

Abstract
Active immunization against the β-amyloid peptide (Αβ) with vaccines or passive immunization with systemic monoclonal anti-Aβ antibodies reduces amyloid deposition and improves cognition in APP transgenic mice. In this report, intracranial administration of anti-Αβ antibodies into frontal cortex and hippocampus of Tg2576 transgenic APP mice is described. The antibody injection resulted initially in a broad distribution of staining for the antibody, which diminished over 7 d. Although no loss of immunostaining for deposited Aβ was apparent at 4 hr, a dramatic reduction in the Αβ load was discernible at 24 hr and was maintained at 3 and 7 d. A reduction in the thioflavine-S-positive compact plaque load was delayed until 3 d, at which time microglial activation also became apparent. At 1 week after the injection, microglial activation returned to control levels, whereas Αβ and thioflavine-S staining remained reduced. The results from this study suggest a two-phase mechanism of anti-Αβ antibody action. The first phase occurs between 4 and 24 hr, clears primarily diffuse Αβ deposits, and is not associated with observable microglial activation. The second phase occurs between 1 and 3 d, is responsible for clearance of compact amyloid deposits, and is associated with microglial activation. The results are discussed in the context of other studies identifying coincident microglial activation and amyloid removal in APP transgenic animals.
Keywords: Alzheimer's disease, Αβ, antibody, immunization, intracranial, phagocytosis
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by progressive cognitive deficits. There are several pathological characteristics to the disease process, including congophilic amyloid plaques that contain the β-amyloid peptide (Αβ) and intracellular inclusions of neurofibrillary tangles that consist of hyperphosphorylated tau protein. Another characteristic of AD is the initiation and proliferation of a brain-specific inflammatory response that consists of activated microglia and astrocytes. Amyloid deposition is thought to be the key step in the pathogenesis of AD (Selkoe, 1991; Hardy and Selkoe, 2002); this is why development of potential therapies focuses on clearance of amyloid.
Vaccination with Αβ1–42 was first described by Schenk et al. (1999). Their report demonstrated that active immunization with Αβ1–42 in the PDAPP transgenic mouse reduced levels of Αβ deposits dramatically. This immunization protected APP+PS1 transgenic mice (Morgan et al., 2000) and TgCRND8 transgenic mice (Janus et al., 2000) from memory deficits. More recent studies showed that treatment with a passive immunization regimen that consisted of anti-Αβ antibodies resulted in a dramatic reduction in Αβ (Bard et al., 2000; DeMattos et al., 2001) and reversal of memory deficits (Dodart et al., 2002; Kotilinek et al., 2002) in the PDAPP mouse.
In this experiment, we show that intracranially administered anti-Αβ antibodies have both an early microglia-independent and a later, possibly microglia-dependent mechanism of action. Αβ levels were dramatically reduced 24 hr after administration in the absence of microglial activation. However, 72 hr after antibody administration, thioflavine-S-positive compact plaques were reduced concomitant with a striking activation of microglia.
Materials and Methods
Transgenic Tg2576 APP mice (Hsiao et al., 1996) were obtained after breeding of Tg2576 APP mice with line 5.1 PS1 mice (Duff et al., 1996), which yielded four different genotypes: nontransgenic, transgenic APP, transgenic PS1, and doubly transgenic APP+PS1 mice. Animals were provided food and water ad libitum and were kept on a 12 hr light/dark cycle; they were housed in groups if possible until before the surgery, when they were all singly housed until they were killed. We used two cohorts of mice in this study, the first cohort of 19-month-old APP mice (n = 16) and the second cohort of 16-month-old APP mice (n = 22).
Mice from the first cohort all received anti-Αβ antibodies (Biosource, Camarillo, CA; mouse anti-Αβ IgG1, recognizing amino acids 1–16). Mice from the second group were assigned to groups that received anti-Αβ antibodies, control antibody (anti-HIV, ID6; K. Ugen, Department of Medical Microbiology and Immunology, University of South Florida, Tampa, FL) (n = 5), or vehicle (0.02% thimerosal in PBS; Sigma-Aldrich, St. Louis, MO) (n = 5). All mice were injected in both the frontal cortex and hippocampus of the right hemisphere, whereas the left hemisphere remained untreated as an internal control. Those mice that received anti-Αβ antibodies were assigned survival times of 4 (n = 5), 24 (n = 7), 72 (n = 8), or 168 (n = 6) hr. Mice receiving either control antibody or vehicle were examined after a 72 hr survival time. A third group of untreated 17-month-old APP mice (n = 5) were killed without having been injected or manipulated to assess differences between the right and left sides of the brain.
On the day of surgery, the mice were weighed, anesthetized with isoflurane, and placed in a stereotaxic apparatus (51603 dual manipulator laboratory standard; Stoelting, Wood Dale, IL). A midsagittal incision was made to expose the cranium, and two burr holes were drilled with a dental drill over the right frontal cortex and hippocampus to the following coordinates: cortex, anteroposterior, +1.5 mm; lateral, –2.0 mm; hippocampus, anteroposterior, –2.7 mm; lateral, –2.5 mm, all taken from bregma. A 26 gauge needle attached to a 10 μl syringe (Hamilton, Reno, NV) was lowered 3 mm ventral to bregma, and a 2 μl injection was made over a 2 min period. The incision was cleaned with saline and closed with surgical staples.
