

Abstract
After traumatic brain injury (TBI), substantial extracellular heme is released from hemoproteins during hemorrhage and cell injury. Heme oxygenase (HO) isozymes are thought to detoxify the pro-oxidant heme to the potent antioxidant, bilirubin. HO-1, the inducible isozyme, is expressed in glial populations after injury and may play a protective role. However, the role of HO-2, the predominant and constitutively expressed isozyme in the brain, remains unclear after TBI. We used a controlled cortical impact injury model to determine the extent and mechanism of damage between HO-2 knock-out (KO) (−/−) and wild-type (WT) (+/+) mice. The specific cellular and temporal expressions of HO-2 and HO-1 were characterized by immunocytochemistry and Western blots. HO-2 was immunolocalized in neurons both before and after TBI, whereas HO-1 was highly upregulated in glia only after TBI. HO activity determined by gas chromatography using brain sonicates from injured HO-2 KO mice was significantly less than that of HO-2 wild types, despite the induction of HO-1 expression after TBI. Cell loss was significantly greater in KO mice in areas including the cortex, the CA3 region of hippocampus, and the lateral dorsal thalamus. Furthermore, motor recovery after injury, as measured by the rotarod assay and an inclined beam-walking task, was compromised in the KO mice. Finally, brain tissue from injured HO-2 KO mice exhibited decreased ability to reduce oxidative stress, as measured with an Fe2+/ascorbic acid-mediated carbon monoxide generation assay for lipid peroxidation susceptibility. These findings demonstrate that HO-2 expression protects neurons against TBI by reducing lipid peroxidation via the catabolism of free heme.
Keywords: heme oxygenase-2, traumatic brain injury, lipid peroxidation, oxidative stress, controlled cortical impact, heme-oxygenase-1
Introduction
Heme is a ubiquitous molecule found in mammalian tissues, with a central role in the binding and delivering of molecular oxygen and in effecting oxidative reactions for cellular metabolism (Maines, 1988). Free heme, however, is not found in normal tissue; it is deposited only under pathological conditions. For example, in traumatic brain injury (TBI), heme is released from red blood cells during hemorrhage and from dying or injured cells, such as neurons, glia, and endothelia. In addition, intravascular free heme can access brain parenchyma through compromises in the blood–brain barrier.
The degradation of heme is catalyzed by heme oxygenase (HO) isozymes, producing equimolar quantities of carbon monoxide (CO), ferrous iron (Fe2+), and biliverdin, which is converted rapidly to bilirubin (Maines, 1997). HO-1 and HO-2 are the two principal isozymes of heme oxygenase. HO-1 is a heat shock protein induced under numerous conditions of cellular stress (Ewing and Maines, 1993; Fukuda et al., 1996; Geddes et al., 1996; Turner et al., 1998;Ferris et al., 1999). The constitutively expressed HO-2, in contrast, is the predominant isozyme found in the adult rodent brain and testes and is regulated only by adrenal glucocorticoids (McCoubrey and Maines, 1994; Maines et al., 1996). A third isozyme, HO-3, has been identified with high amino acid similarity to HO-2, but was found to be a poor heme catalyst (McCoubrey et al., 1997).
The precise role that each HO isozyme plays in TBI and other pathophysiological conditions still remains unclear (Platt and Nath, 1998). Increased HO activity may be protective because it metabolizes pro-oxidant heme into biliverdin and bilirubin. The latter has been shown to exhibit antioxidant properties at low concentrations (Dore and Snyder, 1999; Dennery, 2000). A protective effect has not been uniformly observed, however, and inhibition of HO activity has been found to attenuate cell injury in some models. This cell injury is speculated to be mediated through the generation of free Fe2+ during heme degradation, which may potentiate free radical-induced damage and through the production of bilirubin generate toxicity at high concentrations. Despite much interest in its putative neuromodulatory functions, CO still has an unclear role in neuroprotection (Maines, 1997; Baranano and Snyder, 2001).
We hypothesized that HO-2 affords neuroprotection against oxidative stress in TBI. A standard controlled cortical impact (CCI) model of TBI was used in mutant mice with a targeted deletion for HO-2. We first characterized the expression of both HO-1 and HO-2 isozymes by immunocytochemistry and measured HO activity in HO-2 wild-type and knock-out (KO) animals after TBI. We found that HO-2 and HO-1 are expressed in different populations of cells and that total HO activity is substantially reduced in HO-2 KO mice, despite induction of HO-1. We further demonstrated that HO-2 KO mice sustain increased cell loss, reduced motor recovery, and a greater susceptibility to lipid peroxidation as compared with HO-2 wild-type mice after TBI.
Materials and Methods
Animals. Adult male HO-2 KO and wild-type littermate mice from the founder stock originally described by Poss and Tonegawa were used for the study (Poss et al., 1995). HO-2 KO mice have documented impairments in the central signaling of nociception (Li and Clark, 2000) and acclimation of neurons to odorants (Zufall and Leinders-Zufall, 1997) and in the peripheral signaling in intestinal smooth muscle relaxation (Zakhary et al., 1997) and also ejaculatory reflexes (Burnett et al., 1998). HO-2 KO mice otherwise appear normal and have no other phenotypic differences from wild types, including life span, gross appearance, and activity level. All experiments were performed following an institutionally approved protocol in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All procedures described below were performed blinded with respect to animal genotype.
Surgical procedures and CCI model of injury. Mice (4–6 months of age) were anesthetized with an intraperitoneal injection of 0.02 ml of 2.5% (w/v) 2,2,2-tribromoethanol (Avertin) per gram of body weight. Each animal was then placed in a stereotaxic frame for surgery. After a midline incision, a circular craniectomy, 5 mm in diameter, was made over the left parietal cortex between bregma and lambda with the medial edge 0.5 mm lateral to the midline. Care was taken to keep the dura intact. The animal was positioned in a head holder within the injury device and then subjected to a moderate CCI injury, using a 3 mm convex impactor tip oriented perpendicular to the cortical surface (Smith et al., 1995; Fox et al., 1998). The injury parameters were set to 4 m/sec velocity, 1 mm depth, and 150 msec dwell time. After injury, the scalp was resutured over the craniectomy. Animals were maintained at normal body temperature (36.5–37.5°C) on a circulating water heating pad and injected subcutaneously with 1 ml of isotonic saline before and after the operation to prevent dehydration.
