

Abstract
The initiation of mammalian puberty requires the activation of hypothalamic neurons secreting the neuropeptide luteinizing hormone-releasing hormone (LHRH). It is thought that this activation is caused by changes in trans-synaptic input to LHRH neurons. More recently, it has been postulated that the pubertal increase in LHRH secretion in female animals also requires neuron–glia signaling mediated by growth factors of the epidermal growth factor (EGF) family and their astrocytic erbB receptors. Although it appears clear that functional astrocytic erbB1 receptors are necessary for the timely advent of puberty, the physiological contribution that erbB4 receptors may make to this process has not been established. To address this issue, we generated transgenic mice expressing a dominant-negative erbB4 receptor (DN–erbB4) under the control of the GFAP promoter, which targets transgene expression to astrocytes. DN–erbB4 expression is most abundant in hypothalamic astrocytes, where it blocks the ligand-dependent activation of glial erbB4 and erbB2 receptors, without affecting erbB1 (EGF) receptor signaling. Mice carrying the transgene exhibit delayed sexual maturation and a diminished reproductive capacity in early adulthood. These abnormalities are related to a deficiency in pituitary gonadotropin hormone secretion, caused by impaired release of LHRH, the hypothalamic neuropeptide that controls sexual development. In turn, the reduction in LHRH release is caused by the inability of hypothalamic astrocytes to respond to neuregulin (NRG) with production of prostaglandin E2, which in wild-type animals mediates the stimulatory effect of astroglial erbB receptor activation on neuronal LHRH release. Thus, neuron–astroglia communication via NRG-erbB4/2 receptor signaling appears to be essential for the timely unfolding of the developmental program by which the brain controls mammalian sexual maturation.
Keywords: neuregulin, neuroendocrine, mammalian puberty, hypothalamus, astrocytes, neuron–glia interactions
Introduction
The onset of puberty, one of the most critical developmental events in postnatal mammalian life, is determined by changes that occur within the CNS and, in particular, the hypothalamus. The pubertal activation of the hypothalamic–pituitary–gonadal axis requires an increased secretion of luteinizing hormone-releasing hormone (LHRH) by neuroendocrine neurons, which in rodents are located in the preoptic region of the hypothalamus. These neurons extend their neurosecretory axons to the median eminence, where LHRH is released into the portal vasculature for delivery to the adenohypophysis. There, LHRH induces the secretion of pituitary gonadotropin hormones that promote gonadal development and support reproductive physiology. In this way, LHRH is essential for both sexual development and mature reproductive function.
The mechanisms responsible for the timely release of LHRH during puberty are incompletely understood. Early studies showed that the increase in LHRH release is not caused by events intrinsic to the LHRH neurons themselves but occurs in response to changing inputs from synaptically connected neuronal networks (Terasawa, 1999; Ojeda et al., 2001). However, other experiments suggested that, in addition to trans-synaptic communication, neuron–glia signaling in the hypothalamus might be of critical importance for the activation of LHRH secretion during female sexual development (for review, see Ojeda et al., 2000).
These studies indicated that receptors of the epidermal growth factor receptor (EGFR) family, also known as erbB receptors, may play important roles in the hypothalamic control of puberty. There are four known members of the erbB receptor family. Three of them, erbB1, erbB3, and erbB4, bind and are activated by cognate ligands. In contrast, erbB2 acts as a coreceptor or auxiliary subunit recruited by ligand binding to the other receptors (for review, see Yarden and Sliwkowski, 2001). Hypothalamic astrocytes express the erbB1, erbB2, and erbB4 receptors but not erbB3. In addition, cells in the hypothalamus express the erbB1 ligand transforming growth factor α (TGFα) and several forms of the erbB4 ligand neuregulin (NRG) (Ma et al., 1992, 1994,1999; Chen et al., 1994; Corfas et al., 1995).
Pharmacological inhibition of EGFRs (erbB1) targeted to the median eminence of the hypothalamus (Ma et al., 1992) or inhibition of erbB2 synthesis via the intracerebral administration of antisense oligodeoxynucleotides (Ma et al., 1999) delayed the onset of female puberty in rats. Moreover, in vitro studies using astrocytes and an LHRH-producing neuronal cell line showed that erbB receptor ligands can stimulate LHRH release from the neuronal cells, but do so indirectly, by inducing astrocytes to secrete prostaglandin E2 (PGE2) (Ma et al., 1997,1999), which can then elicit release of the neuropeptide from the neuronal cell line (Voigt et al., 1996). These observations led to the hypothesis that erbB receptors are components of the cell-to-cell communication system used by astrocytes to regulate LHRH secretion during development and hence the initiation of puberty. However, despite of the insights offered by these studies, the physiological contribution of astrocytic erbB4 receptor signaling to normal sexual development has not been demonstrated, primarily because of the unavailability of an animal model in which the function of these receptors is disrupted in a cell-specific manner. Mice lacking the genes encoding NRG or any of its erbB receptors (erbB2, erbB3, or erbB4) die during embryonic life or at birth (Gassmann et al., 1995;Lee et al., 1995; Erickson et al., 1997; Riethmacher et al., 1997;Wolpowitz et al., 2000), precluding their use to study the involvement of erbB receptors in postnatal events. Furthermore, although the importance of erbB2 receptors in the control of puberty has been defined by the ability of intracerebral administration of erbB2 antisense oligodeoxynucleotides to delay puberty (Ma et al., 1999), these results identified neither the receptor subtype involved (erbB1 or erbB4) nor the cell type in which an intact receptor complement must be present to ensure normal function. To define the role of the NRG receptors erbB2, erbB3, and erbB4 in astrocytes, we generated transgenic mice in which the function of these receptors is specifically disrupted in these cells by overexpression of a dominant-negative erbB4 receptor (DN–erbB4) using the promoter for the GFAP.
Here we report that DN–erbB4 expression abolishes astrocytic erbB4–erbB2 receptor signaling without affecting that mediated by erbB1. In keeping with the pattern of GFAP expression in the normal brain, the mutant receptor is expressed most abundantly in hypothalamic astrocytes. Mutant female mice have delayed sexual development and delayed initiation of reproductive capacity. Transgenic female mice also show a selective reduction in NRG-induced LHRH release from the median eminence and in NRG-induced astrocytic PGE2 production, whereas responses to TGFα are normal. Thus, the results define the physiological relevance of the NRG–astrocytic erbB4–erbB2 receptor system in the hypothalamic control of female sexual development. They also establish the concept that this neuron–glia communication system is a core component of the cell-to-cell signaling mechanism used by the hypothalamus to control the initiation of mammalian puberty.
Materials and Methods
Generation of transgenic mice. The LacZ gene was removed from the pGfa2–Lac-1 plasmid (Brenner et al., 1994) byBamHI restriction. The 5.4 kb plasmid backbone containing the Gfa2 promoter was blunt-ended and dephosphorylated. The blunt-ended 2.2 kb DN-erbB4-FLAG DNA was ligated to the Gfa2-containing plasmid with T4 DNA ligase. Several clones were obtained and characterized by restriction mapping and sequencing of the junction region. The resulting pGfa2–DN–erbB4 plasmid was excised with BglII, and the 5 kb Gfa2–DN–erbB4 DNA was gel purified (JETsorb; Genomed, Bad Oeymhausen, Germany) and used for the generation of transgenic mice.
Transgenic mice were generated in an FVB background using standard procedures (Hogan et al., 1994). Animals carrying the transgene were identified by PCR. Tail clips were incubated in 500 μl of lysis buffer (100 mm Tris HCl, pH 8.5, 5 mmEDTA, 0.2% SDS, 200 mm NaCl) with 5 μg/ml Proteinase K at 58°C overnight. The supernatant was subjected to isopropanol precipitation, and the genomic DNA was resuspended in 100 μl of TE buffer. One microliter aliquots of DNA were amplified in a 25 μl PCR with the 5′ primer TGCTGAAGGAATGGTGTGC and the 3′ primer CTTGTCGTCATCGTCTTTG, and the PCR products were analyzed by agarose gel electrophoresis. Hemizygous mice of each line were intercrossed, and putative homozygous mice were identified by Southern blot using a DN–erbB4 probe. The genotypes of homozygous mice were confirmed by analyzing the progeny of presumptive homozygote mice backcrossed to wild-type mice.
