

Abstract
Voltage-gated Ca2+ channels of chromaffin cells are modulated by locally released neurotransmitters through autoreceptor-activated G-proteins. Clear evidence exists in favor of a Ca2+ channel gating inhibition mediated by purinergic, opioidergic, and α-adrenergic autoreceptors. Few and contradictory data suggest also a role of β-adrenergic autoreceptors (β-ARs), the action of which, however, remains obscure. Here, using patch-perforated recordings, we show that rat chromaffin cells respond to the β-AR agonist isoprenaline (ISO) by either upmodulating or downmodulating the amplitude of Ca2+ currents through two distinct modulatory pathways. ISO (1 μm) could cause either fast inhibition (∼25%) or slow potentiation (∼25%), or a combination of the two actions. Both effects were completely prevented by propranolol. Slow potentiation was more evident in cells pretreated with pertussis toxin (PTX) or when β1-ARs were selectively stimulated with ISO + ICI118,551. Potentiation was absent when the β2-AR-selective agonist zinterol (1 μm), the protein kinase A (PKA) inhibitor H89, or nifedipine was applied, suggesting that potentiation is associated with a PKA-mediated phosphorylation of L-channels (∼40% L-current increase) through β1-ARs. The ISO-induced inhibition was fast and reversible, preserved in cell treated with H89, and mimicked by zinterol. The action of zinterol was mostly on L-channels (38% inhibition). Zinterol action preserved the channel activation kinetics, the voltage-dependence of theI–V characteristic, and was removed by PTX, suggesting that β2AR-mediated channel inhibition was mainly voltage independent and coupled to Gi/Go-proteins. Sequential application of zinterol and ISO mimicked the dual action (inhibition/potentiation) of ISO alone. The two kinetically and pharmacologically distinct β-ARs signaling uncover alternative pathways, which may serve the autocrine control of Ca2+-dependent exocytosis and other related functions of rat chromaffin cells.
Keywords: β2-adrenergic receptor, β1-adrenergic receptor, voltage-gated calcium channel, cAMP/PKA signaling, G-protein-coupled receptors, adrenal medullas, zinterol
Introduction
Voltage-gated Ca2+ channels are crucial for Ca2+ signaling during secretion. In adrenal chromaffin cells, released neurotransmitters act autocrinally on Ca2+ channel gating (Albillos et al., 1996a; Currie and Fox, 1996) and the associated Ca2+-dependent secretion (Lim et al., 1997; Powell et al., 2000; Ulate et al., 2000). N- and P/Q-channels are inhibited by G-protein-coupled receptors (GPCRs) (Kleppisch et al., 1992; Gandía et al., 1993; Albillos et al., 1996b) by a mechanism resolved in membrane micropatches, suggesting close localization of Ca2+ channels, GPCRs, and secretory events (Carabelli et al., 1998). Also Cav1 L-channels can be downregulated through membrane-delimited interactions with GPCRs (Hernández-Guijo et al., 1999; Carabelli et al., 2001) or by cGMP/protein kinase G-mediated pathways (Carabelli et al., 2002). Conversely, L-channels are upregulated by cAMP elevations or by D1- and β-adrenergic autoreceptor (AR) stimulation (Artalejo et al., 1990; Doupnik and Pun, 1992; Carabelli et al., 2001; Carbone et al., 2001), thus furnishing a wide range of autocrine Ca2+-current control.
The most intriguing issue regarding the GPCRs modulating L-channel gating concerns the existence of a β-AR-mediated modulation, which can inhibit (Hernández-Guijo et al., 1999) or potentiate (Carbone et al., 2001) L-channel activity of rat chromaffin cells (RCCs). This peculiarity is of relevance because adrenaline (A) and noradrenaline (NA) are stored in chromaffin granules and released at concentrations of 0.5–1 m (Jankowski et al., 1993), and β-AR stimulation increases the basal release of catecholamines in bovine chromaffin cells (BCCs) (Parramon et al., 1995). The existence of two distinct β-AR-mediated responses may derive from the activation of two different signaling cascades. (1) β1-ARs act exclusively via Gs-proteins coupled to adenylyl cyclase (AC), cAMP production, protein kinase A (PKA) activation, and increased L-channel activity (Bean et al., 1984); (2) β2-ARs couple to both Gs- and Gi-proteins and give rise to various specific β-AR-mediated pathways (Xiao et al., 1995; Daaka et al., 1997; Kilts et al., 2000). In rodent cardiac cells, β2-ARs are preferentially localized to caveole and coupled to PTX-sensitive Gi-proteins, thus confining cAMP signaling to compartmentalized regions (Xiao et al., 1995, Zhou et al., 1999). A localized action after β2-AR stimulation has been reported recently in hippocampus, ensuring highly localized and rapid cell signaling (Davare et al., 2001). Thus, β2-ARs activate membrane-delimited pathways in clear contrast to the remote β1-AR stimulation, which is diffusive and involves distal effectors.
Here, using recording conditions close to physiological (10 mm Ca2+ and perforated-patch recordings in place of Ba2+ and whole-cell recording conditions), we show that RCCs possess two distinct β1- and β2-AR signaling pathways. The β1-AR cascade acts by selectively upregulating the L-channel through a PKA-mediated pathway and develops slowly according to its diffusive characteristics. On the contrary, the β2-AR signaling is fast and primarily coupled to PTX-sensitive G-proteins. It produces marked depression on the L-channel (∼40%), which represents the dominant Ca2+ channel subtype in RCCs. The two β-ARs act in parallel, giving rise to considerable Ca2+ current modifications, which may be relevant both for the control of catecholamine secretion and for the opposing effects that the two receptors exert on cell apoptosis (Communal et al., 1999; Zaugg et al., 2000).
Materials and Methods
Isolation and culture of rat adrenal medulla chromaffin cells. All experiments were performed in accordance with the guidelines established by the National Council on Animal Care and were approved by the local Animal Care Committee of Turin University. Chromaffin cells were obtained from adult female Sprague Dawley rats (200–300 gm), which were killed by cervical dislocation. Cell isolation and plating were achieved as described previously (Hernández-Guijo et al., 1999). Briefly, after removal, the adrenal glands were placed in Ca2+- and Mg2+-free Locke's buffer containing (in mm): 154 NaCl, 3.6 KCl, 5.6 NaHCO3, 5.6 glucose, and 10 HEPES, pH 7.2, at room temperature. The glands were decapsulated, and the medullas were precisely separated from the cortical tissue. Medulla digestion was achieved for 30 min at 37°C in Ca2+- and Mg2+-free Locke's buffer containing 1.5 mg/ml collagenase, 3 mg/ml bovine serum albumin. Gentle agitation every 10 min with a Pasteur pipette facilitated cell separation. The cell suspension was then centrifuged for 10 min at 120 × g, washed two times, and resuspended in 1 ml DMEM. Cells were plated in four-well plastic dishes treated with poly-l-ornithine (1 mg/ml) and laminin (5 μg/ml in L-15 carbonate) by placing a drop of concentrated cell suspension in the center of each well. After 1 hr, 1 ml of DMEM supplemented with 5% fetal calf serum (Invitrogen, Grand Island, NY), 50 IU/ml penicillin, and 50 μg/ml streptomycin (Invitrogen) was added to the wells. Cells were then incubated at 37°C in a water-saturated atmosphere with 5% CO2 and used within 2–6 d after plating.
Current recordings, solutions, and data analysis. Ca2+ currents were recorded by the perforated-patch whole-cell recording technique (Korn et al., 1991) using patch pipettes of thin borosilicate glass (Kimax 51; Witz Scientific, Holland, OH). For stimulation and acquisition we used an EPC-9 amplifier and PULSE software (HEKA-Elektronik, Lambrecht, Germany).
The perforated patch was obtained using pipettes containing 50–100 μg/ml amphotericin B (Sigma, St. Louis, MO) and a pipette-filling solution containing (in mm): 135 CsMeSO3, 8 NaCl, 2 MgCl2, 20 HEPES, pH 7.3 with CsOH. The external bath contained (in mm): 135 NaCl, 2.8 KCl, 10 CaCl2, 2 MgCl2, 20 glucose, 10 HEPES, pH 7.4 with NaOH. Amphotericin B was dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C in stock aliquots of 50 mg/ml. Fresh pipette solution was prepared every 2 hr. To facilitate the sealing, the pipette was first dipped in a beaker containing the internal solution and then back-filled with the same solution containing amphotericin B. Pipettes with series resistance of 2–3 MΩ were used to form gigaseals. Recording of Ca2+ currents started when the access resistance decreased below 15 MΩ, which usually happened within 10 min after sealing (Rae et al., 1991). Series resistance was compensated by 80% and monitored throughout the experiment. Because the drugs that were applied to the external solution did not affect the liquid junction potential (LJP), the indicated voltages were not corrected for the LJP at the interface between the pipette solution (135 CsMeSO3) and the bath (135 NaCl), which was −13 mV (Barry and Lynch, 1991). The estimated Donnan equilibrium potential between the cell interior and the pipette was below −2 mV. Thus, the voltage bias for the present measurements was between −13 and −15 mV. Indeed, when comparing the present measurements with previous recordings on whole-cell clamped RCCs, in which LJPs were not compensated, the voltage bias was approximately −10 mV (Gandía et al., 1995; Hernández-Guijo et al., 1999) (see Results).
Ca2+ currents were evoked by step depolarizations of 25–50 msec to a fixed potential (+10 mV) or by ramp commands from −40 to +70 mV with a slope of 2.2 V/sec. The holding potential was −40 mV throughout the experiments, which should correspond to approximately −63 mV when compared with Ca2+ currents measured in 5 mmBa2+ and whole-cell recordings (see Results). Fast capacitative transients during step depolarizations were minimized on-line by the patch-clamp analog compensation. Uncompensated capacitative currents were further reduced by subtracting the averaged currents in response to P/4 hyperpolarizing pulses. All of the experiments were performed at room temperature (22–24°C). Data are given as mean ± SEM for n = number of cells. Statistical significance was calculated using Student's pairedt test, and p < 0.05 was considered significant.
External solutions were exchanged using a gravity system consisting of a multibarreled pipette with a single outlet and five inlets controlled by solenoid electrovalves (The Lee Company, Westbrook, CO). The tip of the perfusion pipette (40–50 μm) was placed ∼40 μm from the cell. In this way the perfusion solution could be changed within 50 msec (Pollo et al., 1993). Nifedipine, isoprenaline, propranolol, ICI 118,551, and H7 were purchased from Sigma. H89 was obtained from CN Biosciences Inc. (Darmstadt, Germany). Zinterol (kindly provided by Bristol-Meyers Squibb, Wallingford, CT) was prepared as a 1 mm stock solution in DMSO and dissolved to a final concentration (0.3–10 μm) just before the experiments. Protein kinase inhibitors (H89 and H7) were dissolved in distilled water and kept frozen in aliquots. Toxins ω-conotoxin (CTx)-GVIA, ω-agatoxin (Aga)-IVA, and ω-conotoxin (CTx)-MVIIC were purchased from Peptide Institute (Osaka, Japan) and prepared to the final concentration as described previously (Magnelli et al., 1998).
