

Abstract
The axonal microtubule stabilizing protein tau is hyperphosphorylated in several neurodegenerative conditions, including Alzheimer's disease, yet the genes that regulate tau phosphorylation are largely unknown. Disabled-1 (Dab1) is a cytoplasmic adapter protein that interacts with apolipoprotein E (ApoE) receptors and controls neuronal positioning during embryonic brain development. We have investigated the role of Dab1 in tau phosphorylation. We found that wild-type Dab1, but not a mutant lacking tyrosine phosphorylation sites, protects mice from the hyperphosphorylation of tau. However, the absence of Dab1 is not sufficient to cause tau hyperphosphorylation, because hyperphosphorylation is manifested only when Dab1 is mutated in specific mouse strain backgrounds. Tau hyperphosphorylation correlates with early death in susceptible mouse strains, and it occurs in the neurons of the hippocampus and dentate gyrus. By quantitative trait locus (QTL) analysis of Dab1-deficient mice on a hybrid strain background, we uncovered one significant and three suggestive chromosomal loci that modulate tau phosphorylation. Two of these QTL regions contain genes that are defective in early onset Alzheimer's disease. Our findings suggest that Dab1 gene disruption sensitizes mice to tau hyperphosphorylation contingent on specific haplotypes that are linked to Alzheimer's disease loci. Dab1 mutant mice provide an animal model for studying the relationships between ApoE receptors, tau hyperphosphorylation, and Alzheimer's disease.
Keywords: Reeler, Disabled-1, tau hyperphosphorylation, quantitative trait locus analysis, Alzheimer's disease, genetic interactions
Introduction
The microtubule-associated protein tau regulates microtubule assembly and disassembly during neuronal differentiation (Mandelkow and Mandelkow, 1995). Tau functions are regulated by phosphorylation at many different sites, and overall tau phosphorylation is high during brain development but declines after birth. Abnormally high levels of tau phosphorylation in adult brains are associated with various neurological pathologies (Lee, 1996; Lee et al., 2001). Some inherited diseases characterized by tau hyperphosphorylation and neurodegeneration are directly attributable to mutations in the tau gene (Hong et al., 1998), whereas in others, such as Alzheimer's disease (AD), tau hyperphosphorylation occurs in the absence of tau mutations (Hardy et al., 1998;Lee et al., 2001). Mutations that increase the risk of early onset AD, and the attendant tau hyperphosphorylation, have been identified in genes involved in forming extracellular amyloid plaque. These genes encode amyloid precursor protein (APP), which is the precursor for the amyloid plaque Aβ protein, and for the multiple membrane-spanning proteins presenilin-1 (PS-1) and PS-2, which process APP (Hardy, 1996;Price et al., 1998). Thus, it is likely that either tau gene mutation or the accumulation of amyloid plaque can trigger the accumulation of phosphorylated tau protein. Late-onset AD is genetically associated with the ε4 isoform of apolipoprotein E (ApoE) (Schmechel et al., 1993). The ApoE-4 allele is a dose-dependent risk factor with incomplete penetrance, but the biochemical basis for this remains largely unknown. The mechanism by which altered APP processing and amyloid plaque formation leads to tau hyperphosphorylation, and the role of ApoE, are unclear.
There is great interest in developing mouse model systems to study tau hyperphosphorylation (Gotz, 2001; Hutton et al., 2001). Mice expressing mutant human APP and presenilins form plaque, but, unlike humans, such mice do not show tau hyperphosphorylation (Holcomb et al., 1998). However, the overexpression of mutant alleles of human tau in transgenic mice leads to increased tau phosphorylation (Lewis et al., 2000). The phenotype is exacerbated if mutant Aβ is injected into the brains of mice that are transgenic for human tau or if double transgenics for tau and APP are prepared (Gotz et al., 2001; Lewis et al., 2001). Despite these transgenic models, mutations in mouse genes that lead to the hyperphosphorylation of endogenous mouse tau have been described only recently.
We reported previously that genetic deficiency of two ApoE receptors (ApoERs), known as very-low-density lipoprotein receptor (VLDLR) and ApoER2, causes tau hyperphosphorylation that is readily detectable at weaning (Hiesberger et al., 1999). VLDLR and ApoER2 are also receptors for Reelin (Reln), a protein that controls neuronal positioning during brain development (Rice and Curran, 1999; Gupta et al., 2002). Mice that were mutant for Reln also had high levels of tau phosphorylation (Hiesberger et al., 1999). This suggested that defective Reelin signaling, or the resultant brain developmental defects, led to tau hyperphosphorylation.
