

Abstract
In cultured chick ciliary neurons, when ATP synthesis is inhibited, ATP depletion is reduced ∼50% by slowing actin filament turnover with jasplakinolide or latrunculin A. Jasplakinolide inhibits actin disassembly, and latrunculin A prevents actin assembly by sequestering actin monomers. Cytochalasin D, which allows assembly–disassembly, but only at pointed ends, is less effective in conserving ATP. Ouabain, an Na+–K+-ATPase inhibitor, and jasplakinolide both prevent ∼50% of the ATP loss. When applied together, they completely prevent ATP loss over a period of 20 min, suggesting that filament stabilization reduces ATP consumption by decreasing actin-ATP hydrolysis directly rather than indirectly by modulating the activity of Na+–K+-ATPase, a major energy consumer.
Keywords: ischemia, actin filament treadmilling, intracellular ATP, jasplakinolide, latrunculin, cytoskeleton
Introduction
The brain constitutes ∼2% of total body weight but consumes a disproportionate 25% of total oxygen (Magistretti, 1999), most of which is used to produce ATP for electrical activity. Glutamate-generated electrical activity alone accounts for ∼80% of total brain ATP consumption (Attwell and Laughlin, 2001). This large energy consumption is assumed to be needed for restoring transmembrane ionic gradients via pumps. Often overlooked is the abundance of actin (Kabsch and Vandekerckhove, 1992) and the ATP hydrolysis required for the vital role that actin dynamics play in presynaptic and postsynaptic regions during electrical activation (Bernstein et al., 1998; Fischer et al., 1998). Here, we provide evidence that the energy consumed by actin dynamics is probably great enough to contribute significantly to the permanent neurological deficits that occur when accidents or strokes deprive the brain of oxygen even briefly.
Actin dynamics have been implicated in neuronal protection in a different context. Gelsolin, a protein that severs the actin filament and caps its fast-growing end, was reported to protect ischemically challenged neurons by attenuating Ca2+influx through channels requiring cytoskeletal integrity (Furukawa et al., 1997; Endres et al., 1999). Our present data support the idea that slowing actin dynamics may protect such neurons via a more direct mechanism: preserving ATP.
Cycles of actin polymerization–depolymerization normally occur continuously even in resting cells and require hydrolysis of ATP (Belmont and Drubin, 1998). Actin cycling is needed presynaptically and postsynaptically during stimulated transmission (Bernstein et al., 1998; Fischer et al., 1998). Actin monomers with ATP bound are added to the barbed (plus) end of the filament, and, after this addition, the terminal phosphate is hydrolyzed and inorganic phosphate is released, leaving ADP trapped in the actin subunit. This phosphate loss alters the actin subunit conformation within the filament (Moraczewska et al., 1999), weakening the subunit interactions and thus promoting subunit release. The conformation change induced by ATP hydrolysis results in different critical concentrations for subunit assembly at the two filament ends. This difference causes actin subunits to treadmill through the filament under steady-state conditions and consume ATP at a rate proportional to that of filament turnover. ATP is exchanged for ADP on the freed subunit at a relatively slow rate unless enhanced by other proteins.
It was estimated in a nucleotide-exchange study that as much as 50% of the total ATP use of resting platelets is needed merely to maintain the actin cytoskeleton (Daniel et al., 1986). However, the significance of the cytoskeleton as an energy drain is not generally appreciated. The bulk of the ATP consumption by the cell is credited to energetically unfavorable chemical reactions, such as synthesis of biological molecules, the active transport of molecules (particularly ions) across cell membranes, and the generation of force and movement (Alberts et al., 1994). We monitored ATP depletion after blockage of its synthesis. Slowing filament turnover reduces by ∼50% the ATP consumed by all neuronal processes during the first few minutes after synthesis is blocked. We show that this preservation of ATP is independent of the energy used by Na+–K+-ATPase, the largest energy consumer in ionic homeostasis, and that blocking both the actin filament turnover and Na+–K+-ATPase preserves ATP levels during prolonged ischemic insult.
Materials and Methods
Materials. All fluorescent dyes, phalloidin, latrunculin A, jasplakinolide, and cytochalasin D were purchased from Molecular Probes (Eugene, OR). Other reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Cell culture and filamentous actin staining. Ciliary ganglia of 10- to 11-d-old chick embryos were dissociated acutely by trypsinization (10 min, 37°C in 0.1% trypsin in HBSS without Ca2+ and Mg2+) and trituration (six to eight ganglia) in 80 μl of Neurobasal medium–B27 supplement (Invitrogen, San Diego, CA)–2 mmglutamine. Cells (10 μl/chamber) were plated on a number 1 glass coverslip coated with Matrigel (Becton Dickinson, Franklin Lakes, NJ) to which a CoverWell silicone rubber perfusion chamber (total capacity, 70 μl; Grace Bio-Labs, Bend, OR) had been sealed. After an additional 30 μl of medium had been added, cells were cultured overnight in a humidified 5% CO2 incubator at 37°C before each experiment. Cultures contained growth cones and synapses.
