

Abstract
Recent studies have indicated that exogenously administered neurotrophins produce antidepressant-like behavioral effects. We have here investigated the role of endogenous brain-derived neurotrophic factor (BDNF) and its receptor trkB in the mechanism of action of antidepressant drugs. We found that trkB.T1-overexpressing transgenic mice, which show reduced trkB activation in brain, as well as heterozygous BDNF null (BDNF+/−) mice, were resistant to the effects of antidepressants in the forced swim test, indicating that normal trkB signaling is required for the behavioral effects typically produced by antidepressants. In contrast, neurotrophin-3+/− mice showed a normal behavioral response to antidepressants. Furthermore, acute as well as chronic antidepressant treatment induced autophosphorylation and activation of trkB in cerebral cortex, particularly in the prefrontal and anterior cingulate cortex and hippocampus. Tyrosines in the trkB autophosphorylation site were phosphorylated in response to antidepressants, but phosphorylation of the shc binding site was not observed. Nevertheless, phosphorylation of cAMP response element-binding protein was increased by antidepressants in the prefrontal cortex concomitantly with trkB phosphorylation and this response was reduced in trkB.T1-overexpressing mice. Our data suggest that antidepressants acutely increase trkB signaling in a BDNF-dependent manner in cerebral cortex and that this signaling is required for the behavioral effects typical of antidepressant drugs. Neurotrophin signaling increased by antidepressants may induce formation and stabilization of synaptic connectivity, which gradually leads to the clinical antidepressive effects and mood recovery.
Keywords: TrkB, neurotrophin receptor, antidepressant, BDNF, CREB, cerebral cortex
Introduction
Antidepressant drugs facilitate signaling of serotonin or norepinephrine either by inhibiting their reuptake to presynaptic terminals or by inhibiting their catabolism or binding to serotonin autoreceptors (for review, see Duman et al., 1997; Skolnick, 1999; Manji et al., 2001; Nestler et al., 2002). However, alterations in monoamine metabolism take place soon after drug administration, but the clinical antidepressant effect develops slowly during several weeks of continuous treatment (Nestler et al., 2002). Electroconvulsive shock therapy, the most effective antidepressant therapy known, also requires repeated administration before the patient recovers (Duman and Vaidya, 1998). These observations suggest that alterations in monoamine metabolism alone cannot explain the entire antidepressant effect (Nestler et al., 2002) and indicate that antidepressants may act by evoking adaptive changes in intracellular signal transduction and synaptic connectivity (Duman et al., 1997;Altar, 1999; Skolnick, 1999; Manji et al., 2001; Reid and Stewart, 2001; Nestler et al., 2002).
Neurotrophins, and particularly brain-derived neurotrophic factor (BDNF), have been shown to function as a key regulator of neurite outgrowth, synaptic plasticity, and the selection of functional neuronal connections in the CNS (Katz and Shatz, 1996; McAllister et al., 1999; Mamounas et al., 2000; Huang and Reichardt, 2001; Poo, 2001), which makes neurotrophins potential mediators of the plastic changes induced by antidepressants (Duman et al., 1997; Altar, 1999;Manji et al., 2001; Nestler et al., 2002). Indeed, electroconvulsive shock treatment rapidly and robustly increases mRNA levels of BDNF and its receptor trkB in hippocampus and cortex, and chronic (but not acute) treatment with antidepressants increases BDNF mRNA in the hippocampus (Nibuya et al., 1995; Russo-Neustadt et al., 2000). Furthermore, infusion of exogenous BDNF into hippocampus or brain stem has antidepressant-like behavioral effects (Siuciak et al., 1997;Shirayama et al., 2002), and BDNF administration increases serotonergic innervation (Mamounas et al., 2000) as well as the levels of serotonin and its metabolites in forebrain (Siuciak et al., 1994, 1996). Moreover, the reduction in BDNF expression induced by stress (Smith et al., 1995) and behavioral abnormalities found in heterozygous BDNF knock-out (BDNF+/−) mice have been reported to be counteracted by antidepressants (Smith et al., 1995; Lyons et al., 1999). These data lead to the suggestion that depression may actually represent an impairment of neuronal plasticity, which could be reversed by increasing the levels of neurotrophic factors, such as BDNF (Duman et al., 1997; Altar, 1999; Skolnick, 1999;Manji et al., 2001; Nestler et al., 2002). However, direct evidence for the role of endogenous BDNF in the mechanism of antidepressant drug action is lacking.
We have here addressed the question of the role of endogenous BDNF signaling in antidepressant action. By investigating transgenic mice with reduced BDNF levels or reduced trkB signaling in brain, we provide evidence that trkB activation is required for a behavioral response typically induced by antidepressants. Furthermore, acute as well as chronic antidepressant treatment induces phosphorylation and activation of trkB in cortex in a BDNF-dependent manner.
Materials and Methods
Male mice of between 3 and 6 months of age were used in all experiments, except in the forced swim test for BDNF+/−mice, in which all of the transgenic and corresponding wild-type mice were females. TrkB.T1 cDNA was tagged N-terminally with an eight amino acid FLAG epitope and inserted into a murine Thy 1.2 expression cassette, which directs transgene expression to postnatal neurons, as described previously (Saarelainen et al., 2000b). Transgenic mice were generated by pronucleus injection of this construct into embryos from CD2F1 (BALB/c x DBA/2) females mated with CD2F1 males. Transgenic founder mice were identified using Southern blot analysis and PCR. Heterozygous mice and their wild-type littermates were used in all of these experiments. In situhybridization was performed according to Wisden and Morris (1994), with an oligonucleotide probe (GGCACTTGTCATCGTCGTCTTTGTAGTCGGCA) that specifically recognizes the FLAG-epitope sequence. Production of BDNF+/− and neurotrophin-3 (NT-3)+/−mice has been described previously (Ernfors et al., 1994a,b); the genetic background of both strains and their wild-type littermates was 129Sv x BALB/c. Litters of mice were selected at the animal house such that they contained approximately the same number of transgenic and wild-type mice. Litters were then randomly assigned to various treatment groups, and the person performing the tests was always blind to the genotype. The production of transgenic mice and all of the animal experiments were done in accordance to the guidelines of The Society for Neuroscience and were accepted by the experimental animal ethics committee of the University of Kuopio.
For the analysis of behavioral effects of antidepressant drugs, we used the forced swim test, which has a high predictive value for antidepressant activity that is applicable to mice (Porsolt et al., 1977; Cryan et al., 2002). Adult male transgenic mice and wild-type littermates were allowed to adapt to the test room for several days and then randomly submitted to a forced swim test without a pre-swim. Saline, imipramine (30 mg/kg; Sigma, St. Louis, MO), or fluoxetine (20 mg/kg; Orion Pharma, Turku, Finland) was injected intraperitoneally, and after 30 min the mice were placed in a clear glass cylinder with a diameter of 16 cm, half-filled with clear water at 24°C (water depth of 14 cm did not allow the mice to reach the bottom of the cylinder; water was changed after each mouse) for a total of 6 min, and immobility was recorded live during the last 4 min by an investigator blind to the genotype and treatment.