Results
Ca2+ currents in perforated-patch conditions
In 10 mm Ca2+ and perforated-patch conditions, the voltage-gated Ca2+ currents of RCCs were stable and could be recorded for long periods of time (15–20 min) without significant rundown (Fig.1A). During brief depolarizations (50 msec), Ca2+ currents activated at approximately −30 mV, reached maximal amplitude at approximately +10 mV, and decreased at higher potentials (Fig.1B, ●). Compared with previous measurements in 5 mm Ba2+ and whole-cell recording conditions (Hernández-Guijo et al., 1999), the I–V characteristics in 10 mm Ca2+ and perforated-patch conditions peaked 23 mV toward more positive voltages (Fig. 1C, ●). This was caused by the sum of the uncompensated liquid junction potential between pipette and bath solution (approximately +10 mV; see Materials and Methods) and the voltage shift associated with the more effective screening of Ca2+ with respect to Ba2+ on membrane negative surface charges (approximately +13 mV).
Fig. 1.
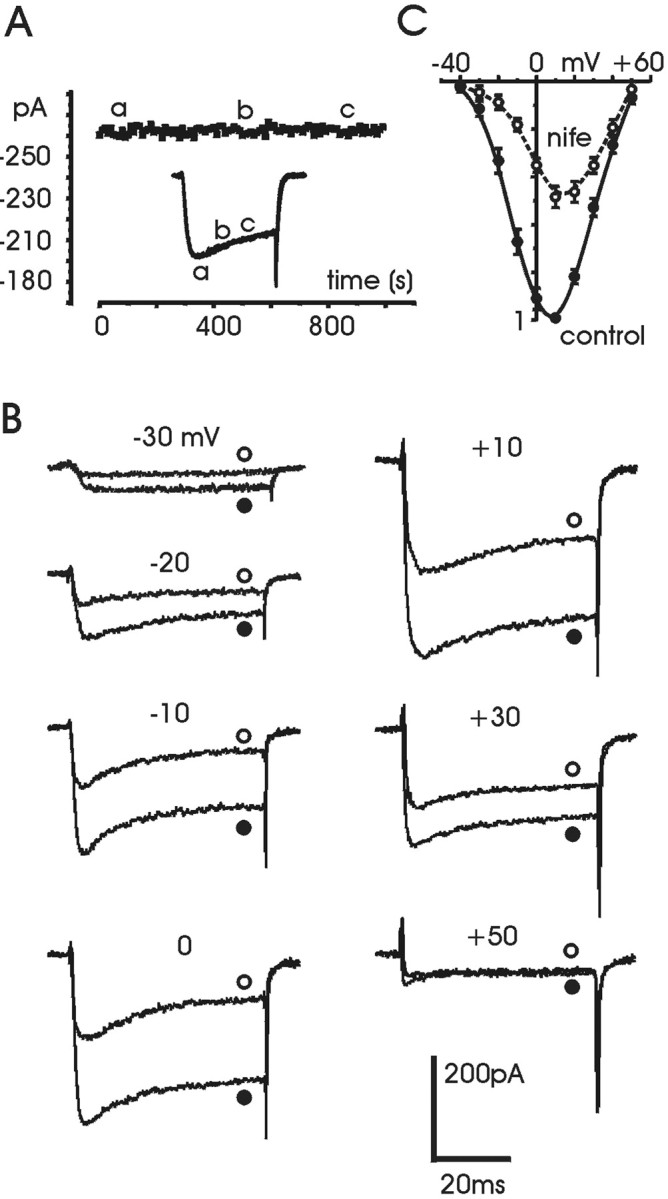
Ca2+ current of RCCs in perforated-patch recording conditions. A, Time course of Ca2+ current amplitude recorded in perforated-patch conditions from an RCC superfused with control external solution. Eachsymbol represents the peak amplitude of Ca2+ current evoked by depolarizing the cell every 10 sec to +10 mV for 25 msec from a holding potential (Vh) of −40 mV. Return potential (Vr) was equal to holding potential. The three current traces shown in the inset were recorded at the time indicated by the correspondingletters in the graph. B, Ca2+ currents recorded before (●) and during (○) application of 3 μm nifedipine at the potentials indicated. Holding potential and return potential are as inA. C, Normalized meanI–V characteristics calculated at the end of the 50 msec pulse from seven cells, before (●) and during (○) application of nifedipine. Notice the slight shift to the right of the I–V curve with nifedipine caused by the block of L-channels.
RCCs express a large percentage (40–50%) of L-type currents (Gandía et al., 1995; Hollins and Ikeda, 1996). This was evident from the block induced by adding 3 μm nifedipine to the bath solution (Fig. 1B,C). The dihydropyridine (DHP) blocked quickly and reversibly ∼50% of the total currents, in good agreement with previous reports. Notice that in control conditions, the percentage of inactivation, estimated by measuring the ratio between peak and steady-state current at the end of the pulse of 50 msec, changed greatly from cell to cell: from a minimum of 15% to a maximum of 55%. As shown below, this variability did not influence the quantification of ISO action on Ca2+ currents.
The dual action of isoprenaline on Ca2+ currents
Addition of 1 μm ISO to the bath caused a potentiation of the total current in 40 of 215 tested cells (18.6%) (Fig.2A,E), an inhibition in 72 cells (33.5%) (Fig.2B,E), a combination of the two actions in 66 cells (30.7%) (Fig. 2C,E), or no effects at all in 37 cells (17.2%) (Fig.2D,E). The onset of potentiation required ∼4 min to be complete, whereas the offset was significantly faster (∼1 min). After a lag of 20–30 sec the current raised slowly and mono-exponentially with a mean τonof 90.3 sec (n = 8) (Fig.3A1), whereas the offset started with no delay and developed approximately fivefold faster (mean τoff = 21.6 sec). The onset of inhibition was significantly faster than potentiation. In most cases the inhibition was complete in <1 min, and washout required a comparable time. A fit with an exponential function for the onset and offset of the inhibition gave average time constants of 8.3 sec (τon) and 12.6 sec (τoff), respectively (n = 10) (Fig. 3B1). When the two effects occurred on the same cell, the fast inhibition always anticipated the slower setting of potentiation. On average, the current amplitude first decreased by 26.3 ± 1.3% (n = 35) and then increased by 26.0 ± 1.7% (n = 35) to reach the original control levels or, in some case, even higher values (Fig.2C). On the other hand, potentiation and inhibition were comparable when they occurred in isolation (25.2 ± 1.9 vs 24.1 ± 1.2%) (Fig. 2F), suggesting that potentiation did not overwhelm the ISO-induced inhibition and that the two modulations were more or less additive with a large degree of independence. The inhibition caused little or no changes to theI–V relationships (Fig.3B2), indicating no sign of voltage-dependent effects on voltage-gated Ca2+ channels. The potentiation, on the contrary, shifted by ∼7 mV to the left the potential of half-maximal activation (Fig. 3A2, crosses), very likely because of the selective potentiation of L-channels, which activate at more negative voltages compared with the other high-threshold Ca2+ channels (Pollo et al., 1993). Inhibition and potentiation also caused few or no changes to the activation–inactivation time course of Ca2+ currents. In most cases, depressed or potentiated currents could nicely overlap by simple amplitude scaling, as illustrated in Fig. 3, A3 andB3.
Fig. 2.
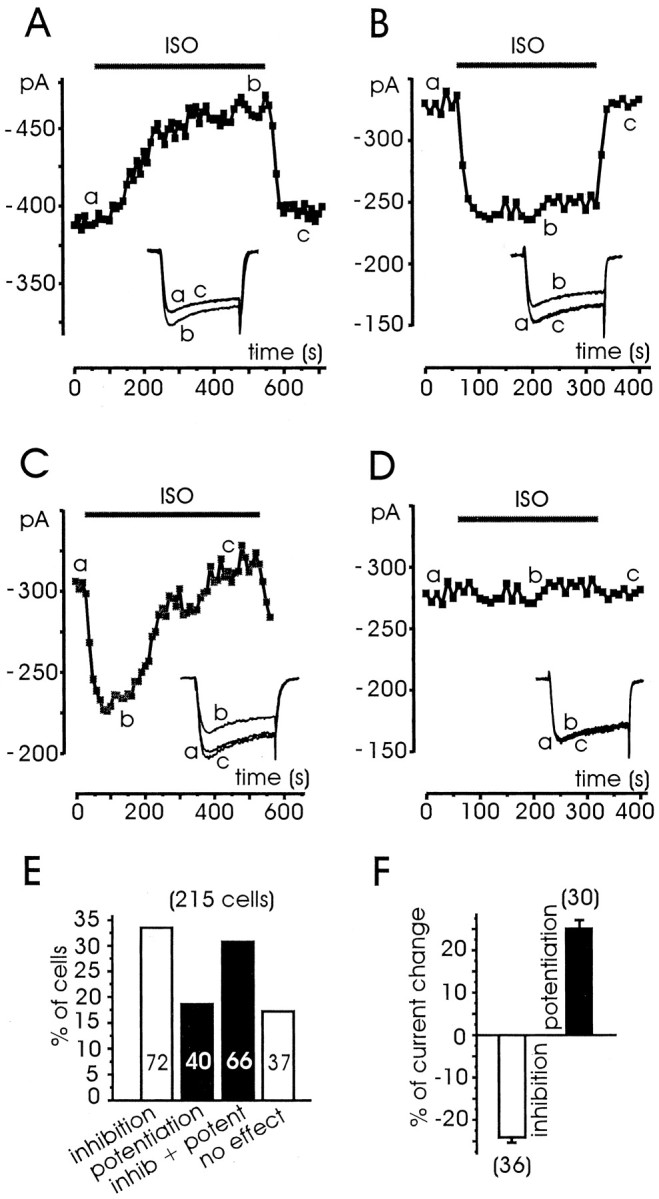
Differential effects of ISO on Ca2+ currents of RCCs. Examples of potentiation (A), inhibition (B), inhibition + potentiation (C), and no action (D) on Ca2+ currents induced by 1 μm ISO on different RCCs. Each cell was initially superfused with control external solution and successively exposed to ISO for the period indicated by the bar. Thesymbols represent peak current amplitudes measured on step depolarization to +10 mV for 25 msec and repeated every 10 sec.Vh and Vr, −40 mV. In the insets are shown the original recordings taken at the time indicated by the letters.E, Percentage of cells exhibiting potentiation, inhibition, inhibition + potentiation, or no action, derived from a total of 215 cells. F, Mean percentage of Ca2+ current inhibition (open bar) and potentiation (filled bar) observed in isolation. Cells were selected among those behaving like the examples illustrated in A and B, with complete recovery after ISO exposure.
Fig. 3.

