

Abstract
Na,K-ATPase plays a critical role in energy metabolism and ion fluxes. Its loss was investigated in the G93A mouse model of amyotrophic lateral sclerosis (ALS) in which the mutation of Cu/Zn superoxide dismutase (SOD1) is thought to lead to aberrant oxidative damage. Observed losses in spinal cord Na,K-ATPase activity exceeded all expectations. All three catalytic subunit isoforms (α1, α2, α3) were reduced, and the global α subunit loss affected not just neurons, glia, and myelinated axon tracts but even ependymal and pial membranes. Decreases in Na,K-ATPase activity were greater than losses of protein, and there were losses of Na,K-ATPase α, but not β, subunits. Together, these observations are consistent with selective degradation of the α subunit after damage. Overexpression of normal SOD1 does not cause ALS-like symptoms, but it has other known pathological effects. In transgenic mice overexpressed normal human SOD1 had a smaller but still considerable effect on Na,K-ATPase. Furthermore, the nitric oxide-mediated regulatory pathway for Na,K-ATPase inhibition was undetectable in spinal cord tissue slices from mice overexpressing either mutant or normal human SOD1. Na,K-ATPase activity did not respond to nitric oxide donors, and the free radical-dependent step of the pathway could not be bypassed by the addition of the downstream protein kinase G activator, 8-Br-cGMP. The data demonstrate that Na,K-ATPase is vulnerable to aberrant SOD1 activity, making it a potential contributing factor in disease pathology. Moreover, the global cellular distribution of Na,K-ATPase loss indicates that SOD1 overexpression is far-reaching in its pathological effects.
Keywords: Na,K-ATPase; SOD1; amyotrophic lateral sclerosis; neurodegeneration; spinal cord; nitric oxide
Introduction
Amyotrophic lateral sclerosis (ALS) is an age-dependent motor neuron disease. Certain familial ALS cases are inherited as an autosomal dominant trait with mutations in cytosolic Cu/Zn superoxide dismutase 1 (SOD1) (Rosen et al., 1993;Brown, 1995). SOD1 normally converts superoxide, a by-product of mitochondrial metabolism, to water and hydrogen peroxide. Simple loss of SOD1 activity has been ruled out as a cause of the disease. Instead, there is evidence for “gain of toxic function,” such as increases in copper-, free radical-, or oxidative damage, which is not alleviated by increases or decreases in the level of normal SOD1 activity (Cleveland and Rothstein, 2001; Julien, 2001).
The Na,K-ATPase consumes 50% of the energy supply in the CNS (Ames, 2000). Its catalytic (α) subunit is sensitive to damage by free radicals and other oxidative stress (Kim and Akera, 1987; Xie et al., 1995; Mense et al., 1997), and the oxidized Na,K-ATPase α subunit can be degraded by calpain, proteosomal, and lysosomal pathways (Zolotarjova et al., 1994; Thevenod and Friedmann, 1999). It thus may be one of the links between alterations in free radical homeostasis and ALS pathology. In other circumstances the inhibition of Na,K-ATPase increases the sensitivity of neurons to glutamate excitotoxicity because of complementary effects on neurons (enhancing glutamate effects and Ca2+ accumulation) and astrocytes (reducing the driving force for Na+-dependent glutamate clearance) (Lees et al., 1990; Brines and Robbins, 1992; Brines et al., 1995; Calabresi et al., 1995; Lees and Leong, 1996; Stelmashook et al., 1999). Furthermore, the free radical nitric oxide (NO·) normally regulates the Na,K-ATPase via the activation of soluble guanylate cyclase and cGMP (McKee et al., 1994), a pathway that is shared by glutamate and oxygen free radicals in the CNS. This pathway forms a convergence point for the action of several intercellular and intracellular molecular messengers that have been implicated in neuronal viability under stress (Dawson et al., 1991; Nathanson et al., 1995). Together, these suggest that either loss or excessive inhibition of Na,K-ATPase could contribute to motor neuron death via direct oxidative damage or via the enhancement of NO· and other free radical effects.
The Na,K-ATPase has two required subunits, α and β. There are three α isoforms (α1, α2, and α3) and three β isoforms (β1, β2, and β3) in the CNS (Sweadner, 1989; Blanco and Mercer, 1998), which have different kinetic properties and are likely to be regulated differently (Blanco and Mercer, 1998; Crambert et al., 2000). Although there are many exceptions to the rule, neurons have predominantly α3β1 and astrocytes have predominantly α2β2, whereas both neurons and glia can express α1.
To test the hypothesis that Na,K-ATPase loss occurs in a mouse model of ALS and in SOD1 overexpression, we used transgenic mice that express either human SOD1 with the G93A missense mutation or normal human SOD1 over and above normal mouse SOD1 (Gurney et al., 1994).
Materials and Methods
Reagents. Routine reagents, sodium nitroprusside (SNP), superoxide dismutase, and ouabain were purchased from Sigma (St. Louis, MO).1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), diethylenetriamine NO (DETA-NO), 8-Br-cGMP, and okadaic acid were obtained from Sigma-RBI (Natick, MA). The cGMP enzyme immunoassay system was purchased from Amersham Biosciences (Piscataway, NJ). Cy3-conjugated goat-anti mouse IgG was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). The Puregene DNA Isolation Kit was purchased from Gentra (Minneapolis, MN).
Transgenic SOD1 mice. The mice were obtained from Dr. Robert H. Brown Jr. (Massachusetts General Hospital, Charlestown, MA) or The Jackson Laboratory (Bar Harbor, ME) and were strains originally developed by Dr. Mark Gurney (Gurney et al., 1994). The G1H strain of G93A mice carries high copy numbers of human SOD1, with a missense mutation substituting glycine with alanine at codon 93, and develops ALS-like symptoms between 3 and 4 months of age. This strain is maintained as a hemizygote hybrid line, progeny of a cross between C57BL/6J and SJL mice. Transgenic males were crossed with nontransgenic B6SJL-F1 females. Animals were genotyped by purifying mouse tail DNA with a Puregene DNA Isolation Kit; PCR products were separated on a 2% agarose gel. The controls were transgenic normal human SOD1 overexpressors of the B5SJL strain and nontransgenic littermates.
Na,K-ATPase activity and cGMP measurements. Whole spinal cord tissue was dissected, and tissue slices (0.4 × 0.4 × 1 mm) were prepared on a Brinkmann chopper cooled to 4°C and suspended (25–30 mg/ml wet weight) in microdissection buffer containing (in mm): 137 NaCl, 5 KCl, 0.8 MgSO4, 0.25 CaCl2, 1.0 MgCl2, 10 HEPES, and 2 NaOH, pH-adjusted to 7.4 at 34°C.
SNP, DETA-NO, or 8-Br-cGMP, when used, was added to tubes that contained 1 ml aliquots of slice suspension. Tubes were incubated for 15 min at 34°C with rocking and then frozen at –80°C. In studies that used ODQ or okadaic acid, the inhibitor was added 3 min before the addition of the other drug. Tubes were thawed and centrifuged (1700 × g for 15 min at 4°C), and the supernatant was removed and assayed for cGMP by using an enzyme immunoassay according to the manufacturer's instructions. The tissue slice pellets were resuspended in resuspension buffer containing (in mm): 85 NaCl, 20 KCl, 4 MgCl2, 0.2 EGTA, and 30 histidine, pH-adjusted to 7.2.