On the day they were killed, the mice were overdosed with pentobarbital 100 mg/kg (Nembutal sodium solution; Abbott Laboratories, North Chicago, IL) and perfused intracardially with 25 ml of 0.9% sodium chloride and 50 ml of freshly prepared 4% paraformaldehyde, pH 7.4. The brains were collected and postfixed for 24 hr in 4% paraformaldehyde. The brains were then incubated for 24 hr in 10, 20, and 30% sucrose sequentially to cryoprotect them. Horizontal sections of 25 μm thickness were then collected with a sliding microtome and stored at 4°C in Dulbecco's PBS buffer with sodium azide to prevent microbial growth. Six to eight sections ∼100 μm apart were selected that spanned the injection site and were stained by free-floating immunohistochemistry methods for total Αβ (rabbit antiserum primarily reacting with the N terminus of the Αβ peptide; 1:10,000), CD45 (Serotec, Raleigh, NC; 1:3000), and major histocompatibility complex class II (MHC-II; BD PharMingen, Palo Alto, CA; 1:3000) as described previously (Gordon et al., 2002). For immunostaining, some sections were omitted from the primary antibody to assess nonspecific immunohistochemical reactions. Immunohistochemical methods were used to stain for the injected antibody with anti-mouse IgG conjugated to horseradish peroxidase (Sigma-Aldrich; 1:1000). Adjacent sections were mounted on slides and stained with 4% thioflavine-S (Sigma-Aldrich) for 10 min. Selected sections stained for CD45 were counterstained for Congo red (Sigma-Aldrich) to detect amyloid deposits on these sections.
The immunohistochemical reaction product on all stained sections was measured with a videometric V150 image analysis system (Oncor, San Diego, CA) in the injected area of cortex and hippocampus and corresponding regions on the contralateral side of the brain. Data are presented as the average ratio of injected side to noninjected side for Αβ, thioflavine-S, and CD45, whereas data for MHC-II are expressed as area occupied by positive stain, because many values on the contralateral side were close to zero.
To assess possible treatment-related differences, the measurement for either cortex or hippocampus of each subject were analyzed by ANOVA with StatView software version 5.0.1 (SAS Institute Inc., Raleigh, NC) followed by Fisher's least significant difference means comparisons.
Results
Immunohistochemistry against mouse IgG was performed to trace the diffusion of anti-Aβ antibodies after injection into the hilus of the dentate gyrus. The injected anti-Αβ antibody showed diffuse distribution throughout the entire hippocampus at 4 hr, with a focal concentration in the outer molecular areas of the dentate and Ammons' horn near the hippocampal fissure (Fig.1A). By 24 hr, the diffuse pattern remained broad, but the focal concentration began shifting toward the granule cell layers of the dentate gyrus (Fig.1B). At 72 hr, staining for the injected antibody was lighter and became concentrated at the granule cell layer of the dentate gyrus (Fig. 1C). Interestingly, by the 1 week time point, the injected antibody staining had cleared for the most part, with some residual staining in the outer molecular layer of the ventral (ventricular) blade of the dentate gyrus and the glial limitans. A similar time course of staining was seen in the frontal cortex (data not shown).
Fig. 1.

Time course of injected anti-Αβ antibody distribution in the hippocampus from 4 hr to 7 d. Immunohistochemical staining is shown for the injected antibody in the hippocampus at 4 (A), 24 (B), 72 (C), and 168 (D) hr. Orientation and locations of hippocampal subregions are as in Figure 2D. Magnification, 40×. Scale bar, 120 μm. A high-resolution color version of this micrograph can be obtained by e-mail from D. Morgan (dmorgan@hsc.usf.edu).
Αβ immunohistochemistry in APP transgenic mice resembled that reported previously by others and ourselves (Hsiao et al., 1996; Gordon et al., 2002). In both cortex (Fig.2A) and hippocampus (Fig. 2C), there were a few intensely stained deposits and a number of smaller, less intensely stained deposits. In previous work, we found the intensely stained Aβ deposits were usually also stained with thioflavine-S or Congo red (Holcomb et al., 1998; Gordon et al., 2001), which indicates they were analogous to compact deposits containing fibrillar amyloid, whereas the less intense deposits were analogous to diffuse, nonfibrillar deposits observed commonly in AD tissue. Whereas the deposits were distributed fairly uniformly within the cortex, in the hippocampus they were concentrated in the outer molecular layers of the dentate gyrus and Ammon's horn (Fig.2C). The subiculum also appeared more rich in deposits than other areas.
Fig. 2.
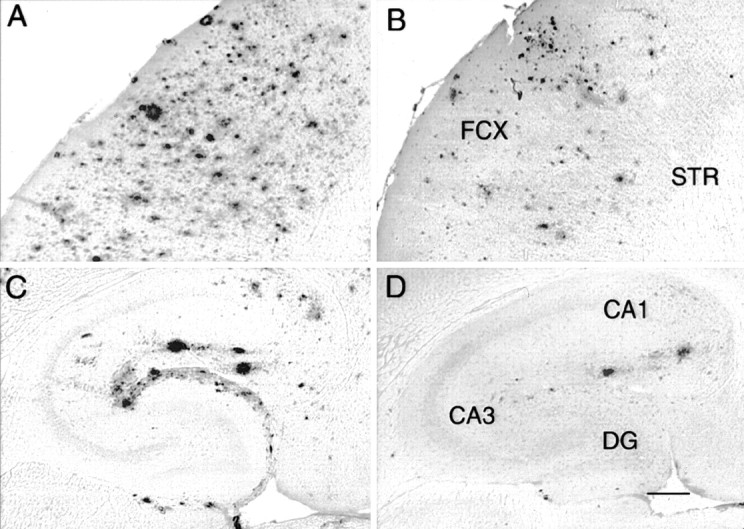
Reduction in Αβ immunohistochemistry 1 d after anti-Αβ antibody injections. Immunohistochemical staining is shown for Αβ in the frontal cortex (A,B) and hippocampus (C,D).A and C are from an animal injected with control antibody, whereas animals for which stains are shown inB and D received the anti-Αβ antibody. Magnification, 40×. Scale bar, 120 μm. B, FCX, frontal cortex; STR, striatum. D, CA1, cornu ammonis 1; CA3, cornu ammonis 3; DG, dentate gyrus. A high-resolution color version of this micrograph can be obtained by e-mail from D. Morgan (dmorgan@hsc.usf.edu).