We have now investigated the role of Disabled-1(Dab1), another gene in the Reln-dependent developmental pathway (Rice and Curran, 1999; Gupta et al., 2002), in regulating tau phosphorylation. Dab1 encodes an adapter protein (Dab1), that binds to the cytoplasmic tails of ApoER2 and VLDLR and is tyrosine-phosphorylated in neurons responding to Reln (Trommsdorff et al., 1998; D'Arcangelo et al., 1999; Hiesberger et al., 1999; Howell et al., 1999b). Dab1 tyrosine phosphorylation is required for Reln signaling during brain development (Howell et al., 2000). However, it was not evident whether Dab1 would be involved in the regulation of tau phosphorylation, because VLDLR and ApoER2 have Dab1-independent binding functions (Stockinger et al., 2000). Here we report that tau phosphorylation, assayed at weaning, is elevated in Dab1knock-out or in Dab1 mutants that cannot undergo tyrosine phosphorylation, dependent on the genetic background. Hyperphosphorylated tau is detected in the hippocampus, dentate gyrus, and certain fiber tracts. Tau hyperphosphorylation depends on the genetic background, and it correlates with death at 3–5 weeks of age. We have used this animal model to map genes that modify the cellular response to Dab1 gene deletion. This animal model system appears to be useful to obtain novel insights into the genetic basis for pathological tau phosphorylation in mice and, by extension, in humans.
Materials and Methods
Western blotting. Postnatal day 18 (P18) to P20 brains were extracted, and heat-soluble proteins were purified as described previously (Matsuo et al., 1994; Hiesberger et al., 1999). Equal amounts of protein were analyzed by SDS 10% PAGE and blotted to nitrocellulose. Phosphorylated tau was routinely detected using AT-8 (Polymedco, Cortlandt Manor, NY) (Goedert et al., 1993), nonphosphorylated tau was detected using Tau-1 (Binder et al., 1985), and total tau was detected with 5E2 (Upstate Biotechnology, Lake Placid, NY). Additional antibodies used were AT-180 (Goedert et al., 1994), AT-270 (Goedert et al., 1994), PHF1 (gift from I. Vincent, University of Washington, Seattle, WA) (Lang et al., 1992), and Tau46 (gift from V. Lee, University of Pennsylvania, Philadelphia, PA) (Kosik et al., 1988). These publications report the epitopes that these antibodies recognize in human tau, as listed in Figures 1 and 3A.
Fig. 1.

Tau phosphorylation at several sites is regulated by genes of the Reln–Dab1 pathway. Brain extracts were prepared at P18–P20, and equal quantities of heat-stable protein were analyzed by SDS PAGE and Western blotting. A, Samples from various mutant mice of different strain backgrounds were analyzed simultaneously with antibodies recognizing phosphorylated (top) and dephosphorylated (bottom) tau.B, Selected samples were reanalyzed with antibodies recognizing additional epitopes on tau. Antibody specificity was determined previously using human tau as the antigen:+P, Phosphorylation dependent; −P, dephosphorylation dependent; Pi, phosphorylation independent (see Materials and Methods for references). Note the altered migration of tau from mutant brains (asterisks), which is indicative of increased phosphorylation.
Fig. 3.

Identification of genetic modifiers of tau phosphorylation. A,Dab1−/+ BALB/cByJ (CC) mice were bred to C57BL/6J (BB) mice, andDab1−/+ F1(BC) offspring were intercrossed. F2offspring that were Dab1−/− were identified, and the brain samples were analyzed by Western blotting. The figure shows data on the first 12 F2 brains analyzed. Phenotypes were scored as 0 (lowest quartile of tau phosphorylation), 1 (intermediate), or 2 (highest). Mice with phenotype 0 or 2 were genotyped. +P, Phosphorylation dependent;−P, dephosphorylation dependent; Pi, phosphorylation independent (see Materials and Methods for references).B, Table of genotypes. Rows correspond to individual mice, grouped according to phenotype; columnscorrespond to markers analyzed, grouped by chromosome (see Materials and Methods for a list of markers). Genotypes are color-coded asCC, BC, or BB. Black indicates no data. Note predominance ofCC (red) on chromosome 4, whereDab1 is located. Markers at the right end of chromosome 1 are predominantly BB (blue) in phenotype 2 mice (high tau phosphorylation), whereas markers on chromosome 16 are predominantly BB in phenotype 0 mice (low tau phosphorylation). C, Genome scan for linkage between genotype and phenotype. Results are plotted as LOD scores compared with the LOD scores required for significant linkage (top dotted line) and suggestive linkage (bottom dotted line). A locus on chromosome 16 (60 cm) is significantly linked to phenotype. D, Direction of effects. Tau phosphorylation is increased by a dominant Ballele on chromosome 16, by a recessive B allele on 1, and by a dominant C allele on 12.
