

Abstract
Neuronal cell death in response to oxidative stress may reflect the failure of endogenous adaptive mechanisms. However, the transcriptional activators induced by oxidative stress in neurons that trigger adaptive genetic responses have yet to be fully elucidated. We report that basal DNA binding of the zinc finger transcription factors Sp1 and Sp3 is unexpectedly low in cortical neurons in vitro and is significantly induced by glutathione depletion-induced or hydrogen peroxide-induced oxidative stress in these cells. The increases in Sp1/Sp3 DNA binding reflect, in part, increased levels of Sp1 and Sp3 protein in the nuclei of cortical neurons. Similar induction of Sp1 and Sp3 protein is also observed in neurons in vivo in a chemical or a genetic model of Huntington's disease, two rodent models in which neuronal loss has been attributed to oxidative stress. Sustained high-level expression of full-length Sp1 or full-length Sp3, but not the Sp1 zinc finger DNA-binding domain alone, prevents death in response to oxidative stress, DNA damage, or both. Taken together, these results establish Sp1 and Sp3 as oxidative stress-induced transcription factors in cortical neurons that positively regulate neuronal survival.
Keywords: Sp1, oxidative stress, neurons, antioxidants, Huntington's disease, DNA damage
Introduction
Oxidative damage to proteins, lipids, or DNA is increased in human autopsy tissue and rodent models of a host of neurodegenerative conditions, including Alzheimer's disease, Parkinson's disease, Friedreich's ataxia, Huntington's disease (HD), multiple sclerosis, and stroke (for review, seeSagara et al., 1998; Atwood et al., 1999; Browne et al., 1999; Castagne et al., 1999; Estevez et al., 1999; Floyd, 1999; Mattson et al., 1999;Ratan, 1999, Albers and Beal, 2000). Free radical damage to cell constituents in disease states may reflect a failure of endogenous compensatory mechanisms to oxidative stress. These compensatory mechanisms include enzymes involved in detoxifying reactive oxygen species (ROS) and repairing oxidative damage to DNA and protein. Therefore, further understanding of the nature and regulation of compensatory responses to oxidative stress may provide novel insights into the pathogenesis of as well as therapy for neurological diseases.
Adaptive responses to oxidative stress are most well defined in prokaryotic systems (Mongkolsuk and Helmann, 2002). Bacteria respond to toxic levels of ROS by increasing the expression of antioxidant and repair genes. ROS-mediated changes in gene expression are coordinated by transcription factors. In prokaryotes, these transcription factors are directly and transiently modified by oxidants. For example, changes in the redox state of one transcription factor (OxyR) lead to enhanced interaction of this factor with the basal transcription machinery and increased expression of genes involved in detoxification (Zheng and Storz, 2000). In contrast, oxidation of the transcription factor organic hydroperoxide resistance (OhR) results in the oxidation of a single cysteine residue (Cys-15) and loss of DNA binding. The loss of OhR DNA binding leads to the derepression of genes involved in detoxification (Fuangthong and Helmann, 2002).
Although it has been established that eukaryotic cells, including neurons, also use transcription factors to respond to ROS and activate protective responses (Lezoualc'h and Behl, 1998; Post et al., 1998;Bijur et al., 1999; Scortegagna et al., 1999), our understanding of the nature of these proteins and their modes of activation in neurons is limited. Primary cultures of cortical neurons provide a convenientin vitro preparation for examining transcription factors that are induced by oxidative stress (Murphy et al., 1989, 1990). Early in their development in culture, cortical neurons exposed continuously to glutamate succumb through a mechanism independent of ionotropic glutamate receptors. Degeneration occurs subsequent to the depletion of glutathione, an important antioxidant. Cell death attributable to glutathione depletion can be completely prevented by a host of classical antioxidants (Murphy et al., 1990) and has many features of apoptosis (Ratan et al., 1994b; Tan et al., 1998; Zaman et al., 1999).
Herein, we use this in vitro model to examine the role of the Sp1 transcription factor family in oxidative stress-induced cell death in neurons. Sp1 is a member of an extended family of DNA-binding proteins that have three zinc finger motifs and bind to guanosine- and cytosine-rich DNA (Briggs et al., 1986; Lania et al., 1997;Suske, 1999; Yang et al., 2000). This family includes members homologous to Sp1, such as Sp2–Sp4. Sp1 activities have been shown to change in response to apoptosis-inducing stimuli. For example, the induction of apoptosis in B-cells after treatment with anti-IgM is associated with the caspase-dependent cleavage of Sp1 (Rickers et al., 1999). Similarly, apoptotic retinoids induce the caspase-dependent cleavage of Sp1 in T-cells (Piedrafita and Pfahl, 1997). Indeed, polyglutamine expansions in the huntingtin protein can induce neuronal toxicity, in part, by sequestering Sp1 and one of its coactivators, TATA binding protein-associated factor (TAF)II130, suggesting a role for Sp1 in neuronal survival (Dunah et al., 2002; Li et al., 2002). Of note, Sp1 has been shown to regulate prosurvival proteins [e.g., the inhibitor of apoptosis (IAP) protein, survivin (Li and Altieri, 1999), and manganese superoxide dismutase (Tanaka et al., 2000)] as well as prodeath proteins [e.g., Fas ligand (Xiao et al., 1999; Chou et al., 2000) and 12-lipoxygenase (Liu et al., 1997)]. Like other transcription factors, the role of Sp1 in regulating cell death may depend on a number of factors, such as the cell type and the death stimulus (Lin et al., 1998).
Herein, we demonstrate that oxidative stress significantly induces Sp1 and Sp3 protein levels and DNA binding in neurons in vitroand in vivo. Moreover, we demonstrate that the enforced expression of Sp1 or Sp3 is neuroprotective. These findings establish Sp1 and Sp3 as redox-regulated transcriptional activators that enhance survival in cortical neurons.
Materials and Methods
Primary neuronal culture. Cell cultures were obtained from the cerebral cortex of Sprague Dawley rats (day 17 of gestation) as described previously (Murphy et al., 1990). All experiments were initiated 24–72 hr after plating. Under these conditions, the cells are not susceptible to glutamate-mediated excitotoxicity. For cytotoxicity studies, cells were rinsed with warm PBS and then placed in Minimum Essential Medium (MEM; Invitrogen, Gaithersburg, MD) with 5.5 gm/l glucose, 10% FCS, 2 mml-glutamine, and 100 μmcystine, containing the glutamate analog homocysteate (HCA; 1 mm). HCA was diluted from 100-fold concentrated solutions that were adjusted to pH 7.5.
Sp1 and Sp3 expression in mouse model of HD. Male transgenic HD mice of the R6/2 strain were originally obtained from The Jackson Laboratory (Bar Harbor, ME). The male R6/2 mice and wild-type littermate control mice used in this study were from the Bedford Veterans Affairs facility and bred with females from their background strain (B6CBAFI/J). 3-Nitropropionic acid (3-NP; Sigma, St. Louis, MO) was dissolved in PBS, pH adjusted to 7.4, and injected intraperitoneally nine times at 12 hr intervals, using 50 mg/kg per injection in wild-type control mice. The mice were killed 3–5 hr after the last 3-NP injection. Both R6/2 and 3-NP-treated mice were deeply anesthetized and transcardially perfused with buffered 4% paraformaldehyde (PFA) and processed for histopathologic evaluation. Glycerol-cryoprotected brains were frozen-sectioned at 50 μm and immunostained with Sp1 and Sp3 antibodies (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) using a previously described conjugated second antibody method (Ferrante et al., 2002).
Generation of recombinant herpes simplex virus vectors. Human Sp1, Sp3, and the Sp1 zinc finger DNA binding domain alone were each tagged with an N-terminal FLAG epitope and were individually subcloned into replication-defective herpes simplex virus (HSV) vectors (pHSVPrpUC) as described previously (Neve and Geller, 1999). Cortical neurons (1 d in vitro) were infected with recombinant HSV 1 d after plating. Multiplicities of infection (MOIs) of 2 were used because this MOI resulted in the transduction of 20–25% of neurons in the dish, with no cytotoxicity as measured by MTT reduction, phase contrast microscopy, or 4′,6′-diamidino-2-phenylindole (DAPI) staining. Neurons were infected in serum-free medium for 3–6 hr; the medium was removed; and standard, serum-containing culture medium was added. In parallel cultures, cells were treated in the presence and absence of the glutamate analog, and after 14–18 hr, they were fixed with 4% PFA in preparation for immunocytochemistry, DAPI staining, and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL).