Two similar ATPase assays were used. For most experiments with tissue from 4-month-old animals the Na,K-ATPase activity was determined by using the pyruvate kinase/lactate dehydrogenase assay that couples the generation of ADP and oxidation of NADH as described previously (Ellis et al., 2000). Treated tissue slices were homogenized with a ground glass homogenizer. Na,K-ATPase activity was calculated from the difference between the slopes in the time course of absorption change at 340 nm in the absence and the presence of 3 mmouabain. For most experiments with tissue from 2-month-old animals the activity was determined by the colorimetric ATPase assay: ATP was hydrolyzed, and the released Pi was measured by forming a complex with molybdate. The pelleted tissue slices were resuspended and refrozen for at least 20 min at –80°C in 1 ml of resuspension buffer. Tubes were thawed on ice water. For further permeabilization saponin (20 μg/ml) was added, and the slices were incubated for 10 min at 34°C. Aliquots of tissue slices (∼10–15 μg; 7.5–10 μl) were added to 300 μl of ATPase buffer containing (in mm): 3 ATP, 140 NaCl, 20 KCl, 3 MgCl2, and 30 histidine, pH 7.2, with or without 3 mm ouabain. In this and previous work we verified the equivalence of the two assays that were used (Ellis et al., 2000).
Immunoblots. Spinal cord tissue was dissected from transgenic mutant SOD1 mice, transgenic normal human SOD1 overexpressors, and nontransgenic littermate controls and then homogenized at 4°C in microdissection buffer. Protein concentrations were determined by the Lowry method, and samples (50 μg of protein) were separated by gel electrophoresis on Laemmli gels and transferred to nitrocellulose membrane electrophoretically. For detection of Na,K-ATPase subunits the isoform-specific monoclonal antibodies used were 6F (for α1; Developmental Studies Hybridoma Bank, Iowa City, IA), McB2 (for α2), and XVI-F9G10 (for α3; Affinity BioReagents, Neshanic Station, NJ), all of which bind in the first 60 residues of the respective α subunits; rabbit polyclonal antibodies SpETb1 and SpETb2 (gift of P. Martin-Vasallo, University of Tenerife, Spain) were used for β1 and β2, respectively. Anti-KETYY against the α C terminus (gift of Dr. J. Kyte, University of California, San Diego, CA) was used to detect all α isoforms together. Anti-superoxide dismutase (Cu/Zn) polyclonal antibody specific to human SOD1 (Calbiochem-Novabiochem, San Diego, CA) was used to quantify the level of overexpressed enzyme. Blots subsequently were stained with horseradish peroxidase-conjugated secondary antibody, developed with luminol reagent, and quantified with a Molecular Dynamics scanning densitometer (Sunnyvale, CA).
Immunofluorescence. Nontransgenic controls and transgenic mutant SOD1 mice were anesthetized and perfused with PBS, followed by periodate-lysine-paraformaldehyde fixative for 20 min (McLean and Nakane, 1974). The spinal cords encased in the vertebral columns were harvested and postfixed for 48 hr. The spinal cords were removed and washed in PBS overnight and then soaked in 25% sucrose in 0.1 m PBS for 24 hr before sectioning. Cryostat sections (10–14 μm) were cut at −20°C and collected on positively charged slides (ProbeOn Plus, Fisher Scientific, Durham, NC). Slides were washed in 0.1 m PBS and incubated in blocking buffer containing 0.1% Triton X-100 and 5% normal goat serum in PBS. Tissue sections were incubated with primary monoclonal antibodies for the α subunits described above, washed, and stained with secondary antibody Cy3-conjugated goat-anti mouse IgG. For β1, monoclonal antibody BSP-3 was used, a gift of Dr. C. Goridis (INSERM-CNRS, Marseille Luminy, France). For β2, monoclonal antibody 426 was used, a gift of Dr. Melitta Schachner (University of Hamburg, Germany). The images were viewed with a Zeiss confocal microscope (Oberkochen, Germany).
Statistics. Statistical comparisons were performed by ANOVA, followed by Fisher's protected least significant difference (PLSD) and Scheffé's F test for comparison of significant difference among different means.
Results
Decreased ouabain-sensitive Na,K-ATPase activity in transgenic mutant SOD1 mice
Ouabain-sensitive Na,K-ATPase activity was measured in spinal cord tissue slice homogenates of transgenic mice expressing mutant or normal human SOD1 and wild-type controls. Because the assay is performed in vitro with saturating levels of ATP and ions, the measured activity reflects the maximal velocity of the enzyme itself and not ATP depletion secondary to mitochondrial damage in vivo. Figure 1Ashows that Na,K-ATPase activity was decreased remarkably (70–75%) in the severely impaired 4-month-old mutant SOD1 mice compared with nontransgenic animals. To determine whether the decreases were a result of the mutation or of overexpression of SOD1, we measured activity in samples from transgenic mice overexpressing normal human SOD1. There was a large but less dramatic decrease (40–45%) in ouabain-sensitive Na,K-ATPase activity in the overexpressors. Immunoblot and densitometric analysis with a species-specific antibody for human SOD1 showed that the total level of exogenous SOD1, compared with a sample of purified enzyme, was not statistically different in mice overexpressing mutant and normal human SOD1 (Fig.1B,C). The greater Na,K-ATPase activity loss in the mutant is consistent with “gain of function” consequences of excess SOD1 that are exacerbated by the mutation.
Fig. 1.