Growth factors and prostaglandins. Human recombinant NRGβ1 was initially obtained from Genentech (San Francisco, CA) and then purchased from Neomarkers (Union City, CA). TGFα was supplied by Becton Dickinson Biosciences (Bedford, MA), and PGE2 was obtained from Sigma (St. Louis, MO).
Cell culture. Astrocytes were isolated from the hypothalamus or the whole brain of 1- to 2-d-old wild-type and transgenic mice and cultured as described previously (Ma et al., 1994, 1999). After a growth period of 8–10 d in 75 cm culture flasks containing DMEM–F12 medium supplemented with 10% calf serum, the astrocytes were isolated from contaminant cells by overnight shaking at 250 rpm and were replated in either 15 cm dishes for immunoblot analysis or six well plates for prostaglandin release experiments. After reaching 80–90% confluence, the medium was replaced with a serum-free, astrocyte-defined medium consisting of DMEM devoid of phenol red, supplemented with 2 mml-glutamine, 15 mm HEPES, 5 μg/ml insulin, and 100 μm putrescine (Ma et al., 1999). The cells were used 2 d later for the experiments. To examine the effect of NRGβ1 and TGFα on PGE2release, the astrocytes were incubated in the presence of either peptide (at 100 ng/ml each) for 16 hr at 37°C.
Immunoprecipitation and Western blots. Cell cultures were lysed in radioimmunoprecipitation assay buffer, and the resulting protein extracts were size-fractionated by SDS-PAGE, as described previously (Rio et al., 1997; Ma et al., 1999). The separated proteins were then transferred to polyvinylidene difluoride (PVDF) membranes and immunoblotted with different antibodies (see below) to identify the DN–erbB4 transgene, GFAP, and the different erbB receptors. Immunoprecipitations were performed using polyclonal anti-erbB1 and anti-erbB2 antibodies directed against epitopes contained within the intracellular domain of the receptors (sc-03-G and sc-284, respectively; Santa Cruz Biotechnology, Santa Cruz, CA), and a monoclonal anti-erbB4 antibody (Ab-1, Neomarkers) that recognizes an epitope present in the extracellular domain of erbB4. Cell lysates (750–1000 μg of protein) were incubated for 90 min at 4°C with 1.5 μg of each antibody. Thereafter, the receptor–antibody complexes were incubated with slurry of protein A–Sepharose (30 μl of Sepharose beads per 750 μl immunoreaction for 45 min at 4°C). The Sepharose beads were collected by centrifugation, washed two times with lysis buffer, resuspended in 2× sample buffer, and boiled for 5 min before loading onto 8% polyacrylamide–SDS gels. After electrophoresis, the size-fractionated proteins were transferred to PVDF membranes and subjected to immunoblotting using the monoclonal phosphotyrosine antibody 4G10 (1.5 μg/ml; Upstate Biotechnology, Lake Placid, NY). To develop the immunoreaction, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), developed using enhanced chemiluminescence (Renaissance; NEN, Boston, MA), and exposed to film. After stripping (62.5 mm Tris HCl, pH 6.7, 2% SDS, 100 mm β-mercaptoethanol, 30 min at 60°C), the membranes were reprobed with the same erbB1 or erbB2 antibodies used for immunoprecipitation or with a polyclonal anti-erbB4 antibody (sc283; Santa Cruz Biotechnology) that recognizes an amino acid sequence contained within the C terminus of erbB4. The DN–erbB4 protein was detected in tissues or cultured cell lysates using an antibody against the FLAG epitope (10 μg/ml, sc-807; Santa Cruz Biotechnology); GFAP was detected with a mouse monoclonal antibody (MAB360; Chemicon, Temecula, CA), and β-actin was detected with a mouse monoclonal antibody (Sigma).
RNase protection assays. Detection of mutant mRNA transcripts was performed using RNase protection assays as reported previously (Ma et al., 1996). Total RNA (15 μg) from brain tissues of 12-d-old mice was hybridized to a [32P]UTP-labeled erbB4–FLAG cRNA complementary to nucleotides 1684–2217 in the extracellular domain of human erbB4 mRNA (HER4 JM-b) (Plowman et al., 1993; Elenius et al., 1997) and to the FLAG encoding sequence (CAAAGACGATGACGACAAG). For normalization purposes, the hybridization mixture also included a [32P]UTP-labeled mouse cyclophilin probe. After completion of the hybridization, the samples were treated with RNase A and T1 to digest unhybridized RNA species and were size-fractionated by PAGE. The hybridization signals were visualized by exposing the dried gels to Reflection x-ray film (NEN).
Immunostaining. The brains of 12-d-old female mice were fixed by transcardiac perfusion with Zamboni's fixative and subjected to double immunohistofluorescence confocal microscopy as described previously (Ma et al., 1999), but using 16 μm cryostat sections. DN–erbB4 was detected with a monoclonal antibody raised against the extracellular domain of human erbB4 (1:200, Ab-1; Neomarkers). Astrocytes were identified with a rabbit polyclonal antibody to GFAP (1:500, Dako, Carpinteria, CA), and LHRH neurons were identified with the antiserum HU60 (1:3000) (Urbanski et al., 1990). Confocal images were acquired using a Leica (Nussloch, Germany) TCS NT confocal system, as described previously (Jung et al., 1999), using the 488 and 568 nm lines of argon and krypton gas lasers to detect the FITC and Texas Red fluorochromes used to visualize each immunoreaction. Single-plane images were processed and merged using Photoshop 5.0 (Adobe Systems, San Jose, CA).
Incubation of median eminence tissue. The median eminence of 12-d-old mice was dissected as described previously (Negro-Vilar et al., 1979) and incubated at 37°C in 250 μl of Krebs–Ringer bicarbonate buffer, pH 7.4, containing 4.5 mg/mld-dextrose, under an atmosphere of 95% O2 and 5% CO2 with shaking (60 cycles per minute). In all experiments, the tissues were preincubated for 30 min, followed by a 2 hr incubation period to determine the basal release of LHRH. At this time, the medium was replaced by fresh medium containing either NRGβ1 or TGFα (at 100 ng/ml each), or PGE2 (at 1 μm), and the incubation was extended for an additional 2 hr in the presence of each secretagogue.
Evaluation of sexual maturation and adult reproductive function. To determine whether the astrocytic expression of DN–erbB4 mutant receptors affects the release of pituitary gonadotropins, groups of wild-type and transgenic animals were killed every 4 d starting on postnatal day 4, and trunk blood was collected for luteinizing hormone (LH) and follicle-stimulating hormone (FSH) measurement. The uterine weights were also recorded to determine whether uterine growth (a physiological index of estrogen secretion) is affected in mutant animals. Starting on postnatal day 26, the mice were inspected daily for inperforation of the vaginal membrane (“vaginal opening”). Thereafter, vaginal lavages were performed daily to detect the appearance of cornified cells, which identify the estrous phase of the rodent estrous cycle. Both vaginal opening and cornification of the vaginal epithelium (estrus) result from the rise in estrogen secretion that accompanies the onset of puberty in rodents (Ojeda and Urbanski, 1994). Ovulation normally occurs on the day of estrus, but in mice it cannot be assumed to occur unless vaginal cornification is followed by the appearance of a predominance of leukocytes in the vaginal lavage (Nelson et al., 1990). This abundance of leukocytes defines the diestrous phase of the estrous cycle and indicates that a functional corpus luteum was formed after ovulation. Therefore, both the age at vaginal opening and the age at first estrus were recorded, the latter being considered as a true first estrus (and thus the age at first ovulation) only when the cornified cells in the vaginal lavages were replaced by at least 2 d of lavages containing primarily leukocytes.
To determine whether the astrocytic blockade of NGR signaling affects early postpubertal reproductive capacity, young adult (50-d-old) wild-type and mutant mice were exposed to a fertile male (one male per female), and the interval between the initial exposure and the birth of the first litter was recorded.
Ovariectomy. Ovariectomy was performed on postnatal day 12. The ovaries were aseptically removed from animals anesthetized with isofluorane via a single dorsal skin incision followed by blunt separation of the underlying muscle–aponeurosis interfase, and different groups of mice were killed 2 and 4 d later.
Measurements of LHRH, PGE2, and serum gonadotropin hormones. LHRH released from the median eminence or present in hypothalamic tissue and PGE2 released from astrocytes were detected by radioimmunoassay, as described previously (Ojeda et al., 1986). In the case of LHRH, we used 125I-labeled LHRH and the rabbit polyclonal antibody HU60, which recognizes the fully processed, mature decapeptide (Urbanski et al., 1990), at a 1:25,000 dilution. The sensitivity of this assay is 0.4 pg per tube. PGE2 was measured using antibody SC10-11/23 (Campbell and Ojeda, 1987) at a 1:8000 dilution and using tritiated PGE2([5,6,8,11,14,15-N-3H] PGE2; NEN) as the trace. The sensitivity of this assay was 3.6 pg per tube.