For F-actin staining, cells were fixed in 0.1% glutaraldehyde in PBS for 15 min, washed three times, permeabilized for 5 min in 0.1% Triton X-100 containing 1 mg/ml NaBH4, incubated in 7.5 U/ml rhodamine phalloidin for 3 hr in the dark, rinsed, and mounted with ProLong Antifade (Molecular Probes).
Monitoring of intracellular ATP, Na+, and Ca2+. Cells were incubated (30 min, humidified 5% CO2at 37°C) in freshly made 10 μm AM ester of magnesium green in PBS (MgGr; 475 nm excitation, 530 nm emission). This dye increases emission as a function of free intracellular Mg2+([Mg2+]i) without shifting emission wavelength (Haugland, 1996). Cells were washed two times with PBS and incubated in growth medium with experimental reagent or its buffer. These included jasplakinolide, latrunculin A, cytochalasin D, and ouabain (Kimelberg et al., 1979). Cells were washed two times with PBS and transferred to a heated stage (35°C) of a Nikon (Tokyo, Japan) Diaphot microscope with a Nikon 20× objective for live cell fluorescence microscopy and a Nikon 60× oil immersion objective and oil immersion condenser for differential interference contrast (DIC) microscopy. Typically, images at three time points were acquired automatically before addition of ATP-synthesis inhibitors (1 vol of 20 mmNaN3–12 mm 2-deoxyglucose in PBS) and during and after addition of inhibitors.
The cells were treated similarly for experiments with sodium green and AM forms of Mag-fura-2 and fura-2, and the same general procedure was followed as outlined above for cells loaded with MgGr. A filter cube (480 ± 15 nm bandpass excitation filter, 510 nm dichroic mirror, and 535 ± 20 nm emission filter) was used for MgGr and sodium green. For fura dyes, 340 ± 7.5 and 380 ± 7.5 nm excitation filters in a computer-controlled filter wheel, a 400 nm dichroic mirror, and a 460 ± 25 nm bandpass emission filter were used. Calcium levels were determined (Grynkiewicz et al., 1985) with a calibration kit (Molecular Probes).
Image acquisition and analysis. Metamorph software (version 4.6; Universal Imaging Corporation, West Chester, PA) was used to control camera settings, store images, and drive all shutters [xenon lamp, high-pressure mercury lamp, and PXL PhotoMetrics (Tucson, AZ) cooled CCD camera fitted with a Kodak 1400 chip (Eastman Kodak, Rochester, NY)], a programmable microscope stage, focus control, and an excitation filter wheel. In the typical experiment, four stage positions were stored, and images were taken of 5–10 microscope fields to the right of each stored position. Stacks consisting of 10–15 images of each microscope field taken at ∼2 min intervals were analyzed by thresholding the fluorescent neuronal somata, transferring the average intensity of the somata and neighboring noncell areas (background) to an Excel (version 97; Microsoft, Redmond, WA) electronic spreadsheet, and reducing those data to intensity increases as a function of time before and after addition of ATP-synthesis inhibitors.
Results
Two membrane-permeable marine natural products, jasplakinolide and latrunculin A, were used to determine the effects of reducing cycles of actin assembly–disassembly on ATP consumption. Both bind with 1:1 stoichiometry to actin, latrunculin A to monomeric actin and jasplakinolide to actin subunits in filamentous actin (F-actin) (Spector et al., 1999). Jasplakinolide reduces turnover rate primarily by inhibiting the release of subunits from the filament pointed end (Bubb et al., 2000). Latrunculin A sequesters monomers as they are released from filaments and prevents their reassembly (Morton et al., 2000), thereby reducing turnover. It may also preserve ATP by slowing nucleotide exchange when it complexes with G-actin (Belmont et al., 1999). Cytochalasin D, also used and also membrane permeable, interacts very differently with actin: it caps the barbed end, preventing assembly–disassembly there but still allowing it at the pointed end (Goddette and Frieden, 1986).