Immunohistochemistry. P18–P20 brains were fixed in 4% paraformaldehyde and cryoprotected in sucrose, and frozen sections were prepared. Sections were reacted with antibody TG-3 (Jicha et al., 1997), stained with diaminobenzidine, and counterstained with hematoxylin.
Quantitative trait locus analysis. Dab1−/− F2 progeny were identified by behavior and killed at P18–P20. DNA was extracted and analyzed by PCR to confirm the Dab1 genotype. Brain samples were analyzed as described above to determine tau phosphorylation. Animals with high and low levels of tau phosphorylation were analyzed further. PCR primers for marker analysis were designed according to information athttp://www.informatics.jax.org/mgihome/(Blake et al., 2001, 2002). The following markers were analyzed: D1Mit66, 1Mit003, 1Mit211, 1Mit478, 1Mit302, 1Mit215, 1Mit365, 1Mit90, 2Mit243, 2Mit037, 2Mit304, 2Mit229, 3Mit164, 3Mit028, 3Mit116, 4Mit288, 4Mit175, 4Mit148, 5Mit233, 5Mit240, 5Mit291, 6Mit188, 6Mit036, 6Mit201, 7Mit117, 7Mit238, 7Mit046, 8Mit293, 8Mit248, 8Mit013, 9Mit191, 9Mit196, 9Mit279, 10Mit80, 10Mit044, 10Mit180, 11Mit227, 11Mit041, 11Mit050, 12Mit012, 12Mit014, 12Mit018, 13Mit003, 13Mit147, 13Mit151, 14Mit133, 14Mit034, 14Mit075, 15Mit265, 15Mit239, 15Mit171, 16Mit34, 16Mit04, 16Mit125, 16Mit185, 16Mit139, 16Mit189, 16Mit94, 16Mit224, 16Mit86, 16Mit106, 17Mit028, 17Mit139, 17Mit142, 18Mit017, 18Mit184, 18Mit049, 19Mit019, 19Mit010, and 19Mit071. Genotype information for these markers is arranged as columns from left to right in Figure 3B.
Data were analyzed by a whole genome scan for linkage as described previously (Lander and Kruglyak, 1995; Sen and Churchill, 2001). A significant quantitative trait locus (QTL) was found at chromosome 16 marker D16Mit224, together with several suggestive QTLs. A second genome scan was then performed with D16Mit224 used as a covariate to fix the chromosome 16 effect. This did not change the significance of the suggestive QTLs. We also performed a genome-wide scan for pairwise interactions. There were no significant interacting pairs. A multiple regression containing the significant and suggestive main effect loci was fitted to the data. The model explains 38% of the variance in the phenotype.
Criteria for significant and suggestive QTLs. Criteria for significant and suggestive thresholds were determined by performing 1000 analyses on permuted data sets. The maximum likelihood log odds (LOD) score exceeds the threshold for significant linkage in 5% of the permuted data analyses, and it exceeds the suggestive threshold in 37%, in accordance with accepted standards for significance in genome-wide QTL scans (Lander and Kruglyak, 1995; Sen and Churchill, 2001).