Serum levels of LH and FSH were measured by radioimmunoassay using reagents for rat hormones provided by the National Hormone and Pituitary program of the National Institute of Diabetes and Digestive and Kidney Diseases. FSH was measured in duplicate (15 μl) samples from individual mice; LH was measured in single samples (25 μl). All samples from a given experiment were measured in the same assay.
Statistics. The differences between several groups were analyzed by ANOVA followed by the Student–Newman–Keuls' multiple comparison test for unequal replications. The Student'st test was used to compare two groups. When comparing percentages, groups were subjected to arc–sine transformation before statistical analysis to convert them from a binomial to a normal distribution (Zar, 1984).
Results
The GFAP promoter drives expression of DN–erbB4 to hypothalamic astrocytes
To specifically disrupt the function of all NRG receptors (erbB2, erbB3, and erbB4) in astrocytes, we engineered transgenic mice carrying DN–erbB4 under the control of the human GFAP promoter (gfa2) (Besnard et al., 1991). This mutant receptor contains the extracellular and transmembrane domains of human erbB4 but lacks most of the intracellular domain, including the entire tyrosine kinase domain and all tyrosine phosphorylation sites (Rio et al., 1997). We have shown previously that high expression of DN–erbB4 blocks NRG-induced activation of erbB receptors in muscle and astroglial cells in culture (Rio et al., 1997). The gfa2promoter, consisting of 2.2 kb of the 5′-flanking region from the human GFAP gene, directs astrocyte-specific transcription in both cell cultures (Besnard et al., 1991) and transgenic mice (Brenner et al., 1994; Delaney et al., 1996). Although in vivo transgene expression driven by this promoter starts at late embryonic stages, high levels of expression are achieved only after birth (Brenner et al., 1994; Delaney et al., 1996). Several founder mice were obtained, and two independent lines (lines 18 and 34), shown by Western blot analysis to express DN–erbB4 with the proper cell specificity (see below), were bred to homozygosity to achieve the highest possible level of DN–erbB4 expression. The mice appeared normal, and no overt disruptions in CNS histology were observed.
Western blot and RNase protection assays showed that the transgenic animals express DN–erbB4 in the brain, with the highest level of expression in the hypothalamus (Fig.1A,B). This was consistent with the detection of higher levels of GFAP protein in the hypothalamus than in the cerebral cortex (Fig. 1A), and indicated that the pattern and level of transgene expression was appropriate. Importantly, the DN–erbB4 protein was undetectable in non-neural tissues involved in reproductive control, such as the ovaries and uterus (Fig. 1A), or in other organs such as the liver (data not shown). Trace amounts of DN–erbB4 immunoreactivity were detected in a pituitary gland sample, which included both the endocrine and neural components of the gland.
Fig. 1.
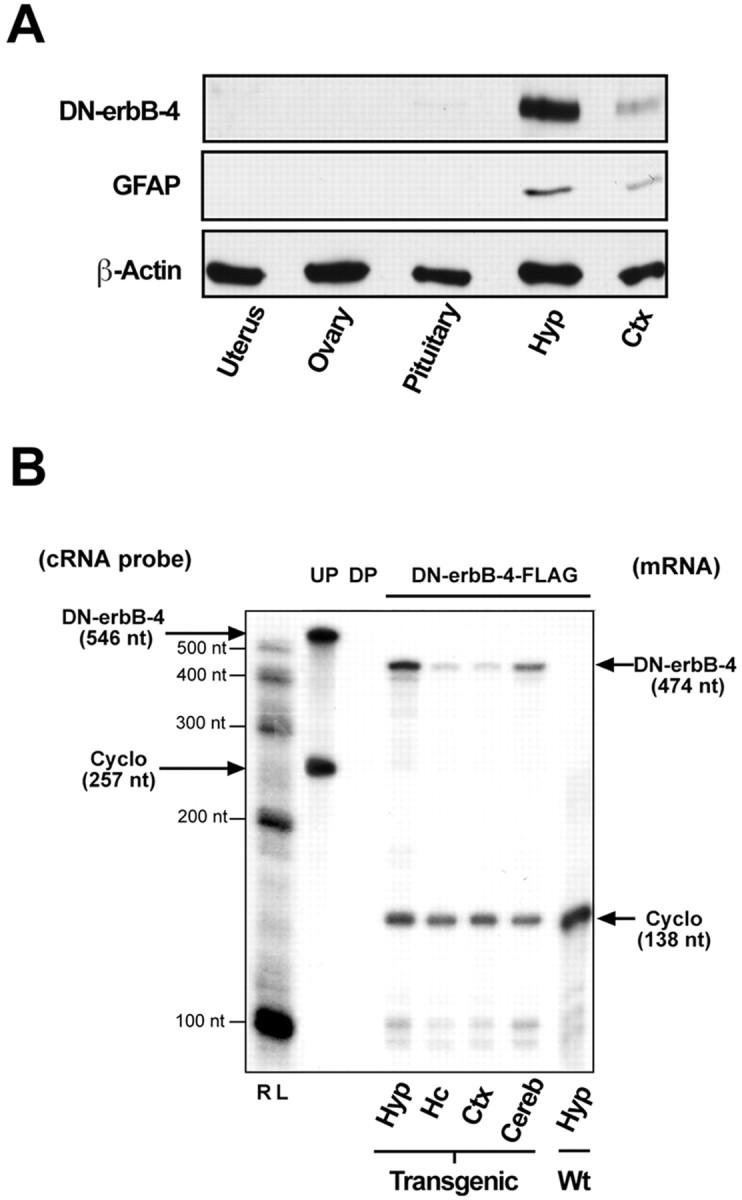
Analysis of transgene expression by Western blot and RNase protection assays. A, DN–erbB4 protein is expressed in both hypothalamic and cortical tissues but not in peripheral organs involved in reproductive function. Notice that the relative abundance of both DN–erbB4 and GFAP proteins is much greater in the hypothalamus than in the cerebral cortex. β-Actin was used as a loading control. Hyp, Hypothalamus;Ctx, cerebral cortex. B, The content of DN–erbB4 mRNA detected by RNase protection assay is greater in the hypothalamus than in other brain regions. RL, RNA ladder; UP, undigested probe; DP, digested probe; Cyclo, cyclophilin; Hc, hippocampus; Cereb, cerebellum; Wt, wild type.
To determine the pattern of expression of DN–erbB4 at the cellular level, tissue sections were stained with anti-GFAP antibodies and with antibodies that recognize the extracellular domain of erbB4. These anti-erbB4 antibodies are directed against the human erbB4 but can also recognize the murine receptor. Confocal microscopy demonstrated that the DN–erbB4 transgene is specifically expressed in astrocytes and verified that the highest level of expression occurs in hypothalamic astrocytes (Fig. 2). erbB4 immunoreactivity in astrocytes of the median eminence, the main terminal field of neuroendocrine LHRH neurons, was conspicuously higher in mice carrying the transgene (Fig. 2D–F) than wild-type animals (Fig. 2A–C). erbB4 immunostaining was also much more abundant in astroglial-like cells surrounding LHRH cell bodies in the preoptic region of transgenic animals than in wild-type mice, but no expression was detected in LHRH neurons (Fig. 2G,H). As predicted by the results of the Western blot analysis (Fig. 1), hypothalamic astrocytes had a much higher content of immunoreactive DN–erbB4 than astrocytes of the cerebral cortex (Fig. 2I–N). No overt differences in the density of astrocytes or neurons of either region were noted between wild-type and transgenic mice.
Fig. 2.
