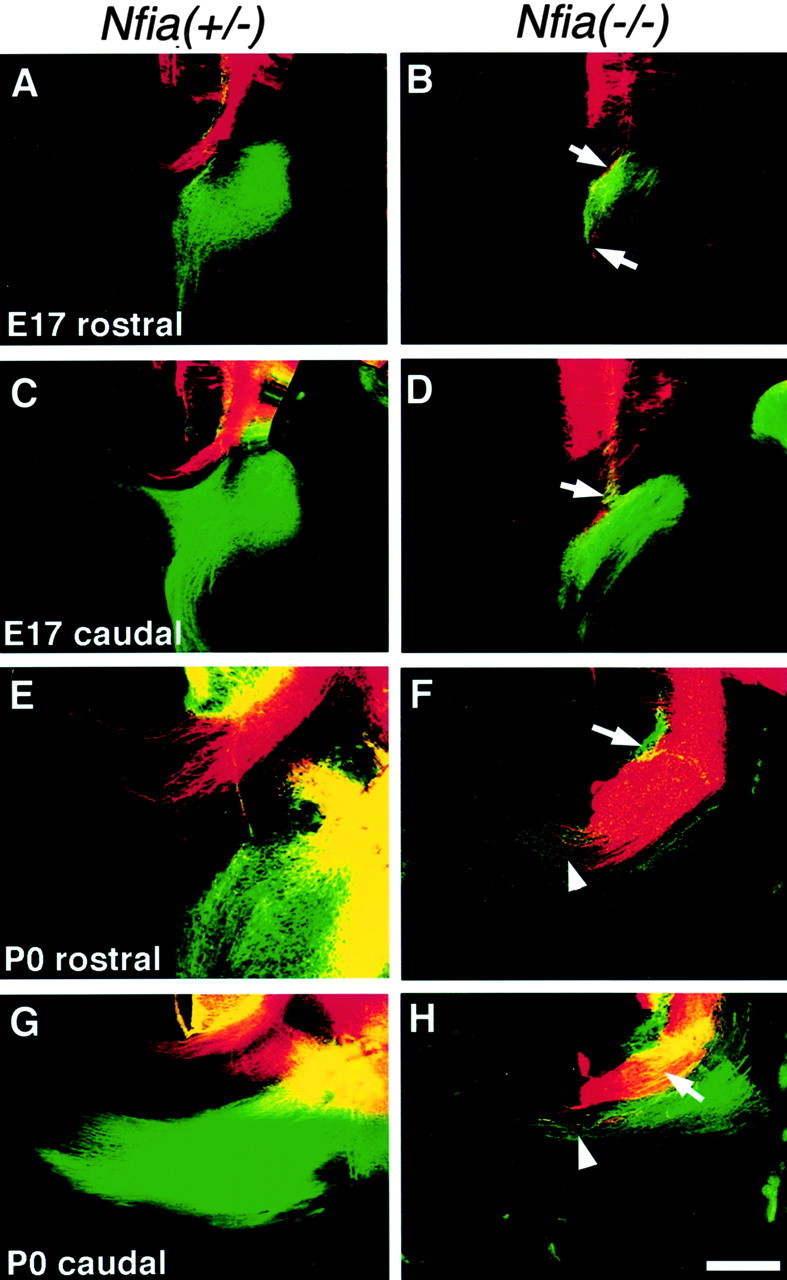
Development of the caudal corpus callosum and the hippocampal commissure inNfia−/−mice. Fixed brains at either E17 (A–D) or P0 (E–H) were injected with both DiI to label the corpus callosum (red axons) and DiA to label the fornix and the hippocampal commissure (green axons). The caudal regions of the corpus callosum form a separate axon tract overlying the more ventrally located hippocampal commissure. The hippocampal commissure is shown crossing the midline (C) in the heterozygote; the fornix is present underlying the caudal corpus callosum in a slightly more rostral section (A). At E17 in the knock-out, axons of both the corpus callosum and the hippocampal commissure fail to cross the midline at both rostral and caudal levels. Instead, the axons of the two commissures aberrantly mix together (B,D, arrows). At P0, both commissures remain as separate bundles inNfia+/−mice in both rostral (fornix region) (E) and caudal regions where the hippocampal commissure is clearly evident (G). In some knock-outs the mixed axon bundle (F, arrow), containing both hippocampal and callosal axons, crosses the midline (F,arrowhead). In other areas the callosal axons do not cross (H, arrow), but the hippocampal commissure crosses alone (H, arrowhead). Scale bar: (in H) A–D, 300 μm; E, G, 100 μm;F, H, 200 μm.