Onset, offset, and voltage dependence of ISO-induced potentiation and inhibition.A1, Onset and offset of ISO-induced potentiation calculated by averaging the Ca2+current increment in eight cells exposed to 1 μmISO. Control currents before ISO were scaled and normalized to their maximum. The solid curves are the result of least-squares fit with single exponential functions with time constants τon = 90.3 sec and τoff = 21.6 sec. A2,I–V curves before and during an ISO-induced potentiation. The crosses indicate the voltage at which the I–V curve reached half its maximal amplitude (V1/2): −4.1 mV (control) and −11.3 mV (ISO). A3, Recordings taken from Figure 2C. Trace b was scaled by a factor of 1.39 to overlap trace c. B1, Onset and offset of ISO-induced inhibition calculated by averaging the current depression of 10 cells, using the same procedure as A1. The fit with single exponentials gave τon = 8.3 sec and τoff = 12.6 sec.B2, I–Vcurves before and during an ISO-induced inhibition.V1/2 was −2.2 mV (control) and −2.9 mV (ISO), indicated by the crosses. B3, Current recordings derived from Figure 2B. Trace b was scaled by a factor of 1.30 to overlap trace c.
The results of Figures 2 and 3 are in qualitative agreement with recent findings in which mixtures of A and NA were found to produce a 34% inhibition of total Ba2+ current in whole-cell clamped RCCs (Hernández-Guijo et al., 1999). As reported, the combined action of NA and A caused almost no changes to the activation kinetics and was insignificantly voltage dependent compared with the pronounced voltage-dependent inhibition by ATP and opioids. The action of the catecholamines was larger than that reported here (25%). This could be attributed to various causes. First, in whole-cell clamp recordings, the action of NA + A was mediated by α- and β-ARs, whereas in the case of Figure 2 the action is limited to β-ARs. Stimulation of α-ARs produces a sizeable reduction of Ca2+ currents in bovine chromaffin cells (Kleppisch et al., 1992). Second, the A- and NA-induced inhibition is mediated by Gi/Go-proteins (Albillos et al., 1996a); it is thus probable that whole-cell recordings made in the presence of high concentrations of GTP (300 μm) enhance the inhibitory effects of the neurotransmitters with respect to the perforated-patch conditions, in which the basal levels of GTP may be significantly lower. A final consideration concerns the existence of the ISO-induced potentiation, which was overlooked in whole-cell recording conditions in either RCCs (Hernández-Guijo et al., 1999) or BCCs (Albillos et al., 1996a). This modulation is usually associated with the presence of PKA catalytic subunits responsible for L-channel phosphorylation, which may be lost during cell dialysis but preserved in cell-attached (Carbone et al., 2001) or perforated-patch conditions (Fig. 2).
The dual action of isoprenaline is mediated by β-ARs
The dual action of ISO was fully prevented by propranolol (1 μm). The unspecific β-AR inhibitor blocked both the ISO-induced inhibition and potentiation. Figure4 shows two examples of the antagonistic action of propranolol pooled from 11 cells in which the two effects were observed either in isolation or superimposed. In one case, ISO alone caused only a marked potentiation (Fig. 4A). In the other case, ISO produced an inhibition followed by a potentiation (Fig. 4B). The dual action of ISO was also fully antagonized by adding mixtures of the β1- and β2-selective receptor antagonists CGP 20712A (0.3 μm) and ICI 118,551 (0.1 μm) (n = 4; data not shown), indicating that both potentiation and inhibition were mediated by β-ARs.
Fig. 4.
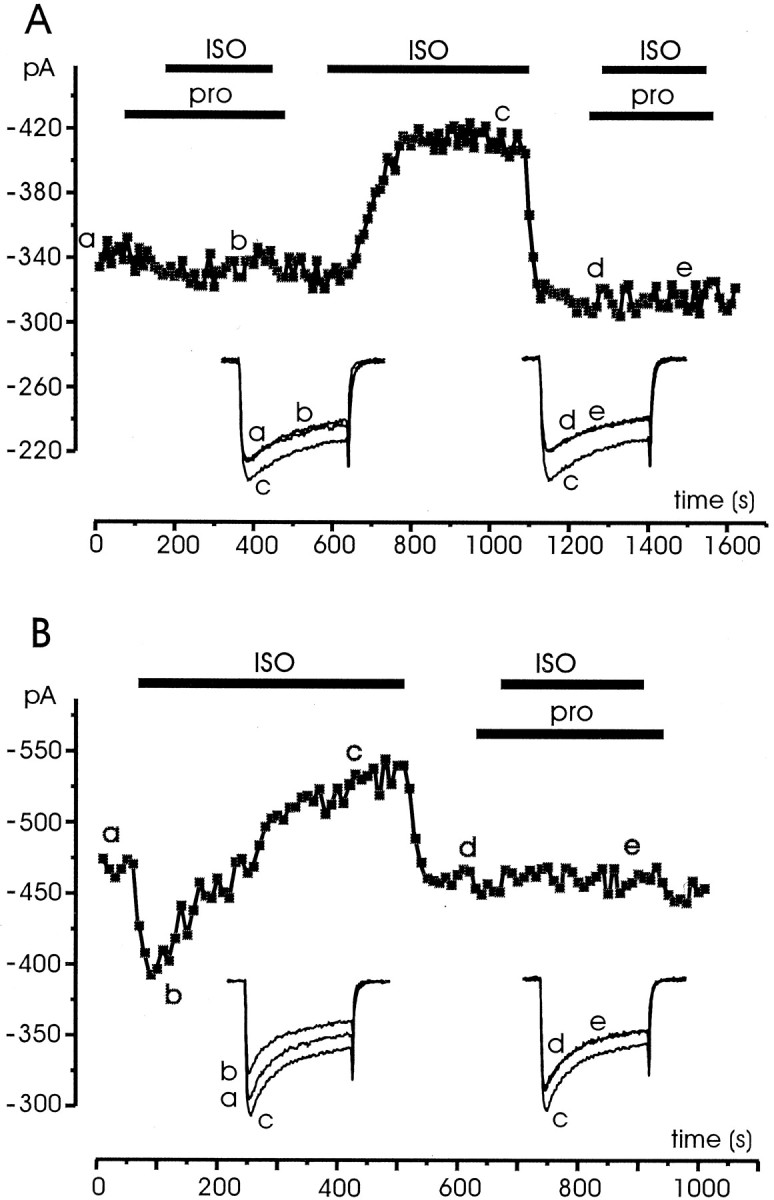
Propranolol prevents the effects of ISO stimulation regardless of type of response. A, The cell was initially superfused with control external solution and successively exposed to solutions containing propranolol (1 μm) and ISO (1 μm) for the periods indicated by the bars. B, Same recording conditions as in A except that the cell responded to ISO stimulation with a fast inhibition followed by a slow potentiation, both prevented by propranolol. Symbols,lettering, and traces have the same meaning as in previous figures.
Potentiation acts on L-channels whereas inhibition affects L- and non-L-channels
The presence of a Ca2+ current potentiation induced by ISO is indicative of the presence of a β-AR modulation on L-type channels mediated by cAMP/PKA. To verify this, we first tested whether the Ca2+ current potentiation observed in ∼50% of the cells persisted in the presence of nifedipine or after application of a PKA inhibitor, such as H89 and H7. As shown in Figure 5A, the DHP nifedipine (3 μm) completely prevented the slow potentiating effects of ISO observed before application of the L-channel blocker (n = 5). The action of the β-AR agonist on nifedipine-resistant currents was exclusively inhibitory. In 17 cells pretreated with nifedipine, the ISO-induced inhibition was 12.3 ± 2.2% after 1 min and 13.1 ± 1.9% after 5 min of the agonist application, suggesting that as for other autoreceptor-mediated pathways (Gandía et al., 1993; Albillos et al., 1996b), the non-L-type channels of RCCs (N, P/Q, R) are negatively coupled to β-ARs.
Fig. 5.
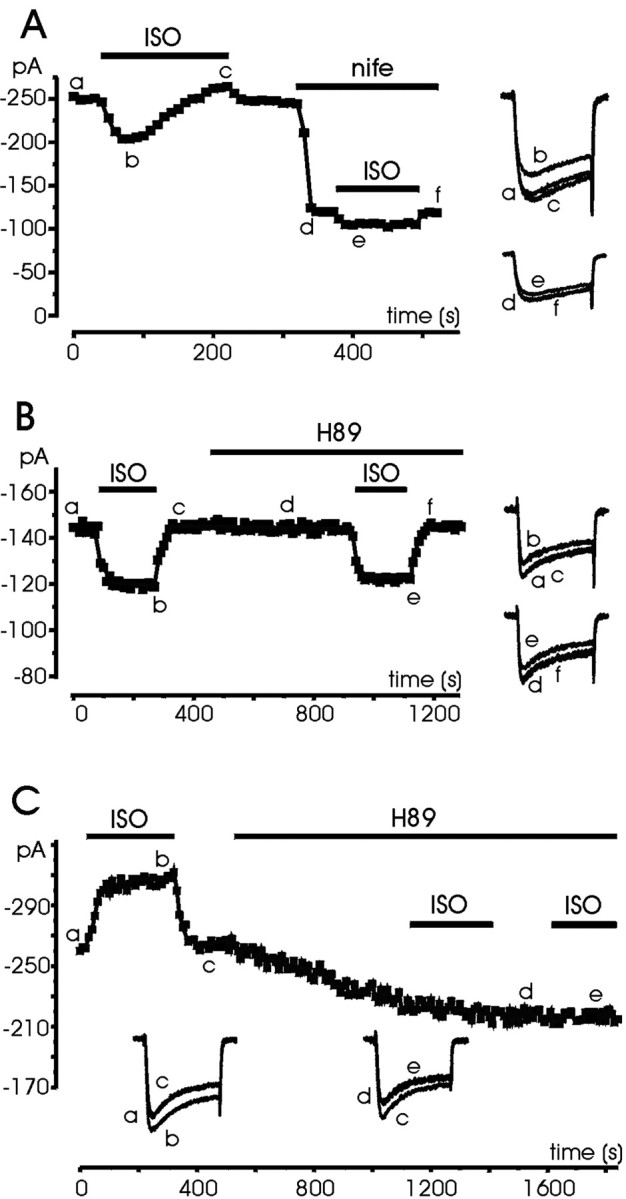
Nifedipine and H89 selectively prevent the Ca2+ current potentiation induced by ISO.A, The cell was first superfused with 1 μmISO in control external solution and then exposed to a solution containing 3 μm nifedipine and ISO for the periods indicated. Notice that after DHP application the cell responded with a weak inhibition and no potentiation. B, The cell was first tested for the effect of ISO, which was inhibitory. Subsequently the cell was superfused with a solution containing H89 (5 μm) and retested for the inhibitory effect of ISO.C, The cell first responded to ISO with a clear potentiation. Subsequent exposure to H89 (5 μm) reduced the Ca2+ current amplitude and removed the effects of ISO. Symbols, lettering, andtraces have the same meaning as in previous figures.