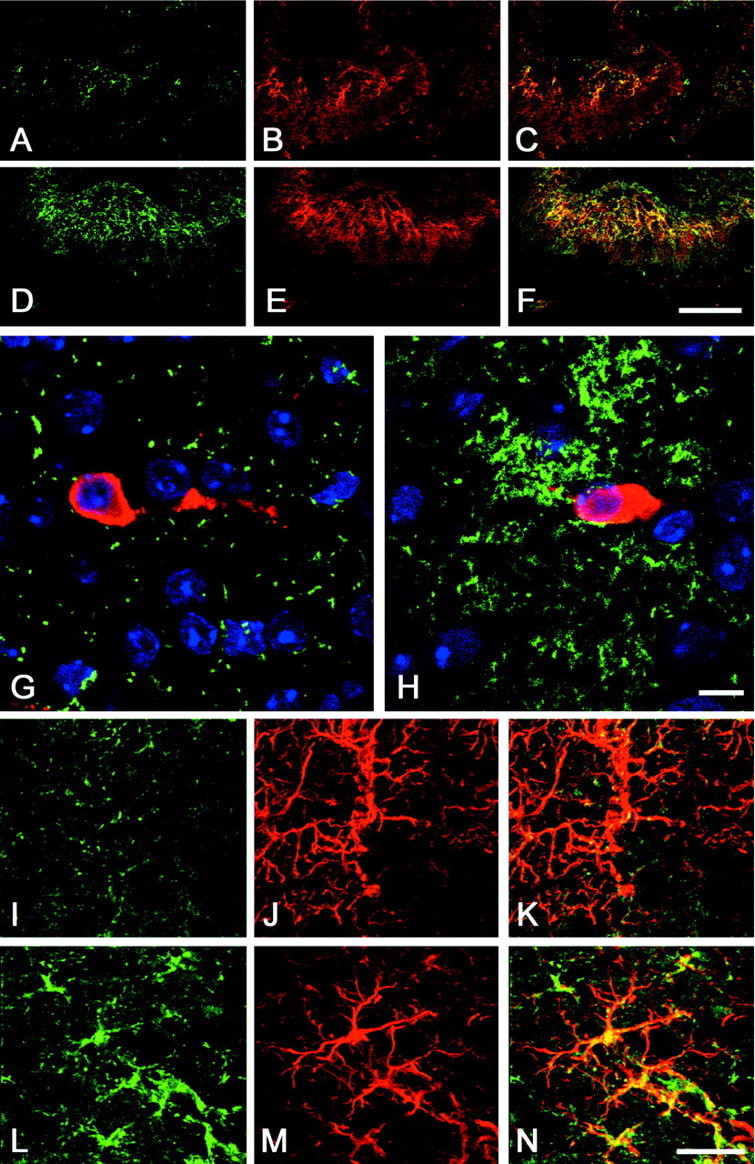
Analysis of DN–erbB4 expression in the hypothalamus of 12-d-old mice by confocal microscopy.A–F, erbB4 immunoreactivity in astrocytes of the median eminence–medial basal hypothalamus of wild-type mice (A–C) and DN–erbB4 transgenic mice (D–F). A, D, erbB4 staining (green). B, E, GFAP staining (red). C, F, Merged images of erbB4–GFAP immunoreactive cells. G, H, Astrocytes of the preoptic area, which contains the cell bodies of LHRH neurons (red), show a much greater abundance of immunoreactive erbB4 (green) in DN–erbB4 mutant mice (H) than in wild-type animals (G). I–N, In DN–erbB4 mice, erbB4 immunoreactivity (green) is more abundant in hypothalamic astrocytes (L–N) than in astrocytes of the cerebral cortex (I–K). Astrocytes are identified by their GFAP immunoreactivity (red; J, M). Merged images for each brain area are shown in K (cerebral cortex) andN (hypothalamus). Scale bars: A–F, 100 μm; G, H, 10 μm; I–N, 5 μm.
DN–erbB4 expression disrupts astrocytic erbB4 and erbB2 but not EGF receptor signaling
Previous observations have shown that in situdifferences in GFAP expression among astrocytes from different brain regions are no longer apparent in vitro. In keeping with these findings, the mutant receptor was abundantly expressed in cultured astrocytes from either the whole brain or the hypothalamus (Fig. 3A). Therefore, astrocytes from the whole brain were used to assess the ability of the mutant receptor to disrupt erbB-mediated signaling. Western blot analysis combined with immunoprecipitation showed that NRG-induced phosphorylation of astrocytic wild-type erbB4 receptors is abolished in astrocytes obtained from mutant mice (Fig. 3B).
Fig. 3.

DN–erbB4 expression in astrocytes blocks ligand-induced erbB4 and erbB2 receptor phosphorylation without affecting erbB1-mediated signaling. A, DN–erbB4 is expressed in cultured astrocytes isolated from either the whole brain (top panel) or the hypothalamus (bottom panel). Cell homogenates prepared from primary astrocyte cultures or COS-7 cells transfected with DN–erbB4 FLAG were subjected to SDS-PAGE and then immunoblotted (IB) with FLAG, erbB4, or GFAP antibodies.B, Inhibition of erbB4 receptor phosphorylation by the transgenic expression of DN–erbB4 in whole brain astrocyte cultures. Cells were treated with NRGβ1 (50 ng/ml for 5 min), lysed, immunoprecipitated (IPP) with erbB4 antibodies, subjected to SDS-PAGE separation, and blotted with 4G10 anti-phosphotyrosine antibodies (top panel). The membrane was stripped and incubated with an antibody that recognizes the intracellular domain of erbB4, showing the relative amount of receptor immunoprecipitated in each sample (middle panel). After stripping again, the membrane was incubated with an anti-FLAG antibody, showing that DN–erbB4 was only expressed in mutant cells, and that the level of expression was similar in all conditions. C, DN–erbB4 expression does not affect ligand-induced erbB1 receptor phosphorylation but abolishes erbB2 phosphorylation. Brain astrocyte cultures were treated with betacellulin (50 ng/ml for 5 min), lysed, and immunoprecipitated with either erbB1 (left panel) or erbB2 (right panel) antibodies, before SDS-PAGE and immunoblotting with 4G10 antiphosphotyrosine antibodies, followed by stripping and immunoblotting with either erbB1 or erbB2 antibodies to ensure that each receptor had been immunoprecipitated. Wt, Wild type.
In addition to erbB4, hypothalamic astrocytes express erbB2 and EGF receptors (erbB1) but do not contain erbB3 receptors (Ma et al., 1999). Because it is clear that erbB4 can heterodimerize with erbB2, and because experiments suggest that it may also dimerize with erbB1 (Tzahar et al., 1997; Pinkas-Kramarski et al., 1998), we tested the possibility that expression of DN–erbB4 would affect astrocytic erbB2 and/or erbB1 signaling. Mutant and wild-type astrocytes were exposed to betacellulin, an EGF family member that binds and activates both erbB1 and erbB4 homodimers and all possible heterodimeric erbB receptor complexes (Dunbar and Goddard, 2000). Then the extent of erbB1 and erbB2 tyrosine phosphorylation was determined by immunoprecipitation with antireceptor antibodies followed by immunoblotting with anti-phosphotyrosine antibodies. Although betacellulin-induced erbB1 phosphorylation was not affected in DN–erbB4 expressing astrocytes (Fig. 3C, left panels), tyrosine phosphorylation of erbB2 receptors was abolished (Fig. 3C, right panels). These results show that erbB2 receptors, presumably forming unproductive heterodimers with DN–erbB4, cannot be activated by erbB4 ligands. They also demonstrate that DN–erbB4 specifically blocks astrocytic erbB4–erbB2 signaling but not that of erbB1.
Expression of DN–erbB4 in astrocytes blocks the NRG-induced release of PGE2 and LHRH from hypothalamic cells
We have shown previously that NRG-induced erbB4–erbB2 activation in cultured astrocytes results in the production of PGE2, and that PGE2 induces LHRH secretion from an immortalized LHRH-producing cell line (Ma et al., 1999) and from the median eminence of the hypothalamus (Ojeda et al., 1990). These results suggested that NRG regulates LHRH production and/or secretion through its effects on astrocytic PGE2 production. Therefore, we tested whether astrocytic expression of DN–erbB4 alters the production and/or secretion of PGE2 and LHRH production by hypothalamic cells. Although both NRGβ and TGFα, a member of the EGF family that signals via erbB1 receptors, induced PGE2 release from wild-type hypothalamic astrocytes, NRGβ1 was completely ineffective in cells from DN–erbB4 mutant animals (Fig.4A). In contrast, TGFα induced PGE2 release from DN–erbB4-expressing astrocytes as efficiently as in wild-type cells, also demonstrating that erbB1 signaling is not affected by DN–erbB4 expression. The presence of DN–erbB4 in astrocytes also disrupted NRGβ1-induced release of LHRH from median eminence fragments containing the neurosecretory terminals of LHRH neurons, without affecting the stimulatory effect of TGFα (Fig. 4B). These results suggested that the inability of NRG to induce LHRH release could be attributable to a lack of PGE2 production in DN–erbB4-expressing astrocytes. To test this possibility we determined whether LHRH release from median eminence nerve terminals could be induced by PGE2. Treatment with the prostaglandin (1 μm) stimulated LHRH release equally well in both normal and mutant tissues (Fig. 4C). Thus, the reduced secretory response of LHRH neurons to NRGβ1observed in DN–erbB4 mutant mice is the direct result of impaired astrocytic erbB4–erbB2 signaling and not the consequence of a neuronal defect resulting from the astrocytic expression of mutant erbB4 receptors.