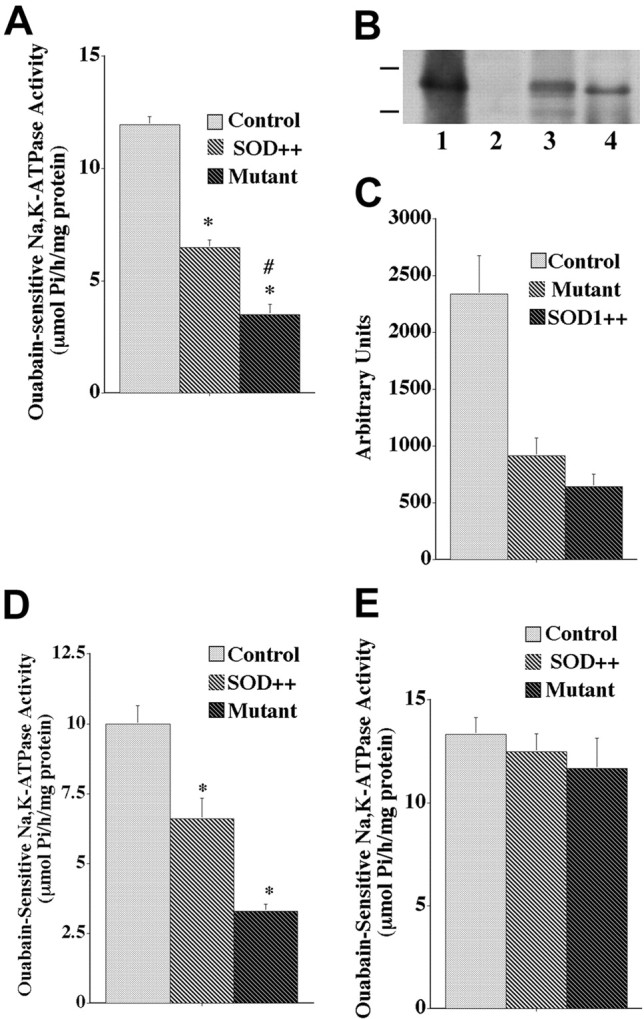
Ouabain-sensitive Na,K-ATPase activity in spinal cord and cerebellum of nontransgenic controls (Control), transgenic normal human SOD1 overexpressors (SOD++), and transgenic SOD1 mice (Mutant), and expression of normal and mutant human SOD1. For A and D, activity is expressed as μmol Pi/hr per milligram of protein.A, Spinal cord tissue slices from 4-month-old animals were homogenized, and ouabain-sensitive Na,K-dependent hydrolysis of ATP was determined. Values for activity represent the means ± SEM for an average of three samples in five experiments. *Significantly different from control at p < 0.05 (by ANOVA, Fisher's PLSD, and Scheffé's F test).#Significantly different from transgenic normal human SOD1 overexpressors at p < 0.05 (by ANOVA and Fisher's PLSD). B, Immunoblot detection of human SOD1 in nontransgenic controls (lane 2), transgenic mutant SOD1 mice (lane 3), normal human SOD1 overexpressors (lane 4), and purified human SOD1 as a positive gel control (lane 1). C, Densitometric analysis of human SOD1 expression levels in transgenic mutant SOD1 mice (Mutant), transgenic normal human SOD1 overexpressors (SOD++), and purified human SOD1 as a positive gel control. Values are expressed as arbitrary units and represent the means ± SEM for an average of three experiments.D, Na,K-ATPase activity was measured from cerebellar tissue slices from 4-month-old animals. Values represent the means ± SEM for an average of three samples in six experiments. *Significantly different from control at p < 0.05 (by ANOVA, Fisher's PLSD, and Scheffé's F test).E, Na,K-ATPase activity was measured in spinal cord tissue slice preparations from 2-month-old animals. Values for ouabain-sensitive Na,K-ATPase activity represent the means ± SEM for an average of three samples in three experiments.
Cerebellum is not implicated in neurodegenerative processes in this ALS mouse model (Almer et al., 1999), but comparable losses in Na,K-ATPase activity also were seen in cerebellum in 4-month-old animals (Fig.1D). In agreement with Almer et al. (1999), we did not see any indication of major neuron loss in cerebellar tissue sections (data not shown). This is an initial indication that the losses of activity were not attributable simply to a loss of neurons. Figure 1E shows that there were no significant reductions in Na,K-ATPase activity in spinal cord of transgenic mutant mice or transgenic overexpressors at 2 months of age, before the onset of obvious neurological symptoms.
Decreased Na,K-ATPase α subunits in transgenic mutant SOD1 mice
Losses in Na,K-ATPase activity could be attributable to enzyme inactivation, protein degradation, changes in gene expression, failure to transport newly synthesized protein to the axon, loss of neurons, or a combination of these. Quantitative detection of Na,K-ATPase subunits makes it possible to assess whether protein levels were changed as much as activity. It also allows some conclusions to be made about whether the effect is on a particular Na,K-ATPase isoform or a particular cell type, because the α3 subunit is all in neurons and the myelinated axon tracts, whereas most of α2 is in astrocytes (Hieber et al., 1991; McGrail et al., 1991; Watts et al., 1991; Peng et al., 1997). The relative content of Na,K-ATPase α1, α2, and α3 subunits was determined in homogenates of spinal cord of transgenic mutant SOD1 mice, transgenic normal human SOD1 overexpressors, and nontransgenic controls by using isoform-specific monoclonal antibodies. Immunoblots (Fig. 2A) and densitometric analysis (Fig. 2B) showed that there were decreases in α1 (65%), α2 (50%), and α3 (50%) in transgenic mutant SOD1 mice compared with nontransgenic controls. Although there were also decreases in the three α isoforms in transgenic normal human SOD1 overexpressors, the decreases were smaller compared with transgenic mutant mice (Fig. 2B). Total Na,K-ATPase α subunit level was quantified with a polyclonal antibody, anti-KETYY, that recognizes all Na,K-ATPase isoforms. Figure2, C and D, shows that there were close to 50% decreases in total α protein in transgenic mutant SOD1 mice when compared with nontransgenic controls and 20–25% decreases in normal SOD1 overexpressors. Isoform-specific antibodies for the β1 and β2 Na,K-ATPase subunits were used to examine β levels. In contrast to the α subunit, no differences were detected in β1 (mostly in neurons) or β2 (mostly in astrocytes) in spinal cord samples from transgenic mutant SOD1, transgenic normal human SOD1 overexpressors, or nontransgenic control animals (Fig. 2E). β3, which is found only in oligodendrocytes and at a low level (Martin-Vasallo et al., 2000), was not examined.
Fig. 2.

Immunoblot detection and quantitative analysis of Na,K-ATPase isoforms in nontransgenic controls (C, control), transgenic mutant SOD1 mice (M, mutant) and transgenic normal human SOD1 overexpressors (O, SOD++).A, B, Immunoblot and densitometric analysis of α1, α2, α3. For all immunoblots 50 μg of spinal cord homogenates was used. A, Separated protein was stained with monoclonal antibodies to α1 (6F), α2 (McB2), and α3 (XVIF9G10). Crude homogenates from rat brain served as positive controls for the antibodies (data not shown). Molecular weight markers are shown in kDa.B, Data are expressed as arbitrary units and represent the means ± SEM for an average of three experiments.C, Immunoblot stained with KETYY, an antibody that recognizes all Na,K-ATPase α subunits. D, Densitometric analysis of KETYY expressed in arbitrary units and representing the means ± SEM for an average of three experiments.E, Spinal cord homogenates were stained for β1 (SpETb1) and β2 (SpETb2) polyclonal antibodies. The data that are shown are representative of multiple experiments.
The reduction in Na,K-ATPase activity (Fig. 1A) exceeded the reduction in total Na,K-ATPase α subunit (Fig.2D) in both mutant SOD1 mice and normal human SOD1 overexpressors by a substantial amount. Both measures were expressed per milligram of protein, and so the losses were over and above any generalized loss of tissue mass, which does occur with the loss of motor neurons. The greater loss of ATP hydrolysis than α subunit indicates that perturbations of SOD1 activity have an acute effect on Na,K-ATPase apart from any effect on gene expression, tissue or axon atrophy, or cell loss.