All results presented are from Nfia mice on a C57BL/6 background in which theNfia−/−mice die at birth (das Neves et al., 1999). The variability in the hippocampal commissure phenotype on the C57BL/6 background prompted us to review our previous data from adultNfia−/−mice on a mixed (129/C57BL/6/Swiss) background, of which ∼5% survive to adulthood. We found that the corpus callosum was absent in four of fourNfia−/−mice on the mixed background and that the hippocampal commissure was absent in three of four animals and severely reduced in one of four animals (R. M. Gronostajski, unpublished observation). We did not determine, in the few cases in which a hippocampal commissure did form in adults, whether there were callosal axons growing within this bundle.
Slit2 and Robo1 and 2 expression inNfia−/−mice
Our previous results showed that Slit2, expressed by the glial wedge, could act as a chemorepellent molecule for callosal axons (Shu and Richards, 2001). Furthermore, in Slit2−/−mice, callosal axons dive into the septum and ignore the glial wedge (Bagri et al., 2002). Given that the phenotype of theNfia−/−mice in some ways resembled that of the Slit2−/−mice and that glial wedge formation was disrupted in these mice, we examined whether the phenotype may be caused by alterations in the expression of Slit2 or Robo1 or Robo2 in these mice. We performed both real time quantitative PCR on whole brains and in situhybridization using probes against Slit2, Robo1, or Robo2. Using real time quantitative PCR we could detect no discernable difference in expression among Nfia+/+,+/−, and−/−mice (data not shown). By in situ hybridization, however, we found a slight decrease in Slit2 expression at the midline in the region of the glial wedge and a loss of expression in the indusium griseum (Fig. 7, compare A,C with B, D). Robo1 and Robo2 expression appeared unchanged in the cortical plate (Fig. 7, compareE, F for Robo1, and G, Hfor Robo2).
The anterior commissure and the perforating pathway are present inNfia−/−mice
Neurofilament immunohistochemistry allowed us to observe a number of midline axonal pathways in the same sections. We found that the perforating pathway (Hankin and Silver, 1988; Shu et al., 2001) appeared relatively normal in theNfia−/−mice. The perforating pathway is primarily derived from neurons in the medial septum and the diagonal band of Broca but has a small reciprocal projection from the ventral cingulate cortex (Shu et al., 2001). The axons form a fasciculated bundle on either side of the midline and project ipsilaterally (Fig.8A,E,arrowhead). When the perforating axons reach the corticoseptal boundary, they leave the midline, turning laterally and then medially, avoiding the glial wedge. They perforate the corpus callosum and project across it to the ventral cingulate cortex (Figs.8B,F, arrow). A precallosal component of the perforating pathway projects in a similar manner but rostral to the genu of the corpus callosum (Fig.8A,E, arrow). Although the corpus callosum did not form, the perforating pathway still crossed over the tangle of callosal axons on either side of the midline (Fig.8F, arrow). The perforating axons did project more laterally than in control mice before turning medially, probably because of the malformation of the glial wedge. As noted previously, at E17 the hippocampal commissure was not present in theNfia−/−mice, and this was also evident by neurofilament labeling (Fig. 8, compare D, H, arrowheads). In addition, the fornix was greatly reduced in size in theNfia−/−mice (Fig. 8G, arrow) compared with the heterozygote littermate (Fig. 8C, arrow). The anterior commissure (Fig. 8D,H,arrow) still crossed the midline in theNfia−/−mice, despite the fragility of this region caused by the absence of the other midline commissures. As previous analysis showed with GFAP labeling, however, the glial tunnel was greatly reduced surrounding a smaller anterior commissure, particularly in the rostral–caudal dimension (Fig. 2H,arrow).