Fig. 4.
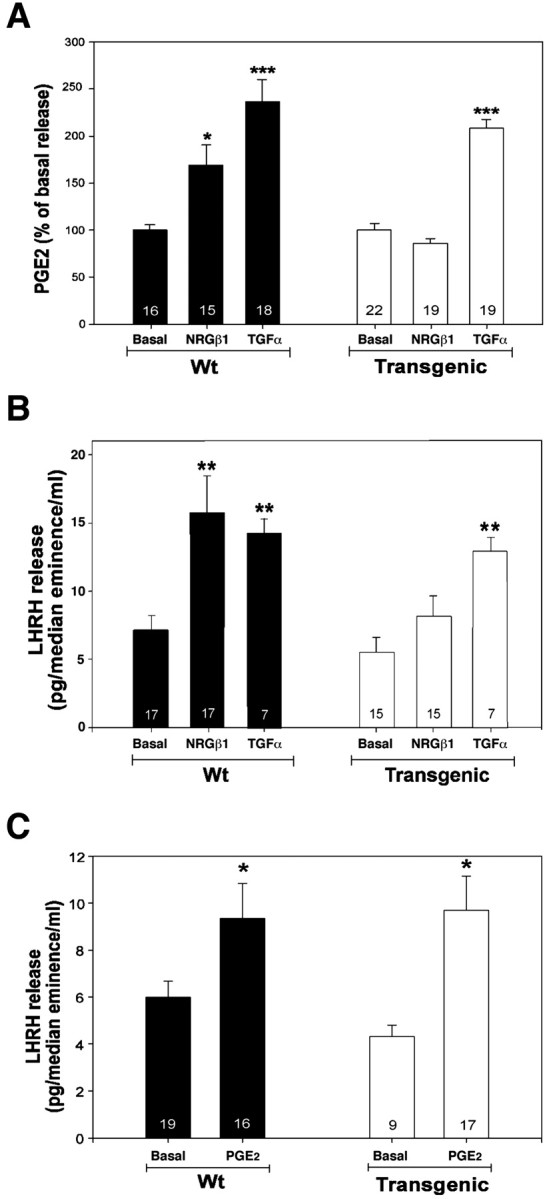
DN–erbB4 expression suppresses NRG-induced release of LHRH from median eminence nerve terminals and PGE2 from hypothalamic astrocytes. A, NRGβ1-induced PGE2 release from primary cultures of hypothalamic astrocytes is abolished in DN–erbB4 mutants, whereas TGFα-stimulated PGE2 release is not altered [basal PGE2 release was similar in wild-type and DN–erbB4 cultures (238 ± 13 vs 223 ± 16 pg/ml, respectively)].B, DN–erbB4 expression suppresses NRGβ1-induced but not TGFα-induced LHRH release from the median eminence. C, PGE2 treatment (1 μm for 30 min) stimulates LHRH release from the median eminence of both wild-type and transgenic mice incubated in Krebs–Ringer bicarbonate buffer. In all panels, *p < 0.05, **p < 0.01, ***p < 0.001 versus basal release.Bars are means, vertical lines are the SEM, and the numbers inside the bars are the number of independent observations per group. Wt, Wild type.
Expression of DN–erbB4 in hypothalamic astrocytes results in hormonal defects in vivo
The in vitro experiments described above indicated that the GFAP–DN–erbB4 transgenic mice could have defects in LHRH production or secretion. Measurements of total hypothalamic LHRH content during prepubertal development showed no differences between transgenic and wild-type mice (Fig.5A), suggesting that LHRH production is normal. During early mammalian sexual development, a transient activation of LHRH secretion (Hompes et al., 1982) induces an elevation in serum levels of pituitary gonadotropin hormones, in particular those of FSH (Ojeda and Urbanski, 1994; Grumbach and Styne, 1998). In rodents, this increase occurs during the second week of postnatal life (Ojeda and Urbanski, 1994). In wild-type mice, plasma FSH levels rose markedly during the first 2 weeks of life, reaching maximum values by day 12, and decreased abruptly thereafter (Fig.5B). In contrast, in mutant mice, the FSH peak was blunted by 50% (Fig. 5B, top panel). Interestingly, the low mean LH levels observed normally at this stage of development were not different between wild-type and mutant mice (Fig. 5B, bottom panel). It thus appears that the physiological requirement for erbB4–erbB2 signaling in NRG-induced LHRH release from median eminence explants in vitro has the appropriate in vivo correlate (i.e., an attenuated infantile surge of FSH that likely reflects a reduced LHRH release in the face of a normal hypothalamic LHRH content).
Fig. 5.
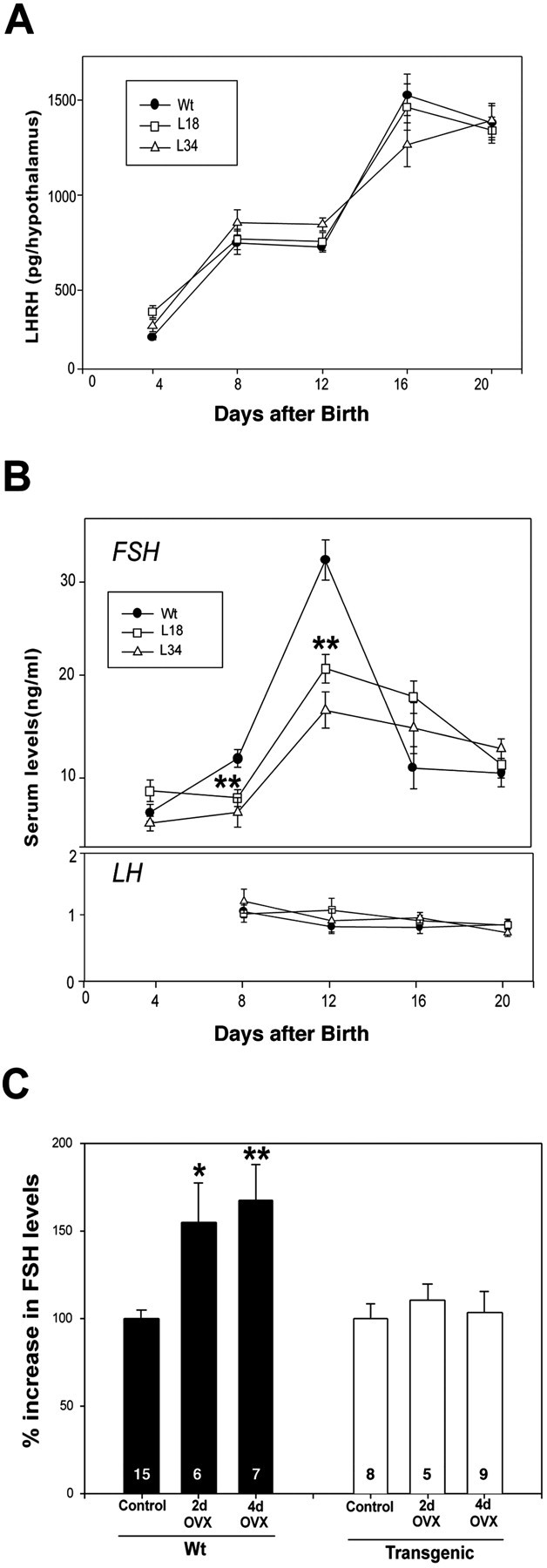
Transgenic mice exhibit a normal content of LHRH in the hypothalamus but have reduced serum FSH levels and a reduced FSH secretory response to removal of ovarian steroid inhibitory control during the infantile period of development. A, Hypothalamic LHRH content measured by radioimmunoassay is similar in wild-type and mutant mice throughout neonatal–infantile development.B, The infantile increase in serum FSH levels that characterizes early postnatal development of the hypothalamic–pituitary unit in rodents is blunted in transgenic mice (top panel), whereas the low serum LH levels observed at this time remain unchanged (bottom panel) (10–20 mice per point). C, Ovariectomy (OVX) on postnatal day 12 results in a significant increase in serum FSH levels 2 and 4 d later in wild-type mice but not in mutant mice. Numbers inside the bars are the number of mice per group. Vertical lines are the SEM. In B, **p < 0.01 versus the wild-type group. InC, *p < 0.05 and **p < 0.01 between ovariectomized and nonovariectomized wild-type groups. Wt, Wild type.