To monitor the loss of ATP in live cells, we took advantage of the fact that the affinity of Mg2+ for ATP (Kd = 50 ± 10 μm) (Gupta et al., 1983) is ∼10-fold higher than for ADP or AMP (Leyssens et al., 1996). As ATP is hydrolyzed to ADP and AMP, [Mg2+]i rises (Budinger et al., 1998). Figure1a shows that the rate of ATP depletion in neuronal soma of cells loaded with the fluorescent dye MgGr immediately after ATP synthesis is blocked by the oxidative phosphorylation inhibitor NaN3 and the glycolysis inhibitor 2-deoxyglucose. In these chemically ischemic cells, preincubation in either 10 nm jasplakinolide or 1 μm latrunculin A for 45 min significantly attenuates the rate of ATP consumption. Neither of these compounds, used in the absence of ATP depletion, affects MgGr fluorescence (data not shown). Cells treated with 5 μm to 10 mm cytochalasin D show only a slight reduction in the rate of ATP depletion compared with control cells (only 10 μm data shown in Fig. 1a). The phase micrograph in Figure 1b shows large ciliary neuron somata and extensive process outgrowth typical of these cultures; 40 min of ATP depletion causes somata to shrivel and processes to retract dramatically (Fig. 1c).
Fig. 1.

a, Slowing filament turnover conserves ATP. Time-lapse imaging of ciliary neurons, loaded with MgGr, was used to monitor ATP depletion after a block in ATP synthesis. As ATP is depleted, [Mg2+]i increases. Analysis includes all cells in 20 fields. Similar results were seen in two replicas of this experiment; mean ± SEM of >100 cells.b, c, Phase micrographs of fixed neurons before (b) and 40 min after (c) application of ATP-synthesis block (10 mm NaN3 to 6 mm 2-deoxyglucose). Chemically ischemic cells show extensive neurite retraction, somata shrinking, and rounding, lysed cell debris (arrowheads), and vacuoles (arrows). Scale bar, 30 μm.jas, Jasplakinolide; lat A, latrunculin A; cyto D, cytochalasin D.
We chose to use 10 nm jasplakinolide because, as seen in Figure 2a–c, higher levels, rather than merely slowing disassembly and stabilizing existing F-actin, induce abnormal amounts of assembled actin. This effect is consistent with the inhibition of disassembly and reduction of nuclei needed for actin assembly by jasplakinolide (Cramer, 1999; Bubb et al., 2000). In these ciliary neuron growth cones, the jasplakinolide-induced assembly is manifested by the elaboration of filopodia that extend as a result of bundled F-actin elongation (Fig.2a–c); filopodia contain parallel bundles of F-actin (Forscher and Smith, 1988). We avoided inducing assembly that would perturb multiple aspects of cell physiology. To determine the minimum concentration of jasplakinolide needed to slow actin filament turnover (Fig. 2d–g), we used FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] to monitor a process that depends on F-actin assembly–disassembly, the recycling of transmitter vesicles (Bernstein et al., 1998). Vesicle recycling indicates that 6 nm is too low, and the growth cone morphology indicates that 20 nm is too high. Morphological evidence for F-actin stabilization by jasplakinolide is seen in the DIC micrographs of Figure 3, a andb. Figure 3a shows the promotion of actin disassembly by latrunculin A when jasplakinolide is absent. The dose-dependent nature of this effect of jasplakinolide is plotted in Figure 3c.
Fig. 2.
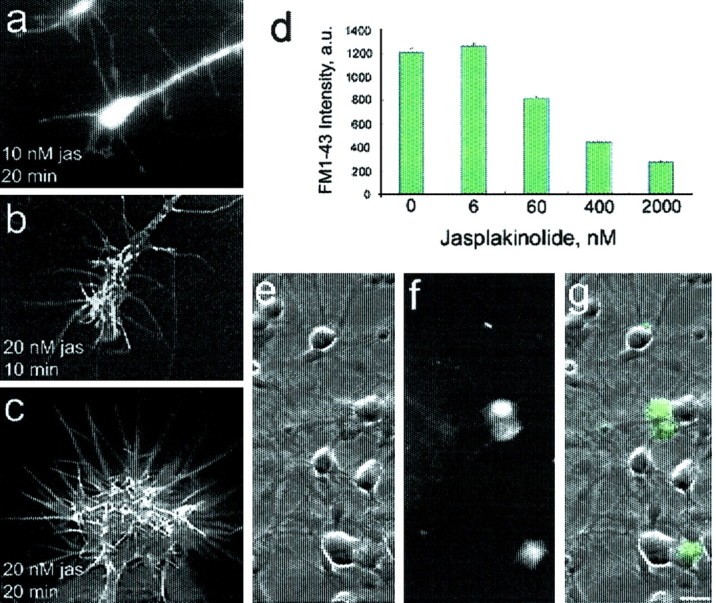
The minimum jasplakinolide concentration needed to stabilize filaments is >6 nm. a–c, Incubation in as little as 20 nm jasplakinolide (jas) induces a filopodial expansion, indicating stimulation of actin assembly rather than mere stabilization of F-actin, here stained with rhodamine phalloidin. Scale bar, 10 μm.d, Transmitter vesicle recycling depends on filament turnover rate (Bernstein et al., 1998) and is attenuated by preincubation in jasplakinolide >6 nm. Cells were depolarized in a 75 mm K+ buffer containing the fluorescent styryl dye FM1-43 (10 μm), which is used to monitor depolarization-induced vesicle cycling (Cochilla et al., 1999). g, Overlay of DIC (e) and fluorescence (f) images, showing depolarization-induced FM1-43 uptake in ciliary calyx. Scale bar, 20 μm.