Acute application of H89 (5 μm;n = 12) or 30 min pretreatment with H7 (200 μm;n = 7) always prevented the potentiating action of ISO. Figure 5, B and C, shows the action of the PKA inhibitor in two representative cases. In the first case (Fig.5B), ISO had an inhibitory effect. H89 did not cause any significant decrease, and reapplication of ISO induced a comparable inhibition. In the second case (Fig. 5C), ISO induced a Ca2+ current potentiation, whereas H89 caused a partial decrease of control currents, which required several minutes to settle, suggesting the existence of a robust percentage of PKA-phosphorylated Ca2+channels under basal conditions. Subsequent applications of ISO had no effect. These data suggest that potentiation proceeds through a cAMP/PKA pathway and is limited to L-channels (Fig. 5A), whereas the inhibition proceeds regardless of the availability of PKA.
The ability of H89 and H7 to completely abolish the potentiating effect allowed a better quantification of the inhibitory effects of ISO on L- and non-L-channels. In seven cells pretreated with H7, we tested the effects of ISO before and after nifedipine application (Fig.6A) and observed no sign of potentiation. Although the two Ca2+ channel families contributed almost equally to the total current (52.4 ± 1.9% L and 46.9 ± 1.5% non-L), the former were more inhibited than the latter: 33.2 ± 5.8% (Fig. 6B, black bars) and 10.5 ± 4.5% (gray bars), respectively. Onset of inhibition was fast for both families of Ca2+ channels, and the same was true for the offset.
Fig. 6.

Percentage inhibition of L- and non-L channels in cells pretreated with H7. A, The H7-treated cell was first tested for the inhibitory effect of ISO and subsequently exposed to nifedipine (3 μm) and ISO (1 μm) for the periods indicated. The percentage inhibition of L-channels was estimated by subtracting the inhibition of the total current from that of non-L-currents properly weighted for the contribution to the total current. Symbols, lettering, and traces have the same meaning as in previous figures. B, Mean percentage inhibition induced by ISO on L-currents, non L-currents, and total currents obtained from six cells pretreated with H7.
PTX prevents Ca2+ channel inhibition favoring the potentiation through β1-ARs
An increasing number of reports on cardiac and neuronal L-type channels of rodent support the view of an effective coupling between β2-ARs and PTX-sensitive Gi/Go-proteins (Xiao et al., 1999; Davare et al., 2001). PTX treatment of rat heart cells was shown to potentiate the responses to ISO, very likely because of the removal of a negative coupling mediated by Gi/Go-proteins on L-channels (Xiao et al., 1995, 1999; Zhang et al., 2000). In chromaffin cells, PTX-sensitive G-proteins are negatively coupled to L- and non-L-type channels (Gandía et al., 1993;Albillos et al., 1996a,b; Currie and Fox, 1996; Carabelli et al., 1998;Hernández-Guijo et al., 1999); thus we hypothesized that the inhibitory effects induced by ISO might have been related to an effective coupling between β2-ARs and Gi/Go-proteins to downregulate Ca2+ channel gating, whereas β1-ARs might have been responsible for the opposite potentiating action mediated by cAMP/PKA. If this were the case, cell pretreatment with PTX would be expected to remove the inhibition and uncover the potentiating action of ISO through β1-ARs. Indeed, in 60% of cells treated for 12 hr with 100 ng/ml PTX (9 of 15 cells), we observed increases in current amplitude similar to control cells (25.9 ± 1.6% with PTX vs 25.2 ± 1.9% in control) (Fig.7A) but a higher probability of success with respect to the potentiation of control cells (60 vs 18.6%). In the remaining six cells, ISO had no effect. Thus, PTX treatment was sufficient to abolish the inhibitory action of ISO, suggesting that as for other neurotransmitters (ATP and opioids), Gi/Go-proteins mediated the inhibition of Ca2+ channels by β-ARs.
Fig. 7.
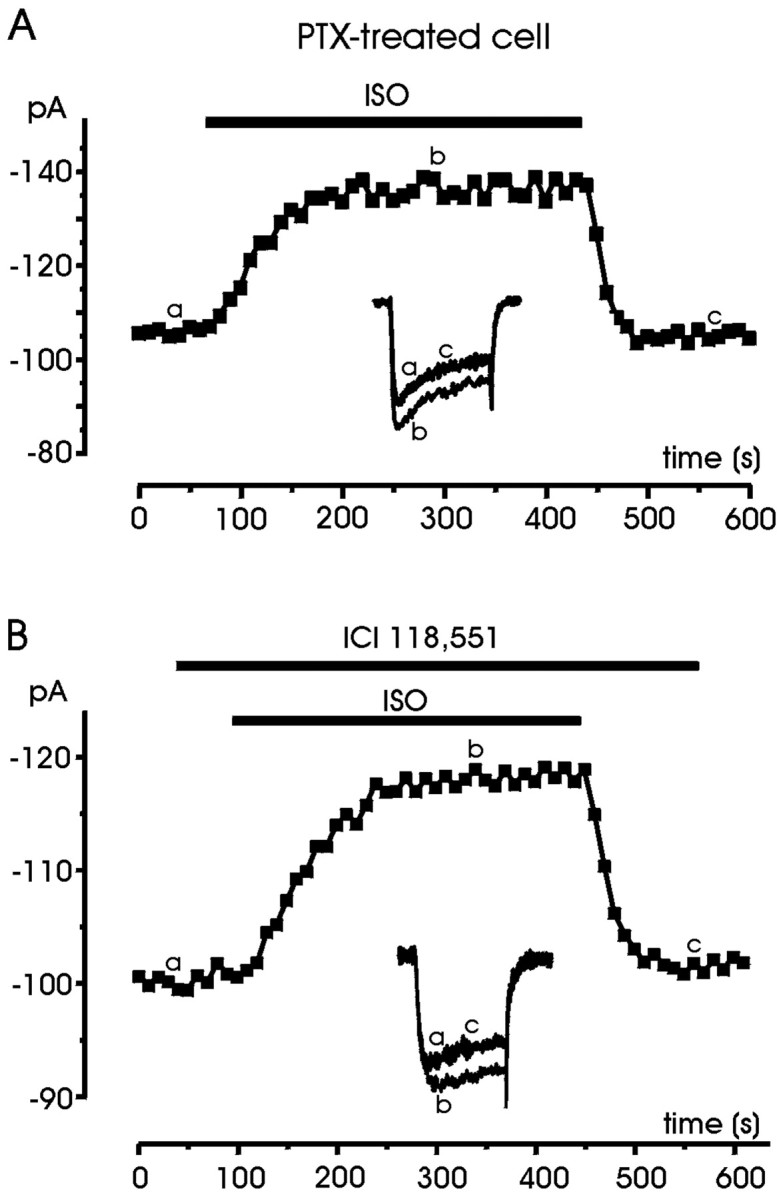
PTX treatment and ICI 118,551 remove the inhibitory action of ISO. A, The PTX-treated cell was first superfused with control external solution and then exposed to 1 μm ISO for the period indicated. B, The cell was first superfused with control external solution and then exposed to the β2-AR-selective antagonist ICI 118,551 (0.1 μm) and ISO (1 μm) for the periods indicated. Symbols, lettering, andtraces have the same meaning as in previous figures.
Further support for the hypothesis that inhibition is mediated by β2-AR and potentiation is controlled by β1-AR stimulation came by testing the effects of ISO (1 μm) on the presence of the specific β2-AR antagonist ICI 118,551 (0.1 μm) (β1-AR stimulation). In 10 cells perfused with ISO + ICI 118,551, we never observed inhibitory effects. β1-AR stimulation caused a clear potentiation in eight cells (21.3 ± 1.8%) (Fig. 7B), whereas it had no action in the remaining two (data not shown).
Ca2+ channel inhibition is mediated by β2-ARs
Zinterol is an agonist with a 25-fold higher affinity for β2-ARs over β1-ARs and is often used as a selective β2-AR agonist, in the range between 0.1 and 1 μm (Minneman et al., 1979). We therefore tested whether zinterol was able to mimic the inhibitory effects of ISO, which we expected to be selectively mediated by β2-AR. In agreement with this hypothesis, we found that in all the cells tested (n = 56), short (40 sec) or long (5 min) applications of 1 μmzinterol produced no sign of Ca2+ current potentiation (Fig.8A,B). In 75% of the cells, zinterol (1 μm) caused an average inhibition of 21.7 ± 1.0% (n = 42), which was comparable with that induced by ISO alone (mean 24.1%;p > 0.05), whereas in the remaining 25% of cells zinterol had no effect. The action of zinterol was fast and reversible. The onset was comparable with that of ISO (mean τon 6.5 sec; n = 10) (Fig.8C), whereas washout was particularly fast (mean τoff 6.6 sec; n = 7) when the agonist was applied for short periods of 40 sec (Fig. 8C,solid line) but became sixfold slower (mean τoff = 38.1 sec, n = 7) when applied for longer periods of 5 min (Fig. 8C, dashed line). A slow τoff for zinterol with respect to ISO is consistent with a high affinity of the agonist for β2-ARs and is in agreement with previous observations of β2-AR stimulation in frog myocytes in which the washout of zinterol was particularly slow (Skeberdis et al., 1997). The inhibitory action was mainly independent of voltage as shown by the down-scaling on Ca2+ currents, which occurred with nearly no changes on the activation kinetics (Fig. 8B), and by the unaltered shape and voltage of half-maximal activation of theI–V characteristics (Fig. 8D). Increasing concentrations of zinterol from 0.3 to 10 μm caused the same degree of block (21–23%) (Fig. 8E). The inhibition decreased to 9.2 ± 1.7% at 0.1 μm (n = 4) and to even lower values (not clearly measurable) at lower concentrations. All this suggests that 1 μm is a saturating concentration for maximal activation of β2-ARs in RCCs.
Fig. 8.