Tyrosine phosphorylation was assayed in cortical samples of male mice that were rapidly dissected at indicated times after saline or antidepressant injection. Tissue lysis, protein precipitation with wheat germ agglutinin (WGA) or pan-trk antibody, and the following Western blot analysis from these samples were performed according to previously described methods (Aloyz et al., 1999) with slight modifications. Briefly, tissue samples were lysed in buffer containing 137 mm NaCl, 20 mm Tris, pH 8.0, 1% NP-40, 10% glycerol, 50 mm sodium fluoride, 2× Complete Mini (Roche Diagnostics, Hertforshire, UK), and 2 mmsodium vanadate. After homogenization, tissues were incubated at +4°C for 20 min and centrifuged at 13,000 rpm for 15 min. TrkB was precipitated using either 50 μl of WGA (Amersham Biosciences, Arlington Heights, IL) or 10 μl of anti-trk antibody (sc-11; Santa Cruz Biotechnology, Santa Cruz, CA) and collected with protein-A Sepharose (Amersham Biosciences). Electrophoresis was performed with 7.5% SDS-polyacrylamide gels. Anti-phosphotyrosine (4G10; Upstate Biochemicals, Waltham, MA; dilution 1:10,000), anti-phospho-trk pY490 (New England Biolabs, Beverly, MA), pY674/675 (New England Biolabs), and anti-trkBout (generously provided by Dr. David Kaplan, McGill University, Montreal, Canada; dilution 1:5000) antibodies were used to detect phosphorylated and total trkB, respectively. Western blots were scanned and quantitated with image analysis. Data are presented as mean percentages of the ratio of phosphorylated trk (4G10, pY674/675, pY490) to full-length trkB (anti-trkBout) signal intensity levels found in saline-treated animals.
For phospho-cAMP response element-binding protein (CREB) immunostaining, mice were injected intraperitoneally with saline, fluoxetine (20 mg/kg), or imipramine (30 mg/kg), deeply anesthetized by pentobarbital 30 min after the injection, and perfused with 4% paraformaldehyde. Floating sections were stained with anti-phospho-CREB-specific antibody (New England Biolabs) and developed with diaminobenzidine. Phospho-Ser131-antibody recognizes not only CREB but also phosphorylated forms of other CREB family members; therefore, the increased staining observed after antidepressant treatments may also indicate changes in the phosphorylation status of other family members as well. Stained cells in the anterior cingulate–prefrontal cortex were counted using an unbiased stereological method (StereoInvestigator, MicroBrightField Inc., Colchester, VA).
All of the data are presented as means ± SEM. Student'st test and one-way ANOVA together with Bonferroni as apost hoc test were used for statistical analysis.
Results
Behavioral response to antidepressants requires normal trkB signaling
Although there is evidence that chronic antidepressant treatment increases BDNF mRNA in hippocampus (Nibuya et al., 1995; Russo-Neustadt et al., 2000) and infusion of BDNF into hippocampus or midbrain area produces behavioral effects that resemble those produced by antidepressant drugs (Siuciak et al., 1997; Shirayama et al., 2002), the role of endogenous BDNF and trkB activation in the behavioral responses to antidepressant drugs remains unclear. We have investigated this question by submitting trkB.T1 transgenic mice to a forced swim test, which is widely used for screening of potential antidepressant drugs and has a high predictive value for antidepressant activity (Porsolt et al., 1977; Cryan et al., 2002). These mice overexpress the dominant-negative trkB.T1 isoform in neurons in hippocampus and cortex, including anterior cingulate and prefrontal cortices, and show reduced trkB signaling in brain (Saarelainen et al., 2000b) (Fig.1). When we previously tested these mice in the Morris water maze, there were no differences in the swimming speed between wild-type and transgenic littermates, although the long-term memory of transgenic mice was impaired (Saarelainen et al., 2000a). These data suggest that any differences in the behavior of these mice in the forced swim test are not simply a reflection of altered swimming performance. Consistent with this, no differences were observed in the immobility time between transgenic and wild-type littermates injected with saline 30 min before the 6 min forced swim test (Fig. 2) (p = 0.28; Mann–Whitney test). In the forced swim assay, mice respond to antidepressants by reducing their immobility time (Porsolt et al., 1977). Indeed, both imipramine and fluoxetine, two widely used antidepressants, significantly reduced immobility in wild-type mice (Fig. 2) (ANOVA: F = 8.52; p = 0.002). In contrast, neither imipramine nor fluoxetine had any significant effect on the immobility time of the trkB.T1 transgenic mice (Fig. 2) (ANOVA: F = 0.098; p = 0.91), indicating that inhibition of trkB signaling rendered transgenic mice insensitive to the behavioral effects of antidepressant drugs. The response to both antidepressants was significantly reduced in transgenic mice when compared with wild-type littermates (imipramine,p = 0.012; fluoxetine, p = 0.004; Bonferroni's test)
Fig. 1.
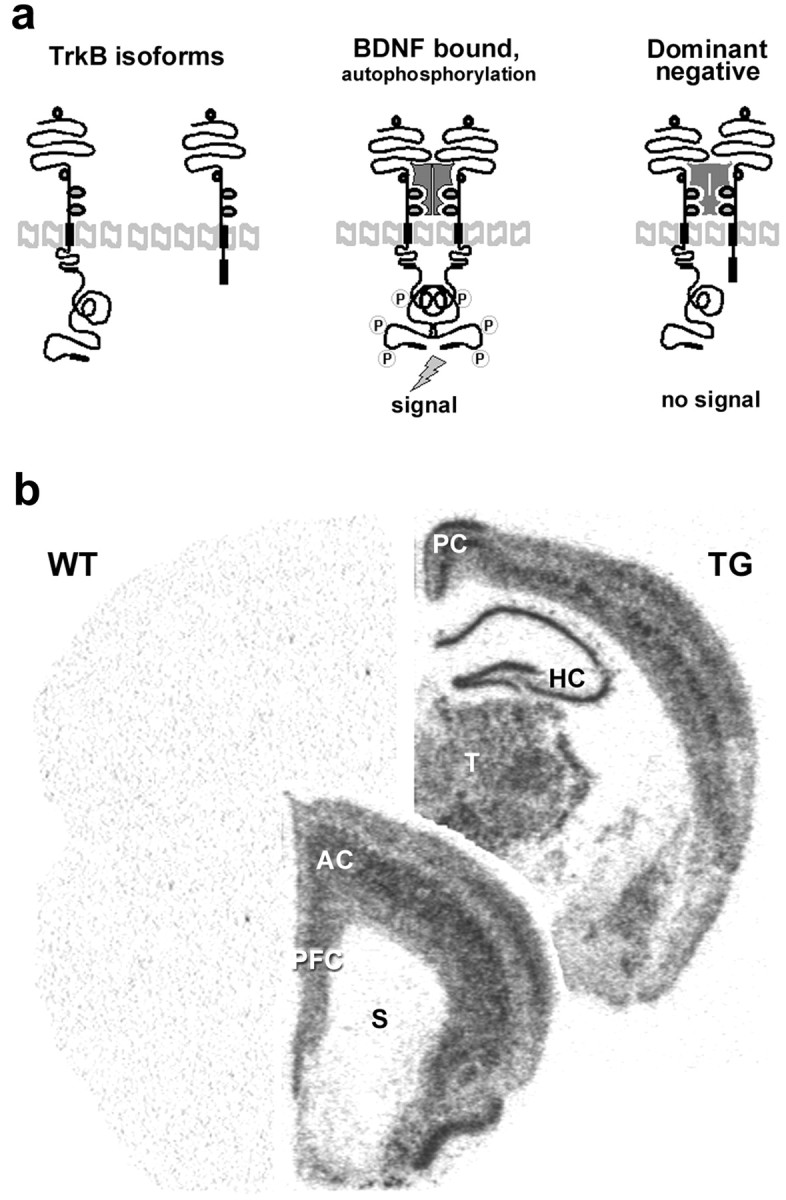
Transgenic mice overexpressing the truncated trkB.T1 isoform in neurons. a, Schematic representation of trkB activation by BDNF and of the dominant-negative role of the truncated trkB.T1 isoform. b, TrkB.T1 transgene expression in adult transgenic mouse brain. In situhybridization of a wild-type (WT) and transgenic (TG) mouse at two different rostrocaudal levels with an oligonucleotide probe that specifically recognizes the FLAG-epitope sequence inserted into the transgene.AC, Anterior cingulate cortex; HC, hippocampus; PC, posterior cingulate cortex;PFC, prefrontal cortex; S, striatum;T, thalamus.