Cell damage and death detection. Nuclear fragmentation was assessed by staining nuclear DNA using DAPI. Neuronal cells were washed with PBS. Cells were fixed in 4% PFA in PBS for 10 min at room temperature. Cells were washed with PBS and stained with DAPI for 15 min at room temperature. More than 300 cells per slide were scored for the incidence of nuclear fragmentation using a fluorescence microscope. Cells with two or more chromatin fragments or shrunken condensed nuclei were considered apoptotic.
In situ detection of nuclear DNA fragmentation by TUNEL. The specimens were rehydrated according to a standard protocol and incubated with proteinase K (Roche Diagnostics, Mannheim, Germany; 20 gm/ml) for 15 min at room temperature with intervening washes in PBS. The TUNEL reaction mixture was prepared by adding 5 μl of terminal deoxynucleotidyl transferase to 45 μl of a fluorescein-labeled nucleotide mixture per section. After it was washed with PBS, the TUNEL reaction mixture was applied to each section, covered with a coverslip, and incubated in a moist chamber for 60 min at 37°C. In control sections, the terminal deoxynucleotidyl transferase was omitted.
Population measurements of neuronal cell viability were measured using a nonradioactive CellTiter 96 assay kit (Promega, Madison, WI). Tetrazolium dye solution was added to cortical neurons 16–24 hr after the treatment of various reagents. Cells were incubated at 37°C for 4 hr, and a solubilization/stop solution was added. Cell plates were allowed to stand overnight in a humidified atmosphere, and the absorbance was measured at 570 nm using a Molecular Devices (Menlo Park, CA) tunable 96-well plate reader.
Immunoblot analysis. Cell lysates were obtained by rinsing cortical neurons with cold PBS and adding 100 mmTris buffer, pH 7.4, containing 1% Triton X-100, 150 mm NaCl, 1 mm sodium orthovanadate, 5 mm sodium fluoride, 3 mm PMSF, 3 mm DTT, 0.5 μg/ml leupeptin, and 10 μg/ml aprotinin. Thirty micrograms of protein from cell lysates were boiled in Laemmli buffer, and 5 mg of protein was electrophoresed under reducing conditions on 8% polyacrylamide gels. Proteins were then transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Nonspecific binding was inhibited by incubation in Tris-buffered saline (TBST; 50 mm Tris HCl, pH 8.0, 0.9% NaCl, and 0.1% Tween 20) containing 5% nonfat dried milk for 0.5 hr. Primary antibodies against Sp1 (PEP2), Sp2 (K-20), Sp3 (D-20), or Sp4 (V-20) (all from Santa Cruz Biotechnology) were diluted at 1:1000 in TBST with 1% milk and exposed to membranes overnight at 4°C. Immunoreactive proteins were detected according to the enhanced chemiluminescence protocol (Amersham Biosciences, Arlington Heights, IL).
Immunofluorescence staining. Indirect labeling methods were used to determine the levels of endogenous or heterologous Sp1–Sp3 and β-galactosidase in cortical neuronal cultures. Dissociated cells from the cerebral cortex (3–5 × 105) were seeded onto poly-d-lysine-coated eight-well culture slides (Becton Dickinson Labware, Bedford, MA) and treated with HCA as described above for 4–5 hr. The cells were washed with warm PBS and fixed at room temperature for 15 min with 4% PFA. After washing with PBS, fixed cells were incubated with blocking solution containing 0.3% Triton X-100, 5% BSA, and 3% goat serum for 1 hr, followed by incubation with rabbit Sp1 antibody (1:500 dilution), rabbit anti-Sp2 polyclonal antibody (1:500), or a rabbit anti-Sp3 polyclonal antibody (1:500 dilution). After three washes with PBS, the cells were incubated for 1 hr with FITC-conjugated goat-anti-rabbit IgG antibody (1:200 dilution) and DAPI. All antibodies were diluted in PBS. The slides were washed three times with PBS and mounted with fluorochrome mounting solution (Vector Laboratories, Burlingame, CA). Images were analyzed under a fluorescence microscope (model AX70TRF; Olympus Optical, Tokyo, Japan). Control experiments were performed in the absence of primary antibody.
Electrophoretic mobility shift assays and supershift analysis. We performed electrophoretic mobility shift assays (EMSAs) on nuclear extracts from cortical neurons using a32P-labeled oligonucleotide containing a wild-type (wt) or mutant Sp1 binding site (Santa Cruz Biotechnology). The sense strand sequences of the double-stranded wt and mutant oligonucleotides are 5′-ATTCGATCGGGGCGGGGCGAGC-3′ and 5′-ATTCGATCGGTTCGGGGCGAGC-3′, respectively. Parallel EMSAs were performed using a radiolabeled Oct-1 (5′-TGTCGAATGCAAATGACTAGAA-3′; Santa Cruz Biotechnology) binding site. Embryonic cortical neurons were lightly trypsinized, pelleted, and resuspended in cold PBS. All subsequent steps were performed at 4°C as described previously (Lin et al., 1995,Chatterjee et al., 2001). To evaluate the effects of various agents on Sp1 and Sp3 DNA binding, we added deferoxamine mesylate (10 μm), d-amino acid oxidase (1–5 mU from red yeast), and d-alanine (20 mm).
Promoter activity assay. Cortical neurons were plated onto 24-well culture plates (Nunc, Naperville, IL). The next day, a transfection mixture was prepared by adding 1.5 μg of the reporter expression vector (pSp1-Luc and pmtSp1-Luc, firefly luciferase plasmid) into 150 μl of DMEM with a combination of pRL cytomegalovirus or thymidine kinase vector (1 μg, containing the Renilla luciferase gene) (Hsiao et al., 1997). Twenty minutes after the addition of DMRIE-C reagent (6 μl; Invitrogen), the transfection mixture was combined with 1.0 ml of fresh medium and placed onto the cells in 24-well plates. The cells were incubated for 24 hr, and then cells were infected with HSV-Flag-Sp1, HSV-Sp3, or HSV-Sp1-ZnF (zinc finger) with or without HCA. The next day, cells were washed with PBS and then lysed with luciferase assay buffer (Promega). The cell particulate was removed by microcentrifugation, and the protein concentration was measured using a protein assay kit (Bio-Rad). Twenty microliters of lysate were used for both the firefly and Renilla luciferase readings. Firefly and Renilla luciferase activities were measured using a dual-luciferase reporter assay system (Promega) and a TD-20/20 luminometer. Firefly luciferase values were standardized to Renilla values or total protein concentration.
Results
Oxidative stress induces Sp1 response element-binding activities in cortical neuronal cultures
To determine the effects of glutathione depletion-induced oxidative stress on Sp1 DNA binding in cortical neurons, we performed EMSAs on nuclear extracts from control cells or cells treated with the glutamate analog HCA, using a radiolabeled oligonucleotide containing a consensus Sp1 DNA-binding site. Five hours of HCA treatment, which leads to a 70% reduction in glutathione levels compared with control (Zaman et al., 1999), significantly increased three DNA-binding activities in cortical neurons (Fig.1A; the three DNA-binding activities are designated a–c). The induction of these DNA-binding activities appeared to be related to glutathione depletion because they were also observed in the presence of buthionine sufloximine (100 μm), an agent that depletes glutathione [to <10% of control levels (Ratan et al., 1994a)] by inhibiting one of the enzymes involved in its synthesis rather than by inhibiting the uptake of its rate-limiting amino acid precursor (data not shown). All three DNA-binding activities were inhibited in a concentration-dependent manner by cold Sp1 oligonucleotide, and none was inhibited by an oligonucleotide containing mutations in the Sp1-binding site (data not shown). Enhanced DNA binding could be observed as early as 2 hr after HCA treatment (Fig. 1C), a time when glutathione levels are 50% of levels in mock-treated cultures, permitting sufficient time for genes to be expressed before cell death commitment (∼12 hr after HCA treatment).
Fig. 1.