To further test the function of the hypothalamic–pituitary–gonadal axis, we examined the effects of ovariectomy, which in normal animals relieves the hypothalamic–pituitary unit of steroid-dependent inhibitory feedback control, resulting in increased circulating levels of pituitary gonadotropins (Ojeda and Urbanski, 1994; Grumbach and Styne, 1998). As expected, the ovariectomy of 12-d-old wild-type mice led to a significant increase in serum FSH levels 2 and 4 d later (Fig. 5C). In contrast, mutant mice failed to respond (Fig.5C) within the time frame examined, indicating that the ability of the LHRH neuronal network to increase its secretory output in response to the loss of ovary-dependent inhibitory control is disrupted in these animals.
GFAP–DN–erbB4 female mice show delayed sexual maturation and a diminished reproductive capacity in early adulthood
The deficits in LHRH–FSH secretion seen in ovary-intact mutant mice were accompanied by a reduction in uterine growth in both transgenic lines (Fig.6A), indicating that ovarian estrogen output was compromised in DN–erbB4 mice. This deficiency is likely to result from the reduction in FSH levels, because this hormone is required for both antral follicular growth and ovarian estrogen secretion (Schwartz, 1974; Kumar et al., 1997). Consistent with the hindering of uterine growth, the time of puberty (defined by the age at first ovulation) was significantly delayed in DN–erbB4 animals compared with wild-type mice (Fig.6B). Once the mutant mice achieved puberty, their reproductive performance was also affected; when a wild-type male was introduced in the cage of a sexually mature female, the birth of the first litter was delayed by >10 d in DN–erbB4 mice compared with wild-type females (Fig. 6C). Young-adult gestational capacity did not appear to be affected, because the mutant animals, once they were able to reproduce, delivered a normal number of pups per litter for at least five consecutive pregnancies.
Fig. 6.
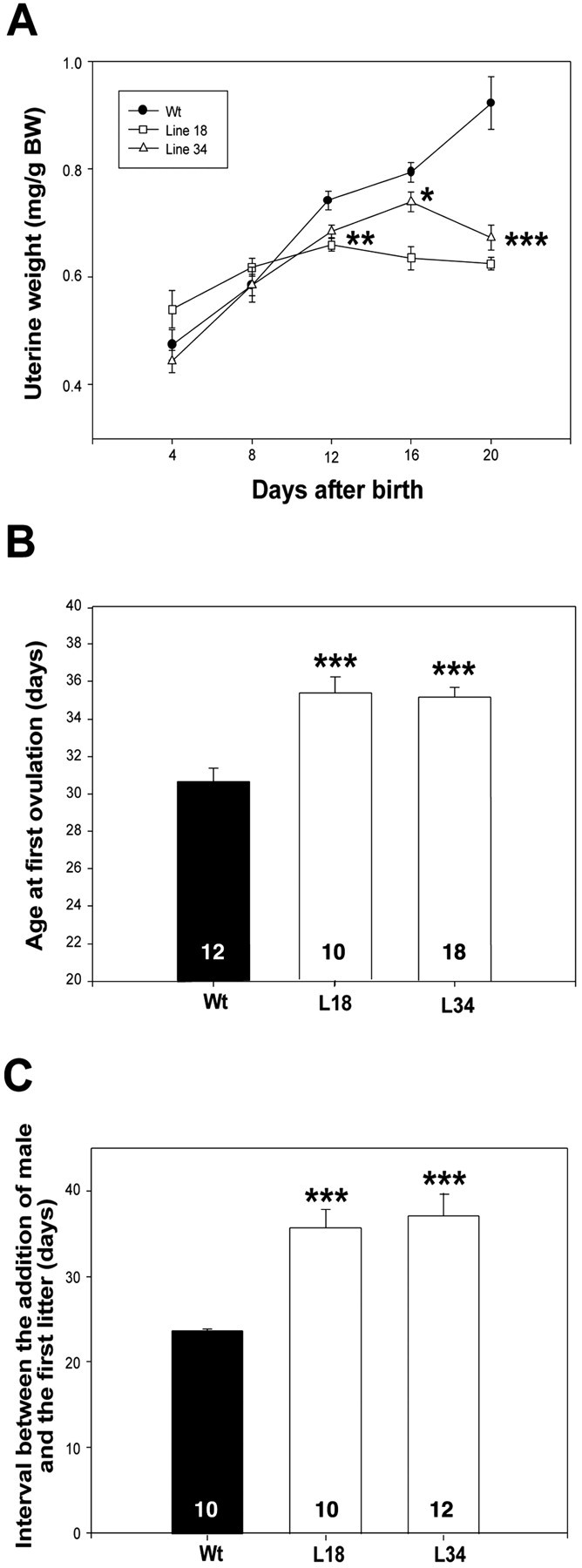
Transgenic mice have retarded uterine growth, delayed sexual development, and reduced reproductive capacity in early adulthood. A, Uterine growth is reduced in mutant mice (8–12 animals per point, except day 12, in which each point has 30–44 mice). B, The age at first ovulation is delayed in transgenic mice. C, The age at first pregnancy is delayed in transgenic mice. In all panels, *p < 0.05, **p < 0.01, and ***p < 0.001 versus wild-type values.Circles, triangles, orbars are means; vertical lines represent the SEM. L18 and L34 are transgenic lines. Wt, Wild-type mice. Numbers inside the bars are the number of animals per group.
Discussion
The present results indicate that erbB4 receptor signaling in hypothalamic astroglial cells is an essential component of the mechanisms by which the brain controls female sexual development and the acquisition of reproductive competence in mammals. By showing that the astrocyte-specific blockade of erbB4–erbB2 receptor function disrupts these landmark events, these results provide for the first time evidence that NRG–erbB signaling is necessary for specific neuron–astrocyte interactions in the normal postnatal brain. Although in addition to hypothalamic astrocytes, DN–erbB4 is expressed in cerebrocortical and cerebellar astrocytes, the high levels of expression required for effective blockade of wild-type receptors appear to be achieved only in the hypothalamus, suggesting that this is the site where fully functional erbB4 receptors are required for normal sexual development. Because DN–erbB4 was abundant in astrocytes of both the preoptic area (where the LHRH cell bodies are located) and the median eminence (which contains the LHRH secretory nerve terminals), it is likely that the astrocytic defect in erbB4 signaling affects LHRH neuronal function at these two neuroanatomical levels. Previous studies have, in fact, shown that both sites are subjected to erbB receptor-mediated regulation (Rage et al., 1997). Although we cannot rule out defects in astrocytes outside the hypothalamus, extensive testing of these mice indicates that other brain functions are not affected by astrocytic DN–erbB4 expression, thus supporting this conclusion. For example, the mutant mice show no overt defects in motor function, coordination, or locomotor activity (data not shown), implying the absence of defects in extrapyramidal circuitries and/or cerebellar function. Moreover, recordings from hippocampal slices showed no differences in basic physiological parameters and in physiological plasticity (long-term potentiation and kindling) (data not shown). Finally, histological analysis of hippocampal and cortical tissues (hematoxylin–eosin staining, silver staining, immunostaining with neuronal and glia markers, electron microscopy) failed to reveal overt abnormalities (data not shown).
Because the mutant erbB4 receptor is highly expressed throughout the hypothalamus, it is possible that the reproductive defects observed in DN–erbB4 animals are the consequences of a generalized alteration in hypothalamic function instead of a specific defect affecting the neuronal–glial system controlling reproductive maturation. However, this is unlikely, because key indicators of various other hypothalamic functions show no alterations in DN–erbB4 mice. The mutant mice have normal body weights, implicating a normal rate of growth hormone secretion; they nurse their litters normally, implying no defects in maternal behavior, milk production (prolactin secretion), or milk ejection (oxytocin secretion); and, finally, routine daily inspection of water bottles showed that water intake was not different from that of wild-type controls, implying no defects in the control of water balance (vasopressin secretion). Although it could also be argued that the reproductive defects may be secondary to defects in sexual behavior, the clear-cut defects in LHRH and gonadotropin secretion observed in prepubertal mutant mice speak against such a possibility. Thus, the present results support the conclusion that the reproductive phenotype observed in DN–erbB4 animals is related to a functional lesion affecting a specific astrocyte–neuronal communication pathway required for normal LHRH release.