Fig. 8.
The perforating pathway and the anterior commissure form normally inNfia−/−mice. E17 brains of eitherNfia+/−(A–D) orNfia−/−(E–H) mice were sectioned and processed for neurofilament immunohistochemistry. Sections shown are in rostral (A, E) to caudal (D, H) order. In rostral regions, axons of the perforating pathway project along the midline (A, E, arrowheads) and then deviate more laterally at the corticoseptal boundary (A, E, arrows). In more caudal regions, the perforating axons cross over and “perforate” the corpus callosum (B, arrow). Even in the knock-out, perforating axons grow around the axon bundles on either side of the midline without significantly deviating from their normal trajectory (F, arrow). In more caudal regions the fornix is evident in the heterozygote (C,arrow) but reduced in the knock-out (G,arrow). Furthermore, the corpus callosum and the hippocampal commissure fail to cross (G,H, arrowheads; arrowhead inD shows the corpus callosum and the hippocampal commissure crossing in caudal regions of the heterozygote) at E17, as seen with carbocyanine dye labeling (Fig.6B,D). The anterior commissure crosses the midline in both wild-type andNfia−/−mice (arrows in D and H, respectively), despite the large cleft formed by the absence of the corpus callosum and the hippocampal commissure in the knock-out. Scale bar: (in H) A–H, 300 μm.
Discussion
We have shown that midline glial populations, the glial wedge, and the indusium griseum glia, fail to develop normally inNfia−/−mice. In addition to the aberrant development of these glial structures, sling cells migrate abnormally into the septum. Together these structures make up the corticoseptal boundary that normally prevents callosal axons from entering the septum. Therefore, inNfia−/−mice, callosal axons fail to cross the midline and in some cases project ventrally into the septum. Furthermore, in more caudal regions, callosal axons and axons of the hippocampal commissure cross within the same region. These data indicate that defects in the formation of midline structures are correlated with defects in midline commissure formation.
In Nfia knock-out mice, GFAP expression is greatly reduced (das Neves et al., 1999). However, Nfia knock-outs display no change in the expression of other glial markers including proteolipid protein (das Neves et al., 1999) and BLBP (see Results). NFI can bind to the GFAP promoter and regulate GFAP expression in vitro (Miura et al., 1990; Krohn et al., 1999) and therefore may play a similar role in vivo. Because GFAP is a major structural component of glial processes, a decrease in GFAP may affect the extension of glial processes inNfia−/−mice. The few glial wedge processes that remain in the knock-out appeared shorter, indicating that they may be unable to adequately extend their processes. BLBP labeling confirmed a reduction or complete absence of the formation of glial processes in the glial wedge and indusium griseum glia. A reduction in GFAP by itself is unlikely to be the cause of ACC in theNfia−/−mutant. Although a reduced size of the corpus callosum attributable to aberrant myelination has been noted in one GFAP knock-out mouse (Liedtke et al., 1996), no callosal changes have been detected in two other GFAP knock-out strains (McCall et al., 1996; Shibuki et al., 1996).