Immunocytochemistry. At 14 d after CCI injury, mice were deeply anesthetized and then transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 PBS. Each brain was removed and fixed in PFA at 4°C for 4 hr and then protected in 20% sucrose for 3 d. Brains were embedded and stored at −20°C. Coronal sections, 50 μm in thickness, were cut on a cryostat and collected in wells containing PBS. Every fourth section was reserved for cell counting and quantification of cortical lesion volume, whereas others were used to immunolocalize HO-2 and HO-1. Sections were first rinsed in 0.05m PBS two times for 5 min each and incubated in 1% H2O2 for 1 min to quench any endogenous peroxidase activity.
To localize HO-2, sections were incubated in the following solutions: 2% goat serum/0.2% Triton X-100/0.1% bovine serum albumin (GS/TX/BSA) for 5 min; 10% GS/TX/BSA for 20 min; rabbit polyclonal HO-2 antibody, 1:500 in 2% GS/TX/BSA, overnight at 4°C (StressGen, Victoria BC, Canada); PBS three times for 5 min each; and biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) in 2% GS/TX/BSA, 1:200 dilution for 1 hr. Sections were incubated in avidin–biotin–horseradish peroxidase complex (Vectastain ABC kit; Vector Laboratories), 1:100 dilution in PBS for 30 min. All sections were rinsed with PBS for three times for 5 min each and reacted with 0.05% 3,3-diaminobenzidine tetrahydrochloride (Invitrogen, Gaithersburg, MD) in 0.02% H2O2 for 5 min. HO-1 was localized using the above procedure with rabbit polyclonal HO-1 antibody, 1:10,000 dilution (StressGen).
Neurons were identified using a modified protocol for a monoclonal antibody to neuronal nuclei (NeuN; Chemicon International, Temecula, CA). A mouse on mouse (MOM) immunodetection kit (Vector Laboratories) was used to avoid nonspecific staining associated with the mouse-derived primary antibody. Sections were incubated in the following solutions: MOM mouse blocking reagent, 1 hr; PBS, three times for 5 min each; MOM diluent, 5 min; NeuN in MOM diluent, 1:1000 dilution, 30 min; PBS, three times for 5 min each; MOM biotinylated anti-mouse IgG reagent, 1:250 dilution, 10 min; PBS, three times for 5 min each; and Vectastain elite ABC reagent (Vector laboratories), 5 min.
Western blot analysis. At 24 hr after injury, mice were deeply anesthetized and transcardially perfused with chilled PBS, pH 7.4. The brains were rapidly removed and homogenized in Laemmli solubilizing buffer (2.5% SDS, 10% glycerol, 62.5 mmol/l Tris-HCl, pH 6.8, 5% 2-mercaptoethanol). After boiling for 10 min to inactivate proteases, equal amounts (55 μg) of protein per sample were separated on 12% SDS-polyacrylamide gels with 4.5% stacking gel. Protein was then electrotransferred onto a nitrocellulose membrane (0.2 mm pore size; Schleicher and Schuell, Keene, NH). Membranes were incubated overnight at 4°C in 0.1m PBS containing 5% nonfat dry milk, 1% BSA, and 0.1% Tween 20. They were rinsed briefly in PBS containing 1% BSA and 0.1% Tween 20 and then incubated for 1.5 hr with a 1:3325 dilution of rabbit polyclonal HO-1 antibody (StressGen). After washes, membranes were incubated with a 1:2500 dilution of anti-rabbit IgG-horseradish peroxidase antibody (Amersham Biosciences, Piscataway, NJ) for 1 hr. Finally, the membranes were washed three times, and the bound antibody was visualized with the ECL chemiluminescence system according to the manufacturer's protocol (Amersham Biosciences). The identical procedure was performed for HO-2 expression, but instead using a 1:1000 dilution of rabbit polyclonal HO-2 antibody (Stressgen).
Determination of total HO activity. At 24 hr after TBI, brains were removed and prepared for determination of total HO activity (KO −/−, n = 6; wild type, n = 6). Baseline levels were also obtained from uninjured control animals (KO −/−, n = 4; KO −/+, n = 4; wild type, n = 4). HO activity was determined through measurements of CO as described previously (Vreman and Stevenson, 1988;Vreman et al., 1988). Tissue (100 ± 2 mg) in 900 μl of iced buffer (0.1 m KPO4 buffer, pH 7.4) was sonicated at 50% power with an ultrasonic cell disruptor with a one-eighth inch microprobe (Model XL2000, Misonics Inc., Farmingdale, NY) in an ice bath. Sonicates were kept on ice and used immediately.
Tissue sonicates were incubated with equal (20 μl) volumes of NADPH (4.5 μm) and methemalbumin (MHA) (50 μm/11.2 μm) for 15 min at 37°C in 2 ml of CO-purged septum-sealed vials. MHA was prepared daily by dissolving heme (9.9 mg) in 2.5 ml 0.4 m sodium phosphate and 100 mg of BSA. After addition of water to 8.0 ml, pH was gradually titrated to 7.40 with 1 m HCl. The volume was then adjusted to 10 ml to yield a stock solution of 1.5 mm. A working solution of 150 μm MHA was prepared by diluting the stock MHA solution 1:10 in phosphate buffer.
The amount of CO generated in the vial headspace was determined by gas chromatography using a 60 × 0.53 cm (internal diameter) stainless-steel column packed with 5A molecular sieve, 60–80 mesh, at a temperature of 150°C and a reduction gas detector (RGA-2, Trace Analytical, Sparks, MD) operated at 270°C. HO activity was expressed as nanomoles of CO per hour per milligram of protein and picomoles of CO per hour per milligram of fresh weight.
Regional cell counting and lesion volume quantification. Cell counting (KO, n = 11; wild type, n= 9) was performed on images captured with a Nikon microscope at 20× magnification with a mounted CCD camera (SPOT-1; Diagnostic Instruments, Sterling Heights, MI). Three consecutive coronal sections, which were centered at the level of the hippocampal and thalamic structures of interest, were chosen for analysis. The number of neurons, exhibiting distinctly stained nuclei, was determined within selected regions shown previously to be vulnerable after TBI: the peritraumatic cortex (cortical area directly lateral to the injury site), hippocampal CA3, lateral dorsal thalamic nucleus, and ventroposterior medial or lateral thalamic nuclei (Igarashi et al., 2001; Sato et al., 2001). Neuronal density was expressed as the ratio of cells counted in the ipsilateral to contralateral hemisphere, with contralateral sites serving as internal controls, and then averaged for each animal.