A previous study showed that intraventricular infusion of an antisense oligodeoxynucleotide that disrupts erbB2 synthesis delays the initiation of female puberty in rats (Ma et al., 1999). Although those experiments demonstrated that erbB2 signaling is important for this process, they did not define the cell type (neuron or glia) where erbB2 receptors are acting, the region of the brain where erbB2 signaling contributes to the control of sexual development, and the erbB receptor subtype (erbB1, erbB3, or erbB4) with which erbB2 heterodimerizes for signaling. The present results provide an answer to these questions. Although the preferential expression of the mutant erbB4 receptors in the hypothalamus establishes this region as the relevant site of erbB receptor action in the control of female puberty, the use of the GFAP promoter to drive expression of DN–erbB4 only to astrocytes demonstrates that this cell type is the site where erbB4 signaling must occur for sexual development to proceed normally. Finally, the finding that DN–erbB4 disrupts both erbB4 and erbB2 function, without compromising erbB1-mediated signaling, identifies erbB4 as the necessary partner used by erbB2 in hypothalamic astrocytes to fulfill its function in the central control of sexual development.
The observation that serum FSH levels were lower in transgenic mice than in wild-type mice at day 12 indicates that regulation of LHRH release by erbB4/2 signaling is important for the regulation of FSH production. Nevertheless, FSH levels still increased during development (day 8 vs day 12) in DN–erbB4 transgenic mice, suggesting that other mechanisms contribute to facilitating the release of LHRH. Based on our previous studies and on the present results, we propose that astrocytes control LHRH release via a dual mechanism involving erbB1 receptor activation by TGFα (which is not affected by overexpression of truncated erbB4 receptors) and erbB4 receptor activation by NRGs (which are inhibited by DN–erbB4). Thus, as suggested by the in vivo results depicted in Figure 4, the changes in serum FSH observed in the mutant animals are likely to reflect, at least in part, the presence of an intact erbB1-mediated signaling system. This could be tested by studying FSH release in animals in which both erbB1 and erbB4/2 signaling are abolished in hypothalamic astrocytes. However, it must also be recognized that LHRH neurons are strongly regulated by trans-synaptic inputs, which are likely to be functional even if both erbB1 and erbB4 astrocytic signaling systems are inhibited, thus contributing to the developmental changes in FSH release observed in the transgenic mice.
In contrast to the receptors, the source and type of the NRG that may be physiologically involved in activating erbB receptors in hypothalamic astrocytes remains undetermined. Ma et al. (1999)originally reported that NRGβ2, but not NRGβ1, was effective in inducing PGE2 release from astrocytes, but the present experiments show that NRGβ1 is active in this system. We believe that the disparity between these results is most likely attributable to a less-than-optimal biological activity of the reagent used in the previous study, and that both of these forms can elicit PGE2 release from hypothalamic astrocytes. Although in situ hybridization studies suggest that NRGs are primarily expressed by neurons (Chen et al., 1994; Corfas et al., 1995), cultured hypothalamic astrocytes also contain the mRNAs encoding NRGs (Ma et al., 1999). Thus, it is possible that the astrocytic erbB receptors that contribute to the control of sexual development are activated in a paracrine or an autocrine manner. We do not know whether adult estrus cyclicity, cycle-related changes in hormone levels, and/or reproductive life span are affected in the transgenic mice. Future experiments are needed to address these issues.
Together, the present results demonstrate that functional integrity of a single astrocytic component involved in neuron–glia signaling is required for normalcy of the centrally driven developmental program that controls sexual maturation in mammals. As such, the results provide important insights into the cell-to-cell communication mechanisms used by the neuroendocrine brain to control the advent of puberty. They also furnish an experimental framework for the concept that a derangement of specific neuron–glia signaling pathways may underlie some of the poorly understood, centrally originated alteration in the onset of human puberty. The delayed sexual development, followed by a transient deficiency in adult reproductive capacity, observed in animals with impaired astrocytic NRG–erbB4/2 receptor signaling is highly reminiscent of the syndrome of idiopathic delayed puberty of central origin related to gonadotropin deficiency in humans (Grumbach and Styne, 1998). A hallmark of this syndrome is that affected individuals have delayed puberty but can reproduce normally after reaching sexual maturity.
V.P. and C.R. contributed equally to this work.
This research was supported in part by National Institute of Neurological Disorders and Stroke Grant R01 NS35884 (G.C.), by the EJLB Foundation (G.C.), by Mental Retardation Research Center Grant NIH P30-HD 18655 (G.C.), and by National Institutes of Health Grants HD25123 (S.R.O.), HD U54 HD18185 (S.R.O.), and RR00163 for the operation of the Oregon Regional Primate Research Center (S.R.O.). We thank Michael Brenner for kindly providing us with the GFAP promoter, Genentech for its generous gift of NRG-β1, Les Dees for doing the radioimmunoassays for LH and FSH, and Henryk F. Urbanski for his generous supply of antibodies to LHRH. We also thank Maria E. Costa for her expert technical assistance, Pieter Dikkes for his help with the histological analysis, and Russell Sanchez, Carl Wang, and Frances Jensen for their help with the electrophysiological tests. We are grateful to Michel Greenberg, Thomas Schwarz, and Zhigang He for their valuable comments on this manuscript.
Correspondence should be addressed to either of the following: Gabriel Corfas, Division of Neuroscience, Children's Hospital, 300 Longwood Avenue, Boston, MA 02115, E-mail, gabriel.corfas@tch.harvard.edu; or Sergio R. Ojeda, Division of Neuroscience, Oregon National Primate Research Center, 505 Northwest 185th Avenue, Beaverton, OR 97006, E-mail: ojedas@ohsu.edu.
G. Cho's present address: Department of Anatomy, College of Medicine, Gyeongsang National University, 92 Chilam-dong, Chinju, Kyungham, 660-751 South Korea.
C. Neville's present address: Department of Surgery, Massachusetts General Hospital, 55 Fruit Street, Warren 1157, Boston, MA 02114.
C. Río's present address: DIATER, C/Soledad 37, 28330 San Martín de la Vega, Madrid, Spain.
N. Rosenthal's present address: European Molecular Biology Laboratory Programme in Mouse Biology, via Ramarini 32 00016, Monterotondo, Rome, Italy.