AlthoughNfia−/−mice display a reduction in midline glial processes, their cell bodies may still be present or they may also be reduced. Slit2 is expressed by these midline glial populations. Therefore, a reduction in the expression of Slit2 at the corticoseptal boundary by in situhybridization may be caused by a reduction in the number of glia within this region. Nfia is not essential for Slit2 expression because we could detect no overall decrease in Slit2 expression by real time PCR between wild-type and Nfia knock-out animals (data not shown). The reduction in Slit2 RNA levels also suggests that reduced levels of Slit2 protein may be presented to the callosal axons. We proposed previously that Slit2 expressed by glia within the indusium griseum above the corpus callosum, and by the glial wedge below the corpus callosum, may provide a surround repulsion mechanism that keeps callosal axons confined to the correct path and repels them toward and across the midline (Shu and Richards, 2001). Because no Slit2 seems to be expressed in the indusium griseum, this surround repulsion mechanism, if it exists, would be lost. These data suggest that the acallosal defect in the Nfia mutant could be caused by defects in the development of midline glia and a reduction in Slit2 expression at the midline.
The formation of the midline zipper glial population (Silver et al., 1993) was also disrupted in theNfia−/−mice. These glia were either absent or their position shifted inNfia−/−mice. Midline zipper glia are proposed to be essential for midline fusion at the corticoseptal boundary (Silver et al., 1993), and therefore a disruption in their development could cause abnormalities in midline fusion and consequently callosal formation. Although we did not observe gross defects in midline fusion by light microscopic analysis,Nfia−/−mice did display small regions along the dorsoventral midline where “spot” adhesions rather than complete fusion may have occurred. Further analysis at the electron microscopic level is required to determine the full extent of midline fusion defects inNfia−/−mice. Furthermore, because very little is known about the molecular mechanisms of how midline fusion actually occurs, it remains to be investigated whether Nfia is directly involved in mediating such processes.
Although it is possible that loss of Nfia results in an intrinsic axonal defect, the findings that Nfia is not expressed in most callosal neurons early in callosal formation and that Robo1 and Robo2 expression are unaffected in Nfia knock-out mice (Fig. 7) makes this less likely. However, further experiments are required to rule out an axon intrinsic defect in the Nfiamutant.
NFI-A is highly expressed in the glial sling, and sling cell migration was greatly disrupted in the knock-out. If sling cells migrate along the glial wedge to the midline, this migration defect could be an indirect effect of the glial wedge malformation. Alternatively,Nfia itself may regulate either the migration or proliferation of sling cells. If the lack of Nfia caused the sling cells to continue to proliferate, then their proliferative state may disrupt their ability to follow and respond to environmental cues as they migrate. Previous studies have shown that in addition to cues within the cellular environment, a critical determinant for layer-specific cell migration in the cerebral cortex is whether a cell has completed S phase of the cell cycle (McConnell and Kaznowski, 1991;Frantz and McConnell, 1996; Desai and McConnell, 2000). Analysis of proliferation and apoptosis rates in the region encompassing the corticoseptal boundary should allow us to distinguish between these hypotheses.
We also observed that the hippocampal commissure failed to form in all embryonic and most of the postnatal mutants. However, this phenotype was variable between animals, indicating that compensatory mechanisms such as the expression of other NFI family members may be involved. In the most caudal regions of the corpus callosum, we found that callosal axons overlapped with axons of the fornix and hippocampal commissure axons, particularly at the border between the two commissures. This overlap of callosal and hippocampal axons could occur either because of defects in glial wedge and sling development, which may help to separate the two commissures, or because Nfia regulates the expression of surface molecules on the hippocampal axons themselves that normally keeps them separated from the callosal axons. We also observed a marked reduction in the size of the fornix and the hippocampal commissure and a reduction in GFAP immunohistochemistry within the fimbria region in theNfia−/−mice. NFI-A is expressed throughout the hippocampus, particularly within the dentate gyrus (Plachez et al., 2001), and therefore may be involved in the development of both neurons and glia within the hippocampus.
In addition to the corpus callosum and the hippocampal commissure defects, we observed a defect in the formation of the glial tunnel and the anterior commissure. Although some axons crossed the midline, even in the absence of other midline commissures, the rostrocaudal size of the anterior commissure was greatly reduced. Again, this may reflect either a role for Nfia in the development of neurons in the olfactory bulb that express Nfia (Plachez et al., 2001) and project through the anterior commissure or an indirect effect via the glial tunnel (Katz et al., 1983; Silver et al., 1993; Cummings et al., 1997; Pires-Neto et al., 1998), which was also malformed. At present it is not known whether the glial tunnel expresses guidance molecules for axons of the anterior commissure. It is possible that similar mechanisms operate in the development of the corpus callosum and the formation of other midline commissures.