Lesion cavity volumes (KO, n = 12; wild type,n = 10) were measured under 1× magnification from three sections in the region of maximal cortical damage. In each section, lesion area (LA) was determined as follows: LA = area of contralateral cortex − area of ipsilateral cortex. The lesion volume was then defined as follows: lesion volume = (LA1 + LA2 + LA3) × 200 μm, where LA1, LA2, and LA3 represent lesion areas of each individual section and 200 μm reflects the sum of the thickness of the section (50 μm) and the distance between the sections (150 μm).
Motor outcome tests. To assess behavioral deficits after TBI, a standard rotarod test was performed at 1, 2, 3, 7, and 14 d after CCI (Hamm et al., 1994). All mice were pretrained on the rotarod for 3 d before the day of trauma. The rotarod was set to start at 4 m/sec and to accelerate to 40 m/sec over 300 sec. Performance score was measured as latency, i.e., the time successfully spent on the rotating rod without falling off.
An inclined beam walking task was also used. Mice were placed at the bottom of 1-m-long wooden beam (6 mm wide) set at 10° above horizontal and were assessed for contralateral hindlimb foot faults while walking the length of the beam. Only mice that could perform this task with less than three foot faults per trial during the 3 d preinjury testing period were included for analysis. The same animals were used in both rotarod and inclined beam motor tasks (KO,n = 11; wild type, n = 9).
Determination of lipid peroxidation activity. Twenty microliters of tissue sonicates (injured KO, n = 6; injured wild type, n = 6; sham-injured wild type,n = 4), prepared as described above, were incubated in the dark for 30 min at 37°C in septum-sealed vials containing ascorbate (100 μm) and Fe2+ (6 μm). Butylated hydroxytoluene (100 μm) was added for the blank reaction. CO produced into the vial headspace was also quantified by gas chromatography as described previously (Vreman et al., 1998). The amount of CO produced in the vial headspace is inversely related to the level of antioxidant protection in the tissue.
Statistical analyses. Data are expressed as the mean ± SEM. Behavioral scores were evaluated using ANOVA followed by Bonferroni exact t test for intergroup comparisons. All other comparisons were performed using an unpaired two-tailedt test (StatView 5.0 software, SAS, Cary, NC). Significance was defined at p < 0.05.
Results
HO-1 and HO-2 exhibit different anatomic and temporal expression after injury
HO-1 and HO-2 immunoexpression were characterized in injured and sham-operated noninjured wild-type mice. HO-2 staining was primarily immunolocalized in neurons throughout the brain (Fig.1). The level of HO-2 expression did not appear to be different between injured and noninjured wild-type mice. As expected, no HO-2 neuronal expression was observed in HO-2 KO mice.
Fig. 1.

Immunolocalization of HO-2 and HO-1 in the uninjured and injured brain. HO-2 is constitutively expressed in cortical neurons in the uninjured brain (A) and after TBI (B) in the HO-2 wild-type mouse. HO-2 is localized primarily to neurons and is unchanged after injury. In contrast, HO-1 expression is virtually undetectable in the uninjured cortex (C) and highly induced in reactive astrocytes and microglia/macrophages after TBI (D). Scale bar, 50 μm.
HO-1 expression in the noninjured wild-type brain was much less than HO-2. HO-1 was expressed in astrocytes at low levels in the hippocampus and was virtually undetectable elsewhere. In contrast, by 14 d after injury, HO-1 was strongly upregulated in microglia/macrophages and reactive astrocytes in areas adjacent to the site of cortical impact (Fig. 2). Low levels of expression in microglia were discernible in the contralateral hemisphere. Activated astrocytes expressing HO-1 were hypertrophied with elaborate processes.
Fig. 2.

Western blots from brain sonicates for HO-1 (bottom) and HO-2 (top) after TBI. Both ipsilateral and contralateral hemispheres are included. HO-1 is highly induced in the ipsilateral hemisphere, but only modestly induced in the contralateral hemisphere. In addition, HO-1 expression appears similar between hemispheres for both HO-2 wild-type and HO-2 KO mice. In contrast, strong HO-2 expression is found in both hemispheres of the HO-2 wild-type mice.
Western blots further confirmed regional HO-2 protein expression (Fig.2). HO-2 expression in wild-type mice was similar in both the ipsilateral and contralateral hemispheres. HO-2 KO mice had no HO-2 expression. HO-1 was strongly upregulated in the injured hemisphere in wild-type mice and was only slightly upregulated on the contralateral hemisphere. The pattern of HO-1 expression was not different between HO-2 KO mice and wild-type mice. These findings demonstrate that HO-2 expression is unchanged after injury, whereas HO-1 is markedly induced. Furthermore, HO-2 expression is primarily restricted to neurons, whereas HO-1 is mostly confined to reactive glia/macrophages.
Total HO activity is reduced in HO-2 KO mice before and after injury
Because HO-1 is primarily induced and HO-3 is a poor heme catalyst, the bulk of HO activity in the uninjured brain presumably comes from the HO-2 isozyme. We found that HO-2 genotype correlated well with total HO enzymatic activity as measured by CO production with gas chromatography (Fig. 3). Wild-type mice showed maximal HO activity, whereas HO-2 heterozygous and HO-2 homozygous KO mice, respectively, exhibited significantly lower activity in the uninjured brain (t test; p< 0.05).
Fig. 3.

Total heme oxygenase (HO) activity by genotype. In uninjured mice (white bars), HO-2 wild-type (+/+) mice demonstrated the highest levels of HO activity. HO-2 heterozygous (+/−) mice showed relatively less activity, and HO-2 homozygous knock-out (−/−) mice showed the least activity (*p ≤ 0.05; ANOVA; Bonferroni t test). Activity in HO-2 KO mice is attributed primarily to the HO-1 isozyme. Total activity is increased at 24 hr after experimental injury in both HO-2 wild-type and HO-2 KO mice (black bars), suggesting induction of HO-1. Total activity, however, is still reduced significantly in HO-2 KO mice compared with HO-2 wild-type mice. Values are shown as the mean ± SEM (*p ≤ 0.05; unpaired t test).