Fig. 2.

TrkB.T1 transgenic mice are resistant to antidepressants in forced swim test. Immobility time in a forced swim test in trkB.T1 transgenic (filled bars) and wild-type mice (open bars) after injection with saline, imipramine (30 mg/kg), or fluoxetine (20 mg/kg; n = 8–12 for each differently treated genotype). *p < 0.05, **p < 0.01, compared with saline-treated wild-type animals. #p < 0.05 and##p < 0.01 show the difference between transgenic and wild-type animals with the same treatment (one-way ANOVA, post hoc analysis; Bonferroni's test). Data are presented as means ± SEM.
Because BDNF signaling influences serotonergic innervation to cortex (Mamounas et al., 2000; Poo, 2001), we examined the tissue contents of 5-HT and 5-hydroxyindoleacetic acid (5-HIAA) from two cortical areas from transgenic and wild-type mice by using HPLC to investigate whether the characteristic neurochemical changes in monoamine metabolism after antidepressant drug administration (Fuller and Wong, 1977) also occur in the brains of transgenic mice (Table1). Basal concentrations of 5-HT were similar in transgenic and wild-type mice in both anterior cingulate and prefrontal cortex. Basal levels of 5-HIAA were similar in anterior cingulate cortex between the different genotypes, but 5-HIAA was reduced in the prefrontal cortex in trkB.T1 transgenic mice, which is consistent with the observation that BDNF increases brain 5-HIAA levels (Siuciak et al., 1994, 1996). Most importantly, treatment with a selective serotonin reuptake inhibitor, fluoxetine, caused a significant increase in serotonin concentrations in the prefrontal and cingulate cortices in both genotypes, demonstrating that the serotonin uptake system was functional and similarly affected by acute administration of fluoxetine in the brains of both wild-type and transgenic mice. The integrity of serotonergic innervation to cortex was further investigated by immunostaining with antibodies against serotonin or serotonin transporter. These stainings revealed no obvious gross differences in cortical serotonin innervation between the trkB.T1 transgenic and wild-type mice (Dr. Laura Mamounas, National Institute of Neurological Disorders and Stroke, Bethesda, MD, personal communication).
Table 1.
Acute effects of fluoxetine on brain indoleamine levels in trkB.T1 transgenic mice
| Wild type | Transgenic | |||
|---|---|---|---|---|
| Saline | Fluoxetine | Saline | Fluoxetine | |
| 5-HT (nmol/gm tissue) | ||||
| Frontal cortex | 2.31 ± 0.12 | 2.86 ± 0.17* | 2.05 ± 0.21 | 2.67 ± 0.11* |
| Cingulate cortex | 1.57 ± 0.07 | 1.88 ± 0.23* | 1.54 ± 0.22 | 1.88 ± 0.21* |
| 5-HIAA (nmol/gm) | ||||
| Frontal cortex | 1.52 ± 0.11 | 1.30 ± 0.04 | 1.20 ± 0.051-159 | 1.33 ± 0.08 |
| Cingulate cortex | 0.77 ± 0.05 | 0.72 ± 0.04 | 0.70 ± 0.05 | 0.70 ± 0.04 |
HPLC analysis of the total tissue contents of 5-HT and 5-HIAA levels in frontal and cingulate cortex from adult wild-type littermates and trkB.T1 transgenic mice 3 hr after fluoxetine (20 mg/kg) or saline intraperitoneal injection. Data are shown as means ± SEM from five to six animals per group.
p < 0.05, indicating the significant difference of the treatment within the same genotype;
F1-159: p < 0.05, significantly different from wild type (Student's t test).
TrkB can be activated by both BDNF and NT-3 (Soppet et al., 1991;Squinto et al., 1991). To investigate which of the trkB ligands is involved in the antidepressant-induced behavioral effects, we submitted mice deficient of BDNF or NT-3 to a forced swim test. Because BDNF−/−and NT-3−/−mice die early during postnatal development (Ernfors et al., 1994a,b), we used heterozygous mice, which produce approximately half of the corresponding neurotrophin in their brain. The swimming behavior of saline-injected BDNF+/− and NT-3+/− mice was indistinguishable from the corresponding saline-treated wild-type mice (Fig. 3). When the mice were treated with imipramine (30 mg/kg; 30 min before the test), the swimming immobility of wild-type as well as NT-3+/− mice was significantly reduced when compared with the corresponding saline-treated mice of the same strain (Fig. 3) (p < 0.01; one-way ANOVA and Bonferroni'spost hoc test). In contrast, the swimming immobility of imipramine-treated BDNF+/− mice was not significantly different from the saline-treated (wild-type or BDNF+/−) mice (Fig. 3). Indeed, the swimming behavior of BDNF+/− mice was similar to that of trkB.T1-overexpressing mice, which suggests that BDNF-mediated trkB activation is required for normal behavioral response to antidepressants in the forced swim test.
Fig. 3.