Glutathione depletion-induced oxidative stress induces Sp1 and Sp3 DNA-binding activity in cortical neurons.A, EMSA performed with increasing amounts of protein from nuclear extracts (NEs) from control (lanes 1, 3, 5) and HCA (a glutamate analog)-treated neurons (lanes 2, 4, 6), 4 hr after the onset of HCA treatment. The three DNA-binding activities induced by oxidative stress are designated a–c. B,Identification of Sp1 in complex a and Sp3 in complexesb and c by supershift analysis using subunit-specific antibodies to Sp1–Sp4. C, Time course of induction of Sp1 and Sp3 DNA-binding activities after HCA (1 mm) treatment. D, The effect of HCA-induced glutathione depletion on Oct-1 DNA-binding activity is shown 4 hr after the onset of HCA treatment. Effects of increasing concentrations of HCA on nuclear Sp1 (E) and Sp3 (F) protein levels 4–5 hr after the onset of HCA treatment are shown. G, Time course of changes of nuclear Sp1 and Sp3 levels after the onset of HCA treatment. Examples are representative of three to five independent experiments.
Identification of Sp1 and Sp3 in nuclear extracts of cortical neurons after treatment with the glutamate analog HCA
To determine whether Sp1 was present in any of the HCA-induced DNA–protein complexes detected by EMSA, we used antibodies specific for the Sp1–Sp4 subunits. Sp2–Sp4 share extensive sequence homology with Sp1, and each of these Sp family members binds specifically to DNA. The addition of individual subunit-specific antibodies during in vitro DNA binding revealed that Sp1 is contained in the slowest migrating complex (Fig. 1B). This complex did not contain other Sp family members, including Sp2–Sp4.
In addition to an Sp1-containing complex, two faster-migrating complexes were also induced by HCA-induced glutathione depletion. These complexes were supershifted by a subunit-specific antibody to Sp3 and were unaffected by Sp1, Sp2, and Sp4 antibodies. Taken together, these results suggest that one Sp1-containing complex and two Sp3-containing complexes are induced by glutathione depletion in cortical neurons. The differences between nuclear Sp1 and Sp3 DNA activity in control and HCA-treated cortical neurons could not be attributed to global differences in nuclear proteins, because levels of Oct-1 DNA binding were similar using 0.5 μg of these two extracts (Fig.1D).
Enhanced DNA-binding activity of Sp1 and Sp3 in response to oxidative stress results, in part, from increased levels of Sp1 and Sp3 protein
Previous studies have established that DNA binding of Sp family members to their cognate sequence can be enhanced by increasing levels of Sp1 protein (Mortensen et al., 1997) or post-transcriptional modifications of the protein such as phosphorylation (Rohlff et al., 1997) or dephosphorylation (Leggett et al., 1995). To determine whether HCA-induced oxidative stress induces changes in Sp1 and Sp3 levels, we performed immunoblot analysis. We found that HCA leads to increases in both Sp1 and Sp3 protein levels (Fig. 1E). Five millimolar HCA leads to more rapid cystine deprivation and glutathione depletion after 5 hr than 1 mm HCA (Ratan et al., 1994a). As expected from these observations, treatment of cortical neurons with 5 mm HCA for 5 hr induced greater levels of Sp1 (Fig. 1E) and Sp3 (Fig.1F) proteins than did 1 mm HCA. A rabbit polyclonal Sp1 antibody detected a dominant band at 95 kDa and a minor band at 105 kDa. This slower-migrating band at 105 kDa likely reflects phosphorylation of the 95 kDa Sp1 polypeptide, because the 95 kDa band completely disappeared in the presence of the phosphatase inhibitor, calyculin A (Ryu et al.) (our unpublished observations). A rabbit polyclonal Sp3 antibody detected a dominant 115 kDa band corresponding to Sp3 previously identified by both immunochemical and molecular genetic techniques (Kennett et al., 1997). A minor, faster-migrating band was observed at 78–80 kDa, a species of Sp3 that has been shown to result from internal translational initiation within Sp3 mRNA (Kennett et al., 1997). The increases in Sp1 and Sp3 proteins were seen as early as 1 hr after HCA treatment, were maximum at 5 hr (Fig. 1G), and could be observed to be returning to baseline levels by 8 hr after HCA treatment (results not shown).
Glutathione depletion-induced oxidative stress induces Sp1 and Sp3 levels in the nucleus of cortical neurons
To verify that Sp1 and Sp3 proteins can be induced in the nuclei of neurons by HCA-induced glutathione depletion, we performed immunocytochemical staining with antibodies to Sp1 (Fig.2a,d), Sp2 (Fig.2g,j), and Sp3 (Fig. 2m,p), using in all cases an FITC-labeled secondary antibody that fluoresces green. In parallel, the same cultures were treated with the nuclear stain DAPI, which fluoresces blue (Molecular Probes, Eugene, OR) (Fig.2b,e,h,i,n,o). When the Sp1, Sp2, and Sp3 antibody fluorescence images were superimposed on the DAPI fluorescence images, most cells that were induced to undergo oxidative stress by treatment with HCA and stained with an Sp1 or Sp3 antibody were found to have blue–green nuclei, indicative of an enhanced protein level in the nucleus (Fig. 2f,r). Moreover, staining with the Sp2 antibody revealed that levels of this protein were not altered in the cytoplasm or nucleus by HCA-induced glutathione depletion and oxidative stress (Fig. 2g–l). Cells that stained positively for Sp1 and Sp3 were determined to be neuronal by parallel immunostaining with a neurofilament antibody (data not shown) as previously described (Zaman et al., 1999).
Fig. 2.
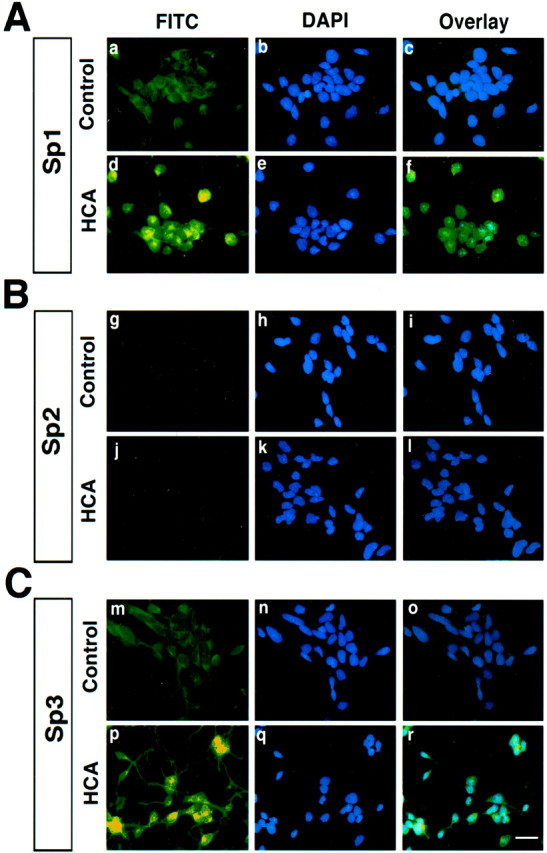
HCA-induced glutathione depletion and oxidative stress induce Sp1 (A) and Sp3 (C), but not Sp2 (B), in the nucleus of embryonic cortical neurons. Immunocytochemical analysis of Sp1 (a, d), Sp2 (i), Sp3 (m, p), and the nuclear stain DAPI (b, e, h, k, n, q) in mock-treated (a, g, m, b, h, n) or HCA-treated (d, j, e, k, q) mixed cortical neuronal cultures. Cells were plated on eight-well chamber slides for 24 hr and treated with HCA (3 mm) for 3 hr. Cells were then fixed with 4% PFA for 15 min, and immunofluorescence staining was performed as described in Materials and Methods. Scale bar, 20 μm.
Antioxidants that prevent glutathione depletion-induced apoptotic death also prevent glutathione depletion-induced Sp1 and Sp3 DNA binding
Oxidative stress is defined as an imbalance between oxidants and antioxidants in favor of oxidants (Sies, 1997; for review, see Ratan, 1999). Thus, if oxidative stress is responsible for the HCA-induced Sp1 and Sp3 DNA binding, then antioxidants that correct the imbalance should diminish or suppress the induction. To address this question, we chose the iron chelator deferoxamine (DFO; 100 μm) and the non-iron-chelating lipid peroxidation inhibitor butylated hydroxyanisole (BHA; 10 μm). We have demonstrated previously that these antioxidants completely abrogate HCA-induced apoptosis in cortical neurons (Ratan et al.,1994b, Zaman et al., 1999). In gel-shift assays, we found that DFO (Fig.3A) and BHA (Fig.3B) suppress HCA-induced Sp1 and Sp3 DNA binding. These results suggest that HCA-induced oxidative stress mediates enhanced DNA binding of Sp1 and Sp3 in cortical neurons. We demonstrated previously that DFO enhances the DNA binding of hypoxia-inducible factor-1 and activating transcription factor-1/cAMP response element-binding protein to the hypoxia response element (Zaman et al., 1999), suggesting that its inhibition of Sp1 and Sp3 activation does not reflect nonspecific inhibition of the DNA binding of all transcription factors.