Twenty-four hours after TBI, total HO activity was significantly reduced in HO-2 KO compared with wild-type mice (∼67%; ttest; p < 0.01). A significant increase in HO activity in the injured brain relative to the noninjured control was observed and, in the context of the Western blots and immunostaining, was interpreted to represent the contribution of HO-1 induction after injury. HO activity was similarly increased in both wild types and KOs compared with noninjured mice (0.35 nmol CO per hour per milligram of protein for wild-type mice vs 0.43 nmol CO per hour per milligram of protein for KOs). Thus, it is unlikely that any additional compensatory upregulation of HO-1 occurred in HO-2 KOs.
Cell loss is greater in HO-2 KO mice
Cortical lesion volume and neuronal density studies were used to histologically quantify injury severity. For both measures, the contralateral hemisphere was used as an internal control. At 14 d after injury, traumatized brains showed a well demarcated cavitation in the cortex (Fig. 4). Occasionally, the lesions involved adjacent hippocampal structures. HO-2 KO mice demonstrated a significantly larger cortical lesion volume than wild-type mice (Fig. 5A) (0.59 ± 0.07 mm3 in HO-2 KO vs 0.35 ± 0.08 mm3 in HO-2 wild type;t test; p < 0.05).
Fig. 4.
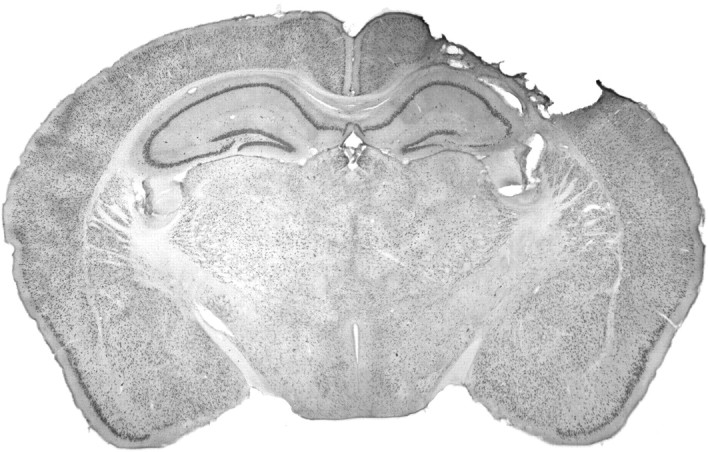
Representative NeuN immunostained coronal section after TBI. At 14 d after injury, a large cortical cavitation at the impact site extends down to the level of the external capsule. Cell loss is discernible in the parenchyma immediately surrounding the cavity, ipsilateral hippocampal CA1 and CA3, and ipsilateral thalamic structures.
Fig. 5.
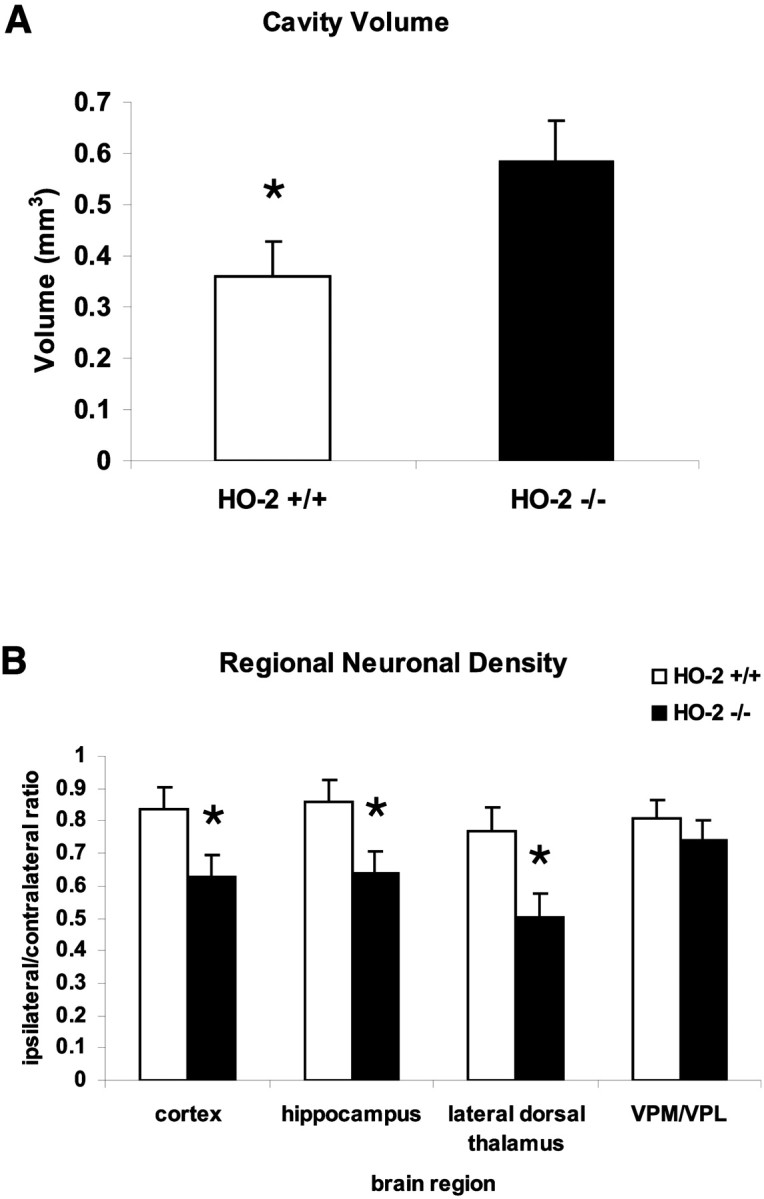
Cell loss after traumatic brain injury.A, Volumetric measurements of cortical cavity on histologic sections. Values are shown as the mean ± SEM (*p ≤ 0.05; unpaired t test).B, Regional neuronal density by NeuN cell counting in adjacent cortex, hippocampal CA3, lateral dorsal thalamus, and medial/lateral ventroposterior nuclei of thalamus. Values are shown as the mean ± SEM (*p ≤ 0.05; unpairedt test).