Na,K-ATPase isoform distribution in normal and mutant spinal cord
The distribution of the various Na,K-ATPase α subunits was determined by using confocal immunofluorescence with isoform-specific monoclonal antibodies. In the dorsal horn of the rat spinal cord some large neurons express both α2 and α3, whereas in the ventral horn some motor neurons express α1 and α3 and the rest just α3 (Mata et al., 1991; McGrail et al., 1991; Watts et al., 1991). In the mice that were investigated here, the pattern of staining for each α isoform was identical in sections from all levels of the spinal cord (cervical, thoracic, lumbar, and sacral; data not shown) except for the underlying structural differences in dorsal and ventral horns and axon tracts at different levels. Figure 3shows immunostaining of tissue sections from the lumbar expansion of the spinal cord in 4-month-old nontransgenic controls and transgenic mutant SOD1 mice. In keeping with the plasma membrane location of the Na,K-ATPase, the cytoplasm of large-diameter neuronal cell bodies was unstained, although there were α3 ring-stained cell bodies in the central and lower regions of the dorsal horn. α2 stain characteristic of astrocytes was brightest in the gray matter, but it extended outward into the myelinated tracts accompanying radiating bundles of axons. It was notable that there were differences in the distribution of Na,K-ATPase isoforms in wild-type mouse spinal cord that have not been described before. The most striking was an apparent complementary difference in predominant α isoforms between dorsal and ventral horn. The parenchyma of dorsal horn stained most brightly for α1, whereas ventral horn stained most brightly for α3. The complementary difference also was observed for axons in the dorsal columns. The dorsal one-half of the dorsal column, the gracile fascicle that consists of ascending sensory axons, was enriched in α1, and the ventral one-half of the dorsal column, a descending corticospinal tract, was enriched in α3 as well as α1. These and the lateral α3-containing axon tracts are presumably the origin of the unidentified α1- versus α3-containing myelinated tracts observed previously in the rat medulla (McGrail et al., 1991). In addition, the endothelial cells that line the central canal were stained brightly for α1, but not for α2 or α3, whereas the ensheathing pial membranes and fragments of dura mater stained for all three isoforms.
Fig. 3.
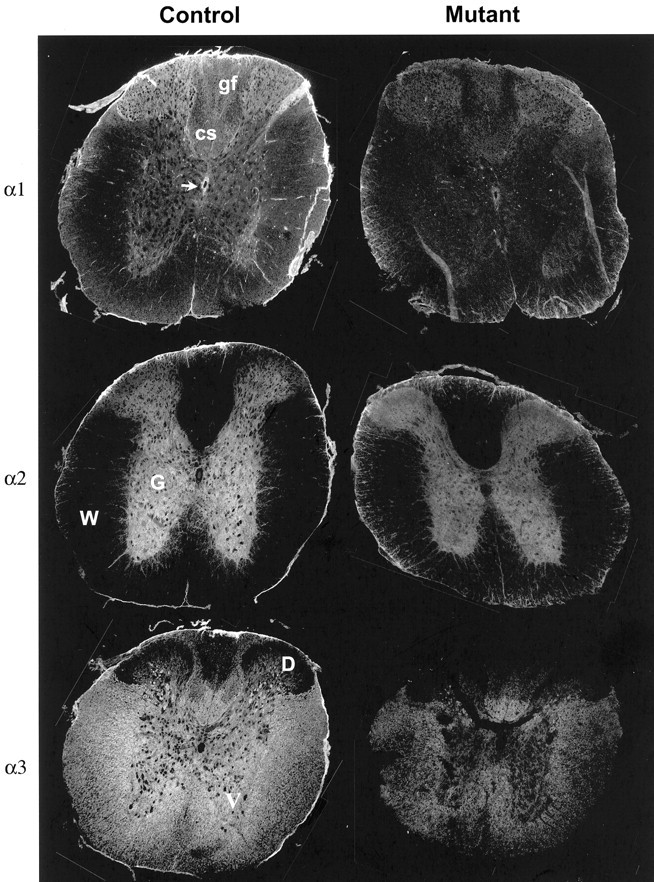
Immunofluorescence detection of Na,K-ATPase isoforms in nontransgenic controls and transgenic mutant SOD1 mice. Tissue sections were labeled with monoclonal antibodies to α1 (6F), α2 (McB2), and α3 (XVIF9G10). Images were taken at 10×, and montages were made to display the entire section. D, Dorsal horn; G, gray matter; W, white matter; V, ventral horn; gf, gracile fascicle of ascending sensory axons; cs, corticospinal tract. The arrow points to the ependyma lining the central canal.
When they were compared with nontransgenic controls, there were markedly lower levels of staining for all three Na,K-ATPase α isoforms in transgenic mutant SOD1 animals (Fig. 3). If losses in Na,K-ATPase had been attributable to specific cell loss or to the atrophy of axons, the remaining cells would have more normal stain intensity. The uniformity of the loss of stain was the most notable feature, extending even to the ependymal lining of the central canal. The pial membranes, which appeared somewhat disrupted in the mutant mice, penetrated the white matter tracts more than they should, and their stain for α3 virtually was abolished.
In contrast, and consistent with the immunoblots shown above, staining patterns for β1 and β2 subunits were unaltered in the transgenic mutant SOD1 mice (Fig. 4). The principal difference that was seen was in the diffusely shrunken appearance of the pathological spinal cord. A lack of effect on β expression suggests a lack of effect on Na,K-ATPase expression and biosynthesis, as discussed below.
Fig. 4.
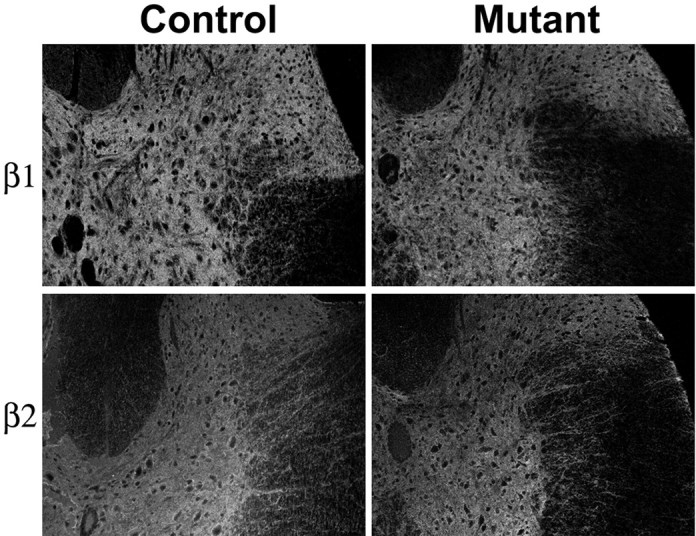
Immunodetection of Na,K-ATPase β isoforms in nontransgenic controls and transgenic mutant SOD1 mice. Tissue sections were labeled with monoclonal antibodies to β1 (BSP-3) and β2 (426).
NO is unable to regulate Na,K-ATPase activity in spinal cord of transgenic mice
In some tissues NO is a normal Na,K-ATPase regulator acting through cGMP. Because free radical homeostasis appears to be perturbed in some forms of ALS, we tested whether regulation of the Na,K-ATPase was affected in this model. Spinal cord tissue slices from nontransgenic control mice were exposed to the NO donors SNP (Garthwaite et al., 1995) and DETA-NO (Diodati et al., 1993) for 15 min. Both donors caused a marked reduction (35–45%) of ouabain-sensitive Na,K-ATPase activity (Fig.5A). The effects were specific to Na,K-ATPase, because no measurable changes were observed in the ouabain-insensitive (Mg-ATPase) activity (data not shown). Many of the physiological actions of NO on Na,K-ATPase activity involve activation of soluble guanylate cyclase (McKee et al., 1994; Nathanson et al., 1995; Scavone et al., 1995; Ellis et al., 2000, 2001). When SNP- or DETA-NO-treated spinal cord tissue slices were exposed to ODQ, an inhibitor selective for soluble guanylate cyclase (Garthwaite et al., 1995), it primarily blocked the SNP- and DETA-NO-induced inhibition of ouabain-sensitive Na,K-ATPase activity (Fig. 5A).