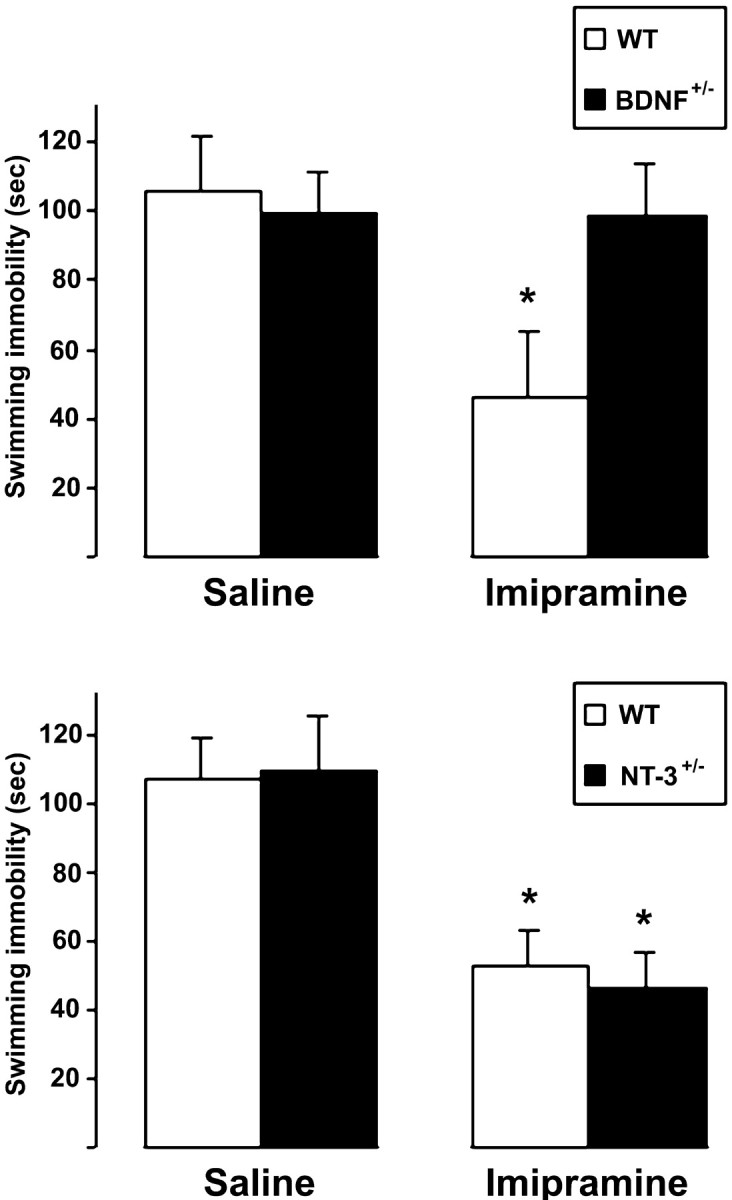
Forced swim test behavior of BDNF+/− (top panel) and NT-3+/− mice (bottom panel). Female BDNF+/− mice and wild-type littermates were injected with saline or imipramine (30 mg/kg;n = 8 and 9 for saline and imipramine, respectively, for both genotypes) and subjected to a forced swim test (6 min, immobility recorded during the last 4 min) 30 min after the injection. Male NT-3+/− mice and wild-type littermates were treated identically (n = 9 and 10 for saline and imipramine, respectively, for both genotypes). Means ± SEM; *p < 0.01; one-way ANOVA and Bonferroni's post hoc test.
Antidepressant treatment induces trkB autophosphorylation
If the behavioral response to antidepressants in the forced swim test requires intact trkB signaling in brain, as our results suggest, then activation of endogenous trkB signaling would be expected to take place within 30 min after antidepressant administration. Previous studies have shown that chronic, but not acute, antidepressant administration increases BDNF mRNA levels in hippocampus and cortex (Nibuya et al., 1995; Russo-Neustadt et al., 2000), but electroconvulsive shock therapy, the most effective antidepressant treatment, increases BDNF mRNA also acutely (Nibuya et al., 1995). We have used phosphorylation of trkB as an assay to investigate activation of neurotrophin signaling in brain. Autophosphorylation of trk receptors is the initial step in the intracellular signal transduction activated by neurotrophins (Kaplan and Miller, 2000), and it has been previously validated as an assay for local neurotrophin release (Aloyz et al., 1999).
Western blots of WGA-precipitated cortical extracts probed with phosphotyrosine (pTyr)-specific 4G10 antibody revealed that imipramine and fluoxetine significantly increased phosphorylation of a protein that migrated at 145 kDa, exactly at the same level as trk receptors (as assayed using trkB-specific antibody) at 30 min after the injection (1.8- and 1.4-fold for imipramine and fluoxetine, respectively), compared with saline-treated animals (Fig.4a); however, total trkB protein levels were not influenced by antidepressants (Fig.4a), suggesting the possibility that these drugs increase trkB phosphorylation but not trkB protein synthesis.
Fig. 4.

Antidepressants induce trkB autophosphorylation in mouse brain. a, Mice were injected intraperitoneally with imipramine (IMI) (30 mg/kg;n = 15) or saline (Sal) (n = 15), or with fluoxetine (FLX) (20 mg/kg, n = 11) or saline (Sal) (n = 11).Top panel, Representative Western blots of WGA-precipitated samples sequentially probed with anti-phosphotyrosine antibody (4G10) and anti-trkBout antibody.TrkB indicates the location of 145 kDa band.Bottom panel, Quantitated levels of the trkB from different treatments. Data are presented as mean ± SEM percentages of the ratio of phosphorylated trk (4G10) to full-length trkB (anti-trkBout) signal intensity levels found in saline-treated animals. **p < 0.01 against saline control; Student's t test.b, Trk immunoprecipitation. Mice received saline (Sal) or imipramine (IMI) (30 mg/kg, i.p.) 30 min before protein extraction. Representative Western blots precipitated with trk antibody and sequentially probed with anti-phosphotyrosine antibody 4G10 and anti-trkBoutantibody are shown. TrkB indicates the location of trkB.IP, Immunoprecipitation; WB, Western blot.
To verify that the phosphorylated 145 kDa protein represents trk receptors, we immunoprecipitated cortical lysates from imipramine-treated mice with a trk receptor-specific antibody. Probing the blot with pTyr antibody revealed that imipramine indeed had induced tyrosine phosphorylation of the trk receptor (Fig. 4b) but had no effect on total trkB protein levels. Because trkB is the predominant trk species in cortex (Aloyz et al., 1999) and the antidepressant-induced trk activation was inhibited by a dominant-negative trkB isoform (see Fig. 9), which inhibits the activation of trkB but not trkA and trkC (Haapasalo et al., 2001), the phosphorylated trk species is most probably trkB. Furthermore, the abnormal forced swim response in BDNF+/− but not in NT-3+/− mice also suggests that trkB is the predominant trk species in response to antidepressants. However, because BDNF also binds to trkC receptors, albeit at a lower affinity than to trkB, these data do not rule out the possibility that trkC is also phosphorylated in response to antidepressants. Blots immunoprecipitated with trk antibody and probed with pTyr antibody revealed two additional bands that were coimmunoprecipitated with trkB and phosphorylated in response to imipramine (Fig. 4b). The identity of these proteins is currently unclear, but they may represent intracellular signaling proteins interacting with trkB and phosphorylated in response to trkB activation.
Fig. 9.

a, TrkB autophosphorylation in trkB.T1 transgenic and wild-type littermate mice after acute fluoxetine treatment and forced swim test (n = 8–16 for each differently treated genotype). There was no significant difference in the extent of trkB phosphorylation induction between saline- and fluoxetine-treated transgenic mice. *p < 0.05, **p < 0.01, compared with saline-treated wild-type animals (Student's t test). Data presented as means ± SEM. b, Representative Western blots corresponding to data shown in a precipitated with WGA and probed first with anti-phosphotyrosine antibody 4G10 and reprobed with anti-trkBout antibody to ascertain that samples contained similar amounts of trkB. Wt, Wild type;Tg, transgenic.