Fig. 3.

Morphological evidence for promotion of actin disassembly by latrunculin (Lat A) and stabilization of F-actin by jasplakinolide (Jas). a, DIC micrographs of live cells without jasplakinolide pretreatment show lysis and loss of cell shape (flattening) after 15 min of incubation in 1 μm latrunculin A. The dose-dependent nature of jasplakinolide stabilization of cell morphology seen inb is plotted in c. A concentration as low as 8 nm has a stabilizing effect. All cells in >20 fields were included in data plotted for each jasplakinolide concentration.
Evidence for the appropriateness of monitoring [Mg2+]i to follow the rate of ATP depletion involved two other studies. Calcium is the most likely species to interfere with Mg2+detection. However, as in the case of cardiomyocytes (Leyssens et al., 1996), it appears not to interfere with [Mg2+]i detection. In ciliary neurons, ATP depletion elevates [Ca2+]i from 350 to 400 nm (Fig.4a), an increase that is six times less than that elicited by 1 mm caffeine (from 350 to 645 nm) (Jha et al., 2002). Because the [Ca2+]iincrease elicited by 1 mm caffeine has no effect on MgGr intensity (data not shown), the far smaller [Ca2+]i increase caused by ATP depletion should not affect MgGr intensity either.
Fig. 4.
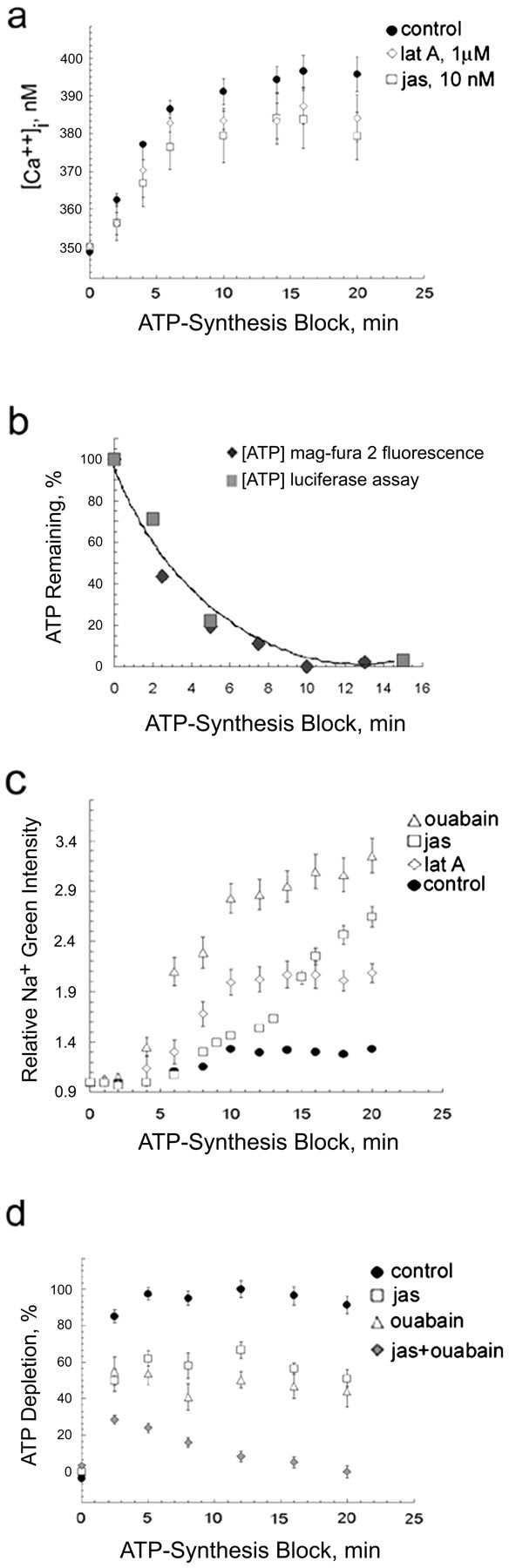
Effects of actin modulators on ATP, Na+, and Ca2+ levels in ischemically stressed cells. a, Reducing filament turnover moderates ischemically induced increase in [Ca2+]i but is not likely to contribute significantly to ATP conservation of jasplakinolide (jas)- and latrunculin A (lat A)-treated cells. b, Similar time courses were observed for ATP depletion in rat embryonic day 18 brain cortical cells measured with either a cell lysate–luciferase assay or an [Mg2+]i fluorescent dye indicator and for ciliary cells using MgGr or Mag-fura-2; exponential curve fit shown. c, d, Actin assembly modulators conserve ATP via a reduction in actin-ATP hydrolysis rather than a reduction in Na+–K+-ATPase activity.c, Preincubation in 10 nm jasplakinolide or 1 μm latrunculin A accelerates [Na+]i increase during ATP depletion. Only in ATP-depleted control cells is [ATP] low enough to reverse Na+–Ca2+ exchanger, pulling Ca2+ in and extruding Na+. Not unexpectedly, PBS–1.5 mm ouabain, an Na+–K+-ATPase inhibitor, causes the fastest rise in Na+. d, Conservation of ATP by ouabain and jasplakinolide is additive.a–d, Mean ± SEM of two or three experiments;n = 200–300 cells in 60 fields.