Neuronal loss in the remaining areas of the brain parenchyma was also characterized (Fig. 5B). Overt neuronal loss was observed in the ipsilateral peritraumatic cortex, hippocampus, and thalamus. There was a significant reduction in neurons in the cortex adjacent to the cavity in HO-2 KO compared with HO-2 wild-type mice (0.63 ± 0.08 vs 0.84 ± 0.07; t test; p < 0.05). The CA3 region of the hippocampus showed a 26% reduction (0.64 ± 0.07 vs 0.86 ± 0.07; t test; p < 0.05). Similarly, the lateral dorsal thalamus showed a loss of 34% over HO-2 wild-type mice (0.50 ± 0.07 vs 0.77 ± 0.08;t test; p < 0.05). Significant differences in neuronal density in the medial and lateral ventroposterior nuclei of the thalamus were not observed between HO-2 wild-type and HO-2 KO mice. Neuronal loss in this region is attributed to delayed corticothalamic degeneration originating from the parietal cortex. The lack of a statistically significant difference here might be attributable to the relatively short post-injury period of 14 d.
HO-2 KO mice have impaired motor recovery
To assess whether neuronal loss correlated with motor behavior, the standard rotarod assay was performed from days 1 to 14 after injury. Latency, the measure of how long the animal can stay on the rotating rod before falling off, reflects the animal's gross motor capacity. To normalize for inter-animal differences before injury, all post-trauma rotarod latencies were plotted as a percentage of preinjury baselines. During the 3 d preinjury period, no significant difference was found between HO-2 wild-type and knock-out mice on the rotarod (latencies: KO = 173 ± 13 sec; WT = 179 ± 16 sec; p = 0.77), suggesting that there was no baseline gross motor deficit in knock-out animals. After TBI, rotarod performance was significantly reduced in all mice. Over time, however, HO-2 KO mice demonstrated impaired ability to remain on the rotarod when compared with HO-2 wild-type mice (ANOVA; p < 0.001) (Fig. 6A). Single time-point comparisons revealed significantly diminished performance at 7, 10, and 14 d after injury in HO-2 KO mice versus HO-2 wild-type mice (Bonferroni t test; p < 0.05).
Fig. 6.

Motor recovery after experimental injury.A, The standard rotarod assay performance was expressed as the percentage of pretrauma latency. A repeated-measures ANOVA shows that overall rotarod performance is significantly worse in HO-2 KO mice compared with HO-2 wild-type mice. Post hoc analysis reveals that significant differences occur on days 7, 10, and 14. Values are shown as the mean ± SEM (*p ≤ 0.05; Bonferroni t test). B, Motor coordination was tested on an inclined beam walking task. Performance was expressed as the ratio of foot faults of the contralateral hindlimb to the number of total steps, subtracted from 1. Motor recovery is significantly worse in HO-2 KO compared with HO-2 wild-type mice (ANOVA), specifically on day 7 after injury. Values are shown as the mean ± SEM (*p ≤ 0.05; Bonferronit test).
The inclined beam task served as a supplementary behavioral assay. Beam walking depends highly on the integrity of sensorimotor cortical processing, and as expected, all animals demonstrated a high percentage of contralateral hindlimb foot faults in the first few days after brain injury. HO-2 KO mice, however, showed a slower recovery from beam walking deficits than HO-2 +/+ mice (ANOVA; p < 0.001) (Fig. 6B). On post hoc analysis, a significant difference was found at 7 d after injury (Bonferronit test; p < 0.05). The shorter duration of impairments detected in this test compared with the rotarod might be related to differences in the aspects of behavior tested. The rotarod is involved in gross motor skills and learning, whereas the beam walking task is involved in more subtle motor skills such as fine coordination (Fox et al., 1998, 2001). Furthermore, it is notable that animals continue to improve with rotarod testing, even beyond preinjury levels, whereas many animals on the beam walking task did not achieve preinjury levels by 14 d after injury.
HO-2 KO mice are more susceptible to lipid peroxidation after injury
An assay to determine susceptibility to lipid peroxidation was used to understand the mechanisms of neuroprotection offered by HO-2 activity. When Fe2+ and ascorbate are added to brain sonicates in vitro, CO is produced. The production of CO has been shown to closely parallel other markers of lipid peroxidation, such as thiobarbituric acid reactive substances, conjugated dienes, and lipid hydroperoxides (Vreman et al., 1998). At 24 hr after TBI, lipid peroxidation was found to be ∼56% higher in HO-2 KO than in wild-type mice (Fig. 7) (unpaired t test; p < 0.05). Similarly, a greater susceptibility to lipid peroxidation was found in injured HO-2 KO mice compared with sham uninjured HO-2 wild-type mice (unpairedt test; p < 0.05). These results suggest that both the loss of HO-2 activity and the pathophysiology of traumatic brain injury contribute individually to the increased susceptibility to lipid peroxidation.
Fig. 7.

Susceptibility to lipid peroxidation. An Fe2+- and ascorbate-containing in vitro lipid peroxidation assay measuring the generation of carbon monoxide (CO) was used to determine susceptibility of brain tissue to lipid peroxidation at 24 hr after experimental injury. Lipid peroxidation is higher in HO-2 KO mice compared with both HO-2 wild-type mice and sham-injured HO-2 KO mice. Values are shown as the mean ± SEM (*p ≤ 0.05; unpairedt test).
Discussion
HO isozymes have been implicated in the regulation of oxidative stress in various tissues throughout the body, including the lung, testes, liver, spleen, and CNS. Oxidative stress is a fundamental contributor to secondary pathophysiology in the progression of injury after trauma (Povlishock and Kontos, 1992; Shohami et al., 1997;McIntosh et al., 1998). We show that after TBI, HO-1 and HO-2 isozymes have markedly different and specific cellular and temporal expression. In the uninjured brain, HO-2 is the predominant contributor to total HO activity and was found mostly in neuronal populations. After injury, HO-1, induced in glia and macrophages, contributes to total HO activity. Nevertheless, HO-2 KO mice still demonstrate an overall significant reduction of activity. The later observations emphasize the dominant contributions of HO-2 to HO activity. We further found that HO-2 protects against cellular injury, especially in areas of the brain that are selectively vulnerable to oxidative stress and associated with greater impairments in behavioral recovery. Finally, we show that there is a specific protection from lipid peroxidation that is conferred by HO-2 in the setting of TBI.