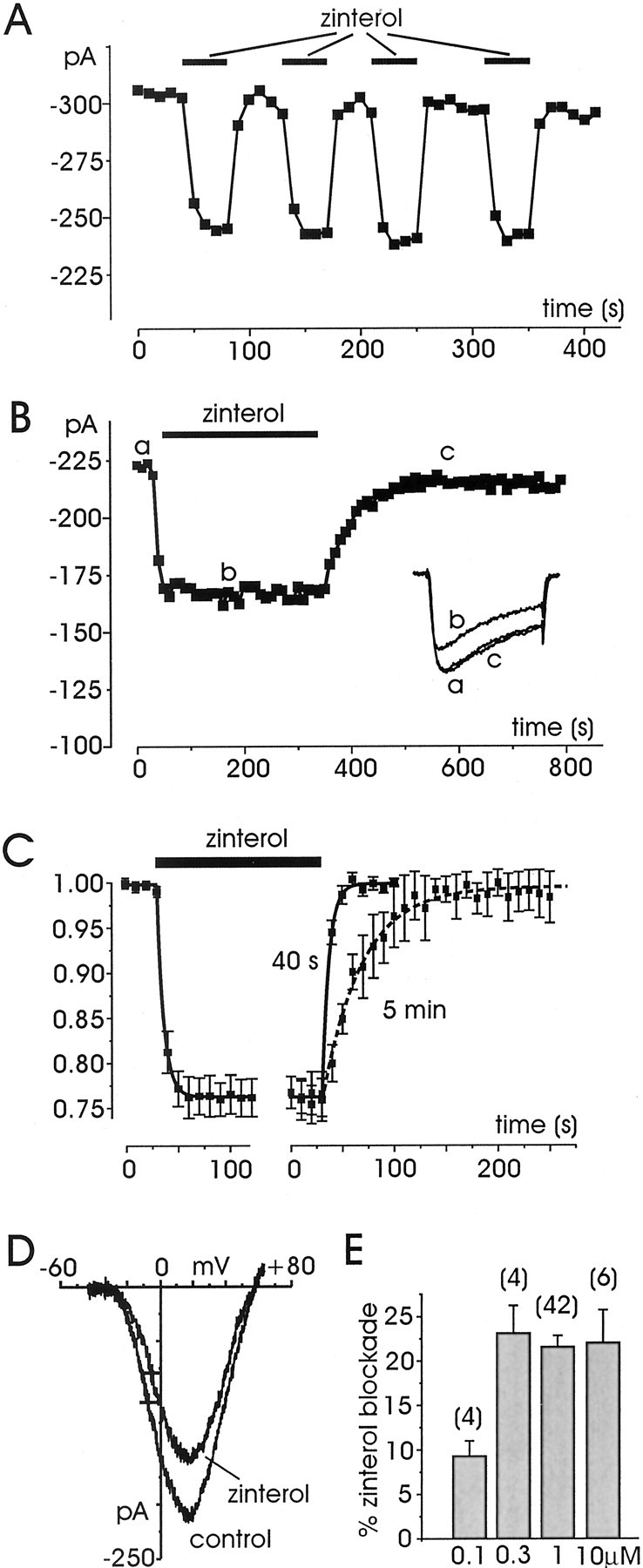
Zinterol induces only Ca2+current inhibitions. A, The cell was first superfused with control external solution and then exposed to four brief applications (40 sec) of the β2-AR-selective agonist zinterol (1 μm). Zinterol produced repeated reversible inhibitions of Ca2+ currents with rapid onset and offset. B, Longer applications of zinterol (5 min) produced the same degree of inhibition but slower offset. ForA and B, symbols,lettering, and traces have the same meaning as that in previous figures. C, Onset and offset of zinterol-induced inhibition calculated by averaging the Ca2+ current depression in cells sequentially exposed for 40 sec or 5 min to 1 μm zinterol. Control currents before zinterol were scaled and normalized to their maximum. The solid curves are the result of a fit with single exponential functions with time constantsτon = 6.5 sec (n = 10) and τoff= 6.6 sec (after 40 sec; n = 7) andτoff = 38.1 sec (after 5 min;n = 7). τoff values were calculated from cells on which zinterol was sequentially applied for short and long periods. D,I–V curves recorded before and during application of zinterol (1 μm). Voltages of half-maximal activation indicated by the crosses(V1/2) were −7.4 mV (control) and −5.3 mV (zinterol). E, Percentage of inhibition at different zinterol concentrations, showing that above 300 nm the percentage of inhibition is saturating.
Consistent with the idea that the inhibitory effects of zinterol were mediated by β2-ARs coupled to PTX-sensitive Gi/Go-proteins, the action of the agonist was fully prevented by application of 0.1 μm ICI 118,551 or cell treatment with PTX (Fig.9). In all the cells in which 1 μm zinterol produced a sizeable inhibition (22.8 ± 1.5%; n = 15), the β2-AR antagonist abolished the action of the agonist and caused no detectable changes on Ca2+ currents (Fig.9A). In PTX-treated cells (n = 8), even long exposures to zinterol had no effect (Fig. 9B). There was neither inhibition nor potentiation, suggesting that β2-AR stimulation in RCCs is strictly coupled to Gi/Go-proteins, and at variance with other cell preparations (Xiao et al., 1995; Skeberdis et al., 1997; Zhang et al., 2000; Davare et al., 2001), zinterol does not induce any measurable Ca2+ current potentiation. Consistent with this, the action of zinterol was not affected by cell pretreatment with H89 (Fig. 9C,left). Short-term (30 sec) and long-term (5 min) applications of the β2-AR agonist in H89-pretreated cells produced inhibitions comparable to those of control cells (21.3 ± 1.7% after 30 sec and 24.2 ± 1.7% after 5 min) (Fig. 9C).
Fig. 9.
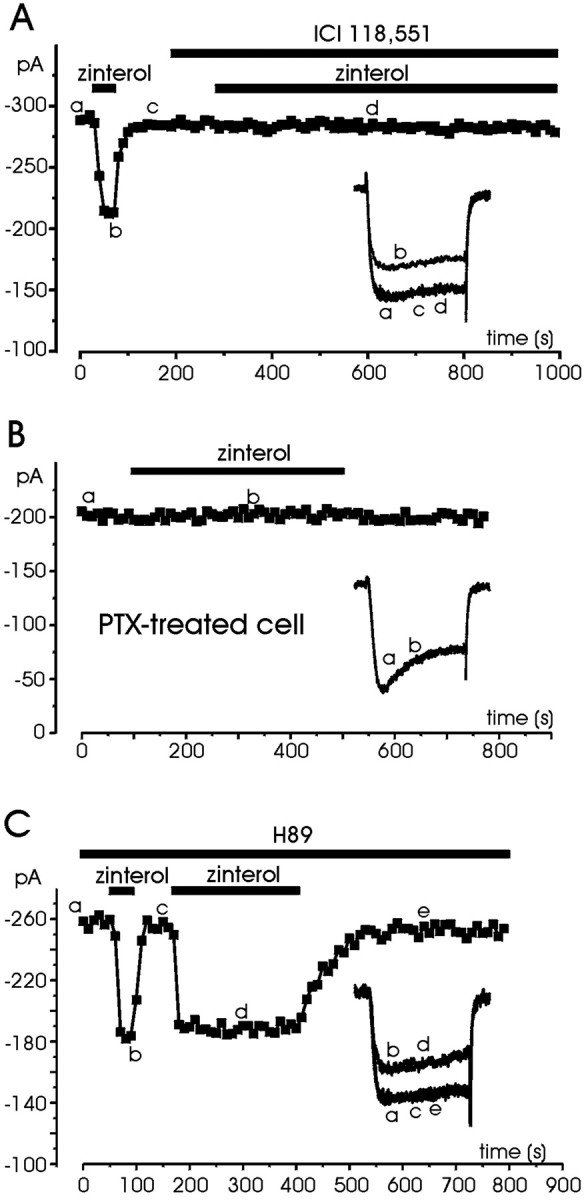
Pharmacology of zinterol action. A, The cell was tested for the effect of zinterol (1 μm) before and during exposure to the β2-AR-selective antagonist ICI 118,551. The antagonists could prevent the zinterol-induced inhibition. B, The PTX-treated cell was superfused with control external solution and tested for the action of zinterol (1 μm) during the period indicated. In eight cells pretreated with PTX, zinterol had practically no action.C, The cell was initially superfused with a solution containing H89 (5 μm) and then exposed to zinterol (1 μm) for the periods indicated. H89 was unable to prevent the inhibitory effects of zinterol. Symbols,lettering, and traces have the same meaning as that in previous figures.
As for ISO, nifedipine blocked a significant fraction of zinterol action (Fig. 10A), suggesting that the β2-AR-mediated action was larger on L- than on non-L-channels. The mean depression of total currents was 22.7%, whereas that of L- and non-L-currents was comparable with the values given in Figure 6B (33.4 and 12.2%, respectively) (Fig. 10B). To better evaluate the inhibitory effects of zinterol on L-channels, we tested the action of the agonists on RCCs in which N- and P/Q-currents were minimized by applying mixtures of ω-CTx-GVIA (1 μm), ω-Aga-IVA (200 nm), and ω-CTx-MVIIC (2 μm). Figure 10C shows that zinterol produces more or less the same degree of inhibition before and after ω-toxin treatment, implying a marked action on ω-toxin-resistant currents. In 12 cells, zinterol blocked by 23.2 ± 1.6% the control currents and by 43.9 ± 2.3% the toxin-resistant ones (Fig. 10C, inset). Given that ω-toxin-resistant currents in RCCs are mainly carried by L-channels and that R-currents stay in a ratio of 1:5 with respect to L-types, the true inhibition of L-currents is likely to be ∼38%, i.e., comparable with that induced by ISO in H7-treated cells (33.2%) (Fig. 6A) and slightly smaller than that induced by NA and A in whole-cell-clamped RCCs (48%) (Hernández-Guijo et al., 1999).
Fig. 10.
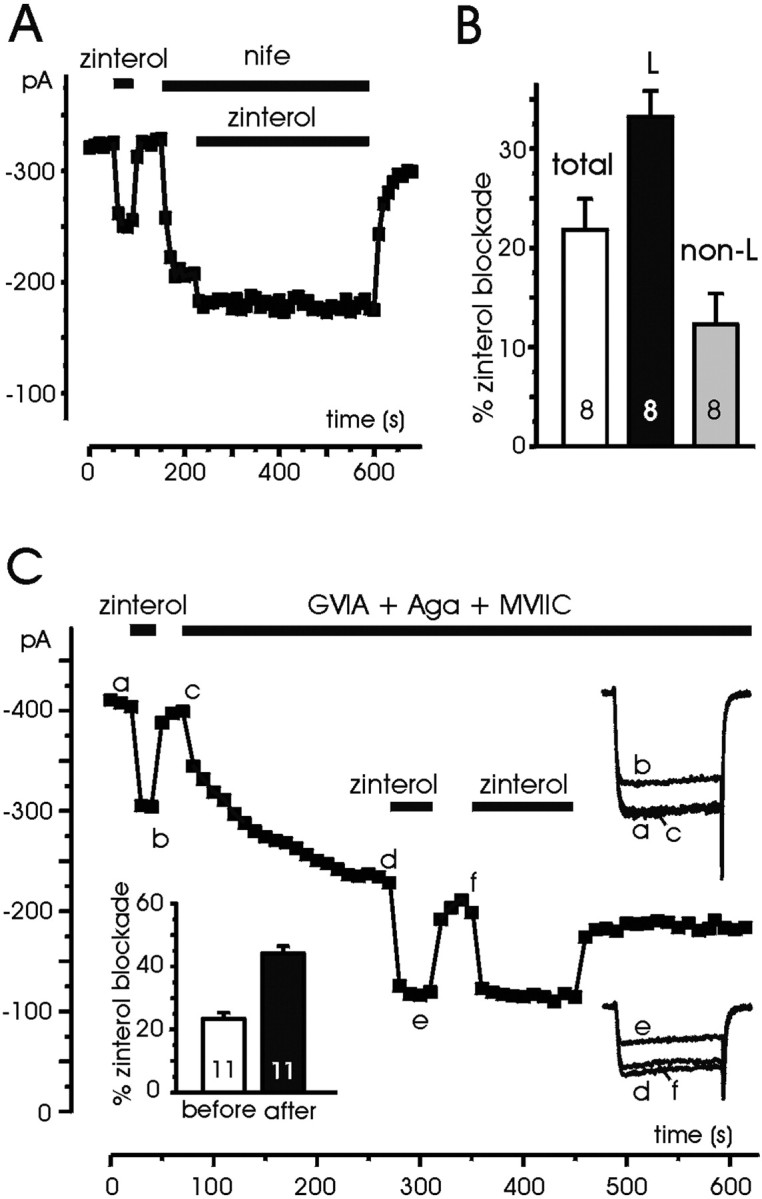
Zinterol has a preferential action on L-channels.A, The cell was superfused with control external solution and then exposed to zinterol (1 μm) before and during nifedipine application (3 μm) for the periods indicated. Notice the small action of zinterol on non-L-currents.B, Percentage of zinterol inhibition on L- and non-L-currents estimated from the blocking action of nifedipine (3 μm) on eight cells as illustrated in A.C, The cell was superfused with control external solution and then exposed to zinterol (1 μm) before and during addition of ω-CTx-GVIA (1 μm), ω-Aga-IVA (200 nm), and ω-CTx-MVIIC (2 μm) for the periods indicated. The action of zinterol is minimally affected by the toxins, suggesting that most of the effects of the β2-AR agonist are on toxin-resistant currents, which are carried mainly by L-channels. Symbols, lettering, andtraces have the same meaning as in previous figures.