Fig. 5.

SNP- and DETA-NO-induced inhibition of ouabain-sensitive Na,K-ATPase was abolished in transgenic mutant SOD1 mice and transgenic normal human SOD1 overexpressors. Spinal cord tissue slices were incubated with or without ODQ (1 μm) for 3 min at 34°C, followed by incubation with DETA-NO (100 μm) or SNP (100 μm) for 15 min at 34°C. Drugs were removed, tissue slices were homogenized, and ouabain-sensitive Na,K-ATPase activity was measured. For all graphs that are shown, activity is expressed as μmol Pi/hr per milligram of protein, and values represent the means ± SEM for experiments on three animals done in triplicate. A, Ouabain-sensitive Na,K-ATPase activity in nontransgenic controls. *Significantly different from the control atp < 0.05 (by ANOVA and Fisher's PLSD).##Significantly different from DETA-NO- and SNP-treated samples at p < 0.05 (by ANOVA and Fisher's PLSD).B, C, Ouabain-sensitive Na,K-ATPase activity in transgenic mutant SOD1 and transgenic SOD1 normal human SOD1 overexpressors.
The effects of NO donors on activity in tissue slices from spinal cord of transgenic mutant SOD1 mice and of transgenic normal human SOD1 overexpressors are shown in Figure 5, B and C. The basal Na,K-ATPase activity was low, and neither SNP nor DETA-NO further inhibited it. The addition of ODQ in the presence of SNP or DETA-NO, or alone, failed to alter ouabain-sensitive Na,K-ATPase activity in spinal cord tissue slices of transgenic mutant SOD1 mice (Fig. 5B) or transgenic normal human SOD1 overexpressors (Fig. 5C). Because ODQ treatment did not restore the lost activity, the data argue against a high basal level of otherwise-normal NO-mediated regulation as the cause of Na,K-ATPase inhibition in the transgenic mice. These results indicate a surprisingly complete perturbation, in normal SOD1 overexpressors as well as mutants, of a regulatory pathway that in physiological conditions depends on the diffusion of NO from its site of synthesis to soluble guanylate cyclase.
The ability of NO donors to increase levels of cGMP by activating soluble guanylate cyclase was tested in the three groups of mice. In nontransgenic control mice the addition of DETA-NO to spinal cord tissue slices caused a 40% increase in cGMP levels that, as expected, was abolished by the guanylate cyclase inhibitor ODQ (Fig.6A). In contrast, there were no measurable changes in cGMP levels in either transgenic mutant SOD1 mice or transgenic normal human SOD1 overexpressors treated with DETA-NO, ODQ, or DETA-NO plus ODQ (Fig. 6B,C). This could mean that soluble guanylate cyclase, like Na,K-ATPase, is inactivated in these mice, possibly targeted by its specific binding site for a free radical, although such losses were not reported in the G1L strain of G93A mice, which expresses mutant SOD1 at a lower level (Facchinetti et al., 1999). Alternatively, the NO generated by the artificial donors could be consumed rapidly in reactions catalyzed by the aberrant elevated levels of SOD1.
Fig. 6.

cGMP levels are unaltered in transgenic SOD1 mice in response to DETA-NO treatment. Spinal cord tissue slices from nontransgenic controls (A), transgenic mutant SOD1 mice (B), or transgenic normal human SOD1 overexpressors (C) were incubated for 3 min at 34°C with ODQ (1 μm), followed by incubation for 15 min at 34°C with DETA-NO (100 μm). After centrifugation the supernatant was removed and assayed for cGMP, expressed as pmol/mg protein. *Significantly different from the control group atp < 0.05 (by ANOVA, Fisher's PLSD, and Scheffé's F test). Values for cGMP levels represent the means ± SEM for three experiments.
The pathway downstream of guanylate cyclase was tested by exposure of spinal cord tissue slices to the permeable protein kinase G activator, 8-Br-cGMP. Figure 7A shows that in nontransgenic controls this caused an inhibition of Na,K-ATPase activity as effective as DETA-NO treatment. However, there were no changes in activity in tissue from transgenic mutant SOD1 mice (Fig.7B). Evidence for the involvement of protein phosphorylation in mediating the normal NO-induced regulation of Na,K-ATPase activity is shown in Figure 7 also. The addition of okadaic acid (400 nm) at concentrations known to inhibit protein phosphatases type 1 and type 2A mimicked the effects of DETA-NO and 8-Br-cGMP in inhibiting ouabain-sensitive Na,K-ATPase activity in nontransgenic SOD1 controls (Fig. 7A). Okadaic acid had no effect, however, on transgenic mutant SOD1 mice (Fig. 7B). The inability to bypass any step of the NO-mediated regulatory pathway is evidence either for the broadly compromised state of the transgenic spinal cord or for the resistance of the residual Na,K-ATPase activity to any further regulation.
Fig. 7.

Ouabain-sensitive Na+,K-ATPase activity in spinal cord of nontransgenic control mice (A) and transgenic mutant SOD1 mice (B) after incubation with DETA-NO (100 μm), 8-Br-cGMP (2 mm), or okadaic acid (OKA; 400 nm). Results are expressed as μmol Pi/hr per milligram of protein, and values represent the means ± SEM for three animals done in triplicate. *Significantly different from the control at p < 0.05 (by ANOVA and Fisher's PLSD).
Discussion
The mechanisms of motor neuron death in ALS are unknown, but several theories have been proposed, including that mutations in SOD1 may result in oxidative stress (Beckman et al., 1993; Crow et al., 1997; Andrus et al., 1998), increases in NO synthase and reactivity of astrocytes (Almer et al., 1999), and glutamate excitotoxicity secondary to glutamate transporter defects (Rothstein et al., 1993; Bruijn et al., 1997; Trotti et al., 1999). Other possible causes of neuronal degeneration include alterations in mitochondrial function (Beal, 1995;Klivenyi et al., 1999), formation of mutant SOD1 protein aggregates (Bruijn et al., 1998), and improper assembly of intermediate filaments that have particular impact on motor neurons because of their exceptionally long axons (for review, see Cleveland and Rothstein, 2001; Julien, 2001). Although all of these may contribute to the pathology and some may determine the selective vulnerability of motor neurons, perturbation of free radical homeostasis directly related to the SOD1 mutation is the phenomenon that is least likely to be secondary to the neurodegeneration process. Curiously, selective expression of mutant SOD1 in either astrocytes or neurons alone has failed to induce the disease (Gong et al., 2000; Pramatarova et al., 2001; Lino et al., 2002).
Uniform loss of Na,K-ATPase
These studies demonstrate that there are surprisingly large decreases in ouabain-sensitive Na,K-ATPase in spinal cord of transgenic mutant SOD1 mice, as assessed by enzyme activity, α subunit content, and anatomical distribution. Because of the uniformity, the losses are not likely to be a secondary consequence of neuron atrophy or death; this was confirmed by the observation of similar losses of Na,K-ATPase in the cerebellum. There were less severe decreases in Na,K-ATPase in the mice overexpressing transgenic normal human SOD1. SOD1 overexpression, which occurs in Down's syndrome as a direct result of trisomy, also is thought to increase oxidative damage (Ceballos-Picot et al., 1991; Lee et al., 2001), exacerbate excitotoxicity (Bar-Peled et al., 1996), and affect hippocampal ultrastructure and function (Barkats et al., 1993; Gahtan et al., 1998). Because the transgenic normal human SOD1 mice we used do not show hallmarks of ALS pathology such as SOD1 aggregates, intermediate filament aggregates, and mitochondrial defects, the observed Na,K-ATPase losses are more likely to be attributable to a “gain of function” abnormality of free radical or oxidant homeostasis than to ALS-associated neurodegeneration. These findings suggest that decreased Na,K-ATPase activity contributes to SOD1 ALS via inactivation and the loss of all three α subunits.