TBI is associated with a host of pathophysiologic conditions, which include mechanical damage, edema, ischemia, and hemorrhage. Each of these processes contributes to the cumulative, substantial release of free heme from denatured hemoproteins. Potential cellular sources of free heme include all cellular components of brain tissue, that is, injured and dying neurons, glia, endothelial cells, and red blood cells that leak either through the damaged blood–brain barrier or during hemorrhage. Heme and the products of heme catabolism have all been shown to be distinct bioactive molecules and therefore the subject of considerable debate (Platt and Nath, 1998).
Although both HO-1 and HO-2 catalyze the same biochemical reaction, the fact that they demonstrate such different patterns of cellular expression suggests that the two isozymes have individual roles in the setting of brain injury. HO-1 induction, for example, has been shown to selectively protect cultured cortical astrocytes, but not neurons, from oxidative stress resulting from exposure to hemoglobin and hydrogen peroxide (Dwyer et al., 1995; Regan et al., 2000). The beneficial effect of HO-1 is in general agreement with many previous observations in neural and non-neural systems.
HO-2, on the other hand, is constitutively expressed in neurons throughout the brain. HO-2 expression has been shown to be protective against apoptotic cell death in cortical, hippocampal, and cerebellar granule cultures and an in vivo model of ischemic injury (Dore and Snyder, 1999; Dore et al., 1999b, 2000). The neuroprotective effects of HO-2 seem to be attributed increasingly to the generation of bilirubin. Dore et al. (1999a) have reported that these effects occur at the physiologic low nanomolar endogenous levels that occur in the brain, several thousand-fold lower than neurotoxic levels of bilirubin that occur during kernicterus (Stocker et al., 1987). Indeed, bilirubin has been suggested to be one of the most potent endogenous antioxidants in mammalian tissue, accounting for the majority of the antioxidant activity of human serum (Gopinathan et al., 1994). Furthermore, others have found a link to neurodegenerative conditions such as familial Alzheimer's disease. Several single point mutations in amyloid precursor proteins, which bind HO, have been associated with a significant reduction in HO activity, resulting in decreased bilirubin staining and increased neurotoxicity (Takahashi et al., 2000).
One attractive hypothesis regarding the strong antioxidant abilities of bilirubin involves the rapid regeneration of bilirubin via redox cycling (Baranano et al., 2002). Each molecule of bilirubin that acts as an antioxidant is thereby itself oxidized to biliverdin. The high level of biliverdin reductase immediately reduces the biliverdin back to bilirubin. This is supported by preliminary work that demonstrates an increased vulnerability to oxidative stress in cell lines designed to express less biliverdin reductase (Baranano et al., 2002). This would be an ideal endogenous system to neutralize the numerous reactive oxygen species generated after traumatic brain injury.
Our results demonstrate that this observed protection against oxidative stress, offered by HO-2, specifically involves a reduction in lipid peroxidation. The high susceptibility of the brain to oxidative damage after traumatic brain injury is attributed to the abundance of peroxidizable fatty acids and relative scarcity of endogenous antioxidant defense systems (Floyd, 1999). Furthermore, we found that regional protection occurs in areas of the brain that are selectively vulnerable to oxidative stress, such as the hippocampus and thalamus (Lowenstein et al., 1992). The hippocampus, in particular, shows prominent expression of HO-2 (Verma et al., 1993; Ewing and Maines, 1997). Increased cell loss in the thalamus is consistent with the delayed neuronal injury that occurs after cortical injury (Conti et al., 1998; Sato et al., 2001). It is quite possible that delayed thalamic cell loss reflects the greater extent of cortical injury via target deprivation and is exacerbated by oxidative processes generated by dying cells. Nonetheless, lipid peroxidation is increased throughout the brain after TBI (Hsiang et al., 1997; Pratico et al., 2002).
Several other groups have argued that HO is harmful, however, primarily because of the toxicity of generated free iron. The administration of HO inhibitors such as tin-protoporphyrin (SnPP), for example, has been shown to reduce the formation of brain edema and to reduce injury from ischemic, hemorrhagic, and even traumatic injury to CA3 hippocampal slices (Kadoya et al., 1995; Panizzon et al., 1996; Huang et al., 2002). It should be noted, however, that SnPP and other metalloporphyrins are nonspecific inhibitors of heme-binding proteins. They have also been reported to be direct inhibitors of lipid peroxidation (Imai et al., 1990; Wong et al., 2000). Proteins reported to interact with metalloporphyrins include cytochrome P450s, nitric oxide synthase, and others.
The generation of free iron has been raised as an explanation for HO-induced injury. Iron is a strong generator of reactive oxygen species. Iron reacts with hydrogen peroxide to form the hydroxyl radical, or it may decompose membrane lipid peroxides to yield alkoxy and peroxy radicals (Rouault, 2001). All three species are capable of initiating lipid peroxidation chain reactions. However, there are several arguments regarding why generation of iron is not as harmful as free heme. First, unlike iron salts, free heme is lipophilic, which enhances lipid peroxidation. Second, in the adult brain, excess iron can be sequestered rapidly by upregulated iron storage proteins such as ferritin, transferrin, and ceruloplasmin (Rouault, 2001; Patel et al., 2002; Regan et al., 2002). Third, recent evidence suggests a role for HO-1 in facilitating the transport of iron out of cells. Transfection of HO-1 into HO-1-deficient mice protects against stress-induced apoptotic cell death (Ferris et al., 1999).
These arguments raise an important point regarding the complex regulation of iron and heme-derived products in the setting of injury. Thus, a few caveats can be raised in interpreting our results. Because all of our analyses were performed within 2 weeks of the injury, the long-term effects of HO-2 activity are unclear. Sequelae from even mild TBI can progress for months in experimental rodents and for decades in humans (Millis et al., 2001). Also, the adequate sequestration of iron appears to be a crucial part of facilitating HO-2-mediated neuroprotection. Further experiments will need to clarify whether protection still exists in very young or aged animals, in which iron regulation is worse than in adults (Rouault, 2001).
Our results are consistent with a growing number of reports suggesting that HO-2 is neuroprotective. The present study shows that constitutive HO-2 expression in neurons reduces cell loss and morphological damage that takes place after injury. This is also the first demonstration that the protection offered by HO-2 is associated with improved behavioral recovery and is related to a specific reduction of lipid peroxidation after TBI. Taken together, these data establish that HO-2 represents an important endogenous antioxidant generating system and thus plays a critical role in limiting secondary neuronal injury and death after TBI.