Results
Effects of Dab1 mutation on tau phosphorylation
We tested whether Dab1 regulates tau phosphorylation, initially using mice of the 129Sv × C57BL/6 (SB) hybrid strain background that is commonly used for maintaining knock-out strains. As found previously (Hiesberger et al., 1999), mice mutant for Reln or for both VLDLR andApoER2 in the SB strain background showed high tau phosphorylation (Fig.1A) (AT-8 antibody, samples 3–6), whereas the levels were low or undetectable in samples from wild-type mice (sample 7) or mice with single mutations in eitherVLDLR or ApoER2 (samples 1 and 2).Dab1 gene disruption in this strain background also caused increased tau phosphorylation (Fig. 1A, samples 10 and 11). Phosphorylation was extensive, as shown by a commensurate decrease in the amount of hypophosphorylated tau (Fig.1A, Tau1 antibody) and by an electrophoretic mobility shift (Fig. 1B, asterisks). Phosphorylation occurred at several sites that are phosphorylated in AD samples, as detected by antibodies specific for different phosphorylated epitopes on human tau (Fig. 1B). A phenylalanine mutant allele of Dab1(Dab15F), which lacks tyrosine phosphorylation sites, is defective in Reln signaling (Howell et al., 2000). This mutant also caused tau hyperphosphorylation (Fig.1A, samples 8 and 9). This implies that the same mutations that cause a Reeler developmental phenotype also affect the level of tau phosphorylation after birth. Thus, it is possible that tau hyperphosphorylation in the mutants results either from abnormal brain development or from the lack of postnatal activity of the Reln–Dab1 pathway.
Tau hyperphosphorylation in the hippocampus
To localize hyperphosphorylated tau, brain sections from wild-type and Dab1-deficient mice were stained with antibodies that recognize phosphorylated tau. Strong staining was detected in sections from Dab1 deletion and phenylalanine mutants in theSB strain background (Fig.2A–D). Staining was restricted to cell bodies in the hippocampus proper and the dentate gyrus and to fiber tracts of the corpus callosum and fasciculus retroflexus. No staining was detected in the wild type (Fig.2E). Cells in the hippocampus were misplaced, as would be expected when Reln–Dab1 signaling is disrupted (Rice and Curran, 1999). However, other misplaced cells, such as the cortical plate pyramidal cells and Purkinje cells of the cerebellum, did not contain detectable hyperphosphorylated tau.
Fig. 2.
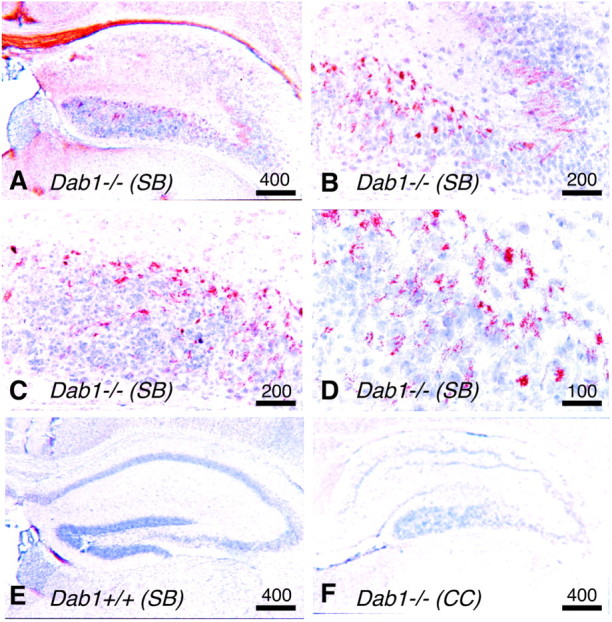
Presence of phosphorylated tau in the hippocampus. All sections were stained for phosphorylated tau, andbrown areas indicate immunoreactivity.A–D, Sections from aDab1−/−SB strain mouse showing the hippocampus (A), CA2 region (B), and dentate gyrus (C, D). E, F, Sections from the hippocampus ofDab1+/+ SB strain mouse (E) andDab1−/− CCstrain mouse (F). Scale bars: A, E, F, 400 μm; B, C, 200 μm; D, 100 μm.
Survival of Dab1 mutants depends on strain background: correlation with tau phosphorylation
We noticed that the viability of Dab1 mutant mice depends on the strain background. Dab1 mutations were originally made in the embryonic stem cells of 129Sv mice (SS), and maintained in pure SS or mixedSB backgrounds (Howell et al., 1997). On these backgrounds,Dab1 mutants and VLDLR/ApoER2 double knock-out mice generally die at 3–5 weeks of age. It was reported that crossing either of two mutant Reln alleles into the CC(BALB/cByJ) background improved viability compared with theBB (C57BL/6) background (Goffinet, 1990). Therefore, we tested whether the death of Dab1−/−homozygotes was also influenced by strain background by repeatedly backcrossing Dab1−/+ to CCstrain mice. CC congenicDab1−/+ mice were then intercrossed.Dab1−/− (CC) animals were found to be uniformly viable, living a normal life-span. Nonetheless,Dab1 deficiency caused a typical Reeler phenotype in theCC background, including a disordered hippocampus (Fig.2F) (data not shown). This suggests that the lethality of the Dab1 mutation depends on strain background and is suppressed in the same strain that suppresses the lethality of aReln mutation (Goffinet, 1990).