The perforating pathway formed normally in the Nfia mutant, although it crossed over the Probst-like bundles to enter the cingulate cortex. NFI-A is not expressed in the medial septum/diagonal band of Broca region where most of the neurons that give rise to the perforating pathway reside (Shu et al., 2001). Perforating axons avoid the glial wedge morphologically in wild-type animals (Shu et al., 2001), but our data in the Nfia mutant suggest that the formation of the glial wedge is not required for the projection of the perforating axons into the cingulate cortex.
Our results indicate that Nfia may play a critical role in regulating genes involved in glial and/or neuronal development and axonal guidance. Given the overlapping expression of Nfiband Nfix in the brain, it is interesting that the mutation of Nfia had such profound effects on these developmental processes. Perhaps this indicates that different family members perform different functions within the same cells and may not provide compensation for each other. Indeed, major differences in transcriptional modulation properties have been seen between different NFI gene products in transient transfection assays (Apt et al., 1994;Chaudhry et al., 1998) (see latter for additional references). Given the complexity of alternatively spliced transcripts of these genes, it will be important to analyze the expression of specific protein isoforms of each NFI gene. It will also be important to identify downstream targets of Nfia to determine at a molecular level how Nfia is involved in the development of glia and neurons and the formation of midline commissures.
Footnotes
This work was supported by March of Dimes Foundation for Birth Defects Grant 5-FY99-842 (L.J.R.) and by National Institutes of Health Grants NS37792 (L.J.R.) and HD34908 (R.M.G.). We thank Kimberley M. Valentino for excellent technical assistance.
Correspondence should be addressed to Dr. Linda J. Richards, Department of Anatomy and Neurobiology, HSF 222, The University of Maryland, Baltimore, 685 West Baltimore Street, Baltimore, MD 21201. Email:lrich001@umaryland.edu.
K. G. Butz's and R. M. Gronostajski's present address: Department of Biochemistry, State University of New York at Buffalo, 140 Farber Hall, 3435 Main Street, Buffalo, NY 14214.
References
- 1.Apt D, Liu Y, Bernard H-U. Cloning and functional analysis of spliced isoforms of human nuclear factor I-X: interference with transcriptional activation by NFI/CTF in a cell-type specific manner. Nucleic Acids Res. 1994;22:3825–3833. doi: 10.1093/nar/22.19.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagri A, Marin O, Plump AS, Mak J, Pleasure SJ, Rubenstein JLR, Tessier-Lavigne M. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002;33:233–248. doi: 10.1016/s0896-6273(02)00561-5. [DOI] [PubMed] [Google Scholar]
- 3.Baumeister H, Gronostajski RM, Lyons GE, Margolis FL. Identification of NFI-binding sites and cloning of NFI-cDNAs suggest a regulatory role for NFI transcription factors in olfactory neuron gene expression. Mol Brain Res. 1999;72:65–79. doi: 10.1016/s0169-328x(99)00210-7. [DOI] [PubMed] [Google Scholar]
- 4.Behrens M, Venkatraman G, Gronostajski RM, Reed RR, Margolis FL. NFI in the development of the olfactory neuroepithelium and the regulation of olfactory marker protein gene expression. Eur J Neurosci. 2000;12:1372–1384. doi: 10.1046/j.1460-9568.2000.00032.x. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhry AZ, Lyons GE, Gronostajski RM. Expression patterns of the four nuclear factor I genes during mouse embryogenesis indicate a potential role in development. Dev Dyn. 1997;208:313–325. doi: 10.1002/(SICI)1097-0177(199703)208:3<313::AID-AJA3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhry AZ, Vitullo AD, Gronostajski RM. Nuclear factor I (NFI) isoforms differentially activate simple versus complex NFI-responsive promoters. J Biol Chem. 1998;273:18538–18546. doi: 10.1074/jbc.273.29.18538. [DOI] [PubMed] [Google Scholar]