Fig. 3.

Increases in Sp1 and Sp3 DNA binding induced by the glutamate analog HCA are inhibited by antioxidants; Sp1 and Sp3 DNA binding in cortical neurons are activated by hydrogen peroxide. Induction of Sp1 and Sp3 DNA binding by HCA-induced glutathione depletion (4 hr) is decreased by the antioxidant iron chelator DFO (100 μm;A) and the lipid peroxidation inhibitor BHA (10 μm;B).C, Addition of exogenous peroxide, generated by the enzyme DAAO and its substrate d-ala (20 mm) for 4 hr increases Sp1 and Sp3 DNA binding in a concentration-dependent manner in cortical neurons. The induction is observed despite no morphological or biochemical evidence of cell death in cortical neurons. D, Addition of catalase abrogates Sp1 and Sp3 DNA binding induced by d-ala (20 mm) and DAAO (5 mU). Examples are representative of three to five independent experiments.
To determine whether the addition of the ROS hydrogen peroxide to cortical neurons is also capable of inducing Sp1 and Sp3 DNA binding, we added increasing concentrations of the enzyme d-amino acid oxidase (DAAO; 1–5 mU) and its substrate d-alanine (d-ala; 20 mm) to the bathing medium of embryonic rat cortical neurons. DAAO catalyzes the stereoselective oxidative deamination of d-amino acids to form hydrogen peroxide via the following reaction: d-amino acid + H2O + O2 → α-keto acid + NH3 + H2O2 (Stegman et al., 1998). As expected, we found that hydrogen peroxide generated from DAAO induces three complexes in cortical neurons in a concentration-dependent manner (Fig. 3C). Supershift analysis confirmed that, like glutathione depletion, the slowest migrating complex contains Sp1, and the two faster-migrating complexes contain Sp3 (data not shown). In addition,d-ala/DAAO-induced Sp1 and Sp3 DNA binding could be completely suppressed by the coapplication of the peroxide-scavenging enzyme catalase (100 U/ml; Fig. 3D) or the nonenzymic peroxide scavenger pyruvate (2 mm; data not shown).
These results establish that Sp1 and Sp3 are oxidative stress-inducible transcription factors in vitro. To determine whether Sp1 and Sp3 protein levels can be similarly induced in vivo in rodent models in which neuronal loss has been attributed, in part, to oxidative stress, we examined Sp1 and Sp3 immunoreactivity in a transgenic mouse model or a chemical mouse model of HD (Fig.4). The R6/2 line is a transgenic mouse line expressing exon 1 of the human HD gene with an expanded CAG repeat (Mangiarini et al., 1996). The mice develop loss of brain and body weight beginning at 6 weeks of age, and at 9–11 weeks, they develop a gait and movement disorder as well as epilepsy. These clinical features, along with striatal atrophy and neuronal intranuclear inclusions, are similar to HD in humans. At 12 weeks of age, Sp1 and Sp3 levels in the brains of R6/2 mice were found to be significantly increased compared with their littermate controls, as monitored by immunohistochemistry (Fig. 4A–F) or Western blot analysis (Fig. 4G). Sp1 and Sp3 were similarly induced in cells in the region adjacent to neuronal loss in response to the 3-NP. 3-NP is a mitochondrial toxin that produces striatal lesions that mimic HD and that are mediated by excitotoxicity-induced oxidative stress mechanisms (Beal et al., 1993; Kim and Chan, 2002). Taken together, these results are consistent with the notion that Sp1 and Sp3 are induced by stimuli that lead to oxidative stress in vivo.
Fig. 4.

Sp1 and Sp3 tissue immunoreactivity in R6/2 transgenic mice and 3-NP-lesioned-mice. Sp1 (A–C) and Sp3 (D–F) immunoreactivities in the neostriatum of 12-week-old wild-type littermate control mice (A, D) and R6/2 transgenic HD mice (B, E) and 3-NP-lesioned wild-type mice (C, F) are shown. Sp1 and Sp3 immunostaining of neurons in wild-type control mice (A, D) is shown. Markedly increased Sp1 and Sp3 immunoreactivity was observed in R6/2 mice compared with wild-type controls, with the greatest increases in Sp3 immunoreactivity. In 3-NP-lesioned mice, Sp1 (C) and Sp3 (F) immunoreactivities were both increased within neurons surrounding the lesion core (asterisk) within the penumbra or transition zone of neuronal injury. Scale bars: (in E) A, B,D, E, 100 μm; (in F), C, F, 200 μm. G, Sp1 and Sp3 protein levels in 12-week-old littermate controls and R6/2 transgenic mouse model of HD. α-Tubulin was used as a loading control. H, Scatter plot of densitometric values (normalized to α-tubulin) for Sp1 and Sp3 protein levels in seven littermate control and nine R6/2 HD mice.
Forced high-level expression of full-length Sp1 or full-length Sp3, but not its zinc finger DNA-binding domain alone, prevents cortical neuronal death attributable to oxidative stress or DNA damage
To determine whether the overexpression of Sp1 is sufficient to abrogate glutathione depletion-induced death in cortical neurons, we generated replication-deficient HSV-1-based vectors that carry the wild-type Sp1 gene (HSV-Sp1-wt) or a truncated form of Sp1 containing the DNA-binding domain alone (HSV-Sp1-ZnF). Sp1-ZnF can bind DNA but does not possess regions of the Sp1 molecule required for activation of transcription (Chapman and Perkins, 2000). As an additional control, we inserted the β-galactosidase (LacZ) gene into the HSV vector (HSV-LacZ). Infection of cortical neurons with HSV-Sp1-wt (Fig.5A) but not HSV-Sp1-ZnF (Fig.5A) or HSV-LacZ (Fig. 5D), resulted in the robust, MOI-dependent expression of heterologous Sp1 in cortical neurons as monitored by an anti-Sp1 antibody (Fig. 5A,D) or by an anti-Flag antibody (Fig. 5D) 6–12 hr after infection. An MOI of 2 was used for the HSV viability experiments, because this MOI resulted in infection of ∼15–25% of the neurons in the dish (Fig. 5C) with no toxicity up to 48 hr after infection. The overexpression of Sp1 by HSV led to a threefold induction of Sp1-dependent luciferase activity compared with HSV-LacZ.
Fig. 5.