A common pool of Gi/Go–coupled receptors mediates the β2-AR-induced inhibition
In a previous study on RCCs it was shown that separate or simultaneous activation of purinergic, opiodergic, and adrenergic autoreceptors (including β-ARs) produce the same degree of inhibition on L-currents by acting on a common pool of Gi/Go-proteins (Hernández-Guijo et al., 1999). This suggests that the β2-AR-mediated inhibition described here may be prevented if mixtures of opioids and ATP, capable of inhibiting the L-currents, already activate the same pool of Gi/Go-proteins. The same cannot be concluded, however, for the potentiating effects mediated by β1-ARs, which may still be able to potentiate the currents in the presence of opioids and ATP. Figure11A shows that the action of ISO on Ca2+ currents, inhibited previously by mixtures of ATP (100 μm) and δ/μ-opioid agonists [1 μm d-[Pen2-Pen5]-enkephalin (DPDPE), 10 μm [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO)], is only stimulatory. Addition of ISO, after the fast inhibition induced by ATP and opioids, does not produce any further depression, but only a slow potentiating effect, which brings the current amplitude slightly above the control level with an onset comparable with that described previously (mean τon 59.8 sec). In 20 similarly treated cells, we observed current increases similar to that of Figure11A. In 11 cells the potentiation was able to fully compensate for the inhibition, whereas in 9 cases there was no action. The same occurred if ISO followed the application of zinterol (Fig.11B), mimicking the dual action illustrated in Figures 2C and 4B. In nine cells, zinterol always produced inhibition of Ca2+currents, whereas subsequent application of ISO caused either a potentiation (six cells) or no effects (three cells). On average, the size of ISO-induced potentiation after zinterol exposure was comparable with the inhibition induced by zinterol, but in some case was even larger, as shown in Figure 11B. In conclusion, the action of ISO on the Ca2+ currents of RCCs appears to be composed of two components: (1) an inhibitory pathway mediated by β2-ARs that shares the same pool of Gi/Go-proteins of purinergic and opioidergic autoreceptors and (2) a stimulatory pathway mediated by β1-ARs that acts regardless of the Gi/Go-induced inhibition.
Fig. 11.
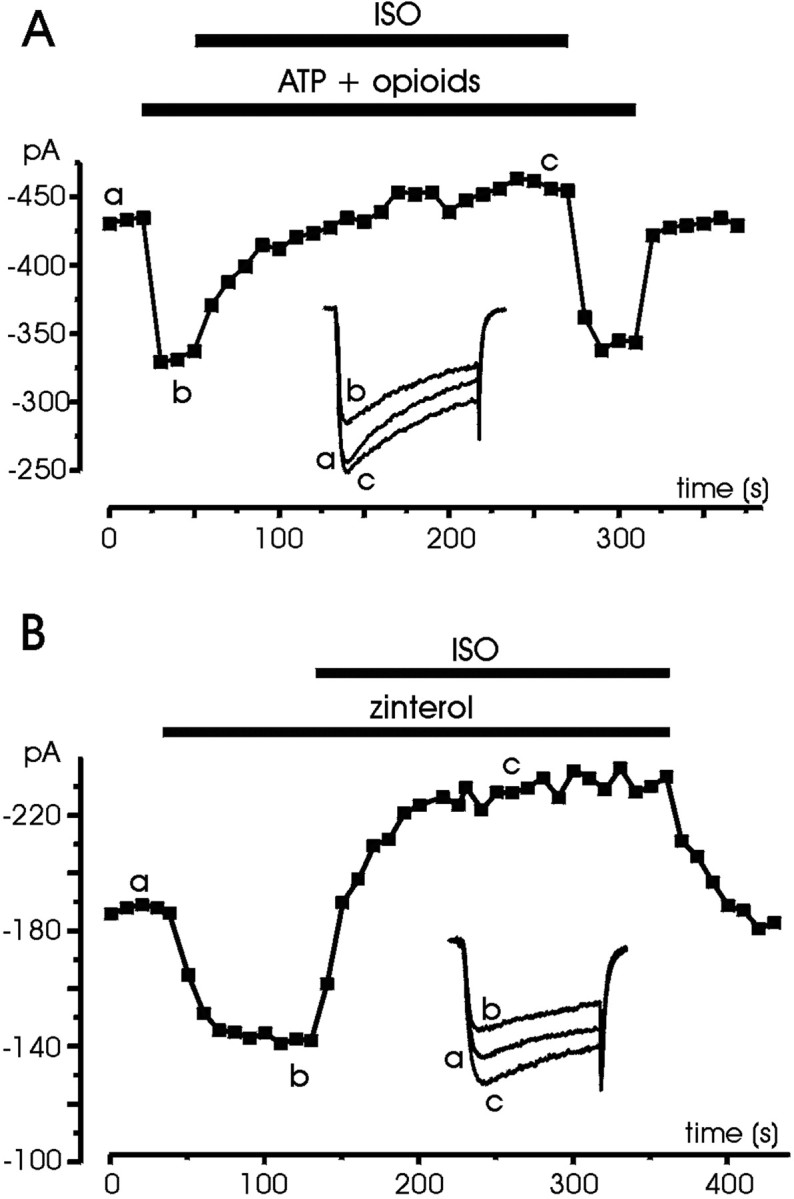
ISO produces only potentiation after the inhibitory effects induced by purinergic, opioidergic, and β2-adrenergic receptors. A, The cell was initially superfused with control external solution and then exposed to ATP (100 μm), DPDPE (1 μm), and DAMGO (10 μm), which caused inhibition. Addition of ISO (1 μm) caused no further depression but only a slow current potentiation. B, Sequential application of zinterol (1 μm) and ISO (1 μm) mimics the dual action of ISO shown in Figures 2C and 4B, proving that inhibition is mediated by β2-ARs, whereas potentiation by ISO proceeds through β1-ARs.Symbols, lettering, andtraces have the same meaning as in previous figures.
Discussion
We have provided evidence that RCCs possess two β-adrenergic signaling pathways exerting opposite effects on L-type channels. The two mechanisms involve distinct β-ARs and second messenger cascades. In one case, the action is mediated by β1-ARs and induces potentiation of L-channel activity through the activation of a cAMP/PKA pathway. In the second case, the action is mediated by β2-ARs and produces fast inhibition of L- and non-L-currents. The two modulations occur either sequentially or in isolation, thus furnishing a wide range of Ca2+ current amplitude variability, depending on the cell. Because adrenaline and noradrenaline are stored and released at high concentrations (Winkler and Westhead, 1980) and L-channels critically control the Ca2+-dependent exocytosis in rat adrenal glands (Nagayama et al., 1999), the present modulations may be crucial for the autocrine control of catecholamine release from adrenal medullas.
The two modulations support the existence of an effective β-AR signaling cascade in RCCs and help to solve the apparent contradiction that L-channels of chromaffin cells may be either inhibited by G-proteins or potentiated by cAMP (Carabelli et al., 2001). In whole-cell recordings of RCCs, β-AR activation by released catecholamines produces marked L-channel inhibition (Hernández-Guijo et al., 1999), whereas in cell-attached patches in which the catalytic subunits of the cAMP/PKA cascade are preserved, stimulation by ISO produces L-channel upregulation (Carbone et al., 2001). These observations find now a rationale in the coexistence of the two opposite signaling cascades, which appear voltage insensitive, i.e., down-modulation by G-proteins and upregulation by PKA do not require any special voltage sensitivity to occur.
The β1-AR-mediated potentiation
An interesting issue of this work is the uncovered β1-AR stimulation in endocrine cells, which increases L-currents through a PKA-mediated pathway. This modulation was overlooked in previous works (Doupnik and Pun, 1994; Albillos et al., 1996b; Hollins and Ikeda, 1996; Hernández-Guijo et al., 1999), for two reasons. First, the L-current increment in RCCs is modest (25% of ICa, corresponding to 35–40% of L-currents) with respect to the sixfold increase in ventricular myocytes (Hartzell, 1988). Second, there may be a loss of catalytic subunits responsible for PKA-mediated channel phosphorylation during cell dialysis in whole-cell recordings, which is preserved in patch-perforated (Figs. 2, 11) or cell-attached configuration (Carbone et al., 2001). The β1-AR stimulation of L-channels in RCCs possesses most of the prerequisites of β1-AR modulation in ventricular myocytes. The action is selective for L-channels and develops slowly. It is prevented by PKA inhibitors and persists after PTX treatment. At variance with cardiac myocytes, the amount of Ca2+current increase is significantly lower and recovers more quickly. This behavior may depend on (1) different β1-AR and adenylate cyclase isoforms and level of expression, (2) density and localization of PKAs, phosphodiesterases, and phosphatases near active sites, and (3) compartmentation and coupling among β1-ARs, signaling molecules, and downstream effectors.
Another interesting issue of β-AR stimulation in RCCs concerns the presence in some cases of basal levels of active PKA that is evident when RCCs are exposed to PKA inhibitors. This was not observed in BCCs (Carabelli et al., 2001) and may be responsible for the larger size of L-currents in RCCs (Gandía et al., 1995). The presence of a sustained basal level of active PKA may also be responsible for a PKA-mediated β1-AR phosphorylation with consequent desensitization (Collins et al., 1992), which could partially explain the smaller ISO-induced potentiation with respect to cardiac cells.
Various reports indicate a role for β1-ARs in catecholamine secretion in BCCs (Greenberg and Zinder, 1982; Wann et al., 1988; Ligier et al., 1994), but data concerning a modulatory action of β1-ARs on L-channels are lacking. The most convincing evidence of β1-AR modulation of L-channels in BCCs is indirect and shows that either ISO stimulation or forskolin stimulation induces cAMP increases and enhanced basal secretion of catecholamines (Parramon et al., 1995). Our findings thus represent the first direct evidence of a β1-AR-mediated potentiation of voltage-gated L-channels through a PKA signaling cascade in RCCs. This is of importance because cAMP production stimulates basal and evoked adrenal secretion (Morita et al., 1987; Przywara et al., 1996), increases the quantal size of exocytotic events (Machado et al., 2001), and upregulates T-type Ca2+ channels and TTX-resistant Na+ channels in chronically exposed RCCs (Novara et al., 2002).