TrkB can be phosphorylated to several intracellular tyrosine residues, and the phosphorylation of particular tyrosines leads to activation of specific signaling pathways. Double tyrosine residues 706/707 (analogous to tyrosines 694/695 in trkA) are situated at the autophosphorylation site, which is required for the activation of trkB. Tyrosine 515 (analogous to Y490 in trkA) is the docking site of the shc adaptor protein, which leads to the activation of the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway (Kaplan and Miller, 2000; Huang and Reichardt, 2001). We used trk phosphotyrosine-specific antibodies to investigate which of the tyrosines are phosphorylated in response to antidepressant treatment. pY694/695 antibody, which recognizes the tyrosine residues 706/707 at the autophosphorylation site, demonstrated a robust increase in phosphorylation in prefrontal cortex 30 min after imipramine treatment (178 ± 23% of control; p < 0.01) (Fig.5). In contrast, pY490 antibody, which recognizes the phosphorylated tyrosine 515 at the shc binding site, failed to show increased phosphorylation in response to antidepressants in prefrontal cortex or hippocampus (108 ± 9% of control;p = 0.53) (Fig. 5), although this antibody detected a robust increase in trk phosphorylation in cultured hippocampal neurons exposed to BDNF (data not shown). These data suggest that antidepressants induce a tyrosine-specific phosphorylation of trkB and indicate that pathways downstream of the pY515 may not be involved in antidepressant-induced trkB signaling.
Fig. 5.

Autophosphorylation of different trkB tyrosines after antidepressant treatment. Top panel, Tissue lysates from prefrontal cortex from mice injected with saline (SAL) or imipramine (IMI) (30 mg/kg) 30 min before dissection were precipitated with anti-pan-trk antibody (IP-trk) or wheat germ agglutinin (WGA) and detected with phosphotyrosine antibody (4G10), pY674/5 antibody (recognized phosphorylated trk autophosphorylation site), pY490antibody (recognizes shc binding site in trk), or panTrkantibody (to verify equal loading). The three WGA-precipitated blots show the same representative lanes on a single filter sequentially probed with different antibodies. Bottom panel, Quantitation of autophosphorylation-different trkB tyrosines. Data are shown as a mean ± SEM; n = 6 for each treatment. The experiment was repeated at least three times for each of the antibodies, with similar results.
We also investigated the effect of antidepressants on total brain BDNF protein levels by BDNF enzyme immunoassay (Nawa et al., 1995). Imipramine did not significantly change total cortical BDNF protein levels in wild-type mice [6.0 ± 0.3 and 6.2 ± 0.3 ng/g of tissue in imipramine (n = 12) and saline-treated animals (n = 11), respectively], which is consistent with the observation that acute treatment with antidepressants does not change BDNF mRNA levels (Nibuya et al., 1995). It should be noted, however, that this assay does not measure changes in BDNF release but detects total cortical BDNF levels. Because a recent study showed that intrahippocampal infusion of neurotrophin-3 produced antidepressant-like behavioral effects (Shirayama et al., 2002), we also investigated the effect of imipramine on cortical neurotrophin-3 protein levels. Also in this case, no significant differences could be observed (94 ± 6.9% of control). These data suggest that if acute antidepressant treatment activates trkB by increasing BDNF release, then the levels of BDNF released, although sufficient to activate trkB signaling, are too low or too local to detectably influence the total pool of cortical BDNF mRNA or protein.
Both acute and chronic antidepressant treatments induce trkB phosphorylation
We next investigated the time course of trkB activation by antidepressants. At 30 min after fluoxetine injection, autophosphorylation of trkB was increased significantly (Fig.6a). In contrast, at 6 hr, trkB phosphorylation was reduced significantly below the control level, presumably reflecting desensitization after stimulation (Fig.6a). At 24 hr, autophosphorylation was back at the baseline. The relatively short duration of trkB autophosphorylation is consistent with the previous observation that the kainic acid-induced increase in trkB phosphorylation was already reduced back to baseline at 2 hr after injection (Aloyz et al., 1999).
Fig. 6.

a, Time course of changes in trkB autophosphorylation in response to fluoxetine administration (20 mg/kg, i.p.). b, Autophosphorylation of trkB after acute and chronic antidepressant treatments. Mice received saline (open bars) or imipramine (30 mg/kg) (filled bars) intraperitoneally once (30 min; n = 18 for both groups) or once daily for 3 weeks (21 d; n = 11 for both groups). Top panel, Representative Western blots precipitated with WGA and sequentially probed with anti-phosphotyrosine antibody 4G10 and anti-trkBout antibody. Bottom panel and a show quantitated levels of phosphorylated trkB from different treatments. Data are presented as means ± SEM percentages of the ratio of phosphorylated trk (4G10) to full-length trkB (anti-trkBout) signal intensity levels found in saline-treated animals. *p < 0.03, **p < 0.01 against saline control; Student'st test.
Because the clinical effects of antidepressants require repeated administration, we investigated whether the induction of trkB phosphorylation persists during chronic treatment. In mice treated with imipramine for 3 weeks, a similar significant increase in trkB autophosphorylation levels was observed 30 min after the last injection as was seen after acute antidepressant administration (Fig.6b). At 6 and 24 hr after chronic imipramine treatment, trkB phosphorylation was not significantly increased (116 ± 13% and 119 ± 16% of controls, respectively). However, no desensitization of the trkB phosphorylation was seen at 6 hr after the chronic treatment, as was observed 6 hr after the acute injection. Thus, trkB autophosphorylation is stimulated to a similar degree by both acute and chronic antidepressant treatments.
TrkB activation is most pronounced in prefrontal cortex
It is unclear in which target brain area antidepressant drugs bring about their action, but hippocampus as well as prefrontal and anterior cingulate cortex are often implicated in this context. The brain area that was included in our analysis of trkB phosphorylation status comprised the anterior cingulate cortex as well as the sensory motor cortices in the frontoparietal cortex. To further investigate whether there are brain area-dependent differences in the responsiveness of trkB phosphorylation to antidepressants, we dissected various cortical areas 30 min after the injection from brains of mice treated acutely (30 min) or chronically (21 d) with imipramine. In mice treated acutely with imipramine, a small but consistent increase in trkB phosphorylation was observed in all the cortical areas examined after acute imipramine injection (Fig.7). However, the trkB autophosphorylation response to antidepressants was by far the most robust in the anterior cingulate–prefrontal cortex area (Fig. 7). After 21 d of imipramine treatment, however, clearly elevated trkB autophosphorylation could be observed also in the hippocampus in addition to the anterior cingulate–prefrontal cortex, but not in other cortical areas examined (Fig. 7). This observation is in agreement with the potential role of the anterior cingulate–prefrontal cortex and hippocampus in the pathophysiology of depression (Drevets, 1998).
Fig. 7.

Effects of acute or chronic antidepressant treatments on trkB phosphorylation in different cortical areas. Lysates of dissected samples from anterior cingulate–prefrontal cortex (AC), hippocampus (HC), posterior cingulate cortex (PC), and parietal cortex (CX) of mice injected once or for 21 d with imipramine (30 mg/kg, 30 min before dissection;n = 6) were precipitated with WGA, and blots were probed with anti-phospho-trk 674/675 antibody. Means ± SEM; percentage of saline-treated control in the same cortical area (n = 6) shown (Cont) is saline control of the AC area. *p < 0.05,#p = 0.053; Student's ttest.