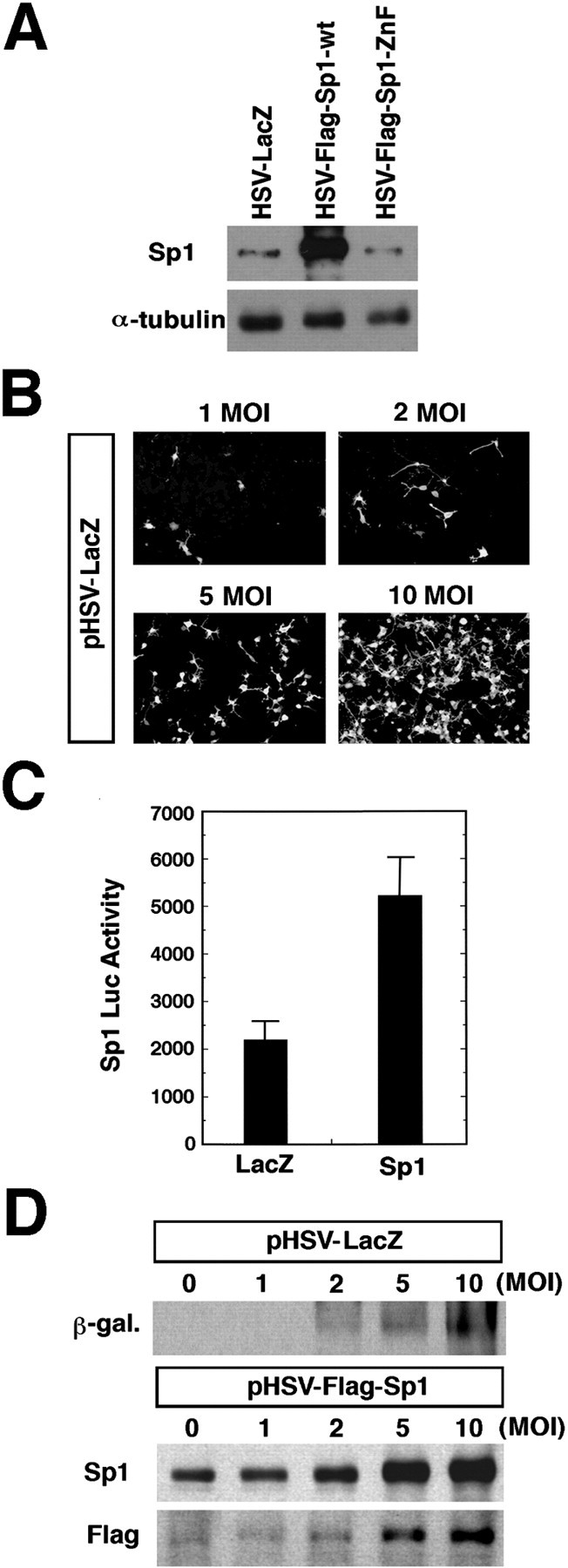
HSV can be used to achieve expression of heterologous Sp1 in cortical neurons. A, Immunoblot demonstrating expression of heterologous Sp1 in cortical neurons 24 hr after infection with HSV vectors. HSV has an MOI of 2.B, HSV-LacZ leads to MOI-dependent β-galactosidase expression in cortical neurons. C, HSV-Sp1 infection of cortical neurons leads to induction of luciferase activity driven by Sp1 response elements compared with HSV-LacZ. Results are means ± SE for three separate experiments. D, Immunoblot analysis confirms MOI-dependent, HSV vector-driven expression of Sp1 (HSV-Sp1) or β-galactosidase. In parallel, immunoblotting with Flag antibody confirms that HSV-Sp1 results in heterologous rather than endogenous Sp1 expression.
To determine the effect of enhancing Sp1 activity using the HSV vector on oxidative stress-induced death in cortical neurons, we monitored the viability of cells infected with HSV-LacZ, HSV-Sp1-wt, and HSV-Sp1-ZnF 17–24 hr after infection. LacZ is expressed diffusely in the cytoplasm (Fig. 6A) and the neurites of neurons, whereas the Sp1-wt and the Sp1-ZnF are found primarily in the nuclear compartment (Fig. 6A). Under control, nonoxidative stress conditions, the nuclei of cells infected with HSV-LacZ, HSV-Sp1-wt, or HSV-Sp1-ZnF and stained with the nuclear probe DAPI appeared oval in shape and homogeneously stained with moderate intensity. These apparently normal nuclei were present in neurons with smooth cell bodies and ramified neurites, as determined by Nomarski differential interference contrast microscopy. Parallel cultures were infected with HSV-LacZ, HSV-Sp1-wt, or HSV-Sp1-dominant negative (dn), exposed to the glutamate analog HCA for 16–18 hr, and stained with DAPI or visualized by Nomarski optics. Cells infected with HSV-Sp1-ZnF or HSV-LacZ and exposed to HCA showed massive changes in chromatin structure (condensation), indicated by greatly enhanced DAPI fluorescence. These changes in chromatin structure are consistent with the apoptotic cell death observed previously in cortical neurons exposed to glutathione depletion (Ratan et al., 1994b). By contrast, cells infected with HSV-Sp1-wt that were exposed to oxidative stress generally retained the oval, moderately intense staining of normal nuclei. Two independent observers who were blinded to the treatment group quantitated the number of LacZ-, Sp1-wt-, and Sp1-ZnF-expressing cells that possessed normal nuclear morphology under conditions of oxidative stress and confirmed that cells expressing the heterologous Sp1-wt were significantly more resistant to glutathione depletion-induced death than cells expressing heterologous LacZ or Sp1-ZnF (Fig. 6B). In addition, TUNEL (a marker of DNA damage resulting from HCA-induced oxidative stress) was performed along with immunfluorescence for Sp1, LacZ, or Sp1-ZnF (Fig.6C). These experiments showed that after 18 hr of HCA (1 mm) treatment, only LacZ- or Sp1-ZnF-expressing neurons were TUNEL-positive, whereas cells expressing Sp1 were not (Fig. 6C).
Fig. 6.

Overexpression of Sp1 (HSV-Flag-Sp1), but not the Sp1 Zn finger DNA binding domain alone (HSV-Flag-Sp1-ZnF) or HSV-LacZ, in cortical neurons inhibits oxidative stress-induced cell death in cortical neurons. A, DAPI staining of LacZ-positive, Flag-Sp1-positive, or Flag-Sp1-ZnF-positive cortical neurons. The FITC column reflects FITC secondary antibody that recognizes LacZ-, Flag-Sp1-wt-, or Flag-Sp1-ZnF-expressing cortical neurons. Note the presence of LacZ in cell bodies as well as neurites, which is typical of cortical neurons in culture. Also note the presence of Sp1-wt and Sp1-ZnF in the nucleus, the expected localization for these transcription factors. The DAPI column shows the DAPI staining of FITC-positive cells in the same row, along with a few surrounding FITC-negative cells. Note that HCA produces an increase in the percentage of cells with pyknotic nuclei that display stronger fluorescence, which is characteristic of apoptotic cells. However, the Flag-Sp1-wt-expressing neurons display dim, diffuse DAPI staining, which is characteristic of normal cells. Cortical neurons were infected with 2–5 MOI of HSV vectors. B, Quantitative analysis. Overexpression of Flag-Sp1-wt but not Flag-Sp1-ZnF or LacZ blocks the increase in apoptotic cells elicited by HCA (LacZ or Sp1-ZnF vs Sp1-wt,p < 0.01). At least 300 cells were counted in each group, and the results are means ± SE for three different slides.C, TUNEL was performed to measure HCA-induced DNA damage of LacZ-positive, Flag-Sp1-positive, and Flag-Sp1-ZnF-positive cortical neurons. Cy3 (red) column reflects Cy3 secondary antibody that recognizes LacZ, Flag-Sp1, Flag-Sp3, or Sp1-ZnF. The overlay column reflects green TUNEL–FITC fluorescence superimposed on red Cy3 secondary antibody fluorescence for β-galactosidase, Sp1, and Sp1-ZnF and blue fluorescence for the nucleus.
To determine whether the overexpression of Sp1 using herpes vectors can inhibit death induced by apoptotic stimuli distinct from glutathione depletion, we examined the effects of HSV-Sp1-wt infection of cortical neurons on apoptosis induced by the DNA-damaging agent camptothecin. Camptothecin is a cytotoxic plant alkaloid that induces cell injury by inhibiting the activity of DNA topoisomerase I, which leads to double-stranded DNA damage (Ng et al., 2001). Previous studies have established that, like glutathione depletion, cortical neuronal death induced by camptothecin has characteristic morphological features of apoptosis. Unlike the case with glutathione depletion-induced apoptosis, camptothecin-induced apoptosis is not associated with increased hydrogen peroxide production and is not blocked by the antioxidant N-acetylcysteine. Cell viability, as measured by MTT reduction, revealed that cortical neurons infected with HSV-Sp1-wt, but not HSV-LacZ or HSV-Sp1-dn, were more resistant to camptothecin-induced death over a range of camptothecin concentrations (Fig.7A). In parallel cultures, TUNEL [a marker of DNA double-strand breaks resulting from camptothecin-induced topoisomerase inhibition (Morris and Geller, 1996)] was performed along with immunfluorescence for Sp1, LacZ, or Sp1-ZnF (Fig. 7B). These studies revealed that after 18 hr of camptothecin treatment, only LacZ- or Sp1-ZnF-expressing neurons were TUNEL-positive, whereas cells expressing Sp1 or Sp3 were not. Taken together, these studies suggest that the overexpression of Sp1 or Sp3 can abrogate neuronal damage and death attributable to oxidative stress or other established apoptosis inducers, including the topoisomerase inhibitor camptothecin (Fig. 7).
Fig. 7.