Second, [Mg2+]ishows the same rise when ATP synthesis is blocked in ciliary cells loaded with the ratiometric fluorescent Mg2+ indicator Mag-fura-2. Moreover, the time course of ATP depletion measured by a cell extract assay on the basis of luciferase (Minamide et al., 2000) is identical to that monitored via fluorescent dye (Fig. 4b).
The observed conservation of ATP obtained by pretreatment with jasplakinolide and latrunculin A (Fig. 1a) could be a result of a reduction in the demand placed on active transport species that maintain ion gradients. If, for instance, slowing filament turnover reduced the [Na+]i, the rate of ATP depletion might be reduced because there is a lighter load on the Na+–K+-ATPase rather than a reduction in the hydrolysis of ATP associated with filament turnover. Conversely, Figure 4c shows that jasplakinolide and latrunculin A accelerate the [Na+]i increase induced by chemical ischemia. One could argue that [Na+]i rises faster and ATP depletion is slowed with actin modulators because the Na+–K+-ATPase activity is blocked by them. If this were true, inhibiting the Na+–K+-ATPase activity of jasplakinolide-treated cells with ouabain should not slow ATP depletion much more than jasplakinolide alone. Yet we see in Figure4d that jasplakinolide plus ouabain, a potent specific inhibitor of Na+–K+-ATPase (Kimelberg et al., 1979), is additive in effect, i.e., in combination, they eliminate most of the ATP depletion. This finding supports the notion that the conservation of ATP seen with inhibitors of actin filament turnover occurs via a reduction in the hydrolysis of ATP associated with dynamically regulated actin.
Conservation of ATP might also be at least partially caused by jasplakinolide and latrunculin A reducing the energy drain involved in regulating [Ca2+]i. Cells pretreated with either jasplakinolide or latrunculin A initially show the same ischemia-induced increase in [Ca2+]i as control cells. After the first few minutes, the rise in [Ca2+]i is somewhat less than that of control cells (Fig. 4a). Regulation of [Ca2+]i in cells treated with actin modulators may contribute slightly to the observed conservation of ATP.
Discussion
More than 15 years have elapsed since the authors of a nucleotide-exchange study estimated that maintenance of the actin cytoskeleton could be responsible for as much as 50% of the total ATP consumption in resting platelets (Daniel et al., 1986). One might expect the fraction of ATP consumed by the cytoskeleton to be higher in platelets than in whole cells, because platelets are enucleate cell fragments derived from megakaryocytes and have much reduced ATP-dependent biosynthetic activity. Here, we provide evidence from live neurons that supports the idea that actin dynamics are a major ATP-consuming process in bona fide cells.
Understanding ATP turnover in neurons is important clinically because neurons are ischemically sensitive and some neurodegenerative diseases are triggered by transient ischemic events (Aliev et al., 2002). One indication of the significance of actin for the physiology of oxidatively stressed neurons is the abundant formation of abnormal actin-containing inclusions (“rods”) within minutes of ischemic insult (Minamide et al., 2000). Rods appear in the axons and dendrites of cultured hippocampal and cortical cells and contain proteins of the actin depolymerizing factor (ADF)–cofilin family that enhance the rapid turnover of actin filaments (Bamburg, 1999). We suggest that the sequestering of proteins during the initial transient formation of rods spares cellular ATP by reducing actin dynamics. The reappearance of rods within 1 d after insult may contribute to pathological neurite degeneration (Minamide et al., 2000).