- 7.Cummings DM, Malun D, Brunjes PC. Development of the anterior commissure in the opossum: midline extracellular space and glia coincide with early axon decussation. J Neurobiol. 1997;32:403–414. [PubMed] [Google Scholar]
- 8.das Neves L, Duchala CS, Godinho F, Haxhiu MA, Colmenares C, Macklin WB, Campbell CE, Butz KG, Gronostajski RM. Disruption of murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc Natl Acad Sci USA. 1999;96:11946–11951. doi: 10.1073/pnas.96.21.11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai AR, McConnell SK. Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Development. 2000;127:2863–2872. doi: 10.1242/dev.127.13.2863. [DOI] [PubMed] [Google Scholar]
- 10.Feng L, Hatten ME, Heinz N. Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron. 1994;12:895–908. doi: 10.1016/0896-6273(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 11.Festing MF, Simpson EM, Davisson MT, Mobraaten LE. Revised nomenclature for strain 129 mice. Mamm Genome. 1999;10:836. doi: 10.1007/s003359901099. [DOI] [PubMed] [Google Scholar]
- 12.Frantz GD, McConnell SK. Restriction of late cerebral cortical progenitors to an upper-layer fate. Neuron. 1996;17:55–61. doi: 10.1016/s0896-6273(00)80280-9. [DOI] [PubMed] [Google Scholar]
- 13.Godement P, Mason CA. Guidance of retinal fibers in the optic chiasm. Perspect Dev Neurobiol. 1993;1:217–225. [PubMed] [Google Scholar]
- 14.Gronostajski RM. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000;249:31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 15.Gronostajski RM, Adhya S, Nagata K, Guggenheimer RA, Hurwitz J. Site-specific DNA binding of nuclear factor I: analysis of cellular binding sites. Mol Cell Biol. 1985;5:964–971. doi: 10.1128/mcb.5.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guillery RW, Walsh C. Changing glial organization relates to changing fiber order in the developing optic nerve of ferrets. J Comp Neurol. 1987;265:203–217. doi: 10.1002/cne.902650205. [DOI] [PubMed] [Google Scholar]
- 17.Hankin MH, Silver J. Development of intersecting CNS fiber tracts: the corpus callosum and its perforating fiber pathway. J Comp Neurol. 1988;272:177–190. doi: 10.1002/cne.902720203. [DOI] [PubMed] [Google Scholar]
- 18.Hennighausen L, Siebenlist U, Danner D, Leder P, Rawlins D, Rosenfeld P, Kelly TJ. High affinity binding site for a specific nuclear protein in the human IgM gene. Nature. 1985;314:289–292. doi: 10.1038/314289a0. [DOI] [PubMed] [Google Scholar]
- 19.Hevner RF, Shi L, Justice N, Hsueh Y-P, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JLR. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- 20.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 21.Katz MJ, Lasek RJ, Silver J. Ontophyletics of the nervous system: development of the corpus callosum and evolution of axon tracts. Proc Natl Acad Sci USA. 1983;80:5936–5940. doi: 10.1073/pnas.80.19.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krohn K, Rozovsky I, Wals P, Teter B, Anderson CP, Finch CE. Glial fibrillary acidic protein transcription responses to transforming growth factor-β1 and interleukin-1 β are mediated by a nuclear factor-I like site in the near-upstream promoter. J Neurochem. 1999;72:1353–1361. doi: 10.1046/j.1471-4159.1999.721353.x. [DOI] [PubMed] [Google Scholar]