The injection of anti-Αβ antibody into brain did not result in a rapid loss of signal in postmortem immunohistochemical reactions, because we did not observe a change in Αβ staining 4 hr after injection in either cortex (Fig.3A) or hippocampus (Fig.3B). However, Αβ staining was reduced at the injection sites in frontal cortex and hippocampus 24 hr after administration of anti-Αβ antibody (Fig. 2B,D, respectively) and remained reduced to roughly the same extent through the 1 week time point (Fig. 3). The reduction in the frontal cortex was 60% compared with both the 4 hr time points and the two control groups of vehicle and anti-HIV antibody (Fig. 3A; p < 0.001). The reduction in the hippocampus was 50% compared with the 4 hr time points and the control groups (Fig. 3B; p < 0.005).
Fig. 3.
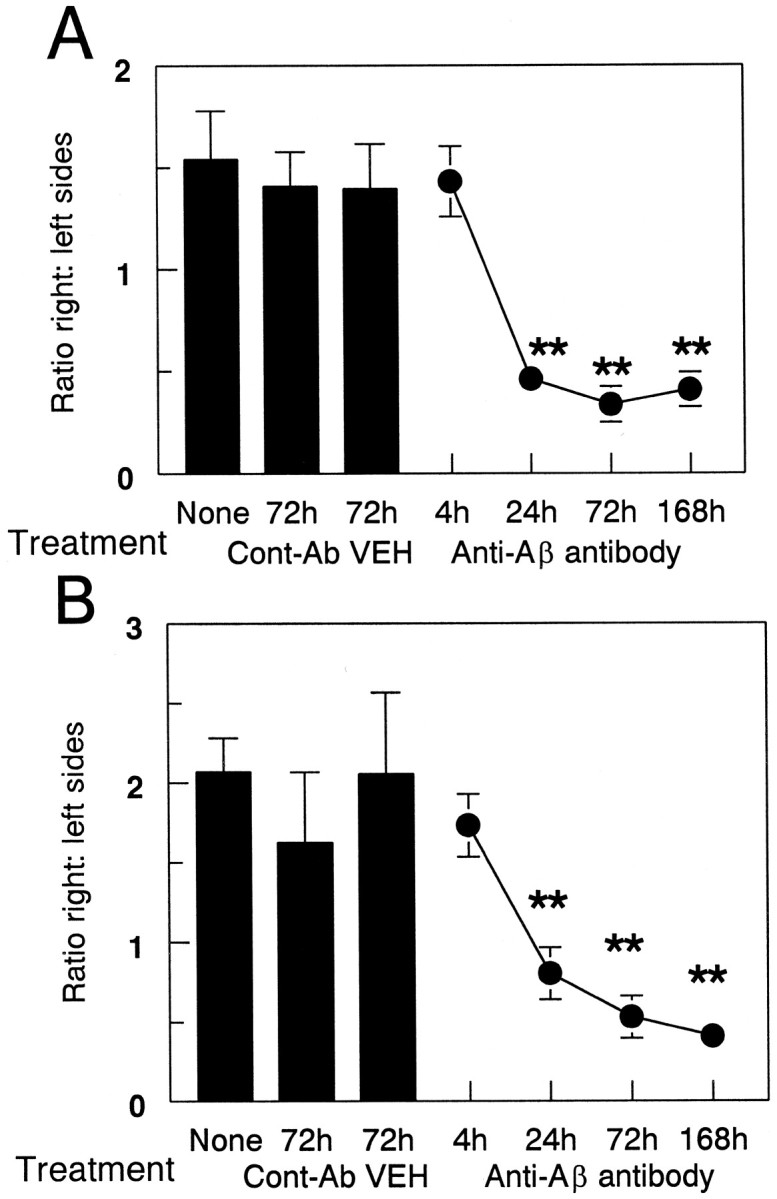
Quantification of reduced Αβ load after anti-Αβ antibody injections. Data are expressed as the ratio of Aβ staining in the injected hemisphere to the control hemisphere. The three bars on the left indicate the Αβ load in the untreated group (None) and the vehicle (VEH) and anti-HIV antibody (Cont-Ab) groups at 72 hr. The line shows the ratio of Αβ immunohistochemical staining at 4, 24, 72, and 168 hr survival times. Reduced Αβ load was observed in the frontal cortex (A) and hippocampus (B) at 24, 72, and 168 hr compared with 4 hr and both control groups (**p < 0.005).