Antidepressants induce trkB-dependent phosphorylation of CREB
Phosphorylation of CREB has been shown to be increased in brain in response to trkB activation (Shaywitz and Greenberg, 1999) as well as after chronic antidepressant administration (Thome et al., 2000). We used an antibody recognizing phosphorylated CREB (as well as other phosphorylated members of the CREB/ATF family) to investigate whether trkB activation in response to antidepressant treatment might induce intracellular signal transduction leading to CREB phosphorylation. In saline-injected mice, a variable staining of nuclei was observed in many cortical areas (Fig. 8). However, in the anterior cingulate and prefrontal cortex, a robust increase in nuclear phospho-CREB staining was observed in five of six mice 30 min after imipramine injection, whereas only few nuclei were stained in this region in six mice injected with saline (1 ± 0.58 and 20.5 ± 6.7 positive cells within the counted area in saline- and imipramine-injected mice, respectively; p < 0.05). A similar, albeit less pronounced response, was also observed in the prefrontal cortex of mice injected with fluoxetine (1 ± 0.58 and 6.25 ± 1.4 positive cells within the counted area in saline- and fluoxetine-injected mice, respectively; p < 0.05). Induction of CREB phosphorylation in anterior cingulate cortex by imipramine was clearly weaker in transgenic mice, which overexpress dominant-negative trkB isoform (Fig. 8), suggesting that antidepressant-induced CREB phosphorylation is at least partially a consequence of increased trkB signaling. Thome et al. (2000) did not observe any significant increase in phospho-CREB levels in hippocampus after acute fluoxetine or desipramine administration; however, they assayed phospho-CREB at 6 hr after injection, at the time when trkB phosphorylation was downregulated (Fig. 6a). Our data suggest that the signal initiated by trkB autophosphorylation in response to antidepressant drugs is further transmitted and conveyed to nucleus by phosphorylated CREB.
Fig. 8.
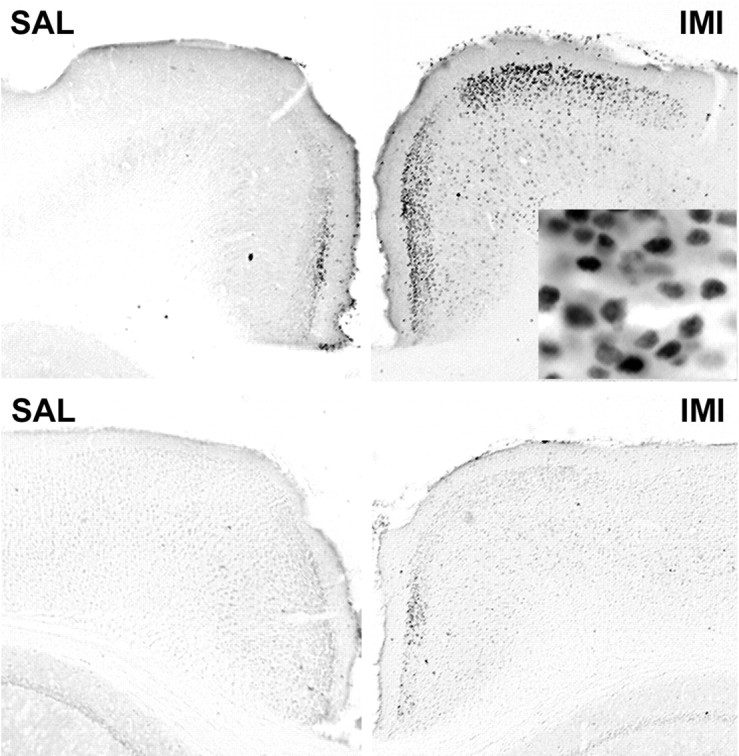
Phosphorylation of CREB in the anterior cingulate cortex 30 min after saline (SAL) or imipramine (IMI) (30 mg/kg, i.p.) administration. Top panel, Wild-type mice; bottom panel, trkB.T1-overexpressing mice; magnification, 40×.Inset shows a larger magnification (400×), demonstrating nuclear localization of the phosphorylated CREB.
TrkB autophosphorylation correlates with the behavioral response to antidepressants
To be able to correlate the lack of behavioral responsiveness to antidepressants in trkB.T1 transgenic mice with the trkB autophosphorylation status, we investigated trkB phosphorylation status in a group of mice subjected to the forced swim test. Exposure to saline injection and forced swim stress alone did not influence the phosphorylation status of trkB when compared with naive mice gently taken directly from their home cage (98 ± 12% of control for trkB; n = 8), indicating that any observed changes in trkB phosphorylation can be attributed to the antidepressant treatment. Fluoxetine injection followed by a forced swim test reduced immobility time (data not shown) and increased trkB phosphorylation at 30 min in wild-type mice (Fig. 9), which was similar to that seen in mice not subjected to forced swim. In trkB.T1 transgenic mice, the basal trkB phosphorylation status was significantly lower than in wild-type mice, consistent with the inhibition of trkB activation by the dominant-negative transgene (Fig.9). More importantly, and in contrast to that observed in wild-type mice, fluoxetine injection did not significantly increase trkB phosphorylation levels in transgenic mice (Fig. 9), which is consistent with the insensitivity of these mice to the behavioral effects of fluoxetine. These data strongly suggest that the trkB activation and signaling observed within 30 min after antidepressant injection is required for the behavioral effects characteristically produced by antidepressants in mice.
Discussion
Previous data show that infusion of BDNF into hippocampus or midbrain area produces behavioral effects similar to those seen after antidepressant treatment (Siuciak et al., 1997; Shirayama et al., 2002). These data have suggested that BDNF may play a role in the long-term effects of antidepressants, but they have not addressed the role of endogenous BDNF in this context. We have here shown that acute as well as chronic antidepressant administration activates neurotrophin signaling in a BDNF-dependent manner in prefrontal cortex and that this signaling is required for the typical behavioral effects produced by these drugs in experimental animals. Together with previous observations of increased BDNF mRNA levels after chronic antidepressant administration (Nibuya et al., 1995; Russo-Neustadt et al., 2000), our data strongly suggest that this clinically widely used class of drugs may act at least partially by activating endogenous BDNF signaling.
Drugs that locally enhance the production and release of endogenous neurotrophins at their normal site of synthesis would have obvious advantages as neurotrophic therapy when compared with infusion of exogenous BDNF into brain or even with potential small molecular weight mimetics of neurotrophin action, which activate neurotrophin receptors all over the body (Thoenen et al., 1994). We show here that antidepressant drugs activate release of endogenous BDNF and induce its signal transduction. Activation of neurotrophin signaling with potential long-term effects on various neuronal systems is consistent with the relatively wide therapeutic efficacy of antidepressants in many CNS disorders. In addition to being used to treat depression, antidepressants are used to treat chronic pain, phobia, obsessive-compulsive disorders, bulimia, and premenstrual syndrome, among others (Nestler et al., 2002). Remarkably, sensory motor activation in stroke patients was recently shown to be improved by a single fluoxetine injection (Pariente et al., 2001), which is consistent with the important role of endogenous BDNF in stroke (Saarelainen et al., 2000b). In addition to antidepressants, antagonists of α2 adrenoreceptors were recently shown to increase BDNF release and trkB activation in cortex (Aloyz et al., 1999). Furthermore, we have recently shown that memantine, a glutamate receptor antagonist that is used clinically to treat mild dementia (Parsons et al., 1999), increases BDNF mRNA in cortex and hippocampus (Marvanova et al., 2001). These data suggest that drugs already in clinical use may act at least partially by locally increasing neurotrophin release.