We suspected that surviving mice may not have hyperphosphorylated tau. Indeed, we found that the Dab1 deletion in the CCstrain background does not cause tau hyperphosphorylation, as detected by Western blotting (Fig. 1A, samples 12–15) or immunohistochemistry (Fig. 2F). When data fromDab1 mutations in various mouse strains were compared, there was a strong relationship between the lack of tau hyperphosphorylation and anticipated survival for >5 weeks (Table1). This correlation suggests either that tau hyperphosphorylation causes death or that tau hyperphosphorylation and early death have a common cause.
Table 1.
Relationship between anticipated survival, tau phosphorylation, Dab1 genotype, and mouse strain background
| Dab1 genotypes | Strain background | Survival1-a | Hyperphosphorylated tau1-b |
|---|---|---|---|
| +/+, +/− | CC | + | 0 /6 |
| −/− | CC | + | 0 /5 |
| +/+, +/−, 5F/+ | SB | + | 1 /9 |
| −/−, 5F/5F | SB | − | 13 /141-c |
Survival for >5 weeks of age.
Number of mice with hyperphosphorylated tau relative to number analyzed.
Probability that survival of mice is not linked to high tau phosphorylation, p < 0.000001 (χ2 test = 26.2; 1 df).
Mapping of genetic loci that modify tau hyperphosphorylation inDab1−/− mice
Because extensive, early onset tau hyperphosphorylation has not been observed previously in mutant mice, we initiated a search for QTLs that modify the phosphorylation of tau in the absence ofDab1. We bred Dab1−/+BALB/cByJ (CC) congenic mice to C57BL/6J (BB).Dab1−/+ F1[BALB/cByJ × C57BL/6J (BC)] offspring were identified and intercrossed. F2 progeny that wereDab1−/− were identified by behavior and confirmed by genotyping. Brain samples were collected at 20 d after birth, and the tau phosphorylation state was assessed (Fig.3A). Tau phosphorylation varied widely, from undetectable to high levels. Some variation may be attributable to altered tau phosphorylation in different brain regions and some to different levels of phosphorylation in the hippocampus and dentate gyrus. We grouped 95 Dab1−/−homozygotes, from ∼400 F2 progeny, according to overall tau phosphorylation levels, separating the 26 highest (phenotype 2) and the 25 lowest (phenotype 0) from 44 with intermediate phosphorylation (phenotype 1). Sixty-nine DNA markers that are polymorphic between CC and BB and cover all autosomes were then analyzed for phenotype 0 and 2 mice (Fig.3B) (see Materials and Methods for markers tested). A genome-wide scan for linkage revealed that a significant risk factor for tau hyperphosphorylation is present at D16Mit224, with suggestive QTLs at D1Mit365, D12Mit014, and D17Mit142 (Fig. 3C). [There is only a 5% chance of finding a significant QTL and a 37% chance of finding a suggestive QTL in a random data set (see Materials and Methods).] Curiously, the main QTL dominantly predisposes to high tau phosphorylation when inherited from the CC parent (Fig.3D), although Dab1 deletion in CC does not cause tau hyperphosphorylation. The most likely explanation for this finding is that a strong predisposing QTL in CC is balanced by one or more other QTLs that are present in BBand that regulate tau phosphorylation in an opposite manner. Indeed, the suggestive QTLs on chromosomes 1, 12, and 17 increase tau phosphorylation dominantly or recessively when inherited from theBB parent (Fig. 3D) and may more than balance the effect of the strong QTL on chromosome 16. Analysis of more mutant mice would be needed to confirm the suggestive QTLs.