An interesting phenomenon was that the ratio of Aβ staining on the right to left sides in untreated mice was >1, which indicates more Αβ deposition on the right side than the left (Fig. 3). It appears that this pattern of Αβ deposition is a consistent property of the APP mice. The Aβ distribution seen in the mice administered control injections at 3 d and anti-Αβ antibody at the 4 hr time point is the typical distribution found in APP transgenic mice of this age.
As expected, considerably fewer deposits were stained with thioflavine-S than by Αβ immunohistochemistry. Nonetheless, the regional distribution of these deposits approximately paralleled that of Αβ-positive deposits in the cortex and hippocampus (Fig.4A,C). In contrast to the Αβ, thioflavine-S-positive staining at the injection site was not reduced until 72 hr after administration of anti-Αβ antibody (Fig. 4B,D) and remained reduced at the 1 week time point (Fig. 5). The reduction in frontal cortex was 80% compared with the 4 and 24 hr time points and the control groups (Fig. 5A; p < 0.001). The reduction in hippocampus was 60% compared with both the 4 and 24 hr time points and the control groups (Fig. 5B;p < 0.005).
Fig. 4.

Reduction in thioflavine-S staining 3 d after anti-Αβ antibody injections. Thioflavine-S staining is shown in frontal cortex (A, B) and hippocampus (C, D). Mice in A andC received control antibody, whereas those inB and D received anti-Αβ antibody. Magnification, 40×. Scale bar, 120 μm. Orientation and locations of major subregions are as in Figure 2, B andD. A high-resolution color version of this micrograph can be obtained by e-mail from D. Morgan (dmorgan@hsc.usf.edu).
Fig. 5.
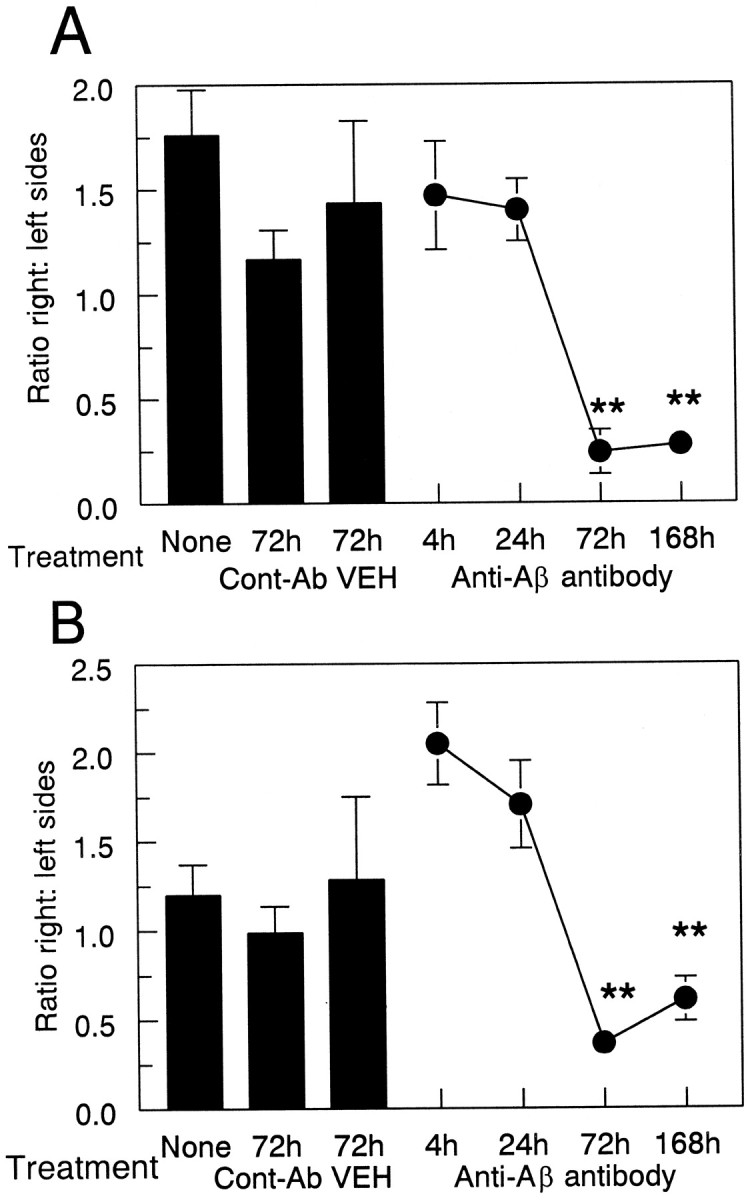
Anti-Αβ antibody injections result in a reduction of thioflavine-S-positive plaques. Data are expressed as the ratio of thioflavine-S staining in the injected hemisphere to the control hemisphere. The three bars show thioflavine-S-positive staining in the untreated group (None) and the vehicle (VEH) and anti-HIV antibody (Cont-Ab) groups at 72 hr. The line shows the ratio of thioflavine-S staining at 4, 24, 72, and 168 hr survival times. Reduced thioflavine-S staining was observed in the frontal cortex (A) and hippocampus (B) at 72 and 168 hr compared with 4 and 24 hr and both control groups (**p < 0.005).
In untreated mice, activated microglia stained with CD45 or MHC-II antibodies were found only in the immediate periphery of compacted plaques. In the injected control groups, some microglial activation was detected at the 72 hr survival time by CD45 antibodies, and this was restricted primarily to the injection site (Fig.6A,C, arrows; quantified in Fig. 7). Very little staining for CD45 was detected on the uninjected side of the brain, which led to inflated right-to-left ratios with relatively small increases in staining. MHC-II had a lower overall level of expression than that of CD45 and was mostly unaffected in mice administered control injections (Fig.8A,C; quantified in Fig. 9).