Our findings also have interesting implications for cell biology because they allow for a new method of estimating filament length in neurons. If one assumes total ATP use of 80 μmol · l−1 · sec−1for brain tissue, which can be derived from human cerebral blood flow and metabolic rates (Sokoloff, 1996), then actin turnover is responsible for 50% of the ATP turnover, or ∼40 μmol · l−1 · sec−1. The actin treadmilling rate in cells is ∼20 sec−1, assuming that it is 200 times faster than the in vitro rate of 0.1 sec−1(Zigmond, 1993), i.e., there is a release of 20 subunits per pointed end per second (Didry et al., 1998). Because the rate-limiting step for actin-associated ATP hydrolysis is the subunit release rate, a concentration of filament ends of 2 μmol/l is required for 20 subunits per pointed end per second for the degradation of 40 μmol · l−1 · sec−1ATP. Assuming that total cellular actin concentration is 100 μm, a filament end concentration of 2 μm means there is an average of only 50 subunits per filament. This is significantly shorter than the estimate for the average filament length in neurons, which is 0.55 μm or 204 subunits (∼370 subunits per micrometer) (Fath and Lasek, 1988). The discrepancy of approximately fourfold is not large given the possible errors in the estimates used here but could also arise from the difficulty of visualizing short filaments by electron microscopy of axoplasm. The filament distribution in neurons might consist of some long, easily visualized filaments and a larger population of short filaments.
The approach we used, pharmacologically slowing filament turnover while blocking ATP synthesis, clearly could have effects on ATP-dependent processes unrelated to actin-ATP hydrolysis. Many proteins involved in ion gradient regulation are modulated by the actin cytoskeleton, are interdependent, and do use ATP (Mills et al., 1994). The active transport protein most likely to contribute to ATP depletion in this study is Na+–K+-ATPase, because it is a major energy consumer and because its activity is stimulated by G-actin (Cantiello, 1995). One might expect slowing filament turnover and preserving ATP to reduce the pathological increase in [Na+]ithat is an early, profoundly injurious effect of ischemia. The [Na+]i overload inhibits the Na+–H+exchanger, thus acidifying ischemically stressed cells, and the [Na+]i overload also inhibits the Na+–Ca2+exchanger, causing Ca2+ overload (Friedman and Haddad, 1994). Surprisingly, although jasplakinolide and latrunculin A preserve ATP and so should enhance the ability of Na+–K+-ATPase to extrude Na+ and minimize its intracellular accumulation, they instead exacerbate [Na+]iaccumulation. A possible explanation for this observation is that modulation of the actin cytoskeleton inhibits Na+–K+-ATPase. However, this appears not to be the case, because cells incubated in ouabain and jasplakinolide show an additive reduction in ATP depletion. This finding suggests that ATP preservation resulting from actin modulation does not occur via inhibition of the Na+–K+-ATPase but rather via attenuation of actin-ATP hydrolysis per se. Actin-ATP hydrolysis may account for <50% of the total ATP consumed by mature neurons for two reasons. First, Na+–K+-ATPase immunostaining, and perhaps activity, increases in rat hippocampus during the first 5 postnatal weeks (Fukuda and Prince, 1992). Second, actin turnover may be faster in growth cones, in which assembly–disassembly underlies motility, than in mature terminals; our cultures contained both growth cones and terminals.
Actin assembly modulators probably amplify the ischemic Na+ overload by a mechanism involving the Na+–Ca2+exchanger. This regulator normally helps to maintain the steep Ca2+ gradient of healthy cells by extruding Ca2+, but when the Na+ load becomes extreme, as it does during ischemia or even tetanic stimulation (Zhong et al., 2001), the exchanger reverses and extrudes Na+. Apparently, the actin modulators maintain sufficiently high ATP levels to prevent attainment of [Na+]i that reverses the Na+–Ca2+exchanger. Hence, cells treated with actin modulators continue to extrude Ca2+ in exchange for Na+ influx, resulting in elevated [Na+]i and somewhat reduced [Ca2+]i relative to control ischemic cells.
Jasplakinolide slows filament turnover by reducing the subunit off-rate from the naked F-actin pointed end (Bubb et al., 2000). If any non-actin-related effects of jasplakinolide occur, they have been minimized in this study by using the lowest concentration necessary for filament stabilization (10 nm). In addition to the already cited off-rate effect on naked filaments, jasplakinolide also slows turnover by competing with ADF for filament binding (Chen, 2001). ADF and cofilin are the major enhancers of actin filament dynamics in most cells; they both sever filaments and promote disassembly (Bamburg, 1999). It is possible that jasplakinolide preserves ATP by stabilizing ATP bound to filament subunits rather than by reducing the release rate of subunits. However, this mechanism would also support our conclusion that the hydrolysis of ATP associated with actin is a process that consumes a major fraction of the total energy of the cell.