References
- 1.Besnard F, Brenner M, Nakatani Y, Chao R, Purohit HJ, Freese E. Multiple interacting sites regulate astrocyte-specific transcription of the human gene for glial fibrillary acidic protein. J Biol Chem. 1991;266:18877–18883. [PubMed] [Google Scholar]
- 2.Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell WB, Ojeda SR. Measurement of prostaglandins by radioimmunoassay. Methods Enzymol. 1987;141:323–350. doi: 10.1016/0076-6879(87)41080-x. [DOI] [PubMed] [Google Scholar]
- 4.Chen MS, Bermingham-McDonogh O, Danehy FT, Jr, Nolan C, Scherer SS. Expression of multiple neuregulin transcripts in postnatal rat brains. J Comp Neurol. 1994;349:389–400. doi: 10.1002/cne.903490306. [DOI] [PubMed] [Google Scholar]
- 5.Corfas G, Rosen KM, Aratake H, Krauss R, Fischbach GD. Differential expression of ARIA isoforms in the rat brain. Neuron. 1995;14:103–115. doi: 10.1016/0896-6273(95)90244-9. [DOI] [PubMed] [Google Scholar]
- 6.Delaney CL, Brenner M, Messing A. A conditional ablation of cerebellar astrocytes in postnatal transgenic mice. J Neurosci. 1996;16:6908–6918. doi: 10.1523/JNEUROSCI.16-21-06908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunbar AJ, Goddard C. Structure-function and biological role of betacellulin. Int J Biochem Cell Biol. 2000;32:805–815. doi: 10.1016/s1357-2725(00)00028-5. [DOI] [PubMed] [Google Scholar]
- 8.Elenius K, Corfas G, Paul S, Choi CJ, Rio C, Plowman GD, Klagsbrun M. A novel juxtamembrane domain isoform of HER4/erbB4. J Biol Chem. 1997;272:26761–26768. doi: 10.1074/jbc.272.42.26761. [DOI] [PubMed] [Google Scholar]
- 9.Erickson SL, O'Shea KS, Ghaboosi N, Loverro L, Frantz G, Bauer M, Lu LH, Moore MW. ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development. 1997;124:4999–5011. doi: 10.1242/dev.124.24.4999. [DOI] [PubMed] [Google Scholar]
- 10.Gassmann M, Casagranda F, Orioll D, Simon H, Lal C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- 11.Grumbach MM, Styne DM. Puberty: ontogeny, neuroendocrinology, physiology, and disorders. In: Williams RH, Foster DW, Kroenenberg H, Larsen PR, Zorab R, editors. Williams textbook of endocrinology, Ed 9. Saunders; Philadelphia: 1998. pp. 1509–1625. [Google Scholar]
- 12.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: a laboratory manual, Ed 2. Cold Spring Harbor Laboratory; New York: 1994. [Google Scholar]
- 13.Hompes PGA, Vermes I, Tilders FJH, Schoemaker J. In vitro release of LHRH from the hypothalamus of female rats during prepubertal development. Neuroendocrinology. 1982;35:8–12. doi: 10.1159/000123347. [DOI] [PubMed] [Google Scholar]
- 14.Jung H, Carmel P, Schwartz MS, Witkin JW, Bentele KHP, Westphal M, Piatt JH, Costa ME, Cornea A, Ma YJ, Ojeda SR. Some hypothalamic hamartomas contain transforming growth factor α, a puberty-inducing growth factor, but not luteinizing hormone-releasing hormone neurons. J Clin Endocrinol Metab. 1999;84:4695–4701. doi: 10.1210/jcem.84.12.6185. [DOI] [PubMed] [Google Scholar]
- 15.Kumar TR, Wang Y, Lu N, Matsuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- 16.Lee K-F, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–398. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- 17.Ma YJ, Junier M-P, Costa ME, Ojeda SR. Transforming growth factor α (TGFα) gene expression in the hypothalamus is developmentally regulated and linked to sexual maturation. Neuron. 1992;9:657–670. doi: 10.1016/0896-6273(92)90029-d. [DOI] [PubMed] [Google Scholar]
- 18.Ma YJ, Berg-von der Emde K, Moholt-Siebert M, Hill DF, Ojeda SR. Region-specific regulation of transforming growth factor α (TGFα) gene expression in astrocytes of the neuroendocrine brain. J Neurosci. 1994;14:5644–5651. doi: 10.1523/JNEUROSCI.14-09-05644.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma YJ, Dissen GA, Rage F, Ojeda SR. RNase protection assay. Methods. 1996;10:273–278. doi: 10.1006/meth.1996.0102. [DOI] [PubMed] [Google Scholar]
- 20.Ma YJ, Berg-von der Emde K, Rage F, Wetsel WC, Ojeda SR. Hypothalamic astrocytes respond to transforming growth factor α with secretion of neuroactive substances that stimulate the release of luteinizing hormone-releasing hormone. Endocrinology. 1997;138:19–25. doi: 10.1210/endo.138.1.4863. [DOI] [PubMed] [Google Scholar]
- 21.Ma YJ, Hill DF, Creswick KE, Costa ME, Ojeda SR. Neuregulins signaling via a glial erbB2/erbB4 receptor complex contribute to the neuroendocrine control of mammalian sexual development. J Neurosci. 1999;19:9913–9927. doi: 10.1523/JNEUROSCI.19-22-09913.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Negro-Vilar A, Ojeda SR, McCann SM. Catecholaminergic nodulation of LHRH release by median eminence terminals in vitro. Endocrinology. 1979;104:1749–1751. doi: 10.1210/endo-104-6-1749. [DOI] [PubMed] [Google Scholar]
- 23.Nelson JF, Karelus K, Felicio LS, Johnson TE. Genetic influences on the timing of puberty in mice. Biol Reprod. 1990;42:649–655. doi: 10.1095/biolreprod42.4.649. [DOI] [PubMed] [Google Scholar]
- 24.Ojeda SR, Urbanski HF. Puberty in the rat. In: Knobil E, Neill JD, editors. The physiology of reproduction, Ed 2, Vol 2. Raven; New York: 1994. pp. 363–409. [Google Scholar]
- 25.Ojeda SR, Urbanski HF, Katz KH, Costa ME. Activation of estradiol-positive feedback at puberty: estradiol sensitizes the LHRH-releasing system at two different biochemical steps. Neuroendocrinology. 1986;43:259–265. doi: 10.1159/000124535. [DOI] [PubMed] [Google Scholar]
- 26.Ojeda SR, Urbanski HF, Costa ME, Hill DF, Moholt-Siebert M. Involvement of transforming growth factor α in the release of luteinizing hormone-releasing hormone from the developing female hypothalamus. Proc Natl Acad Sci USA. 1990;87:9698–9702. doi: 10.1073/pnas.87.24.9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ojeda SR, Ma YJ, Lee BJ, Prevot V. Glia-to-neuron signaling and the neuroendocrine control of female puberty. Recent Prog Horm Res. 2000;55:197–224. [PubMed] [Google Scholar]
- 28.Ojeda SR, Prevot V, Heger S. Regulation of puberty. Curr Opin Endocrinol Diabetes. 2001;8:154–160. [Google Scholar]
- 29.Pinkas-Kramarski R, Shelly M, Guarino BC, Wang LM, Lyass L, Alroy I, Alimandi M, Kuo A, Moyer JD, Lavi S, Eisenstein M, Ratzkin BJ, Seger R, Bacus SS, Pierce JH, Andrews GC, Yarden Y, Alimandi M. ErbB tyrosine kinases and the two neuregulin families constitute a ligand-receptor network. Mol Cell Biol. 1998;18:6090–6101. doi: 10.1128/mcb.18.10.6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plowman GD, Culouscou J-M, Whitney GS, Green JM, Carlton GW, Foy L, Neubauer MG, Shoyab M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc Natl Acad Sci USA. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rage F, Lee BJ, Ma YJ, Ojeda SR. Estradiol enhances prostaglandin E2 receptor gene expression in luteinizing hormone-releasing hormone (LHRH) neurons and facilitates the LHRH response to PGE2 by activating a glia-to-neuron signaling pathway. J Neurosci. 1997;17:9145–9156. doi: 10.1523/JNEUROSCI.17-23-09145.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- 33.Rio C, Rieff HI, Qi P, Khurana TS, Corfas G. Neuregulin and erbB receptors play a critical role in neuronal migration. Neuron. 1997;19:39–50. doi: 10.1016/s0896-6273(00)80346-3. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz NB. The role of FSH and LH and of their antibodies on follicular growth and on ovulation. Biol Reprod. 1974;10:236–272. doi: 10.1095/biolreprod10.2.236. [DOI] [PubMed] [Google Scholar]
- 35.Terasawa E. Hypothalamic control of the onset of puberty. Curr Opin Endocrinol Diabetes. 1999;6:44–49. [Google Scholar]
- 36.Tzahar E, Pinkas-Kramarski R, Moyer JD, Klapper LN, Alroy I, Levkowitz G, Shelly M, Henis S, Eisenstein M, Ratzkin BJ, Sela M, Andrews GC, Yarden Y. Bivalence of EGF-like ligands drives the ErbB signaling network. EMBO J. 1997;16:4938–4950. doi: 10.1093/emboj/16.16.4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urbanski HF, Kim SO, Connolly ML. The influence of photoperiod and 6-methoxybenzoxazolinone on the reproductive axis of inbred LSH/Ss Lak male hamsters. J Reprod Fertil. 1990;90:157–163. doi: 10.1530/jrf.0.0900157. [DOI] [PubMed] [Google Scholar]
- 38.Voigt P, Ma YJ, Gonzalez D, Fahrenbach WH, Wetsel WC, Berg-von der Emde K, Hill DF, Taylor KG, Costa ME, Seidah NG, Ojeda SR. Neural and glial-mediated effects of growth factors acting via tyrosine kinase receptors on LHRH neurons. Endocrinology. 1996;137:2593–2605. doi: 10.1210/endo.137.6.8641214. [DOI] [PubMed] [Google Scholar]
- 39.Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- 40.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 41.Zar JH. Biostatistical analysis, Ed 2. Prentice Hall; Englewood Cliffs, NJ: 1984. [Google Scholar]