Fig. 6.
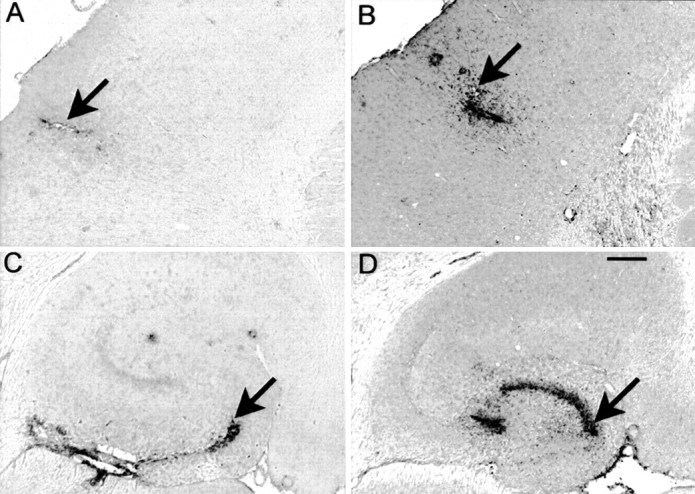
CD45 immunohistochemistry is increased 3 d after anti-Αβ antibody injections. CD45 immunohistochemistry is shown in frontal cortex (A, B) and hippocampus (C, D). Mice inA and C received control antibody, whereas those in B and D received anti-Αβ antibody. Magnification, 40×. Scale bar, 120 μm. Arrows indicate the site of injection identified from the needle tract. A high-resolution color version of this micrograph can be obtained by e-mail from D. Morgan (dmorgan@hsc.usf.edu).
Fig. 7.
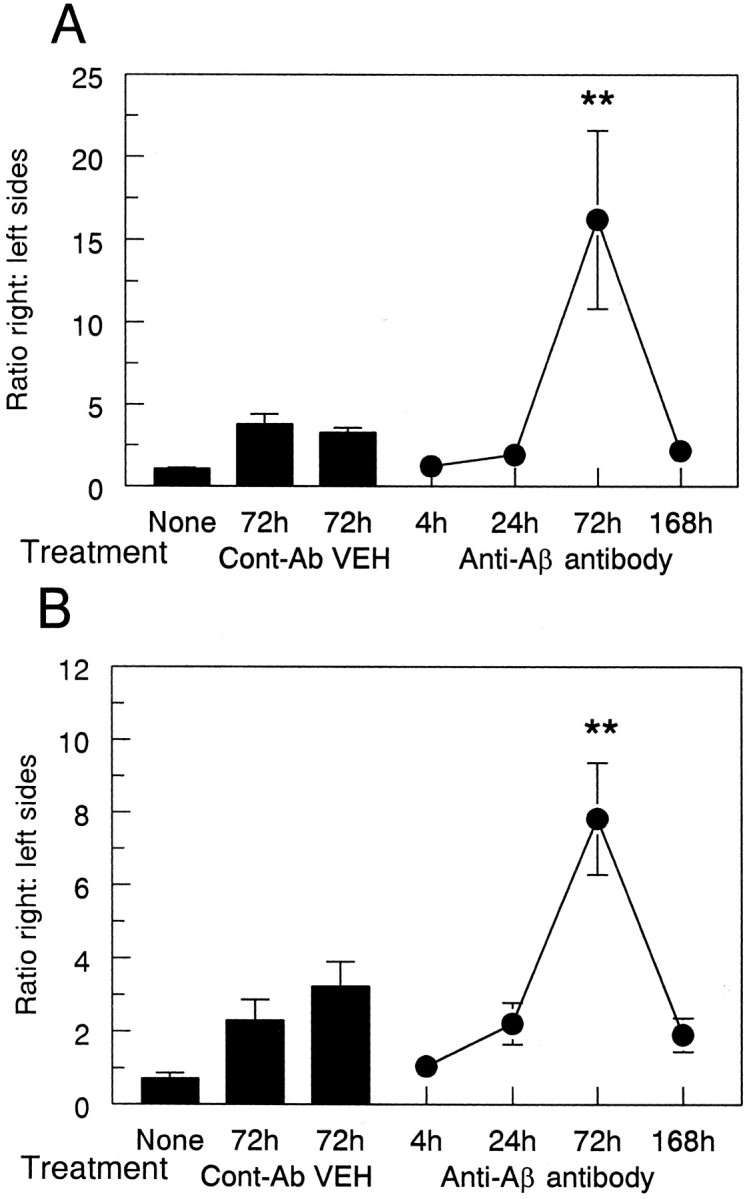
Anti-Αβ antibody injections result in increased CD45 immunohistochemistry 3 d after injection. Data are expressed as the ratio of CD45 staining in the injected hemisphere to the control hemisphere. The three bars indicate CD45 expression in the untreated group (None) and the vehicle (VEH) and anti-HIV antibody (Cont-Ab) groups at 72 hr. The line shows the ratio of CD45 staining at 4, 24, 72, and 168 hr survival times. Increased CD45 staining was observed in the frontal cortex (A) and hippocampus (B) at 72 hr compared with 4, 24, and 168 hr and both control groups (**p < 0.005).
Fig. 8.