Footnotes
This work was supported in part by March of Dimes Birth Defects Foundation Research Grant 6-FY99-627 (B.W.B.), by National Institutes of Health Grants GM-35126 and NS-40371, and by Alzheimer's Association Grant IIRG-01-2730 (J.R.B.).
Correspondence should be addressed to Barbara W. Bernstein, Department of Biochemistry and Molecular Biology, 1870 Campus Delivery, Colorado State University, Fort Collins, CO 80523-1870. E-mail:bwb@lamar.colostate.edu.
References
- 1.Alberts B, Bray D, Raff M, Roberts K, Watson JD. Molecular biology of the cell, pp 41–88. Garland; London: 1994. [Google Scholar]
- 2.Aliev G, Smith MA, Seyidov D, Neal ML, Lamb BT, Nunomura A, Gasimov EK, Vinters HV, Perry G, LaManna JC, Friedland RP. The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer's disease. Brian Pathol. 2002;12:21–35. doi: 10.1111/j.1750-3639.2002.tb00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 5.Belmont LD, Drubin DG. The yeast V159N actin mutant reveals roles for actin dynamics in vivo. J Cell Biol. 1998;142:1289–1299. doi: 10.1083/jcb.142.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belmont LD, Patterson GM, Drubin DG. New actin mutants allow further characterization of the nucleotide binding cleft and drug binding sites. J Cell Sci. 1999;112:1325–1336. doi: 10.1242/jcs.112.9.1325. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein BW, DeWit M, Bamburg JR. Actin disassembles reversibly during electrically induced recycling of synaptic vesicles in cultured neurons. Mol Brain Res. 1998;53:236–250. doi: 10.1016/s0169-328x(97)00319-7. [DOI] [PubMed] [Google Scholar]
- 8.Bubb MR, Spector I, Beyer BB, Fosen KM. Effects of jasplakinolide on the kinetics of actin polymerization: an explanation for certain in vivo observations. J Biol Chem. 2000;275:5163–5170. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- 9.Budinger GR, Duranteau J, Chandel NS, Schumacker PT. Hibernation during hypoxia in cardiomyocytes: role of mitochondria as the O2 sensor. J Biol Chem. 1998;273:3320–3326. doi: 10.1074/jbc.273.6.3320. [DOI] [PubMed] [Google Scholar]
- 10.Cantiello HF. Actin filaments stimulate the Na(+)-K(+)-ATPase. Am J Physiol. 1995;269:F637–F643. doi: 10.1152/ajprenal.1995.269.5.F637. [DOI] [PubMed] [Google Scholar]
- 11.Chen H. PhD thesis. Colorado State University; 2001. In vitro analysis of the functional differences between proteins of the ADF/cofilin family. [Google Scholar]
- 12.Cochilla AJ, Angleson JK, Betz WJ. Monitoring secretory membrane with FM1–43 fluorescence. Annu Rev Neurosci. 1999;22:1–10. doi: 10.1146/annurev.neuro.22.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Cramer LP. Role of actin-filament disassembly in lamellipodium protrusion in motile cells revealed using the drug jasplakinolide. Curr Biol. 1999;9:1095–1105. doi: 10.1016/s0960-9822(99)80478-3. [DOI] [PubMed] [Google Scholar]
- 14.Daniel JL, Molish IR, Robkin L, Holmsen H. Nucleotide exchange between cytosolic ATP and F-actin-bound ADP may be a major energy-utilizing process in unstimulated platelets. Eur J Biochem. 1986;156:677–684. doi: 10.1111/j.1432-1033.1986.tb09631.x. [DOI] [PubMed] [Google Scholar]
- 15.Didry D, Carlier MF, Pantaloni D. Synergy between actin depolymerizing factor/cofilin and profilin in increasing actin filament turnover. J Biol Chem. 1998;273:25602–25611. doi: 10.1074/jbc.273.40.25602. [DOI] [PubMed] [Google Scholar]
- 16.Endres M, Fink K, Zhu J, Stagliano NE, Bondada V, Geddes JW, Azuma T, Mattson MP, Kwiatkowski DJ, Moskowitz MA. Neuroprotective effects of gelsolin during murine stroke. J Clin Invest. 1999;103:347–354. doi: 10.1172/JCI4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fath KR, Lasek RJ. Two classes of actin microfilaments are associated with the inner cytoskeleton of axons. J Cell Biol. 1988;107:613–621. doi: 10.1083/jcb.107.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 19.Forscher P, Smith SJ. Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. J Cell Biol. 1988;107:1505–1516. doi: 10.1083/jcb.107.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman JE, Haddad GG. Removal of extracellular sodium prevents anoxia-induced injury in freshly dissociated rat CA1 hippocampal neurons. Brain Res. 1994;641:57–64. doi: 10.1016/0006-8993(94)91815-5. [DOI] [PubMed] [Google Scholar]