- 23.Kurtz A, Zimmer A, Schnutgen F, Bruning G, Spener F, Muller T. The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development. 1994;120:2637–2649. doi: 10.1242/dev.120.9.2637. [DOI] [PubMed] [Google Scholar]
- 24.Leegwater PAJ, van Driel W, van der Vliet PC. Recognition site of nuclear factor I, a sequence-specific DNA-binding protein from HeLa cells that stimulates adenovirus DNA replication. EMBO J. 1985;4:1515–1521. doi: 10.1002/j.1460-2075.1985.tb03811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 1996;17:607–615. doi: 10.1016/s0896-6273(00)80194-4. [DOI] [PubMed] [Google Scholar]
- 26.Livy DJ, Wahlsten D. Tests of genetic allelism between four inbred mouse strains with absent corpus callosum. J Hered. 1991;82:459–464. doi: 10.1093/oxfordjournals.jhered.a111128. [DOI] [PubMed] [Google Scholar]
- 27.Livy DJ, Wahlsten D. Retarded formation of the hippocampal commissure in embryos from mouse strains lacking a corpus callosum. Hippocampus. 1997;7:2–14. doi: 10.1002/(SICI)1098-1063(1997)7:1<2::AID-HIPO2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 28.Marcus RC, Blazeski R, Godement P, Mason CA. Retinal axon divergence in the optic chiasm: uncrossed axons diverge from crossed axons within a midline glial specialization. J Neurosci. 1995;15:3716–3729. doi: 10.1523/JNEUROSCI.15-05-03716.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A. Targeted deletion in astrocyte intermediate filament (GFAP) alters neuronal physiology. Proc Natl Acad Sci USA. 1996;93:6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McConnell SK, Kaznowski CE. Cell cycle dependence of laminar determination in developing neocortex. Science. 1991;254:282–285. doi: 10.1126/science.254.5029.282. [DOI] [PubMed] [Google Scholar]
- 31.Miura M, Tamura T-A, Mikoshiba K. Cell specific expression of the mouse glial fibrillary acidic protein gene: identification of the cis- and trans-acting promoter elements for astrocyte-specific expression. J Neurochem. 1990;55:1180–1188. doi: 10.1111/j.1471-4159.1990.tb03123.x. [DOI] [PubMed] [Google Scholar]
- 32.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 33.Nagata K, Guggenheimer R, Enomoto T, Lichy J, Hurwitz J. Adenovirus DNA replication in vitro identification of a host factor that stimulates synthesis of the preterminal protein-dCMP complex. Proc Natl Acad Sci USA. 1982;79:6439–6442. doi: 10.1073/pnas.79.21.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagata K, Guggenheimer RA, Hurwitz J. Specific binding of a cellular DNA replication protein to the origin of replication of adenovirus DNA. Proc Natl Acad Sci USA. 1983;80:6177–6181. doi: 10.1073/pnas.80.20.6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nowock J, Borgmeyer U, Puschel AW, Rupp RAW, Sippel AE. The TGGCA protein binds to the MMTV-LTR, the adenovirus origin of replication, and the BK virus enhancer. Nucleic Acids Res. 1985;13:2045–2061. doi: 10.1093/nar/13.6.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pires-Neto M, Braga-De-Souza S, Lent R. Molecular tunnels and boundaries for growing axons in the anterior commissure of hamster embryos. J Comp Neurol. 1998;399:176–188. doi: 10.1002/(sici)1096-9861(19980921)399:2<176::aid-cne3>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 37.Plachez C, Shu T, Margolis FL, Gronostajski RM, Richards LJ. Expression of nuclear factor I-A in developing mouse brain. Soc Neurosci Abstr. 2001;27:794.15. [Google Scholar]
- 38.Preston SL, Shu T, Corte G, Rubenstein JLR, Richards LJ. Emx-1 is required for the development of the glial sling and formation of the corpus callosum. Soc Neurosci Abstr. 2000;13:577. [Google Scholar]