Footnotes
This research was supported by the University of California Los Angeles Brain Trauma Initiative and National Institutes of Health Grant NS 41256. We thank D. Clark for providing additional HO-2 KO and wild-type mice and T. Yamauchi, N. Maida, T. Tjoa, J. Raber, A. Abate, and P. Dennery for technical suggestions.
Correspondence should be addressed to Dr. Linda J. Noble-Haeusslein, Department of Neurological Surgery, University of California, San Francisco, 521 Parnassus Avenue, Room C224, San Francisco, CA 94143-0520. E-mail: noblelj@itsa.ucsf.edu.
References
- 1.Baranano DE, Snyder SH. Neural roles for heme oxygenase: contrasts to nitric oxide synthase. Proc Natl Acad Sci USA. 2001;98:10996–11002. doi: 10.1073/pnas.191351298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA. 2002;99:16093–16098. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burnett AL, Johns DG, Kriegsfeld LJ, Klein SL, Calvin DC, Demas GE, Schramm LP, Tonegawa S, Nelson RJ, Snyder SH, Poss KD. Ejaculatory abnormalities in mice with targeted disruption of the gene for heme oxygenase-2. Nat Med. 1998;4:84–87. doi: 10.1038/nm0198-084. [DOI] [PubMed] [Google Scholar]
- 4.Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennery PA. Regulation and role of heme oxygenase in oxidative injury. Curr Top Cell Regul. 2000;36:181–199. doi: 10.1016/s0070-2137(01)80008-x. [DOI] [PubMed] [Google Scholar]
- 6.Dore S, Snyder SH. Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann NY Acad Sci. 1999;890:167–172. doi: 10.1111/j.1749-6632.1999.tb07991.x. [DOI] [PubMed] [Google Scholar]
- 7.Dore S, Takahashi M, Ferris CD, Zakhary R, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci USA. 1999a;96:2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dore S, Sampei K, Goto S, Alkayed NJ, Guastella D, Blackshaw S, Gallagher M, Traystman RJ, Hurn PD, Koehler RC, Snyder SH. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol Med. 1999b;5:656–663. [PMC free article] [PubMed] [Google Scholar]
- 9.Dore S, Goto S, Sampei K, Blackshaw S, Hester LD, Ingi T, Sawa A, Traystman RJ, Koehler RC, Snyder SH. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience. 2000;99:587–592. doi: 10.1016/s0306-4522(00)00216-5. [DOI] [PubMed] [Google Scholar]
- 10.Dwyer BE, Nishimura RN, Lu SY. Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Brain Res Mol Brain Res. 1995;30:37–47. doi: 10.1016/0169-328x(94)00273-h. [DOI] [PubMed] [Google Scholar]
- 11.Ewing JF, Maines MD. Glutathione depletion induces heme oxygenase-1 (HSP32) mRNA and protein in rat brain. J Neurochem. 1993;60:1512–1519. doi: 10.1111/j.1471-4159.1993.tb03315.x. [DOI] [PubMed] [Google Scholar]
- 12.Ewing JF, Maines MD. Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res Brain Res Protoc. 1997;1:165–174. doi: 10.1016/s1385-299x(96)00027-x. [DOI] [PubMed] [Google Scholar]
- 13.Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, Tysoe SA, Wolosker H, Baranano DE, Dore S, Poss KD, Snyder SH. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol. 1999;1:152–157. doi: 10.1038/11072. [DOI] [PubMed] [Google Scholar]
- 14.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 15.Fox GB, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- 16.Fox GB, Curzon P, Decker MW. The behavioral assessment of sensorimotor processes in the mouse: acoustic startle, locomotor activity, rotarod, and beam walking. In: Buccafusco JJ, editor. Methods of behavior analysis in neuroscience. CRC; New York: 2001. pp. 28–48. [PubMed] [Google Scholar]
- 17.Fukuda K, Richmon JD, Sato M, Sharp FR, Panter SS, Noble LJ. Induction of heme oxygenase-1 (HO-1) in glia after traumatic brain injury. Brain Res. 1996;736:68–75. doi: 10.1016/0006-8993(96)00680-4. [DOI] [PubMed] [Google Scholar]
- 18.Geddes JW, Pettigrew LC, Holtz ML, Craddock SD, Maines MD. Permanent focal and transient global cerebral ischemia increase glial and neuronal expression of heme oxygenase-1, but not heme oxygenase-2, protein in rat brain. Neurosci Lett. 1996;210:205–208. doi: 10.1016/0304-3940(96)12703-8. [DOI] [PubMed] [Google Scholar]
- 19.Gopinathan V, Miller NJ, Milner AD, Rice-Evans CA. Bilirubin and ascorbate antioxidant activity in neonatal plasma. FEBS Lett. 1994;349:197–200. doi: 10.1016/0014-5793(94)00666-0. [DOI] [PubMed] [Google Scholar]
- 20.Hamm RJ, Pike BR, O'Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- 21.Hsiang JN, Wang JY, Ip SM, Ng HK, Stadlin A, Yu AL, Poon WS. The time course and regional variations of lipid peroxidation after diffuse brain injury in rats. Acta Neurochir (Wien) 1997;139:464–468. doi: 10.1007/BF01808884. [DOI] [PubMed] [Google Scholar]
- 22.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 23.Igarashi T, Huang TT, Noble LJ. Regional vulnerability after traumatic brain injury: gender differences in mice that overexpress human copper, zinc superoxide dismutase. Exp Neurol. 2001;172:332–341. doi: 10.1006/exnr.2001.7820. [DOI] [PubMed] [Google Scholar]
- 24.Imai K, Aimoto T, Sato M, Kimura R. Antioxidative effect of several porphyrins on lipid peroxidation in rat liver homogenates. Chem Pharm Bull (Tokyo) 1990;38:258–260. doi: 10.1248/cpb.38.258. [DOI] [PubMed] [Google Scholar]
- 25.Kadoya C, Domino EF, Yang GY, Stern JD, Betz AL. Preischemic but not postischemic zinc protoporphyrin treatment reduces infarct size and edema accumulation after temporary focal cerebral ischemia in rats. Stroke. 1995;26:1035–1038. doi: 10.1161/01.str.26.6.1035. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Clark JD. Heme oxygenase type 2 plays a role in formalin-induced nociception. Pain. 2000;86:75–80. doi: 10.1016/s0304-3959(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 27.Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 29.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 30.Maines MD, Eke BC, Zhao X. Corticosterone promotes increased heme oxygenase-2 protein and transcript expression in the newborn rat brain. Brain Res. 1996;722:83–94. doi: 10.1016/0006-8993(96)00184-9. [DOI] [PubMed] [Google Scholar]