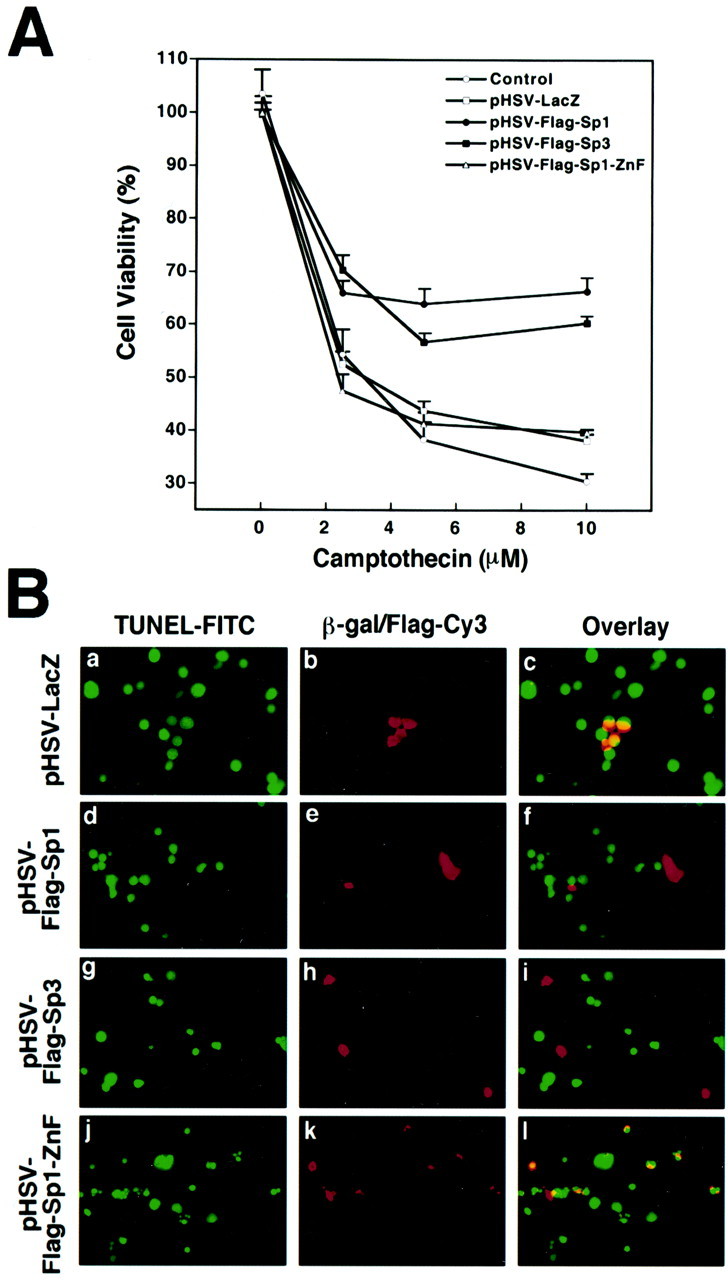
Overexpression of Sp1-wt or Sp3-wt, but not the Sp1-ZnF or LacZ, inhibits neuronal death attributable to the DNA-damaging agent and topoisomerase inhibitor camptothecin.A, Quantitative analysis. Overexpression of Flag-Sp1 or Flag-Sp3 but not LacZ or Flag-Sp1-ZnF diminishes cell death induced by camptothecin (2–10 μm). Cortical neurons were infected with 2–5 MOI of HSV vectors. Cell viability was measured by incubating cortical neurons with MTT for 2 hr and measuring the amount of reduction to blue formazan. Results are means ± SE for three separate experiments. B, TUNEL, a marker of DNA double-strand breaks resulting from camptothecin-induced topoisomerase inhibition (Morris and Geller, 1996) of LacZ-positive, Flag-Sp1-positive, Flag-Sp3-positive, or Flag-Sp1-ZnF-positive cortical neurons. The Cy3 (red) column reflects Cy3 secondary antibody that recognizes LacZ, Flag-Sp1, Flag-Sp3, or Sp1-ZnF. The overlay column reflects green TUNEL–FITC fluorescence superimposed on red Cy3 secondary antibody fluorescence. Note that only cells expressing LacZ or Flag Sp1-ZnF are yellow, whereas Cy3-positive Flag-Sp1- and Flag-Sp3-expressing cells are not TUNEL labeled and are therefore red.
Discussion
Sp1 and Sp3 are redox-regulated transcription factors in neurons
There is abundant evidence linking oxidative stress to the initiation or propagation of neuronal loss in neurological disease (Beal, 2000). This evidence has stimulated a search for transcriptional regulators that are induced by oxidative stress in neurons whose net effect is to prevent neuronal death (Ratan, 1999). Herein, we demonstrate that basal Sp1 DNA-binding activities are unexpectedly low in embryonic cortical neurons and are dramatically induced by agents that induce oxidative stress in these cells (Figs. 1, 2,4A). Second, we used gel-shift assays to show that one of the Sp1 DNA-binding complexes induced by oxidative stress in cortical neurons contains Sp1, and two slower-migrating complexes containin Sp3 (Fig. 1B). Furthermore, we used immunoblot analysis (Fig. 1E–G) and immunocytochemistry (Fig. 1H) to demonstrate that protein levels of these transcription factors are increased in the nuclei of oxidatively stressed cortical neurons and that antioxidants shown previously to inhibit oxidative stress-induced death in cortical neurons also abrogate glutathione depletion-induced Sp1 and Sp3 DNA binding (Fig. 2A,B). Finally, we demonstrate that oxidative stress can enhance the activity of an Sp1-dependent reporter gene (data not shown). These results establish Sp1 and Sp3 as redox-regulated transcription factors in embryonic cortical neurons and add these factors to a small list of transcriptional activators and repressors (e.g., nuclear factor κB, activator protein-1, and heat shock transcription factor-1) known to be induced by oxidative stress in neurons (Lezoualc'h and Behl, 1998; Post et al., 1998; Bijur et al., 1999; Scortegagna et al., 1999).
Sp1 and Sp3 activation appear to be temporally related to the onset of oxidative stress in cortical neurons and not a late event that is a consequence of oxidative stress-induced cell death. Activation of Sp1 and Sp3 DNA binding occurs within the first 2 hr of glutamate or HCA exposure and is maximal by 5 hr. The kinetics of Sp1 and Sp3 activation demonstrate that induction of these factors is an “early” response to cell stress, and their activation is initiated 8–10 hr before the point at which neurons become irreversibly “committed” to the cell death pathway (Ratan et al., 1994b; Zaman et al., 1999; Chatterjee et al., 2001). The close temporal relationship between oxidative stress and Sp1 and Sp3 activation is also supported by the observation that structurally diverse antioxidants known to ameliorate oxidative stress and inhibit oxidative glutamate toxicity can block the activation of Sp1 and Sp3 by glutathione depletion, despite having no effect on glutathione depletion per se (Ratan et al., 1994a; Zaman et al., 1999).
Although classically thought to regulate the constitutive expression of numerous housekeeping genes, Sp1 transcriptional activities have been found to change in association with differentiation (Leggett et al., 1995; Yan and Ziff, 1997; Krainc et al., 1998) and proliferation (Black et al., 1999) and to regulate gene expression in association with these as well as other cellular functions. In agreement with the data presented herein, one of these studies demonstrated nearly absent basal DNA binding of Sp1 and Sp3 in organs such as brain and muscle, in which most cells are postmitotic (Leggett et al., 1995). Two-dimensional electrophoresis revealed that the decreased DNA binding of Sp1 and Sp3 in postmitotic tissues is associated with the phosphorylation of Sp1 and Sp3. Accordingly, in vitro treatment of nuclear extracts from adult brain with a phosphatase abolished Sp1 and Sp3 phosphorylation and increased the affinity of these factors for a canonical Sp1 DNA-binding site by 10-fold. These results are consistent with a model in which the post-translational modification of Sp1 by activation of a phosphatase (dephosphorylation) or inhibition of a histone deacetylase (acetylation) (Ryu et al., 2003) could account for the dramatic increase in DNA binding we observe in response to oxidative stress. Our ability to observe oxidative stress-induced changes in Sp1 and Sp3 DNA binding is likely also enhanced, because we have been careful to verify that the concentration of the nuclear extract we use is in the dynamic range of DNA binding for Sp1 and Sp3 (Fig. 1A).