Any given cell type expresses a particular combination of α and β subunits (McGrail et al., 1991; Watts et al., 1991; Peng et al., 1997;Wetzel et al., 1999; Martin-Vasallo et al., 2000). Many neurons, for example, express α3β1, mature astrocytes express α2β2, and oligodendrocytes express α2β3, but there are exceptions, such as the granule neurons of the cerebellum that express α3β2, Müller glial cells that express α1β2, and neurons that express more than one assembled complex. Decreases in α3 are diagnostic for neuronal defects, because this isoform is not expressed in glia. Decreases in α2 are additional evidence that astrocytes as well as neurons are affected in the disease. Two well established functions of glia are the clearance of potassium and the Na+-dependent uptake of glutamate from the extracellular space after synaptic activation. Na,K-ATPase is activated when [Na+]i rises concomitantly with glutamate uptake (Rose and Ransom, 1996), and this stimulates the uptake of K+. The importance of these observations to ALS is highlighted by reports of selective loss of the astrocyte-specific glutamate transporter EAAT2 (GLT-1) (Rothstein et al., 1995) and of its vulnerability to oxidation by certain mutant SOD1 forms (Trotti et al., 1999). A defect in the glutamate transporter, a defect in the underlying glial sodium pump activity, and a defect in Na,K-ATPase activity in the neurons themselves are compatible with previous proposals that excitotoxicity contributes to the progression of ALS.
Detection of the simultaneous loss of Na,K-ATPase α isoforms specific to all different cell types in the spinal cord paints a compelling picture of a defect that is not confined to the motor neurons that die, however. The implications of the diffuse nature of the Na,K-ATPase alterations are potentially far-reaching. It suggests, for one thing, that the mutant mice have a “sick cord” and that studies of almost any candidate protein could reveal alterations that may contribute to the final pathology. It also suggests that alterations in the barrier organs such as the ependyma and pia may contribute to the disease. The loss of one-half to three-quarters of the enzyme that is the largest consumer of ATP may help to offset the effects of mitochondrial pathology and suggests that further investigation of energy metabolism in ALS would be fruitful (Browne et al., 1998; Klivenyi et al., 1999;Ames, 2000).
Altered free radical homeostasis and regulation of Na,K-ATPase
The NO/soluble guanylate cyclase pathway was altered severely in both transgenic mutant human SOD1 mice and transgenic normal human SOD1 overexpressors, whereas it inhibited ouabain-sensitive Na,K-ATPase activity in nontransgenic control mice. NO-mediated regulation of Na,K-ATPase activity is known in other tissues, including ciliary process and choroid plexus (Ellis et al., 2000, 2001). In the CNS, NO modulates cerebral blood flow and synaptic transmission via the activation of soluble guanylate cyclase and increases in cGMP (Murad, 1998). The loss of NO regulation in transgenic mutant SOD1 mice might have been predicted, considering the severity of the illness. However, the total blockade of the NO/cGMP pathway for Na,K-ATPase regulation in transgenic normal SOD1 overexpressors was unexpected.
The role of NO in CNS-related diseases is not completely clear. Several lines of evidence have emerged that suggest that NO can be either neuroprotective (Lipton et al., 1993; Chiueh, 1999) or neurodestructive (Dawson et al., 1991, 1992; Samdani et al., 1997). NO generated from NO donors or synthesized endogenously after activation of the ionotropic glutamate receptor (Lafon-Cazal et al., 1993) can lead to neurotoxicity in part by reaction with superoxide anion and the subsequent formation of peroxynitrite (Lipton et al., 1993; Beckman and Koppenol, 1996). In contrast, other studies have demonstrated that the neuroprotective effects of NO in CNS may result from nitration or nitrosylation of iron- or thiol-containing proteins (Stamler et al., 1992; Lipton et al., 1993; Chiueh, 1999). Such chemical modifications may alter significantly the biological activity of the protein and minimize the generation of reactive oxygen species and associated oxidative stress. A lack of effect on ALS pathology when G93A mice were crossed with neuronal nitric oxide synthase (nNOS) knock-out mice suggested no involvement, but residual nNOS activity because of the synthesis of truncated forms complicated the interpretation (Facchinetti et al., 1999).
The degree of inhibition of Na,K-ATPase activity in transgenic mutant mice exceeded the decrease in the α subunit. The Na,K-ATPase can be inhibited chemically by oxygen free radicals and their by-products (Mense et al., 1997). For example, superoxide anion, hydrogen peroxide, and hydroxyl radicals inhibited Na,K-ATPase activity, and this decrease correlated with increased lipid peroxidation (Viani et al., 1991; Huang et al., 1992). Of interest is the finding that various NO donors (excluding sodium nitroprusside) caused substantial direct alterations in Na,K-ATPase activity via reaction with free sulfhydryl groups (Boldyrev et al., 1997; Sato et al., 1997). Furthermore, as with other proteins (Stadtman, 1992), exposure of the Na,K-ATPase to free radicals made it more susceptible to degradation by proteolytic enzymes (Huang et al., 1992; Thevenod and Friedmann, 1999). Because α and β subunits normally are found in a 1:1 ratio, the reduction in α, but not β, was unexpected. The data are consistent with greater vulnerability of the α subunit to damage and degradation. This is plausible, considering that the majority of the mass of the β subunit is in the extracellular space where it is adapted to a more oxidizing environment than the cytoplasm, and that its six extracellular sulfhydryl groups are all in buried disulfide bonds. Most of the mass of the α subunit, in contrast, is in the reducing environment on the cytoplasmic side of the membrane, and it has 23 free sulfhydryls and other oxidizable groups.
The widespread defects in Na,K-ATPase, not confined to either neurons or glia, support the original hypothesis that perturbation of free radical homeostasis is the most likely root cause of mutant SOD1 ALS pathology. The abrogation of the NO/cGMP pathway of Na,K-ATPase regulation is an unexpected and potentially important event, whether it is a parallel defect with the same root cause or a causative step in Na,K-ATPase loss.
Footnotes
This work was supported by grants from the Amyotrophic Lateral Sclerosis Association and the Fiftieth Anniversary Scholars Program of Harvard Medical School to D.Z.E. and by National Institutes of Health Grant R01-NS27653 to K.J.S. We thank Dr. Robert H. Brown Jr. (Massachusetts General Hospital) for advice and stimulating discussions. We are also grateful to Drs. J. Kyte, P. Martin-Vasallo, C. Goridis, and M. Schachner for antibodies.
Correspondence should be addressed to Dr. Kathleen J. Sweadner, 149-6118, Massachusetts General Hospital, 149 13th Street, Charlestown, MA 02129. E-mail: sweadner@helix.mgh.harvard.edu.