- 21.Fukuda A, Prince DA. Postnatal development of electrogenic sodium pump activity in rat hippocampal pyramidal neurons. Brain Res Dev Brain Res. 1992;65:101–114. doi: 10.1016/0165-3806(92)90013-m. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa K, Fu WM, Li Y, Witke W, Kwiatkowski DJ, Mattson MP. The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability to excitotoxicity in hippocampal neurons. J Neurosci. 1997;17:8178–8186. doi: 10.1523/JNEUROSCI.17-21-08178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goddette DW, Frieden C. Actin polymerization: the mechanism of action of cytochalasin D. J Biol Chem. 1986;261:15974–15980. [PubMed] [Google Scholar]
- 24.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 25.Gupta RK, Gupta P, Yushok WD, Rose ZB. Measurement of the dissociation constant of MgATP at physiological nucleotide levels by a combination of 31P NMR and optical absorbance spectroscopy. Biochem Biophys Res Commun. 1983;117:210–216. doi: 10.1016/0006-291x(83)91562-0. [DOI] [PubMed] [Google Scholar]
- 26.Haugland RP. Handbook of fluorescent probes and research chemicals, p 528. Molecular Probes; Eugene, OR: 1996. [Google Scholar]
- 27.Jha MN, Bamburg JR, Bernstein BW, Bedford JS. Caffeine eliminates gamma-ray-induced g(2)-phase delay in human tumor cells but not in normal cells. Radiat Res. 2002;157:26–31. doi: 10.1667/0033-7587(2002)157[0026:cegrig]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 28.Kabsch W, Vandekerckhove J. Structure and function of actin. Annu Rev Biophys Biomol Struct. 1992;21:49–76. doi: 10.1146/annurev.bb.21.060192.000405. [DOI] [PubMed] [Google Scholar]
- 29.Kimelberg HK, Bowman C, Biddlecome S, Bourke RS. Cation transport and membrane potential properties of primary astroglial cultures from neonatal rat brains. Brain Res. 1979;177:533–550. doi: 10.1016/0006-8993(79)90470-0. [DOI] [PubMed] [Google Scholar]
- 30.Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J Physiol (Lond) 1996;496:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magistretti PJ. Brain energy metabolism. In: Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire L, editors. Fundamental neuroscience. Academic; San Diego: 1999. p. 389. [Google Scholar]
- 32.Mills JW, Schwiebert EM, Stanton BA. The cytoskeleton and membrane transport. Curr Opin Nephrol Hypertens. 1994;3:529–534. doi: 10.1097/00041552-199409000-00009. [DOI] [PubMed] [Google Scholar]
- 33.Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- 34.Moraczewska J, Wawro B, Seguro K, Strzelecka-Golaszewska H. Divalent cation-, nucleotide-, and polymerization-dependent changes in the conformation of subdomain 2 of actin. Biophys J. 1999;77:373–385. doi: 10.1016/S0006-3495(99)76896-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morton WM, Ayscough KR, McLaughlin PJ. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat Cell Biol. 2000;2:376–378. doi: 10.1038/35014075. [DOI] [PubMed] [Google Scholar]
- 36.Sokoloff L. The metabolism of the central nervous system in vivo. In: Field J, Magoun HW, Hall VE, editors. Handbook of physiology-neurophysiology. American Physiological Society; Washington, DC: 1996. pp. 1843–1864. [Google Scholar]
- 37.Spector I, Braet F, Shochet NR, Bubb MR. New anti-actin drugs in the study of the organization and function of the actin cytoskeleton. Microsc Res Tech. 1999;47:18–37. doi: 10.1002/(SICI)1097-0029(19991001)47:1<18::AID-JEMT3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 38.Zhong N, Beaumont V, Zucker RS. Roles for mitochondrial and reverse mode Na+/Ca2+ exchange and the plasmalemma Ca2+ ATPase in post-tetanic potentiation at crayfish neuromuscular junctions. J Neurosci. 2001;21:9598–9607. doi: 10.1523/JNEUROSCI.21-24-09598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zigmond SH. Recent quantitative studies of actin filament turnover during cell locomotion. Cell Motil Cytoskel. 1993;25:309–316. doi: 10.1002/cm.970250402. [DOI] [PubMed] [Google Scholar]