Sp1 and Sp3 can act as antideath transcription factors
The data herein support the conclusion that Sp1 and Sp3 are sufficient components of the protective, homeostatic response to oxidative stress (Fig. 6) and one potential consequence of oxidative stress, DNA damage (Fig. 7), in neurons. Indeed, recent studies suggest that Sp1 motifs are responsible for the regulation of the IAP protein survivin (Li and Altieri, 1999). Members of the IAP protein family have been shown to suppress apoptosis induced by oxidative stress by directly suppressing the activity of terminal caspase-3 and caspase-7 (Tamm et al., 1998; Suzuki et al., 2000). Interestingly, survivin is expressed not only in common human cancers but also in some types of embryonic neurons (Adida et al., 1998), suggesting that this protein is poised to act as an inhibitor of apoptosis in the cortical neurons used in the present study. Bcl-2 and Bcl-x, two other general inhibitors of apoptosis, also have essential Sp1 sites in their promoters (Grillot et al., 1997; Dong et al., 1999), and p53 has been shown to repress the expression of the antiapoptotic factor telomerase by binding to cognate Sp1 motifs in the telomerase promoter (Kanaya et al., 2000). Finally, Sp1 is cleaved by caspases in IgM-induced apoptosis in B cells (Rickers et al., 1999) and in retinoid-induced apoptosis in T-cells (Piedrafita and Pfahl, 1997). Taken together, these observations support an antiapoptotic role for Sp1.
Inhibition of Sp1 activation in polyglutamine disorders such as HD may account for increased vulnerability to oxidative stress
The data herein are consistent with a model in which Sp1 is a part of a compensatory genetic program to oxidative stress in neurons. A prediction of this model is that neurons with acquired or inherited defects in Sp1 signaling will be more vulnerable to oxidants. Indeed, autopsy tissue from HD patients shows prominent defects in Sp1 DNA binding (despite increased Sp1 levels; Dunah et al., 2002) (Fig. 3) and increased markers of oxidative damage (Browne et al., 1999). Furthermore, toxicity of cultured neurons induced by the forced expression of mutant huntingtin with pathological numbers of polyglutamine repeats can be abrogated by the coexpression of Sp1 and its coactivator TAFII130 (Dunah et al., 2002; Li et al., 2002) or by the exogenous addition of small-molecule antioxidants (Wyttenbach et al., 2002). Future studies will define the precise order of oxidative stress and disrupted Sp1 signaling in HD. Nevertheless, these findings suggest that small-molecule activators of Sp1-dependent gene expression may be propitious therapeutic targets for a host of neurodegenerative conditions, including HD, Parkinson's disease, amyotrophic lateral sclerosis, and stroke, which in some cases have been associated with expanded polyglutamine repeats and in all cases have been associated with oxidative stress.
Footnotes
This work was supported by National Institutes of Health Grants R29 NS34943 and R01 NS39170 to R.R.R. The cDNA for the zinc finger domain of Sp1 was a gift from Dr. Neil Perkins (Department of Biochemistry, University of Dundee, Dundee, UK) and the full-length Sp1 and Sp3 were gifts from Dr. Guntram Suske (Institute of Molecular Biology and Tumor Research, Philipps-University Marburg, Marburg, Germany). We thank Rini Ratan and Donald DeFranco for careful review of this manuscript.
Correspondence should be addressed to Dr. Rajiv R. Ratan, Neurology Laboratories at Beth Israel Deaconess Medical Center, Harvard Institutes of Medicine, Room 857, 77 Avenue Louis Pasteur, Boston, MA 02115. E-mail: rratan@caregroup.harvard.edu.
Dr. Ryu's present address: Department of Neurology, University of Massachusetts Medical School and Memorial Medical Center, Worcester, MA 06105
References
- 1.Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol. 1998;152:43–49. [PMC free article] [PubMed] [Google Scholar]
- 2.Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm [Suppl] 2000;59:133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- 3.Atwood CS, Huang X, Moir RD, Tanzi RE, Bush AI. Role of free radicals and metal ions in the pathogenesis of Alzheimer's disease. Met Ions Biol Syst. 1999;36:309–364. [PubMed] [Google Scholar]
- 4.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histological characterization of striatal excitotoxic lesions produced by the mitochondrial toxin, 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beal MF. Oxidative metabolism. Ann NY Acad Sci. 2000;924:164–169. doi: 10.1111/j.1749-6632.2000.tb05575.x. [DOI] [PubMed] [Google Scholar]
- 6.Bijur GN, Davis RE, Jope RS. Rapid activation of heat shock factor-1 DNA binding by hydrogen peroxide and modulation by glutathione in human neuroblastoma and Alzheimer's disease cybrid cells. Brain Res Mol Brain Res. 1999;71:69–77. doi: 10.1016/s0169-328x(99)00168-0. [DOI] [PubMed] [Google Scholar]
- 7.Black AR, Jensen D, Lin SY, Azizkhan JC. Growth/cell cycle regulation of Sp1 phosphorylation. J Biol Chem. 1999;274:1207–1215. doi: 10.1074/jbc.274.3.1207. [DOI] [PubMed] [Google Scholar]
- 8.Briggs MR, Kadonaga JT, Bell SP, Tjian R. Purification and biochemical characterization of the promoter-specific transcription factor, Sp1. Science. 1986;234:47–52. doi: 10.1126/science.3529394. [DOI] [PubMed] [Google Scholar]
- 9.Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington's disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castagne V, Gautschi M, Lefevre K, Posada A, Clarke PG. Relationships between neuronal death and the cellular redox status: focus on the developing nervous system. Prog Neurobiol. 1999;59:397–423. doi: 10.1016/s0301-0082(99)00012-x. [DOI] [PubMed] [Google Scholar]
- 11.Chapman NR, Perkins ND. Inhibition of the RelA(p65) NF-κB subunit by Egr-1. J Biol Chem. 2000;275:4719–4725. doi: 10.1074/jbc.275.7.4719. [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann Neurol. 2001;49:345–354. [PubMed] [Google Scholar]
- 13.Chou C, Peng H, Wang C, Yang Y, Han S. An Sp1 binding site involves the transcription of the fas ligand gene induced by PMA and ionomycin in jurkat cells. J Biomed Sci. 2000;7:136–143. doi: 10.1007/BF02256620. [DOI] [PubMed] [Google Scholar]
- 14.Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S. Mechanisms of transcriptional activation of bcl-2 gene expression by 17β-estradiol in breast cancer cells. J Biol Chem. 1999;274:32099–32107. doi: 10.1074/jbc.274.45.32099. [DOI] [PubMed] [Google Scholar]
- 15.Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 16.Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barveito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 17.Ferrante RJ, Andreassen O, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 19.Fuangthong M, Helmann JD. The OhR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci USA. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grillot DA, Gonzalez-Garcia M, Ekhterae D, Duan L, Inohara N, Ohta S, Seldin MF, Nunez G. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl-x gene. J Immunol. 1997;158:4750–4757. [PubMed] [Google Scholar]
- 21.Hsiao M, Tse V, Carmel J, Tsai Y, Felgner PL, Haas M, Silverberg GD. Intracavitary liposome-mediated p53 gene transfer into glioblastoma with endogenous wild-type p53 in vivo results in tumor suppression and long-term survival. Biochem Biophys Res Commun. 1997;233:359–364. doi: 10.1006/bbrc.1997.6459. [DOI] [PubMed] [Google Scholar]
- 22.Kanaya T, Kyo S, Hamada K, Takakura M, Kitagawa Y, Harada H, Inoue M. Adenoviral expression of p53 represses telomerase activity through down-regulation of human telomerase reverse transcriptase transcription. Clin Cancer Res. 2000;6:1239–1247. [PubMed] [Google Scholar]