References
- 1.Almer G, Vukosavic S, Romero N, Przedborski S. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;72:2415–2425. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- 2.Ames A., III CNS energy metabolism as related to function. Brain Res Brain Res Rev. 2000;34:42–68. doi: 10.1016/s0165-0173(00)00038-2. [DOI] [PubMed] [Google Scholar]
- 3.Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1998;71:2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- 4.Barkats M, Bertholet J-Y, Venault P, Ceballos-Picot I, Nicole A, Phillips J, Moutier R, Roubertoux P, Sinet PM, Cohen-Salmon M. Hippocampal mossy fiber changes in mice transgenic for the human copper/zinc superoxide dismutase gene. Neurosci Lett. 1993;160:24–28. doi: 10.1016/0304-3940(93)90908-4. [DOI] [PubMed] [Google Scholar]
- 5.Bar-Peled O, Korkotian E, Segal M, Groner Y. Constitutive overexpression of Cu/Zn superoxide dismutase exacerbates kainic acid-induced apoptosis of transgenic Cu/Zn superoxide dismutase neurons. Proc Natl Acad Sci USA. 1996;93:8530–8535. doi: 10.1073/pnas.93.16.8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 7.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 8.Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD, and peroxynitrite. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 9.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 10.Boldyrev AA, Bulygina ER, Kramarenko GG, Vanin AF. Effect of nitroso compounds on Na/K-ATPase. Biochim Biophys Acta. 1997;1321:243–251. doi: 10.1016/s0005-2728(97)00053-4. [DOI] [PubMed] [Google Scholar]
- 11.Brines ML, Robbins RJ. Inhibition of α2/α3 sodium pump isoforms potentiates glutamate neurotoxicity. Brain Res. 1992;591:94–102. doi: 10.1016/0006-8993(92)90982-f. [DOI] [PubMed] [Google Scholar]
- 12.Brines ML, Dare AO, de Lanerolle NC. The cardiac glycoside ouabain potentiates excitotoxic injury of adult neurons in rat hippocampus. Neurosci Lett. 1995;191:145–148. doi: 10.1016/0304-3940(95)11577-j. [DOI] [PubMed] [Google Scholar]
- 13.Brown RH., Jr Amyotrophic lateral sclerosis: recent insights from genetics and transgenic mice. Cell. 1995;80:687–692. doi: 10.1016/0092-8674(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 14.Browne SE, Bowling AC, Baik MJ, Gurney ME, Brown RH, Jr, Beal MF. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J Neurochem. 1998;71:281–287. doi: 10.1046/j.1471-4159.1998.71010281.x. [DOI] [PubMed] [Google Scholar]
- 15.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 16.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 17.Calabresi P, De Murtas M, Pisani A, Stefani A, Sancesario G, Mercuri NB, Bernardi G. Vulnerability of medium spiny striatal neurons to glutamate: role of Na+/K+-ATPase. Eur J Neurosci. 1995;7:1674–1683. doi: 10.1111/j.1460-9568.1995.tb00689.x. [DOI] [PubMed] [Google Scholar]
- 18.Ceballos-Picot I, Nicole A, Briand P, Grimber G, Delacourte A, Defossez A, Javoy-Agid F, Lafon M, Blouin JL, Sinet PM. Neuronal-specific expression of human copper/zinc superoxide dismutase gene in transgenic mice: animal model of gene dosage effects in Down's syndrome. Brain Res. 1991;552:198–214. doi: 10.1016/0006-8993(91)90084-9. [DOI] [PubMed] [Google Scholar]
- 19.Chiueh CC. Neuroprotective properties of nitric oxide. Ann NY Acad Sci. 1999;890:301–311. doi: 10.1111/j.1749-6632.1999.tb08007.x. [DOI] [PubMed] [Google Scholar]
- 20.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 21.Crambert G, Hasler U, Beggah AT, Yu C, Modyanov NN, Horisberger J-D, Lelievre L, Geering K. Transport and pharmacological properties of nine different human Na,K-ATPase isozymes. J Biol Chem. 2000;275:1976–1986. doi: 10.1074/jbc.275.3.1976. [DOI] [PubMed] [Google Scholar]
- 22.Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration of peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 23.Dawson TM, Dawson VL, Snyder SH. A novel neuronal messenger molecule in brain: the free radical, nitric oxide. Ann Neurol. 1992;32:297–311. doi: 10.1002/ana.410320302. [DOI] [PubMed] [Google Scholar]
- 24.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diodati JG, Quyyumi AA, Keefer LK. Complexes of nitric oxide with nucleophiles as agents for the controlled biological release of nitric oxide: hemodynamic effects in rabbit. J Cardiovasc Pharmacol. 1993;22:287–292. doi: 10.1097/00005344-199308000-00018. [DOI] [PubMed] [Google Scholar]
- 26.Ellis DZ, Nathanson JA, Sweadner KJ. Carbachol inhibits Na+-K+-ATPase activity in choroid plexus via stimulation of the NO/cGMP pathway. Am J Physiol. 2000;279:C1685–C1693. doi: 10.1152/ajpcell.2000.279.6.C1685. [DOI] [PubMed] [Google Scholar]
- 27.Ellis DZ, Nathanson JA, Rabe J, Sweadner KJ. Carbachol and NO inhibit Na,K-ATPase in ciliary process via activation of guanylate cyclase and cGMP. Invest Ophthalmol Vis Sci. 2001;42:2625–2631. [PubMed] [Google Scholar]
- 28.Facchinetti F, Sasaki M, Cutting FB, Zhai P, MacDonald JE, Reif D, Beal MF, Huang PL, Dawson TM, Gurney ME, Dawson VL. Lack of involvement of neuronal nitric oxide synthase in the pathogenesis of a transgenic mouse model of familial amyotrophic lateral sclerosis. Neuroscience. 1999;90:1483–1492. doi: 10.1016/s0306-4522(98)00492-8. [DOI] [PubMed] [Google Scholar]
- 29.Gahtan E, Auerbach JM, Groner Y, Segal M. Reversible impairment of long-term potentiation in transgenic Cu/Zn SOD mice. Eur J Neurosci. 1998;10:538–544. doi: 10.1046/j.1460-9568.1998.00058.x. [DOI] [PubMed] [Google Scholar]
- 30.Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 31.Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit L, Siddique T. Motor neuron degeneration in mice that express a human Cu/Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 33.Hieber V, Siegel GJ, Fink DJ, Beaty MW, Mata M. Differential distribution of (Na,K)-ATPase α isoforms in the central nervous system. Cell Mol Neurobiol. 1991;11:253–262. doi: 10.1007/BF00769038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang WH, Wang Y, Askari A. (Na+ + K+)-ATPase: inactivation and degradation induced by oxygen radicals. Int J Biochem. 1992;24:621–626. doi: 10.1016/0020-711x(92)90337-z. [DOI] [PubMed] [Google Scholar]
- 35.Julien J-P. Amyotrophic lateral sclerosis: unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- 36.Kim MS, Akera T. O2 free radicals: cause of ischemia-reperfusion injury to cardiac Na+,K+-ATPase. Am J Physiol. 1987;252:H252–H257. doi: 10.1152/ajpheart.1987.252.2.H252. [DOI] [PubMed] [Google Scholar]