The mechanism through which antidepressants activate trkB signaling remains unclear. The observation that BDNF+/− mice and trkB.T1 mice showed a similar phenotype in the forced swim test suggests that antidepressants might directly or indirectly stimulate BDNF release, which then leads to trkB activation. Antidepressant drugs acutely facilitate signaling of serotonin or norepinephrine by inhibiting their reuptake to presynaptic terminals (tricyclic antidepressants, such as imipramine) or by inhibiting their catabolism (monoamine oxidase inhibitors), or binding to serotonin autoreceptors (for review, see Duman et al., 1997; Charney, 1998; Skolnick, 1999; Manji et al., 2001; Nestler et al., 2002). BDNF is anterogradely transported to nerve terminals and released in response to nerve stimulation in a manner similar to neuropeptides (Altar and DiStefano, 1998; Aloyz et al., 1999; Balkowiec and Katz, 2000). Inhibition of presynaptic α2-adrenoreceptors on noradrenergic nerve terminals by yohimbine has been shown to increase the release not only of norepinephrine but also of BDNF, and this increased release induces trkB autophosphorylation in cortex (Aloyz et al., 1999). However, because long-term antidepressant treatment silences rather than activates monoaminergic neuronal firing (Szabo et al., 1999), it is possible that increased 5-HT and norepinephrine levels may locally induce increased BDNF release. Indeed, exposure of cultured raphe neurons to serotonin increased BDNF release, which in turn enhanced serotonergic differentiation of the neurons (Galter and Unsicker, 2000). Taken together, these data are clear evidence that manipulation of monoamine transmission may induce BDNF release. It is possible that changes in synaptic serotonin and norepinephrine levels induced by antidepressants increase BDNF release through a comparable mechanism, thereby initiating trkB signaling and a neurotrophic response. However, because we failed to observe any changes in total BDNF protein content in response to antidepressants, it is also possible that antidepressants activate trkB through intracellular signal transduction, as has been reported recently for certain G-protein-coupled receptors (Lee and Chao, 2001). The observation that only a subset of tyrosines in trkB is phosphorylated might also favor an intracellular activation mechanism.
We have observed a robust autophosphorylation response to antidepressants in the autophosphorylation site of trkB (tyrosines 706/707), but no response was observed when a phospho-specific antibody to the shc site (tyrosine 515) was used. The lack of specific antibodies to the other phosphorylated tyrosines in trkB prevented us from testing whether the phosphorylation occurs exclusively to tyrosines 706/707 or whether other tyrosines in trkB are also phosphorylated in trkB. These results suggest that trkB signal transduction induced by antidepressants does not involve the shc interaction with trkB. Mice that have tyrosine 515 of trkB gene mutated to phenylalanine are viable and fertile and display defects mainly in systems dependent on NT-4 signaling, suggesting that tyrosine 515 is not absolutely required for BDNF-dependent signaling in vivo(Minichiello et al., 1998). Although tyrosine 515-dependent pathways are robustly activated in vitro and when high concentrations of BDNF are injected into brain in vivo, signaling changes that take place after the release of endogenous BDNF in brain may use, at least partially, other signal transduction pathways.
The response of trkB phosphorylation to antidepressant administration was highest in the region of anterior cingulate and prefrontal cortex after both acute and chronic treatments but was observed in hippocampus only after chronic treatment. Several studies have implicated dysfunction of prefrontal and anterior cingulate cortices in the pathophysiology of depression (for review, see Drevets, 1998). Depression is associated with reduced cortical thickness and neuronal size (Rajkowska et al., 1999) as well as with decreased blood flow and metabolism in prefrontal and anterior cingulate cortex (Drevets et al., 1997; Drevets, 1998), and these effects are reversed when depression is treated successfully (Wu et al., 1992; Nobler et al., 1994). These studies suggest that depression is associated with and perhaps even caused by cortical neuronal atrophy and the accompanying loss of neuronal communication in prefrontal cortex. Because neuronal atrophy has also been observed in cortex of mice lacking trkB in forebrain (Xu et al., 2000) and serotonergic innervation to cortex is gradually lost with age in BDNF+/− mice (Lyons et al., 1999), it is possible that reduced trkB signaling may be one of the factors that leads to decreased neuronal communication and development of depression. However, we did not observe any baseline deficits in the forced swim behavior in trkB.T1 and BDNF+/−mice, and similar findings were recently reported for BDNF+/− mice (MacQueen et al., 2001), which indicates that trkB signaling may be more important for the antidepressant drug effect than for the baseline alterations of depression.
Increase in BDNF release and trkB activation in cortex after antidepressant administration is expected to lead to plastic changes in synaptic function and neuronal connectivity. BDNF stimulates outgrowth and regeneration of dendrites and axons (McAllister et al., 1999;Mamounas et al., 2000; Poo, 2001), promotes synaptogenesis (Poo, 2001), and mediates activity-dependent selection and maturation of functional synapses (Katz and Shatz, 1996). It is conceivable that BDNF-induced plastic changes may take time to develop and mature, which might explain in part why the clinical antidepressive effect develops with a delay after the beginning of the treatment. Although antidepressants induce BDNF release and trkB signaling virtually immediately after administration, the neuronal events that are produced by increased neurotrophin signaling may develop slowly and correlate with the gradual appearance of the clinical response. Our results support the hypothesis that neurotrophin-mediated improvement in neuronal communication (Duman et al., 1997; Altar, 1999; Skolnick, 1999; Manji et al., 2001; Reid and Stewart, 2001), or perhaps regeneration of synaptic connections lost during depression (Drevets, 1998; Rajkowska et al., 1999), may be the underlying mechanism of antidepressant drug action and suggest that stimulation of BDNF release and trkB signaling in response to antidepressant drug treatment may be the critical event that initiates this effect.
T.S. and P.H. contributed equally to this work.
This work was supported by grants from The Academy of Finland, Finnish National Technology Agency, and Sigrid Juselius Foundation to E.C., and from the Swedish Research Council and Göran Gustafssons Foundation to P.E. We thank Dr. D. Kaplan (Montreal Neurological Institute, McGill University, Canada) for the trkB antibodies, Dr. L. Mamounas (National Institute of Neurological Disorders and Stroke, National Institutes of Health) for help with 5-HT and SERT stainings, Drs. H. Thoenen, M. Castrén, M. Saarma, J. Tuomisto, and J. Lehtonen for their comments on this manuscript, Dr. J. Sirviö (Orion-Pharma, Turku, Finland) for the donation of fluoxetine, and L. Kaskela, A.-M. Friis, I. Sipola, O. Kontkanen, and T. Rauramaa for assistance.
Correspondence should be addressed to Eero Castrén, A. I. Virtanen Institute, University of Kuopio, P.O. Box 1627, 70211 Kuopio, Finland. E-mail: Eero.Castren@uku.fi.
R. Aloyz's present address: Lady Davis Institute for Medical Research, Sir Mortimer B. Davis–Jewish General Hospital, and McGill University, Montreal, Quebec, Canada H3A 2B4.