- 39.Rash BG, Richards LJ. A role for cingulate pioneering axons in the development of the corpus callosum. J Comp Neurol. 2001;434:147–157. doi: 10.1002/cne.1170. [DOI] [PubMed] [Google Scholar]
- 40.Reese BE, Maynard TM, Hocking DR. Glial domains and axonal reordering in the chiasmatic region of the developing ferret. J Comp Neurol. 1994;349:303–324. doi: 10.1002/cne.903490211. [DOI] [PubMed] [Google Scholar]
- 41.Shibuki K, Gomi H, Chen L, Bao S, Kim JJ, Wakatsuki H, Fujisaki T, Fujimoto K, Katoh A, Ikeda T, Chen C, Thompson RF, Itohara S. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16:587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- 42.Shu T, Richards LJ. Cortical axon guidance by the glial wedge during the development of the corpus callosum. J Neurosci. 2001;21:2749–2758. doi: 10.1523/JNEUROSCI.21-08-02749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shu T, Valentino KM, Seaman C, Cooper HM, Richards LJ. Expression of the netrin receptor, deleted in colorectal cancer (DCC), is largely confined to projecting neurons in the developing forebrain. J Comp Neurol. 2000;416:201–212. doi: 10.1002/(sici)1096-9861(20000110)416:2<201::aid-cne6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 44.Shu T, Shen W-B, Richards LJ. Development of the perforating pathway: an ipsilaterally projecting pathway between the medial septum/diagonal band of Broca and the cingulate cortex that intersects the corpus callosum. J Comp Neurol. 2001;436:411–422. doi: 10.1002/cne.1077. [DOI] [PubMed] [Google Scholar]
- 45.Silver J. Glia-neuron interactions at the midline of the developing mammalian brain and spinal cord. Perspect Dev Neurobiol. 1993;1:227–236. [PubMed] [Google Scholar]
- 46.Silver J, Ogawa MY. Postnatally induced formation of the corpus callosum in acallosal mice on glia-coated cellulose bridges. Science. 1983;220:1067–1069. doi: 10.1126/science.6844928. [DOI] [PubMed] [Google Scholar]
- 47.Silver J, Lorenz SE, Wahlsten D, Coughlin J. Axonal guidance during development of the great cerebral commissures: descriptive and experimental studies, in vivo, on the role of preformed glial pathways. J Comp Neurol. 1982;210:10–29. doi: 10.1002/cne.902100103. [DOI] [PubMed] [Google Scholar]
- 48.Silver J, Edwards MA, Levitt P. Immunocytochemical demonstration of early appearing astroglial structures that form boundaries and pathways along axon tracts in the fetal brain. J Comp Neurol. 1993;328:415–436. doi: 10.1002/cne.903280308. [DOI] [PubMed] [Google Scholar]
- 49.Super H, Soriano E. The organization of the embryonic and early postnatal murine hippocampus. II. Development of entorhinal, commissural, and septal connections studied with the lipophilic tracer DiI. J Comp Neurol. 1994;344:101–120. doi: 10.1002/cne.903440108. [DOI] [PubMed] [Google Scholar]
- 50.Valentino KL, Jones EG. The early formation of the corpus callosum: a light and electron microscopic study in foetal and neonatal rats. J Neurocytol. 1982;11:583–609. doi: 10.1007/BF01262426. [DOI] [PubMed] [Google Scholar]
- 51.Wahlsten D. Prenatal schedule of appearance of mouse brain commissures. Dev Brain Res. 1981;1:461–473. doi: 10.1016/0165-3806(81)90001-8. [DOI] [PubMed] [Google Scholar]
- 52.Wang LC, Dani J, Godement P, Marcus RC, Mason CA. Crossed and uncrossed retinal axons respond differently to cells of the optic chiasm midline in vitro. Neuron. 1995;15:1349–1364. doi: 10.1016/0896-6273(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 53.Wise SP, Jones EG. The organization and postnatal development of the commissural projection of the rat somatic sensory cortex. J Comp Neurol. 1976;168:313–344. doi: 10.1002/cne.901680302. [DOI] [PubMed] [Google Scholar]