MHC-II immunohistochemistry is increased 3 d after anti-Αβ antibody injections. MHC-II immunohistochemistry is shown in frontal cortex (A, B) and hippocampus (C, D). Mice inA and C received control antibody, whereas those in B and D received anti-Αβ antibody. Magnification, 40×. Scale bar, 120 μm. A high-resolution color version of this micrograph can be obtained by e-mail from D. Morgan (dmorgan@hsc.usf.edu).
Fig. 9.
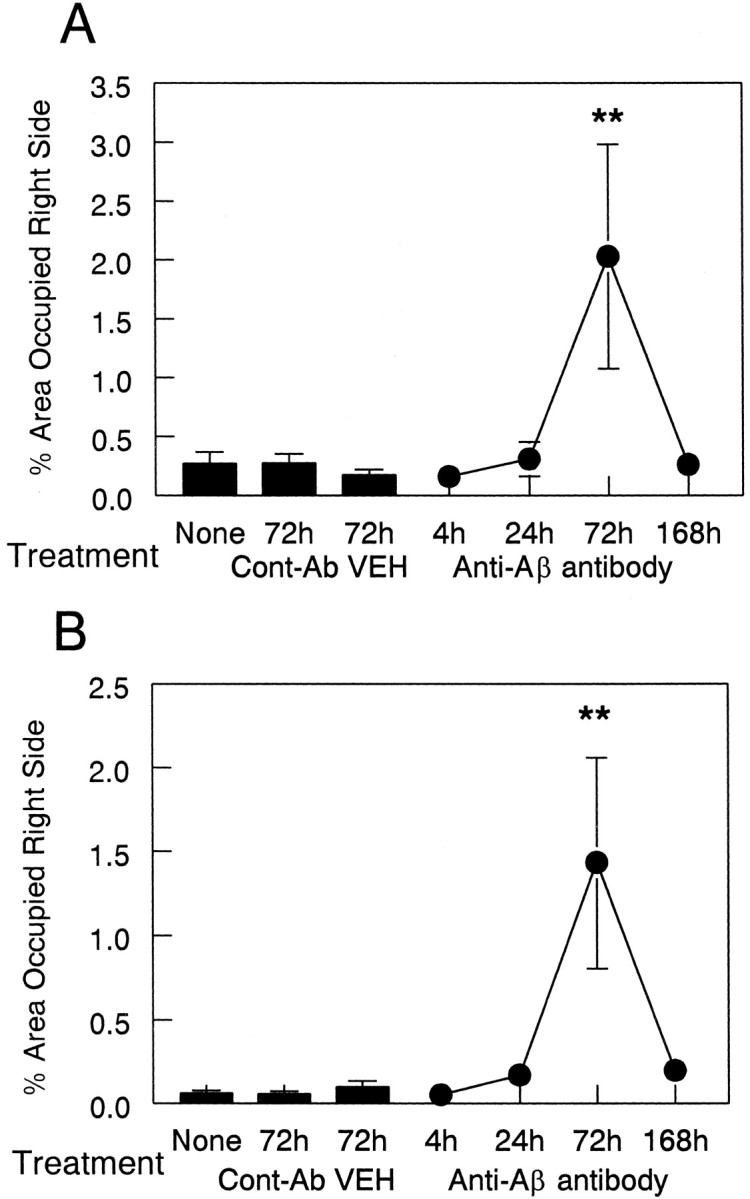
Anti-Αβ antibody injections result in an increase in MHC-II immunohistochemistry 3 d after injection. Data are expressed as percentage area occupied by MHC-II-positive staining in the injected hemisphere. The three bars indicate MHC-II expression in the untreated (None) group and the vehicle (VEH) and anti-HIV antibody (Cont-Ab) groups at 72 hr. The line shows the amount of MHC-II staining at 4, 24, 72, and 168 hr survival times. Increased MHC-II staining was observed in the frontal cortex (A) and hippocampus (B) at 72 hr compared with 4, 24, and 168 hr and both control groups (**p < 0.01).
In contrast, 72 hr after the injection of anti-Aβ antibodies, the microglial activation detected with CD45 antibodies was more widespread, detected not only at the injection site but also away from the injection site in the frontal cortex (Fig.6B) and throughout the dentate gyrus, with a concentration within the granule cell layer at the 72 hr time point (Fig. 6D). MHC-II staining revealed a similar pattern, although not as extensive as that found with CD45 staining (Fig. 8B,C).
Quantification of these results indicated that the injection of anti-Αβ antibodies increased expression of the microglial marker CD45 significantly only at the 72 hr time point compared with all other time points and control groups in both cortex (Fig. 7A;p < 0.005) and hippocampus (Fig. 7B;p < 0.005). Also, the injection of anti-Αβ antibodies increased the expression of the microglial marker MHC-II at the 72 hr time point compared with all other time points and control groups in both cortex (Fig. 9A; p < 0.01) and hippocampus (Fig. 9B; p < 0.005). The expression of CD45 and MHC-II in the frontal cortex at the anti-Αβ injection site increased more than eightfold over that of all other time points, including 1 week, and both of the control groups. The expression of CD45 in the hippocampus at the anti-Αβ injection site increased more than twofold, whereas the increase in expression of MHC-II was more than eightfold. As in our previous work, there was considerable variability among samples with both microglial markers; however, all anti-Αβ-injected animals had values that were greater than the means for the control groups.