- 31.McCoubrey WK, Jr, Maines MD. The structure, organization and differential expression of the gene encoding rat heme oxygenase-2. Gene. 1994;139:155–161. doi: 10.1016/0378-1119(94)90749-8. [DOI] [PubMed] [Google Scholar]
- 32.McCoubrey WK, Jr, Huang TJ, Maines MD. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem. 1997;247:725–732. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- 33.McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ. The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Appl Neurobiol. 1998;24:251–267. doi: 10.1046/j.1365-2990.1998.00121.x. [DOI] [PubMed] [Google Scholar]
- 34.Millis SR, Rosenthal M, Novack TA, Sherer M, Nick TG, Kreutzer JS, High WM, Jr, Ricker JH. Long-term neuropsychological outcome after traumatic brain injury. J Head Trauma Rehabil. 2001;16:343–355. doi: 10.1097/00001199-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 35.Panizzon KL, Dwyer BE, Nishimura RN, Wallis RA. Neuroprotection against CA1 injury with metalloporphyrins. NeuroReport. 1996;7:662–666. doi: 10.1097/00001756-199601310-00067. [DOI] [PubMed] [Google Scholar]
- 36.Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien JP, David S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci. 2002;22:6578–6586. doi: 10.1523/JNEUROSCI.22-15-06578.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Platt JL, Nath KA. Heme oxygenase: protective gene or Trojan horse. Nat Med. 1998;4:1364–1365. doi: 10.1038/3947. [DOI] [PubMed] [Google Scholar]
- 38.Poss KD, Thomas MJ, Ebralidze AK, O'Dell TJ, Tonegawa S. Hippocampal long-term potentiation is normal in heme oxygenase-2 mutant mice. Neuron. 1995;15:867–873. doi: 10.1016/0896-6273(95)90177-9. [DOI] [PubMed] [Google Scholar]
- 39.Povlishock JT, Kontos HA. The role of oxygen radicals in the pathobiology of traumatic brain injury. Hum Cell. 1992;5:345–353. [PubMed] [Google Scholar]
- 40.Pratico D, Reiss P, Tang LX, Sung S, Rokach J, McIntosh TK. Local and systemic increase in lipid peroxidation after moderate experimental traumatic brain injury. J Neurochem. 2002;80:894–898. doi: 10.1046/j.0022-3042.2002.00777.x. [DOI] [PubMed] [Google Scholar]
- 41.Regan R, Kumar N, Gao F, Guo Y. Ferritin induction protects cortical astrocytes from heme-mediated oxidative injury. Neuroscience. 2002;113:985. doi: 10.1016/s0306-4522(02)00243-9. [DOI] [PubMed] [Google Scholar]
- 42.Regan RF, Guo Y, Kumar N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/s0304-3940(00)00817-x. [DOI] [PubMed] [Google Scholar]
- 43.Rouault TA. Systemic iron metabolism: a review and implications for brain iron metabolism. Pediatr Neurol. 2001;25:130–137. doi: 10.1016/s0887-8994(01)00260-0. [DOI] [PubMed] [Google Scholar]
- 44.Sato M, Chang E, Igarashi T, Noble LJ. Neuronal injury and loss after traumatic brain injury: time course and regional variability. Brain Res. 2001;917:45–54. doi: 10.1016/s0006-8993(01)02905-5. [DOI] [PubMed] [Google Scholar]
- 45.Shohami E, Beit-Yannai E, Horowitz M, Kohen R. Oxidative stress in closed-head injury: brain antioxidant capacity as an indicator of functional outcome. J Cereb Blood Flow Metab. 1997;17:1007–1019. doi: 10.1097/00004647-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 47.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi M, Dore S, Ferris CD, Tomita T, Sawa A, Wolosker H, Borchelt DR, Iwatsubo T, Kim SH, Thinakaran G, Sisodia SS, Snyder SH. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer's disease. Neuron. 2000;28:461–473. doi: 10.1016/s0896-6273(00)00125-2. [DOI] [PubMed] [Google Scholar]
- 49.Turner CP, Bergeron M, Matz P, Zegna A, Noble LJ, Panter SS, Sharp FR. Heme oxygenase-1 is induced in glia throughout brain by subarachnoid hemoglobin. J Cereb Blood Flow Metab. 1998;18:257–273. doi: 10.1097/00004647-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259:381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 51.Vreman HJ, Stevenson DK. Heme oxygenase activity as measured by carbon monoxide production. Anal Biochem. 1988;168:31–38. doi: 10.1016/0003-2697(88)90006-1. [DOI] [PubMed] [Google Scholar]
- 52.Vreman HJ, Stevenson DK, Henton D, Rosenthal P. Correlation of carbon monoxide and bilirubin production by tissue homogenates. J Chromatogr. 1988;427:315–319. doi: 10.1016/0378-4347(88)80134-8. [DOI] [PubMed] [Google Scholar]
- 53.Vreman HJ, Wong RJ, Sanesi CA, Dennery PA, Stevenson DK. Simultaneous production of carbon monoxide and thiobarbituric acid reactive substances in rat tissue preparations by an iron-ascorbate system. Can J Physiol Pharmacol. 1998;76:1057–1065. doi: 10.1139/cjpp-76-12-1057. [DOI] [PubMed] [Google Scholar]
- 54.Wong RJ, Vreman HJ, Stevenson DK. Effects of (metallo)porphyrin inhibitors of heme oxygenase on lipid peroxidation. Acta Haematol. 2000;103:4309. [Google Scholar]
- 55.Zakhary R, Poss KD, Jaffrey SR, Ferris CD, Tonegawa S, Snyder SH. Targeted gene deletion of heme oxygenase 2 reveals neural role for carbon monoxide. Proc Natl Acad Sci USA. 1997;94:14848–14853. doi: 10.1073/pnas.94.26.14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zufall F, Leinders-Zufall T. Identification of a long-lasting form of odor adaptation that depends on the carbon monoxide/cGMP second-messenger system. J Neurosci. 1997;17:2703–2712. doi: 10.1523/JNEUROSCI.17-08-02703.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]