- 23.Kennett SB, Udvadia AJ, Horowitz JM. Sp3 encodes multiple proteins that differ in their capacity to stimulate or repress transcription. Nucleic Acids Res. 1997;25:3110–3117. doi: 10.1093/nar/25.15.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;2:798–809. doi: 10.1097/00004647-200207000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Krainc D, Bai G, Okamoto S, Carles M, Kusiak JW, Brent RN, Lipton SA. Synergistic activation of the N-methyl-d-aspartate receptor subunit 1 promoter by myocyte enhancer factor 2C and Sp1. J Biol Chem. 1998;273:26218–26224. doi: 10.1074/jbc.273.40.26218. [DOI] [PubMed] [Google Scholar]
- 26.Lania L, Majello B, De Luca P. Transcriptional regulation by the Sp family proteins. Int J Biochem Cell Biol. 1997;29:1313–1323. doi: 10.1016/s1357-2725(97)00094-0. [DOI] [PubMed] [Google Scholar]
- 27.Leggett RW, Armstrong SA, Barry D, Mueller CR. Sp1 is phosphorylated and its DNA binding activity down-regulated upon terminal differentiation of the liver. J Biol Chem. 1995;270:25879–25884. doi: 10.1074/jbc.270.43.25879. [DOI] [PubMed] [Google Scholar]
- 28.Lezoualc'h F, Behl C. Transcription factor NF-κB: friend or foe of neurons? Mol Psychiatry. 1998;3:15–20. doi: 10.1038/sj.mp.4000295. [DOI] [PubMed] [Google Scholar]
- 29.Li F, Altieri DC. Transcriptional analysis of human survivin gene expression. Biochem J. 1999;344:305–311. doi: 10.1042/0264-6021:3440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. 2002;22:1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin KI, Lee SH, Narayanan R, Baraban JM, Hardwick JM, Ratan RR. Thiol agents and Bcl-2 identify an alphavirus-induced apoptotic pathway that requires activation of the transcription factor NF-kappa B. J Cell Biol. 1995;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin KI, DiDonato JA, Hoffmann A, Hardwick JM, Ratan RR. Suppression of steady-state, but not stimulus-induced NF-κB activity inhibits alphavirus-induced apoptosis. J Cell Biol. 1998;141:1479–1487. doi: 10.1083/jcb.141.7.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu YW, Arakawa T, Yamamoto S, Chang WC. Transcriptional activation of human 12-lipoxygenase gene promoter is mediated through Sp1 consensus sites in A431 cells. Biochem J. 1997;324:133–140. doi: 10.1042/bj3240133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 35.Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandolsa S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann NY Acad Sci. 1999;893:154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x. [DOI] [PubMed] [Google Scholar]
- 36.Mongkolsuk S, Helmann JD. Regulation of inducible peroxide stress responses. Mol Microbiol. 2002;45:9–15. doi: 10.1046/j.1365-2958.2002.03015.x. [DOI] [PubMed] [Google Scholar]
- 37.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mortensen ER, Marks PA, Shiotani A, Merchant JL. Epidermal growth factor and okadaic acid stimulate Sp1 proteolysis. J Biol Chem. 1997;272:16540–16547. doi: 10.1074/jbc.272.26.16540. [DOI] [PubMed] [Google Scholar]
- 39.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 40.Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- 41.Neve RL, Geller AI. Genetic analysis of neuronal physiology with defective herpes simplex virus vectors. Adv Neurol. 1999;79:1027–1032. [PubMed] [Google Scholar]
- 42.Ng CE, Mazaheri K, Payant C, Raaphorst GP. Evaluation of cell survival, DNA double strand breaks, and DNA synthesis during concurrent camptothecin and X-radiation treatments. Int J Cancer. 2001;96:277–285. doi: 10.1002/ijc.1031. [DOI] [PubMed] [Google Scholar]
- 43.Piedrafita FJ, Pfahl M. Retinoid-induced apoptosis and Sp1 cleavage occur independently of transcription and require caspase activation. Mol Cell Biol. 1997;17:6348–6358. doi: 10.1128/mcb.17.11.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Post A, Holsboer F, Behl C. Induction of NF-κB activity during haloperidol-induced oxidative toxicity in clonal hippocampal cells: suppression of NF-κB and neuroprotection by antioxidants. J Neurosci. 1998;18:8236–8246. doi: 10.1523/JNEUROSCI.18-20-08236.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratan RR. Antioxidants and the treatment of neurological disease. In: Koliatsos VE, Ratan RR, editors. Cell death and disease of the nervous system. Humana; Totowa, NJ: 1999. pp. 649–666. [Google Scholar]
- 46.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994a;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994b;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 48.Rickers A, Peters N, Badock V, Beyaert R, Vandenabeele P, Dorken B, Bommert K. Cleavage of transcription factor SP1 by caspases during anti-IgM-induced B-cell apoptosis. Eur J Biochem. 1999;261:269–274. doi: 10.1046/j.1432-1327.1999.00273.x. [DOI] [PubMed] [Google Scholar]
- 49.Rohlff C, Ahmad S, Borellini F, Lei J, Glazer RI. Modulation of transcription factor Sp1 by cAMP-dependent protein kinase. J Biol Chem. 1997;272:21137–21141. doi: 10.1074/jbc.272.34.21137. [DOI] [PubMed] [Google Scholar]
- 50.Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeat via an Sp1-dependent pathway. Proc Natl Acad Sci USA. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sagara Y, Dargusch R, Chambers D, Davis J, Schubert D, Maher P. Cellular mechanisms of resistance to chronic oxidative stress. Free Radic Biol Med. 1998;24:1375–1389. doi: 10.1016/s0891-5849(97)00457-7. [DOI] [PubMed] [Google Scholar]
- 52.Scortegagna M, Galdzicki Z, Rapoport SI, Hanbauer I. Activator protein-1 DNA binding activation by hydrogen peroxide in neuronal and astrocytic primary cultures of trisomy-16 and diploid mice. Brain Res Mol Brain Res. 1999;73:144–150. doi: 10.1016/s0169-328x(99)00257-0. [DOI] [PubMed] [Google Scholar]
- 53.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 54.Stegman LD, Zheng H, Neal ER, Ben-Yoseph O, Pollegioni L, Pilone MS, Ross BD. Induction of cytotoxic oxidative stress by d-alanine in brain tumor cells expressing Rhodotorula gracilisd-amino acid oxidase: a cancer gene therapy strategy. Hum Gene Ther. 1998;9:185–193. doi: 10.1089/hum.1998.9.2-185. [DOI] [PubMed] [Google Scholar]
- 55.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki T, Kimura A, Nagai R, Horikoshi M. Regulation interaction of the acetyltransferase region of p300 and the DNA-binding domain of Sp1 on and through DNA binding. Genes Cells. 2000;5:29–41. doi: 10.1046/j.1365-2443.2000.00302.x. [DOI] [PubMed] [Google Scholar]
- 57.Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspase, and anticancer drugs. Cancer Res. 1998;58:5315–5320. [PubMed] [Google Scholar]
- 58.Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanaka T, Kurabayashi M, Aihara Y, Ohyama Y, Nagai R. Inducible expression of manganese superoxide dismutase by phorbol 12-myristate 13-acetate is mediated by Sp1 in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:392–401. doi: 10.1161/01.atv.20.2.392. [DOI] [PubMed] [Google Scholar]
- 60.Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 61.Xiao S, Matsui K, Fine A, Zhu B, Marshak-Rothstein A, Widom RL, Ju ST. FasL promoter activation by IL-12 through SP1 and NFAT but not Egr-2 and Egr-3. Eur J Immunol. 1999;29:3456–3465. doi: 10.1002/(SICI)1521-4141(199911)29:11<3456::AID-IMMU3456>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 62.Yan GZ, Ziff EB. Nerve growth factor induces transcription of the p21 WAF1/CIP1 and cyclin D1 genes in PC12 cells by activating the Sp1 transcription factor. J Neurosci. 1997;17:6122–6132. doi: 10.1523/JNEUROSCI.17-16-06122.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y, Hwang CK, Junn E, Lee G, Mouradian MM. Zic2 and Sp3 repress Sp1 activation of the human D1A dopamine receptor gene. J Biol Chem. 2000;275:38863–38869. doi: 10.1074/jbc.M007906200. [DOI] [PubMed] [Google Scholar]
- 64.Zaman K, Ryu H, Hall D, O'Donovan K, Lin K, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuron cultures by iron chelators is associated with increased expression of glycolytic enzymes, p21WAF/CIP1, and erythropoietin and enhanced DNA binding of hypoxia inducible factor-1 and ATF-1/CREB. J Neurosci. 1999;19:9821–9830. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng M, Storz G. Redox sensing by prokaryotic transcription factors. Biochem Pharmacol. 2000;59:1–6. doi: 10.1016/s0006-2952(99)00289-0. [DOI] [PubMed] [Google Scholar]