- 37.Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daowk R, Beal MF. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;3:347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- 38.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 39.Lee M, Hyun D-H, Jenner P, Halliwill B. Effect of overexpression of wild-type and mutant Cu/Zn superoxide dismutases on oxidative damage and antioxidant defenses: relevance to Down's syndrome and familial amyotrophic lateral sclerosis. J Neurochem. 2001;76:957–965. doi: 10.1046/j.1471-4159.2001.00107.x. [DOI] [PubMed] [Google Scholar]
- 40.Lees GJ, Leong W. Interactions between excitotoxins and the Na+,K+-ATPase inhibitor ouabain in causing neuronal lesions in the rat hippocampus. Brain Res. 1996;714:145–155. doi: 10.1016/0006-8993(95)01518-3. [DOI] [PubMed] [Google Scholar]
- 41.Lees GJ, Lehmann A, Sandberg M, Hamberger A. The neurotoxicity of ouabain, a sodium/potassium ATPase inhibitor, in the rat hippocampus. Neurosci Lett. 1990;120:159–162. doi: 10.1016/0304-3940(90)90027-7. [DOI] [PubMed] [Google Scholar]
- 42.Lino MM, Schneider C, Caroni P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci. 2002;22:4825–4832. doi: 10.1523/JNEUROSCI.22-12-04825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipton SA, Choi Y-B, Pan Z-H, Lei SZ, Chen H-S, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 44.Martin-Vasallo P, Wetzel RK, Garcia-Segura LM, Molina-Holgado E, Arystarkhova E, Sweadner KJ. Oligodendrocytes in brain and optic nerve express the β3 subunit isoform of Na,K-ATPase. Glia. 2000;31:206–218. doi: 10.1002/1098-1136(200009)31:3<206::aid-glia20>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 45.Mata M, Siegel GJ, Hieber V, Beaty MW, Fink DJ. Differential distribution of (Na,K)-ATPase α isoform mRNAs in the peripheral nervous system. Brain Res. 1991;546:47–54. doi: 10.1016/0006-8993(91)91157-v. [DOI] [PubMed] [Google Scholar]
- 46.McGrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J Neurosci. 1991;11:381–391. doi: 10.1523/JNEUROSCI.11-02-00381.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKee M, Scavone C, Nathanson JA. Nitric oxide, cGMP, and hormone regulation of active sodium transport. Proc Natl Acad Sci USA. 1994;91:12056–12060. doi: 10.1073/pnas.91.25.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixative for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- 49.Mense M, Stark G, Apell H-J. Effects of free radicals on partial reactions of the Na,K-ATPase. J Membr Biol. 1997;156:63–71. doi: 10.1007/s002329900188. [DOI] [PubMed] [Google Scholar]
- 50.Murad F. Nitric oxide signaling: would you believe that a simple free radical could be a second messenger, autacoid, paracrine substance, neurotransmitter, and hormone? Recent Prog Horm Res. 1998;53:43–59. [PubMed] [Google Scholar]
- 51.Nathanson JA, Scavone C, Scanlon C, McKee M. The cellular Na+ pump as a site of action for carbon monoxide and glutamate: a mechanism for long-term modulation of cellular activity. Neuron. 1995;14:781–794. doi: 10.1016/0896-6273(95)90222-8. [DOI] [PubMed] [Google Scholar]
- 52.Peng L, Martin-Vasallo P, Sweadner KJ. Isoforms of Na,K-ATPase α and β subunits in the rat cerebellum and in granule cell cultures. J Neurosci. 1997;17:3488–3502. doi: 10.1523/JNEUROSCI.17-10-03488.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21:3369–3374. doi: 10.1523/JNEUROSCI.21-10-03369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rose CR, Ransom BR. Mechanisms of H+ and Na+ changes induced by glutamate, kainate, and d-aspartate in rat hippocampal astrocytes. J Neurosci. 1996;16:5393–5404. doi: 10.1523/JNEUROSCI.16-17-05393.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Gusella JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 56.Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 58.Samdani AF, Newcamp C, Resink A, Facchinetti F, Hoffman BE, Dawson VL, Dawson TM. Differential susceptibility to neurotoxicity mediated by neurotrophins and neuronal nitric oxide synthase. J Neurosci. 1997;17:4633–4641. doi: 10.1523/JNEUROSCI.17-12-04633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sato T, Kamata Y, Irifune M, Nishikawa T. Inhibitory effect of several nitric oxide-generating compounds on purified Na,K-ATPase activity from porcine cerebral cortex. J Neurochem. 1997;68:1312–1318. doi: 10.1046/j.1471-4159.1997.68031312.x. [DOI] [PubMed] [Google Scholar]
- 60.Scavone C, Scanlon C, McKee M, Nathanson JA. Atrial natriuretic peptide modulates sodium and potassium-activated adenosine triphosphatase through a mechanism involving cyclic GMP and cyclic GMP-dependent protein kinase. J Pharmacol Exp Ther. 1995;272:1036–1043. [PubMed] [Google Scholar]
- 61.Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 62.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 63.Stelmashook EV, Weih M, Zorov D, Vicotrov I, Dirnagl U, Isaev N. Short-term block of Na+/K+-ATPase in neuro-glial cell cultures of cerebellum induces glutamate-dependent damage of granule cells. FEBS Lett. 1999;456:41–44. doi: 10.1016/s0014-5793(99)00922-9. [DOI] [PubMed] [Google Scholar]
- 64.Sweadner KJ. Isozymes of the Na+/K+-ATPase. Biochim Biophys Acta. 1989;988:185–220. doi: 10.1016/0304-4157(89)90019-1. [DOI] [PubMed] [Google Scholar]
- 65.Thevenod F, Friedmann JM. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K+-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 1999;13:1751–1761. doi: 10.1096/fasebj.13.13.1751. [DOI] [PubMed] [Google Scholar]
- 66.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- 67.Viani P, Cervato G, Fiorilli A, Cestaro B. Age-related differences in synaptosomal peroxidative damage and membrane properties. J Neurochem. 1991;56:253–258. doi: 10.1111/j.1471-4159.1991.tb02589.x. [DOI] [PubMed] [Google Scholar]
- 68.Watts AG, Sanchez-Watts G, Emanuel JR, Levenson R. Cell-specific expression of mRNAs encoding Na+,K+-ATPase α- and β-subunit isoforms within the rat central nervous system. Proc Natl Acad Sci USA. 1991;88:7425–7429. doi: 10.1073/pnas.88.16.7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wetzel RK, Arystarkhova E, Sweadner KJ. Cellular and subcellular specification of Na,K-ATPase α and β isoforms in the postnatal development of mouse retina. J Neurosci. 1999;19:9878–9889. doi: 10.1523/JNEUROSCI.19-22-09878.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie Z, Jack-Hays M, Periyasamy SM, Blanco G, Huang W-H, Askari A. Different oxidant sensitivities of the α1 and α2 isoforms of Na+/K+-ATPase expressed in baculovirus-infected insect cells. Biochem Biophys Res Commun. 1995;207:155–159. doi: 10.1006/bbrc.1995.1166. [DOI] [PubMed] [Google Scholar]
- 71.Zolotarjova N, Ho C, Mellgren RL, Askari A, Huang W-H. Different sensitivities of native and oxidized forms of Na,K-ATPase to intracellular proteinases. Biochim Biophys Acta. 1994;1192:125–131. doi: 10.1016/0005-2736(94)90152-x. [DOI] [PubMed] [Google Scholar]