Discussion
Combined with previous results from Reln,VLDLR, and ApoER2 mutants (Hiesberger et al., 1999), our current results show that mutations that prevent the Reln-dependent induction of Dab1 tyrosine phosphorylation cause tau hyperphosphorylation in the hippocampus at an early age. Because all of the mutant alleles that cause tau hyperphosphorylation also cause abnormal brain development, it is likely that the specific abnormalities resulting from defective Reln signaling during development cause tau hyperphosphorylation after birth. However, there is evidence that Reln is involved in various postnatal events, including the regulation of Dab1 protein levels, innervation of the hippocampus, and modulation of hippocampal synapses in culture (Del Rio et al., 1997; Howell et al., 1999a; Weeber et al., 2002). Therefore, it is also possible that continued Reln–Dab1 signaling after birth regulates tau hyperphosphorylation. Definitive determination of whether tau hyperphosphorylation is regulated by Reln, VLDLR, ApoER2, and Dab1 in a normal brain may be possible only when conditional knock-out alleles of Reln, Dab1, or the receptors are available.
We also found that survival of Dab1 mutants for >5 weeks of age depended on the strain background and correlated strongly with the lack of high levels of phosphorylated tau (Table 1). A survey of published Reln and Dab1 mutants reveals variable survival of homozygotes, depending on strain background. Viable alleles and strains include Relned in the B6C3Fe strain (Falconer, 1951), Relntg(B6C3F1 × B6D2) (D'Arcangelo et al., 1995), andDab1scm (C3HeB/FeJ × DC/Le) (Sweet et al., 1996). In contrast, Dab1yot(126Sv × C57BL/6) (Yoneshima et al., 1997) andDab1− (129Sv × C57BL/6) (Howell et al., 1997) are lethal. Here we report that Dab1 mutants are viable in BALB/c but nonviable in 129Sv or in 129Sv × C57BL/6, which resembles the pattern found for two Reln alleles: viable in BALB/c but nonviable in C57BL/6 (Goffinet, 1990). Thus, the same genes that modify the survival of Reln mutants may modify the survival of Dab1 mutants. In both BALB/c and C57BL/6, the Reln mutation caused a similar phenotype in the cortex and the hippocampus, although subtle differences were detected in the cerebellum (Goffinet, 1990). Variation in cerebellar morphology has been documented in wild-type mice from different inbred strains; it is attributable to several loci (Wahlsten and Andison, 1991; Garretson and Neumann, 1993; Neumann et al., 1993). It is possible that the genes that affect cerebellar morphology in wild-type mice contribute to the background effect on cerebellar morphology in Reln mutants. It is also possible that variable survival and tau phosphorylation in C57BL/6 and BALB/c Dab1−/− mice could be secondary to the different severities of neurological effects. However, our results are consistent with the hypothesis that tau hyperphosphorylation is the cause of early death ofDab1−/− mice in susceptible strain backgrounds.
The detection of hyperphosphorylated tau in the hippocampus and the dentate gyrus of Dab1−/− mice resembles findings in human AD (Lee et al., 2001). The accumulation of hyperphosphorylated tau in the corresponding regions of mouseDab1 mutant and human AD brains may indicate a heightened sensitivity of these cells to an imbalance between protein kinases and phosphatases or a common molecular mechanism of tau phosphorylation inDab1 mutants and AD. A common molecular mechanism is also suggested by our identification of QTLs that affect tau hyperphosphorylation in Dab1−/− mice. The main effect of QTL on chromosome 16 (60 cm) includes the genes for APP (at 56 cm) and superoxide dismutase (SOD) (at 61 cm), both factors in inherited neurodegenerative illnesses associated with tau hyperphosphorylation. Not only is APP the precursor of the amyloid plaque that accumulates in AD and a genetic cause of familial AD (Hardy et al., 1998), but the APP protein binds to Dab1 (Homayouni et al., 1999; Howell et al., 1999b). SODmutations cause amyotrophic lateral sclerosis (Deng et al., 1993; Rosen et al., 1993), which can also be caused by tau mutations (Poorkaj et al., 2001). This mouse chromosomal region is syntenic with human chromosome 21. Trisomy 21, or Down syndrome, is a frequent genetic disorder that includes neurodegenerative features resembling AD. One of the suggestive QTLs on chromosome 12 (38 cm) lies close to the gene encoding PS-1 (Psen1, at 37 cm). Mutations inPsen1 alter APP processing and can cause human AD (Hardy et al., 1998).