References
- 1.Aloyz R, Fawcett JP, Kaplan DR, Murphy RA, Miller FD. Activity-dependent activation of TrkB neurotrophin receptors in the adult CNS. Learn Mem. 1999;6:216–231. [PMC free article] [PubMed] [Google Scholar]
- 2.Altar CA. Neurotrophins and depression. Trends Pharmacol Sci. 1999;20:59–61. doi: 10.1016/s0165-6147(99)01309-7. [DOI] [PubMed] [Google Scholar]
- 3.Altar CA, DiStefano PS. Neurotrophin trafficking by anterograde transport. Trends Neurosci. 1998;21:433–437. doi: 10.1016/s0166-2236(98)01273-9. [DOI] [PubMed] [Google Scholar]
- 4.Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998;59:11–14. [PubMed] [Google Scholar]
- 6.Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- 7.Drevets WC. Functional neuroimaging studies of depression: the anatomy of melancholia. Annu Rev Med. 1998;49:341–361. doi: 10.1146/annurev.med.49.1.341. [DOI] [PubMed] [Google Scholar]
- 8.Drevets WC, Price JL, Simpson JR, Jr, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 9.Duman RS, Vaidya VA. Molecular and cellular actions of chronic electroconvulsive seizures. J Ect. 1998;14:181–193. [PubMed] [Google Scholar]
- 10.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 11.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994a;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 12.Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994b;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 13.Fuller RW, Wong DT. Inhibition of serotonin reuptake. Fed Proc. 1977;36:2154–2158. [PubMed] [Google Scholar]
- 14.Galter D, Unsicker K. Sequential activation of the 5-HT1(A) serotonin receptor and TrkB induces the serotonergic neuronal phenotype. Mol Cell Neurosci. 2000;15:446–455. doi: 10.1006/mcne.2000.0841. [DOI] [PubMed] [Google Scholar]
- 15.Haapasalo A, Koponen E, Hoppe E, Wong G, Castrén E. Truncated trkB.T1 is dominant negative inhibitor of trkB.TK+-mediated cell survival. Biochem Biophys Res Commun. 2001;280:1352–1358. doi: 10.1006/bbrc.2001.4296. [DOI] [PubMed] [Google Scholar]
- 16.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 18.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 19.Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci USA. 2001;98:3555–3560. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacQueen GM, Ramakrishnan K, Croll SD, Siuciak JA, Yu G, Young LT, Fahnestock M. Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav Neurosci. 2001;115:1145–1153. doi: 10.1037//0735-7044.115.5.1145. [DOI] [PubMed] [Google Scholar]
- 22.Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci. 2000;20:771–782. doi: 10.1523/JNEUROSCI.20-02-00771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 24.Marvanova M, Lakso M, Pirhonen J, Nawa H, Wong G, Castren E. The neuroprotective agent memantine induces brain-derived neurotrophic factor and trkB receptor expression in rat brain. Mol Cell Neurosci. 2001;18:247–258. doi: 10.1006/mcne.2001.1027. [DOI] [PubMed] [Google Scholar]
- 25.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 26.Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- 27.Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur J Neurosci. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 28.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 29.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nobler MS, Sackeim HA, Prohovnik I, Moeller JR, Mukherjee S, Schnur DB, Prudic J, Devanand DP. Regional cerebral blood flow in mood disorders. III. Treatment and clinical response. Arch Gen Psychiatry. 1994;51:884–897. doi: 10.1001/archpsyc.1994.03950110044007. [DOI] [PubMed] [Google Scholar]
- 31.Pariente J, Loubinoux I, Carel C, Albucher JF, Leger A, Manelfe C, Rascol O, Chollet F. Fluoxetine modulates motor performance and cerebral activation of patients recovering from stroke. Ann Neurol. 2001;50:718–729. doi: 10.1002/ana.1257. [DOI] [PubMed] [Google Scholar]
- 32.Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist–a review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 33.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 34.Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–336. [PubMed] [Google Scholar]
- 35.Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 36.Reid IC, Stewart CA. How antidepressants work: new perspectives on the pathophysiology of depressive disorder. Br J Psychiatry. 2001;178:299–303. doi: 10.1192/bjp.178.4.299. [DOI] [PubMed] [Google Scholar]
- 37.Russo-Neustadt AA, Beard RC, Huang YM, Cotman CW. Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neuroscience. 2000;101:305–312. doi: 10.1016/s0306-4522(00)00349-3. [DOI] [PubMed] [Google Scholar]
- 38.Saarelainen T, Pussinen R, Koponen E, Alhonen L, Wong G, Sirviö J, Castrén E. Transgenic mice overexpressing truncated trkB neurotrophin receptors in neurons have impaired long-term spatial memory but normal hippocampal LTP. Synapse. 2000a;38:102–104. doi: 10.1002/1098-2396(200010)38:1<102::AID-SYN11>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 39.Saarelainen T, Lukkarinen JA, Koponen S, Gröhn OH, Jolkkonen J, Koponen E, Haapasalo A, Alhonen L, Wong G, Koistinaho J, Kauppinen RA, Castrén E. Transgenic mice overexpressing truncated trkB neurotrophin receptors in neurons show increased susceptibility to cortical injury after focal cerebral ischemia. Mol Cell Neurosci. 2000b;16:87–96. doi: 10.1006/mcne.2000.0863. [DOI] [PubMed] [Google Scholar]
- 40.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 41.Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siuciak JA, Altar CA, Wiegand SJ, Lindsay RM. Antinociceptive effect of brain-derived neurotrophic factor and neurotrophin-3. Brain Res. 1994;633:326–330. doi: 10.1016/0006-8993(94)91556-3. [DOI] [PubMed] [Google Scholar]
- 43.Siuciak JA, Boylan C, Fritsche M, Altar CA, Lindsay RM. BDNF increases monoaminergic activity in rat brain following intracerebroventricular or intraparenchymal administration. Brain Res. 1996;710:11–20. doi: 10.1016/0006-8993(95)01289-3. [DOI] [PubMed] [Google Scholar]
- 44.Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 45.Skolnick P. Antidepressants for the new millennium. Eur J Pharmacol. 1999;375:31–40. doi: 10.1016/s0014-2999(99)00330-1. [DOI] [PubMed] [Google Scholar]
- 46.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 48.Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, Glass DJ, Masiakowski P, Furth ME, Valenzuela DM, DiStefano PS, Yancopoulos GD. TrkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell. 1991;65:885–893. doi: 10.1016/0092-8674(91)90395-F. [DOI] [PubMed] [Google Scholar]
- 49.Szabo ST, de Montigny C, Blier P. Modulation of noradrenergic neuronal firing by selective serotonin reuptake blockers. Br J Pharmacol. 1999;126:568–571. doi: 10.1038/sj.bjp.0702343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thoenen H, Castren E, Berzaghi M, Blöchl A, Lindholm D. Neurotrophic factors: possibilities and limitations in the treatment of neurodegenerative disorders. In: Racagni G, Brunello N, Langer SZ, editors. Recent advances in the treatment of neurodegenerative disorders and cognitive dysfunction. Karger; Basel: 1994. pp. 197–203. [Google Scholar]
- 51.Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, Storm D, Duman RS. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wisden W, Morris B. In situ hybridization with synthetic oligonucleotide probes. In: Wisden W, Morris B, editors. In situ hybridization protocols for neurobiology. Academic; London: 1994. pp. 1–34. [Google Scholar]
- 53.Wu JC, Gillin JC, Buchsbaum MS, Hershey T, Johnson JC, Bunney WE., Jr Effect of sleep deprivation on brain metabolism of depressed patients. Am J Psychiatry. 1992;149:538–543. doi: 10.1176/ajp.149.4.538. [DOI] [PubMed] [Google Scholar]
- 54.Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]