There were few remaining amyloid deposits near the injection sites in the anti-Αβ antibody-injected mice at 72 hr (Figs. 4, 5). These residual deposits were relatively faint when stained with Congo red and were contacted by rounded, CD45-positive microglial cells (Fig.10B). In contrast, the more abundant amyloid deposits on the contralateral side and in the control animals were contacted by microglia with long processes that were stained for CD45, whereas the cell body stained only faintly for this marker of microglial activation (Fig. 10A).
Fig. 10.

Anti-Αβ antibody injections result in rounded microglia in association with remaining congophilic amyloid deposits 3 d after injection. CD45 immunostaining counterstained with Congo red is shown in the hippocampus at the 72 hr time point.A, Typical intensely stained congophilic deposit surrounded by CD45 immunostained microglial processes, with faintly stained somata (arrow). B, Faintly stained congophilic deposit in the anti-Αβ antibody-injected hippocampus. Note the two rounded intensely CD45-positive cells in contact with the faintly stained deposit (arrow). Magnification, 600×. Scale bar, 8.33 μm.
Discussion
Intracranial anti-Aβ antibody injections reduced Αβ load substantially in the vicinity of the injection in both anterior cortex and hippocampus over a 7 d time frame. By 4 hr after the injection, there was a broad distribution of injected antibody that filled a volume of ∼0.5 mm3, as estimated from anti-IgG immunohistochemistry. In addition to the broad pattern of diffusion, the antibody was concentrated in the outer molecular layers of Ammon's horn and the dentate gyrus, a zone that mostly overlaps with the distribution of Aβ staining in transgenic mice of this age (Fig. 2C). Thus, it appears the injected antibody was binding to in situ Aβ at this early time point but was also spread throughout the hippocampus. By 24 hr, there was a reduction in the Aβ immunostaining in the vicinity of the antibody injection in both cortex and hippocampus. This reduction in Αβ load is unlikely to be an artifact caused by the injected antibody masking the epitope of the primary antibody used for immunohistochemistry, because the reduced load was not detected 4 hr after administration, and by 24 hr, the injected IgG appeared to be concentrated closer to the granule cell region than the outer molecular layer in the hippocampus. Furthermore, the stoichiometry of injected antibody (13 pmol) to Αβ in deposits (estimated at 250 pmol in 0.5 mg; Chapman et al., 1999) is likely too low to interfere substantially with the histochemical reaction. This early reduction in Aβ load occurred in the absence of the expression of microglial activation markers CD45 and MHC-II. Although this does not preclude some rapid response of the microglia, it does suggest that the role of microglia is qualitatively different at this early postsurvival time from when markers of activation are being expressed.
Between 24 and 72 hr after injection of anti-Aβ antibodies, there were parallel reductions in fibrillar amyloid deposits detected by thioflavine-S and increases in microglial activation evaluated by CD45 and MHC-II staining. Although control injections of anti-HIV antibody and vehicle caused some elevation of the CD45 marker, activation was restricted to the immediate vicinity of the injection site and was likely caused by mechanical injury associated with needle insertion and fluid compression of the tissue. Occasionally, in the anti-Aβ antibody-injected mice, some remaining wisps of amyloid could be found in the vicinity of the antibody injection at 72 hr, and these were in contact with rounded CD45-immunopositive cells suggestive of phagocytic microglia or macrophages. Also at the 72 hr time point, there was a concentration of staining for both the injected antibody and the microglia near the granule cell layer of the dentate gyrus. The temporal association of fibrillar amyloid loss with microglial activation suggests some causal role for microglial activation in this process. One possibility is that between 1 and 3 d, activated microglia near the deposits in the outer molecular layer phagocytosed opsonized amyloid via Fc receptor- or complement-mediated mechanisms and migrated toward the granule cell layer. CD45-positive microglia could be detected readily in the outer molecular layer near the fissure at 3 d, although they were concentrated most heavily near the granule cell layer at that time point (Fig. 6D). A second option is that after dissolution of the Aβ deposits, the antibodies diffused to the granule cell region independently of the microglia. Possibly, the fibrillar deposits simply required more time to dissolve than the more diffuse material. More detailed time course studies of the period between 1 and 3 d coupled with immunoelectron microscopy will likely be required to decide between these options. Remarkably, the microglial activation was terminated rapidly and returned to normal levels by the 1 week time point in conjunction with a significant reduction in staining for the injected IgG and Aβ.
An accumulating body of evidence finds an association between microglial activation and amyloid reductions in transgenic mouse models of amyloid deposition. Schenk et al. (1999) noted in the first study evaluating Aβ vaccines that the clearance of amyloid was associated with enhanced microglial activity around the remaining deposits.Wilcock et al. (2001) mostly confirmed this observation in a different transgenic model. Nakagawa et al. (2000) unexpectedly found that fluid percussion injury activates microglia and results in reduced amyloid deposition as mice grow older. Lim et al. (2001) noted that transgenic mice treated with curcumin had a reduced amyloid load but an increase in the activation state of microglia surrounding plaques. Similarly,Jantzen et al. (2002) found a reduced amyloid load in transgenic mice treated with a nitric oxide-releasing nonsteroidal anti-inflammatory drug, NCX-2216, which was also associated with increased microglial activation. Wyss-Coray et al. (2001) found that the crossing of APP transgenic mice with mice that overexpressed TGF-β led to increased microglial activation and reduced amyloid loads. Conversely,Wyss-Coray et al. (2002) found that blocking of complement activation with soluble complement receptor-related protein Y overexpression diminished the microglial reaction in APP transgenic mice and led to elevated amyloid loads. DiCarlo et al. (2001) attempted to activate microglia directly by injecting lipopolysaccharide and found that this was associated with clearance of Aβ in the vicinity of the injection. However, Qiao et al., (2001) injected lipopolysaccharide chronically into young transgenic mice before normal amyloid deposition and found that it could stimulate Aβ deposition. It is also the case that careful serial section electron microscopy failed to detect internalized amyloid in microglia associated with amyloid deposits in untreated APP23 transgenic mice (Stalder et al., 2001), although mice treated to provoke microglial activation have yet to be examined. Nonetheless, there is a growing body of literature that associates the activation of microglia with a reduction in Aβ deposition in the transgenic mouse models.