The map locations of QTLs that modify tau hyperphosphorylation inDab1−/− mice raise the possibility that polymorphisms in genes that cause neurodegeneration and tau hyperphosphorylation in humans also regulate tau hyperphosphorylation when Reln–Dab1 signaling is defective. If so, Reln pathway defects, inherited tau mutations, and defective APP processing may induce tau phosphorylation through similar mechanisms. However, extensive additional mapping of the QTLs would be required to determine whetherAPP, Psen1, or SOD is directly responsible for modulating tau phosphorylation or whether other nearby genes are functionally involved.
In summary, our results do not distinguish whether the lack of postnatal Reln signals or abnormal brain architecture, secondary to defects in the Reln signaling pathway during development, is responsible for increased tau phosphorylation inDab1−/− mice. However, the genetic modifiers do not detectably alter the severity of the developmental phenotype, only the level of tau phosphorylation and long-term survival. Thus, the genetic modifiers act downstream of Dab1or of the developmental abnormalities. We suggest that an imbalance in the interaction between the Reln signaling pathway and other genes affects tau phosphorylation in mice. The Dab1 mutant mouse provides a useful model for rapidly identifying novel modifier genes involved in neurodegenerative diseases.
Footnotes
This work was supported by grants from the National Institutes of Health (J.A.C., J.H., M.M., E.K.W.), from the Alzheimer Association, and from the Humboldt Foundation (J.H.). J.B. is a fellow of the Ernst and Hedda Wollheim Foundation and a recipient of a Boehringer Ingelheim scholarship. We thank P. K. O'Brien and T. Herrick for expert technical assistance, G. Schellenberg, V. Lee, and I. Vincent for antibodies, and E. Giniger, B. Ballif, and G. Schellenberg for valuable comments on this manuscript.
Correspondence should be addressed to either of the following: Jonathan A. Cooper, Fred Hutchinson Cancer Research Center A2-025, 1100 Fairview Avenue North, Seattle, WA 98109, E-mail: jcooper@fhcrc.org; or Joachim Herz, University of Texas Southwestern Medical Center, Dallas, TX 75390, E-mail: joachim.herz@utsouthwestern.edu.
J. Brich's present address: Department of Clinical Biochemistry and Pathobiochemistry, Julius-Maximilians-University, D-97078 Würzburg, Germany.
B. W. Howell's present address: National Institute of Neurological Disorders and Stroke, National Institutes of Health, Building 10, 3B04, 10 Center Drive, Bethesda, MD 20892
References
- 1.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blake JA, Eppig JT, Richardson JE, Bult CJ, Kadin JA. The Mouse Genome Database (MGD): integration nexus for the laboratory mouse. Nucleic Acids Res. 2001;29:91–94. doi: 10.1093/nar/29.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blake JA, Richardson JE, Bult CJ, Kadin JA, Eppig JT. The Mouse Genome Database (MGD): the model organism database for the laboratory mouse. Nucleic Acids Res. 2002;30:113–115. doi: 10.1093/nar/30.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 5.D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 6.Del Rio JA, Heimrich B, Borrell V, Forster E, Drakew A, Alcantara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E. A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature. 1997;385:70–74. doi: 10.1038/385070a0. [DOI] [PubMed] [Google Scholar]
- 7.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 8.Falconer DS. Two new mutants, “trembler” and “reeler”, with neurological actions in the house mouse. J Genet. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- 9.Garretson JD, Neumann PE. Further evidence that a mouse chromosome 4 locus influences cerebellar folial pattern. Brain Res. 1993;630:221–225. doi: 10.1016/0006-8993(93)90660-f. [DOI] [PubMed] [Google Scholar]
- 10.Goedert M, Jakes R, Crowther RA, Six J, Lubke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VM. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA. 1993;90:5066–5070. doi: 10.1073/pnas.90.11.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J. 1994;301:871–877. doi: 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goffinet AM. Cerebellar phenotype of two alleles of the “reeler” mutation on similar backgrounds. Brain Res. 1990;519:355–357. doi: 10.1016/0006-8993(90)90102-h. [DOI] [PubMed] [Google Scholar]
- 13.Gotz J. Tau and transgenic animal models. Brain Res Brain Res Rev. 2001;35:266–286. doi: 10.1016/s0165-0173(01)00055-8. [DOI] [PubMed] [Google Scholar]
- 14.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 15.Gupta A, Tsai LH, Wynshaw-Boris A. Life is a journey: a genetic look at neocortical development. Nat Rev Genet. 2002;3:342–355. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- 16.Hardy J. New insights into the genetics of Alzheimer's disease. Ann Med. 1996;28:255–258. doi: 10.3109/07853899609033127. [DOI] [PubMed] [Google Scholar]