Our data exclude a functional role for β3-AR in RCCs. Block of ISO stimulation by propranolol or mixtures of ICI 118,551 and CGP 20712A indeed exclude the involvement of β3-ARs in L-channel modulation (Strosberg, 1997). Previous conclusions based on radioligand bindings about the presence of atypical β3-ARs in BCCs (Clarkson et al., 1994) appear doubtful. As suggested recently, commonly used iodinated β-AR ligands exhibit low potency for β3-ARs and bind to β1-ARs, β2-ARs, and other receptors as well (Gauthier et al., 2000).
The β2-AR-mediated inhibition
Our work produces the first evidence for a fast negative coupling between β2-AR and voltage-gated L-channels through a common pool of Gi/Go-proteins shared with purinergic and opioidergic autoreceptors (Hernández-Guijo et al., 1999). Support for this comes from the following observations: (1) the inhibitory action of zinterol is fast (τon 6.5 sec at 1 μm), (2) PTX fully prevents the effects of β2-AR stimulation without uncovering any zinterol-induced potentiation of L-channel activity (Fig.9B), and (3) zinterol action is preserved by H89, thus excluding a possible role of PKA on L-current potentiation via β2-ARs. From this point of view, the β2-AR modulation of L-channels in RCCs is in line with a number of reports regarding adult rat ventricular myocytes in which β2-ARs appear strongly coupled to Gi-proteins and do not detectably elevate cytoplasmic cAMP levels (Kuznetsov et al., 1995; Laflamme and Becker, 1998). In general, it is widely accepted that beside coupling to Gs-proteins, β2-ARs can couple to Gi-proteins to (1) activate a phosphoprotein phosphatase 1 localized near cardiac L-channels (Xiao et al., 1995; Chen-Izu et al., 2000), (2) lead signals to the nucleus through a Ras–MAP kinases pathway (Daaka et al., 1997), (3) ensure rapid and specific activation of L-channels in hippocampus (Davare et al., 2001), or (4) mediate anti-apoptotic nuclear signaling in adult cardiac myocytes (Communal et al., 1999; Chesley et al., 2000;Zhu et al., 2001).
In our case, the β2-AR-mediated modulation is mainly inhibitory. We never observed Ca2+current potentiation with zinterol, even in the presence of PTX. This may be the consequence of subcellular compartmentation, already observed for neuronal and cardiac β2-ARs, in which G-proteins, ACs, PKA-anchoring-proteins, and phosphatases colocalize to form plasmalemma complexes, ensuring rapid activation or deactivation of specific signaling pathways (Xiao et al., 1999; Davare et al., 2001). The possibility that the β2-AR signaling switches from a Gs- to a Gi-mediated cascade after PKA-mediated phosphorylation of the receptor itself is unlikely (Daaka et al., 1997). In fact, L-current inhibition by ISO or zinterol proceeds regardless of the presence of PKA inhibitors. Alternative possibilities are discussed by Steinberg and Brunton (2001) and include subcellular localization of phosphodiesterases in the vicinity of the receptor, maintaining low levels of cAMP independently of the degree of β2-AR stimulation. Alternatively, it is possible that β2-ARs are localized in membrane regions rich in Gi-proteins and poor in Gs-proteins (or ACs). Quick sequestration of β2-ARs by Gi-proteins may prevent Gs-protein cascade, thus causing direct Ca2+ current inhibition.
Relevance of L-channel modulation by β1- and β2-AR stimulation
Ca2+ currents in chromaffin cells are autocrinally modulated by two distinct inhibitory pathways: a voltage-dependent one acting on N- and P/Q-channels and a voltage-independent one acting on L-channels (Carbone et al., 2001). Both mechanisms are fast, membrane delimited, and Gi/Go-protein mediated. The former is removed during strong or repeated depolarizations and may thus contribute to the rapid rise of intracellular Ca2+ and catecholamine secretion during maximal sympathetic stimulation (fight or flight response). The latter may act as a constant negative feedback to limit the outcome of catecholamines during basal and sustained chromaffin cell activity. The slow β1-AR L-channel potentiation described here appears suitable to remove the fast autocrine inhibition of L-channels induced by purinergic, opioidergic, and β2-ARs and to further support Ca2+ rising induced by the quick removal of non-L-type channel inhibition during repeated stimulation. However, the role of β1-ARs may not be linked only to L-channel modulation. cAMP elevation has been shown to act downstream of the Ca2+-entry by enhancing the size of quantal events (Machado et al., 2001), the depolarization-induced exocytosis (V. Carabelli, A. R. Artalejo, E. Carbone, unpublished results), and the density of newly available T-type channels with consequent increased cell excitability (Novara et al., 2002). The role of β2-AR appears less evident but nevertheless interesting. The β2-AR modulation described here resembles that induced by purinergic and opioidergic receptors and would probably ensure Ca2+ channel inhibition in those cells in which either the content of ATP and opioids is particularly low or purinergic and opioid receptors are weakly expressed (Bunn et al., 1988). However, the role of β2-ARs may also be linked to crucial cell functions indirectly related to Ca2+ channels, such as the anti-apoptotic action developed via Gi-proteins to counterbalance the negative action of β1-ARs on cell survival (Communal et al., 1999; Chesley et al., 2000).
T.C. and J.M.H.-G. contributed equally to this work.
This work was supported by the Italian Consiglio Nazionale delle Ricerche (Grant 01.00443.ST97), by the Ministero dell'Istruzione Università e Ricerca (Grant 2001055324-006), and by a Marie Curie Fellowship of the European Community Human Potential Programme to J.M.H.-G. (Contract HPMF-CT-2000-00899).
Correspondence should be addressed to Dr. Emilio Carbone, Department of Neuroscience, Corso Raffaello 30, I-10125 Torino, Italy. E-mail:emilio.carbone@unito.it.
T. Cesetti's present address: Department of Biomedical Science, Via G. Colombo 3, I-35122 Padova, Italy.
References
- 1.Albillos A, Gandía L, Michelena P, Gilabert JA, del Valle M, Carbone E, García AG. The mechanism of calcium channel facilitation in bovine chromaffin cells. J Physiol (Lond) 1996a;494:687–695. doi: 10.1113/jphysiol.1996.sp021524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albillos A, Carbone E, Gandía L, García AG, Pollo A. Opioid inhibition of Ca2+ channel subtypes in bovine chromaffin cells: selectivity of action and voltage-dependence. Eur J Neurosci. 1996b;8:1561–1570. doi: 10.1111/j.1460-9568.1996.tb01301.x. [DOI] [PubMed] [Google Scholar]
- 3.Artalejo CR, Ariano MA, Perlman RA, Fox AP. Activation of facilitation calcium channels in chromaffin cells by D1 dopamine receptors through a cAMP/protein kinase A-dependent mechanism. Nature. 1990;348:239–242. doi: 10.1038/348239a0. [DOI] [PubMed] [Google Scholar]
- 4.Barry PH, Lynch JW. Liquid junction potentials and small cell effects in patch-clamp analysis. J Membr Biol. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- 5.Bean BP, Nowycky MC, Tsien RW. β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375. doi: 10.1038/307371a0. [DOI] [PubMed] [Google Scholar]
- 6.Bunn SJ, Marley PD, Livett BG. The distribution of opioid binding subtypes in the bovine adrenal medulla. Neuroscience. 1988;27:1081–1094. doi: 10.1016/0306-4522(88)90212-6. [DOI] [PubMed] [Google Scholar]
- 7.Carabelli V, Carra I, Carbone E. Localised secretion of ATP and opioids revealed through single Ca2+ channel modulation in bovine chromaffin cells. Neuron. 1998;20:1255–1268. doi: 10.1016/s0896-6273(00)80505-x. [DOI] [PubMed] [Google Scholar]
- 8.Carabelli V, Hernández-Guijo JM, Baldelli P, Carbone E. Direct autocrine inhibition and cAMP-dependent potentiation of single L-type Ca2+ channels in bovine chromaffin cells. J Physiol (Lond) 2001;532:73–90. doi: 10.1111/j.1469-7793.2001.0073g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carabelli V, D'Ascenzo M, Carbone E, Grassi C. Nitric oxide inhibits neuroendocrine Cav1 L-channel gating via cGMP-dependent protein kinase in cell-attached patches of bovine chromaffin cells. J Physiol (Lond) 2002;541:351–366. doi: 10.1113/jphysiol.2002.017749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carbone E, Carabelli V, Cesetti T, Baldelli P, Hernández-Guijo JM, Giusta L. G-protein- and cAMP-dependent L-channel gating modulation: a many fold system to control calcium entry in neurosecretory cells. Pflügers Arch. 2001;442:801–813. doi: 10.1007/s004240100607. [DOI] [PubMed] [Google Scholar]
- 11.Chen-Izu Y, Xiao RP, Izu LT, Cheng H, Zhou YY, Kuschel M, Spurgeon H, Lakatta EG. Gi-dependent localization of β2-adrenergic receptor signaling to L-type Ca2+ channels. Biophys J. 2000;79:2547–2556. doi: 10.1016/S0006-3495(00)76495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG, Crow MT. Gi-dependent localization of β2-adrenergic receptor signaling to L-type Ca2+ channels. Circ Res. 2000;87:1172–1179. doi: 10.1161/01.res.87.12.1172. [DOI] [PubMed] [Google Scholar]
- 13.Clarkson K, Tobin JR, Breslow MJ. Atypical β-adrenoceptor in bovine adrenal medulla. Eur J Pharmacol. 1994;263:R1–2. doi: 10.1016/0014-2999(94)90730-7. [DOI] [PubMed] [Google Scholar]
- 14.Collins S, Caron MG, Lefkowitz RJ. From ligand binding to gene expression: new insights into the regulation of G-protein coupled receptors. Trends Biochem Sci. 1992;17:37–39. doi: 10.1016/0968-0004(92)90425-9. [DOI] [PubMed] [Google Scholar]
- 15.Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocytes apoptosis. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 16.Currie KPM, Fox AP. ATP serves as a negative feedback inhibitor of voltage-gated Ca2+ channel currents in cultured bovine chromaffin cells. Neuron. 1996;16:1027–1036. doi: 10.1016/s0896-6273(00)80126-9. [DOI] [PubMed] [Google Scholar]