A number of studies have demonstrated that cultured microglial cells are capable of internalizing Αβ1–42 aggregates (Paresce et al., 1996; Webster et al., 2001). Aβ can also be cleared from unfixed brain sections by anti-Aβ antibodies in a microglia-dependent manner (Bard et al., 2000). Direct imaging of amyloid deposits in vivo by multiphoton microscopy has shown clearance of plaque after application of an anti-Αβ antibody in association with an upregulation of activated microglia (Bacskai et al., 2001). Suggested alternative mechanisms to microglial phagocytosis include a physical interaction between antibody and Αβ that results in disaggregation of deposits, which was demonstrated in vitro with monoclonal anti-Aβ antibodies (Solomon, 2001). Consistent with this idea,Bacskai et al. (2002) demonstrated recently that F(ab′)2 fragments prepared from an anti-Aβ antibody reduced amyloid deposits as effectively as the intact antibody when applied topically to the cortex of transgenic mice through a craniotomy. Although there was no measurement of microglial activation in this study, it is plausible that this occurred in the absence of microglial involvement. This antibody-mediated dissolution hypothesis is consistent with the early phase of Aβ reduction described here and may still be found to be responsible for the second phase of fibrillar deposit reduction.
A major unresolved question is how this antibody-mediated clearance of Aβ might apply to the human condition. Increasingly, AD has been argued to involve inflammation as a component of its pathogenesis (McGeer and McGeer, 2001). The early stages of the Aβ vaccine trials resulted in a small fraction of patients developing adverse reactions consistent with inflammation of the CNS, presumably including microglial activation (Hock et al., 2002; Schenk and Yednock, 2002). Although adverse reactions to immunotherapy have been rare in the transgenic models (Pfeifer et al., 2002), it remains the best experimental system in which to understand the different components of the immune reactions to vaccines and to identify those components that cause adverse outcomes. Certainly, identification of immunotherapies that avoid the problem of deleterious CNS inflammation will be necessary if this treatment approach is to find use in the clinical setting. A better understanding of the mechanisms of antibody-mediated clearance of Aβ in the transgenic models of amyloid deposition should benefit this effort.
Footnotes
This work was supported by National Institutes of Aging, National Institutes of Health Grants AG15490 (M.N.G.), AG18478 (D.M.), and AG20227(K.E.U.). D.M.W. is a Benjamin Scholar in Alzheimer's Disease Research.
Correspondence should be addressed to Dave Morgan, 12901 Bruce B. Downs Boulevard, MDC 9, Tampa, FL 33612. E-mail: dmorgan@hsc.usf.edu.
References
- 1.Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 2.Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberberg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer's disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 4.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TVP, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 5.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Aβ load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 7.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 8.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 9.Gordon MN, King DL, Diamond DM, Jantzen PT, Boyett KV, Hope CE, Hatcher JM, DiCarlo G, Gottschall WP, Morgan D, Arendash GW. Correlation between cognitive deficits and Abeta deposits in transgenic APP+PS1 mice. Neurobiol Aging. 2001;22:377–385. doi: 10.1016/s0197-4580(00)00249-9. [DOI] [PubMed] [Google Scholar]
- 10.Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O'Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- 11.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 12.Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, Von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–1275. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- 13.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 15.Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 17.Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging. 2001;22:799–809. doi: 10.1016/s0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 20.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VM, Trojanowski JQ. Brain trauma in aged transgenic mice induces regression of established abeta deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 22.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 23.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 24.Qiao X, Cummins DJ, Paul SM. Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur J Neurosci. 2001;14:474–482. doi: 10.1046/j.0953-816x.2001.01666.x. [DOI] [PubMed] [Google Scholar]
- 25.Schenk DB, Yednock T. The role of microglia in Alzheimer's disease: friend or foe? Neurobiol Aging. 2002;23:677–679. doi: 10.1016/s0197-4580(02)00034-9. [DOI] [PubMed] [Google Scholar]
- 26.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 27.Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 28.Solomon B. Immunotherapeutic strategies for prevention and treatment of Alzheimer's disease. DNA Cell Biol. 2001;20:697–703. doi: 10.1089/10445490152717550. [DOI] [PubMed] [Google Scholar]
- 29.Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–434. doi: 10.1016/s0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- 30.Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Antibody-mediated phagocytosis of the amyloid beta-peptide in microglia is differentially modulated by C1q. J Immunol. 2001;166:7496–7503. doi: 10.4049/jimmunol.166.12.7496. [DOI] [PubMed] [Google Scholar]
- 31.Wilcock DM, Gordon MN, Ugen KE, Gottschall PE, DiCarlo G, Dickey C, Boyett KW, Jantzen PT, Connor KE, Melachrino J, Hardy J, Morgan D. Number of Abeta inoculations in APP+PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA Cell Biol. 2001;20:731–736. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- 32.Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 33.Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]