- 17.Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- 18.Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 19.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 20.Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 22.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 23.Howell BW, Herrick TM, Cooper JA. Reelin-induced tyrosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999a;13:643–648. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol Cell Biol. 1999b;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10:877–885. doi: 10.1016/s0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- 26.Hutton M, Lewis J, Dickson D, Yen SH, McGowan E. Analysis of tauopathies with transgenic mice. Trends Mol Med. 2001;7:467–470. doi: 10.1016/s1471-4914(01)02123-2. [DOI] [PubMed] [Google Scholar]
- 27.Jicha GA, Lane E, Vincent I, Otvos L, Jr, Hoffmann R, Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem. 1997;69:2087–2095. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- 28.Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM, Lee G. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1988;1:817–825. doi: 10.1016/0896-6273(88)90129-8. [DOI] [PubMed] [Google Scholar]
- 29.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 30.Lang E, Szendrei GI, Lee VM, Otvos L., Jr Immunological and conformation characterization of a phosphorylated immunodominant epitope on the paired helical filaments found in Alzheimer's disease. Biochem Biophys Res Commun. 1992;187:783–790. doi: 10.1016/0006-291x(92)91264-q. [DOI] [PubMed] [Google Scholar]
- 31.Lee VM. Regulation of tau phosphorylation in Alzheimer's disease. Ann NY Acad Sci. 1996;777:107–113. doi: 10.1111/j.1749-6632.1996.tb34408.x. [DOI] [PubMed] [Google Scholar]
- 32.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 33.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 34.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 35.Mandelkow E, Mandelkow EM. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol. 1995;7:72–81. doi: 10.1016/0955-0674(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 36.Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O'Connor M, Trojanowski JQ, Lee VM. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 37.Neumann PE, Garretson JD, Skabardonis GP, Mueller GG. Genetic analysis of cerebellar folial pattern in crosses of C57BL/6J and DBA/2J inbred mice. Brain Res. 1993;619:81–88. doi: 10.1016/0006-8993(93)91598-m. [DOI] [PubMed] [Google Scholar]
- 38.Poorkaj P, Tsuang D, Wijsman E, Steinbart E, Garruto RM, Craig UK, Chapman NH, Anderson L, Bird TD, Plato CC, Perl DP, Weiderholt W, Galasko D, Schellenberg GD. TAU as a susceptibility gene for amyotrophic lateral sclerosis-parkinsonism dementia complex of Guam. Arch Neurol. 2001;58:1871–1878. doi: 10.1001/archneur.58.11.1871. [DOI] [PubMed] [Google Scholar]
- 39.Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer's disease: genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–493. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- 40.Rice DS, Curran T. Mutant mice with scrambled brains: understanding the signaling pathways that control cell positioning in the CNS. Genes Dev. 1999;13:2758–2773. doi: 10.1101/gad.13.21.2758. [DOI] [PubMed] [Google Scholar]
- 41.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 42.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Periack-Vance MA, Goldgaber D, Roses AD. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stockinger W, Brandes C, Fasching D, Hermann M, Gotthardt M, Herz J, Schneider WJ, Nimpf J. The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J Biol Chem. 2000;275:25625–25632. doi: 10.1074/jbc.M004119200. [DOI] [PubMed] [Google Scholar]
- 45.Sweet HO, Bronson RT, Johnson KR, Cook SA, Davisson MT. Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm Genome. 1996;7:798–802. doi: 10.1007/s003359900240. [DOI] [PubMed] [Google Scholar]
- 46.Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- 47.Wahlsten D, Andison M. Patterns of cerebellar foliation in recombinant inbred mice. Brain Res. 1991;557:184–189. doi: 10.1016/0006-8993(91)90133-g. [DOI] [PubMed] [Google Scholar]
- 48.Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277:39944–39952. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- 49.Yoneshima H, Nagata E, Matsumoto M, Yamada M, Nakajima K, Miyata T, Ogawa M, Mikoshiba K. A novel neurological mutant mouse, yotari, which exhibits reeler-like phenotype but expresses CR-50 antigen/reelin. Neurosci Res. 1997;29:217–223. doi: 10.1016/s0168-0102(97)00088-6. [DOI] [PubMed] [Google Scholar]