- 17.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 18.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinerg RJ, Horne MC, Hoshi T, Hell JW. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel CaV12. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- 19.Doupnik CA, Pun RY. Cyclic AMP-dependent phosphorylation modifies the gating properties of L-type Ca2+ channels in bovine adrenal chromaffin cells. Pflügers Arch. 1992;420:61–71. doi: 10.1007/BF00378642. [DOI] [PubMed] [Google Scholar]
- 20.Doupnik CA, Pun RYK. G-protein activation mediates prepulse facilitation of Ca2+ channel currents in bovine chromaffin cells. J Membr Biol. 1994;140:47–56. doi: 10.1007/BF00234485. [DOI] [PubMed] [Google Scholar]
- 21.Gandía L, García AG, Morad M. ATP modulation of calcium channels in chromaffin cells. J Physiol (Lond) 1993;470:55–72. doi: 10.1113/jphysiol.1993.sp019847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandía L, Borges R, Albillos A, García AG. Multiple calcium channel subtypes in isolated rat chromaffin cells. Pflügers Arch. 1995;430:55–63. doi: 10.1007/BF00373839. [DOI] [PubMed] [Google Scholar]
- 23.Gauthier C, Langin D, Balligand JL. β3-Adrenoceptors in the cardiovascular system. Trends Pharmacol Sci. 2000;21:426–431. doi: 10.1016/s0165-6147(00)01562-5. [DOI] [PubMed] [Google Scholar]
- 24.Greenberg A, Zinder O. Alpha- and beta-receptor control of catecholamine secretion from isolated adrenal medulla cells. Cell Tissue Res. 1982;226:655–665. doi: 10.1007/BF00214792. [DOI] [PubMed] [Google Scholar]
- 25.Hartzell HC. Regulation of cardiac ion channels by catecholamines, acetylcholine and 2nd messenger systems. Prog Biophys Mol Biol. 1988;52:165–247. doi: 10.1016/0079-6107(88)90014-4. [DOI] [PubMed] [Google Scholar]
- 26.Hernández-Guijo JM, Carabelli V, Gandía L, García AG, Carbone E. Voltage-independent autocrine modulation of L-type channels mediated by ATP, opioids and catecholamines in rat chromaffin cells. Eur J Neurosci. 1999;11:3574–3584. doi: 10.1046/j.1460-9568.1999.00775.x. [DOI] [PubMed] [Google Scholar]
- 27.Hollins B, Ikeda SR. Inward currents underlying action potentials in rat adrenal chromaffin cells. J Neurophysiol. 1996;76:1195–1211. doi: 10.1152/jn.1996.76.2.1195. [DOI] [PubMed] [Google Scholar]
- 28.Jankowski JA, Schroeder TJ, Ciolkowski EL, Wightman RM. Temporal characteristics of quantal secretion of catecholamines from adrenal medullary cells. J Biol Chem. 1993;268:14694–14700. [PubMed] [Google Scholar]
- 29.Kilts JD, Gerhardt MA, Richardson MD, Sreeram G, Mackensen GB, Grocott HP, White WD, Davis RD, Newman MF, Reves JG, Schwinn DA, Kwatra MM. β2-Adrenergic and several other G protein-coupled receptors in human atrial membranes activate both Gi and Gs. Circ Res. 2000;87:705–709. doi: 10.1161/01.res.87.8.705. [DOI] [PubMed] [Google Scholar]
- 30.Kleppisch T, Ahnert-Hilger G, Gollasch M, Spicher K, Hescheler J, Schultz G, Rosenthal W. Inhibition of voltage-dependent Ca2+ channels via α2-adrenergic and opioid receptors in cultured bovine chromaffin cells. Pflügers Arch. 1992;421:131–137. doi: 10.1007/BF00374819. [DOI] [PubMed] [Google Scholar]
- 31.Korn SJ, Marty A, Connor JA, Horn R. Perforated patch recording. Methods Neurosci. 1991;4:264–273. [Google Scholar]
- 32.Kuznetsov V, Pak E, Robinson RB, Steinberg SF. β2-Adrenergic receptor actions in neonatal and adult rat ventricular myocytes. Circ Res. 1995;76:40–52. doi: 10.1161/01.res.76.1.40. [DOI] [PubMed] [Google Scholar]
- 33.Laflamme MA, Becker PL. Do β2-adrenergic receptors modulate Ca2+ in adult rat ventricular myocytes? Am J Physiol. 1998;274:H1308–1314. doi: 10.1152/ajpheart.1998.274.4.H1308. [DOI] [PubMed] [Google Scholar]
- 34.Ligier B, Breslow MJ, Clarkson K, Raff H, Traystman RJ. Adrenal blood flow and secretory effects of adrenergic receptor stimulation. Am J Physiol. 1994;266:H220–227. doi: 10.1152/ajpheart.1994.266.1.H220. [DOI] [PubMed] [Google Scholar]
- 35.Lim W, Kim SJ, Yan HD, Kim J. Ca2+-channel-dependent and -independent inhibition of exocytosis by extracellular ATP in voltage-clamped rat adrenal chromaffin cells. Pflügers Arch. 1997;435:34–42. doi: 10.1007/s004240050481. [DOI] [PubMed] [Google Scholar]
- 36.Machado JD, Morales A, Gomez JF, Borges R. cAMP modulates exocytotic kinetics and increases quantal size in chromaffin cells. Mol Pharmacol. 2001;60:514–520. [PubMed] [Google Scholar]
- 37.Magnelli V, Baldelli P, Carbone E. Antagonists-resistant calcium currents in rat embryo motoneurons. Eur J Neurosci. 1998;10:1810–1825. doi: 10.1046/j.1460-9568.1998.00178.x. [DOI] [PubMed] [Google Scholar]
- 38.Minneman KP, Hedberg A, Molinoff PB. Comparison of beta-adrenergic receptor subtypes in mammalian tissues. J Pharmacol Exp Ther. 1979;211:502–508. [PubMed] [Google Scholar]
- 39.Morita K, Dohi T, Kitayama S, Koyama Y, Tsujimoto A. Enhancement of stimulation-evoked catecholamine release from cultured bovine adrenal chromaffin cells by forskolin. J Neurochem. 1987;48:243–247. doi: 10.1111/j.1471-4159.1987.tb13154.x. [DOI] [PubMed] [Google Scholar]
- 40.Nagayama T, Matsumoto T, Kuwakubo F, Fukushima Y, Yoshida M, Suzuki-Kusaba M, Hisa H, Kimura T, Satoh S. Role of calcium channels in catecholamine secretion in the rat adrenal gland. J Physiol (Lond) 1999;520:503–512. doi: 10.1111/j.1469-7793.1999.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novara M, Baldelli P, Hernández-Guijo JM, Giusta L, Carbone E. Chronic exposure to cAMP upregulates T-type Ca2+ channels and TTX-insensitive Na+ channels in cultured rat chromaffin cells. J Physiol (Lond) 2002;543.P:67P. [Google Scholar]
- 42.Parramon M, González MP, Oset-Gasque MJ. A reassessment of the modulatory role of cyclic AMP in catecholamine secretion by chromaffin cells. Br J Pharmacol. 1995;114:517–523. doi: 10.1111/j.1476-5381.1995.tb13257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollo A, Lovallo M, Biancardi E, Sher E, Socci C, Carbone E. Sensitivity to dihydropyridines, ω-conotoxins and noradrenaline reveals multiple high-voltage activated Ca2+ channels in rat insulinoma and human pancreatic β-cells. Pflügers Arch. 1993;423:462–471. doi: 10.1007/BF00374942. [DOI] [PubMed] [Google Scholar]
- 44.Powell AD, Teschemacher AG, Seward EP. P2Y purinoceptors inhibit exocytosis in adrenal chromaffin cells via modulation of voltage-operated calcium channels. J Neurosci. 2000;20:606–616. doi: 10.1523/JNEUROSCI.20-02-00606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Przywara DA, Guo X, Angelilli ML, Wakade TD, Wakade AR. A non-cholinergic transmitter, pituitary adenylate cyclase-activating polypeptide, utilizes a novel mechanism to evoke catecholamines secretion in rat adrenal chromaffin cells. J Biol Chem. 1996;271:10545–10550. doi: 10.1074/jbc.271.18.10545. [DOI] [PubMed] [Google Scholar]
- 46.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 47.Skeberdis VA, Jurevicius J, Fischmeister R. Beta-2 adrenergic activation of L-type Ca2+ current in cardiac myocytes. J Pharmacol Exp Ther. 1997;283:452–461. [PubMed] [Google Scholar]
- 48.Steinberg SF, Brunton LI. Compartmentation of G-protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- 49.Strosberg AD. Structure and function of the β3-adrenergic receptor. Annu Rev Pharmacol Toxicol. 1997;37:421–450. doi: 10.1146/annurev.pharmtox.37.1.421. [DOI] [PubMed] [Google Scholar]
- 50.Ulate G, Scott SR, Gonzalez J, Gilabert JA, Artalejo AR. Extracellular ATP regulates exocytosis in inhibiting multiple Ca2+ channel types in bovine chromaffin cells. Pflügers Arch. 2000;439:304–314. doi: 10.1007/s004249900185. [DOI] [PubMed] [Google Scholar]
- 51.Xiao RP, Xiangwu JI, Lakatta EG. Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol. 1995;47:322–329. [PubMed] [Google Scholar]
- 52.Xiao RP, Cheng H, Zhou YY, Kuschel M, Lakatta EG. Recent advances in cardiac β2-adrenergic signal transduction. Circ Res. 1999;85:1092–1100. doi: 10.1161/01.res.85.11.1092. [DOI] [PubMed] [Google Scholar]
- 53.Wann DC, Powis DA, Marley PD, Livett BG. Effects of alpha- and beta-adrenoceptor agonists and antagonists on ATP and catecholamine release and desensitization of the nicotinic response in bovine adrenal chromaffin cells. Biochem Pharmacol. 1988;37:725–736. doi: 10.1016/0006-2952(88)90147-5. [DOI] [PubMed] [Google Scholar]
- 54.Winkler H, Westhead E. The molecular organisation of adrenal chromaffin granules. Neuroscience. 1980;5:1803–1823. doi: 10.1016/0306-4522(80)90031-7. [DOI] [PubMed] [Google Scholar]
- 55.Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ, Siddiqui MA. β-Adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation. 2000;102:344–350. doi: 10.1161/01.cir.102.3.344. [DOI] [PubMed] [Google Scholar]
- 56.Zhang SJ, Cheng H, Zhou YY, Wang DJ, Zhu W, Ziman B, Spurgoen H, Lefkowitz RJ, Lakatta EG, Koch WJ, Xiao RP. Inhibition of spontaneous β2-adrenergic activation rescues β1-adrenergic contractile response in cardiomyocytes overexpressing β2-adrenoceptors. J Biol Chem. 2000;275:21773–21779. doi: 10.1074/jbc.M909484199. [DOI] [PubMed] [Google Scholar]
- 57.Zhou YY, Cheng H, Song LS, Wang D, Lakatta EG, Xiao RP. Spontaneous β2-adrenergic signaling fails to modulate L-type Ca2+ current in mouse ventricular myocytes. Mol Pharmacol. 1999;56:485–493. doi: 10.1124/mol.56.3.485. [DOI] [PubMed] [Google Scholar]
- 58.Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. Dual modulation of cell survival and cell death by β2-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci USA. 2001;98:1607–1612. doi: 10.1073/pnas.98.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]