

Abstract
A number of recent in vitro studies have described a role for endogenous cannabinoids (“endocannabinoids”) as transsynaptic modulators of neuronal activity in the hippocampus and other brain regions. However, the impact that endocannabinoid signals may have on activity-dependent neural events in vivoremains mostly unknown and technically challenging to address because of the short half-life of these chemical messengers in the brain. Mice lacking the enzyme fatty acid amide hydrolase [FAAH (−/−) mice] are severely impaired in their ability to degrade the endocannabinoid anandamide and therefore represent a unique animal model in which to examine the function of this signaling lipidin vivo. Here, we show that the administration of anandamide dramatically augments the severity of chemically induced seizures in FAAH (−/−) mice but not in wild-type mice. Anandamide-enhanced seizures in FAAH (−/−) mice resulted in significant neuronal damage in the CA1 and CA3 regions of the hippocampus for the bicuculline and kainate models, respectively. Notably, in the absence of anandamide treatment, FAAH (−/−) mice exhibited enhanced seizure responses to high doses of kainate that correlated with greatly elevated endogenous levels of anandamide in the hippocampus of these animals. Collectively, these studies suggest that both exogenously administered and endogenously produced anandamide display FAAH-regulated proconvulsant activity and do not support a general neuroprotective role for this endocannabinoid in response to excitotoxic stimuli in vivo. More generally, these findings demonstrate that the disinhibitory actions of endocannabinoids observed in hippocampal slices in vitro may also occurin vivo.
Keywords: anandamide, bicuculline, CB1 receptor, endocannabinoid, epilepsy, excitotoxicity, fatty acid amide hydrolase, kainate, seizure
Introduction
The primary psychoactive ingredient of Cannabis sativa, Δ9-tetrahydrocannabinol (THC), produces many neuropharmacological effects in mammals primarily by activating a single G-protein-coupled receptor CB1(Ledent et al., 1999; Zimmer et al., 1999). The broad distribution of the CB1 receptor in the CNS, coupled with the identification of two natural CB1 ligands,N-arachidonoyl ethanolamine (anandamide) (Devane et al., 1992) and 2-arachidonoyl glycerol (2-AG) (Mechoulam et al., 1995;Sugiura et al., 1995), has spurred considerable interest in elucidating the function of the endogenous cannabinoid (“endocannabinoid”) system. Studies using CB1 agonists and antagonists, in combination with CB1 (−/−) mice, have indicated a role for this receptor in modulating several behavioral processes, including pain (Zimmer et al., 1999), feeding (Di Marzo et al., 2001), memory (Varvel and Lichtman, 2002), and locomotor activity (Zimmer et al., 1999).
Many of the behavioral effects of cannabinoids could reflect an inhibitory influence of these agents on neuronal firing in the CNS, as has been observed in the cerebellum (Kreitzer and Regehr, 2001a) and nucleus accumbens (Pistis et al., 2002). In the hippocampus, however, slice electrophysiological studies have provided evidence that endocannabinoids mediate a distinct form of transsynaptic communication referred to as “depolarization-induced suppression of inhibition” (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). In this form of retrograde transmission, endocannabinoids disinhibit pyramidal neuron activity by suppressing neurotransmitter release from GABAergic interneurons (Katona et al., 1999), thus providing an example in which the net effect of cannabinoids on neuronal firing is predicted to be stimulatory in vivo (Wilson and Nicoll, 2002). However, the limited number of in vivo studies conducted to date have not described a disinhibitory function for cannabinoids in the hippocampus. For example, THC was found to suppress the firing rates of hippocampal neurons in response to sensory stimuli during a learning test (Hampson and Deadwyler, 2000). These findings suggest that exocannabinoids and, by extension, endocannabinoids may produce rather complex and even contradictory effects on neuronal activity in the hippocampus, possibly depending on the distribution of the CB1 receptor in the circuits under examination. Accordingly, how cannabinoids impact pathological events related to excess hippocampal excitation, such as seizure and neurotoxicity, remains perplexingly difficult to predict. Indeed, although exocannabinoids are generally considered to be anticonvulsant (Adams and Martin, 1996), THC induces seizures in certain strains of rabbits (Martin and Consroe, 1976).
A major technical challenge facing the study of endocannabinoidsin vivo is their rapid rate of catabolism. For example, the half-life of anandamide in the rodent brain is less than a few minutes (Willoughby et al., 1997), primarily attributable to hydrolysis by the membrane-associated enzyme fatty acid amide hydrolase (FAAH) (Cravatt et al., 1996). FAAH is broadly expressed throughout the CNS (Thomas et al., 1997), exhibiting a complementary cellular distribution to CB1 receptors (Egertova et al., 1998; Tsou et al., 1998). Here, we took advantage of a mouse model in which FAAH has been genetically deleted (Cravatt et al., 2001) to examine the role that anandamide plays in regulating limbic seizures and hippocampal excitotoxicity in vivo.
Materials and Methods
Animals. FAAH (+/+) and FAAH (−/−) mice were generated by intercrossing 129vJ–C57BL/6 FAAH (+/−) mice as described previously (Cravatt et al., 2001). Studies were performed on littermate FAAH (+/+) and FAAH (−/−) mice that had been backcrossed into the C57BL/6 strain for at least five generations to limit potential strain-dependent allelic variations that might contribute to behavioral and physiological differences. CB(+/+) and (−/−) mice were bred from CB(+/−) mice backcrossed onto the C75BL/6 strain as described previously (Zimmer et al., 1999). All experiments were performed with mice from both genders, and no significant differences between female and male mice were observed.
Pharmacological agents. Anandamide, N-oleoyl ethanolamine (OEA), and N-palmitoyl ethanolamine (PEA), synthesized as described previously (Cravatt et al., 1996), (R)-(+)-WIN 55,212–2 (Sigma, St. Louis, MO), (S)-(−)-WIN 55,212–3 (Sigma), and SR141716A (kindly provided by National Institute on Drug Abuse) were dissolved in ethanol and diluted in a final ratio of 1:1:18 in ethanol/Emulphor/saline. Kainate (Ocean Produce International, Shelburne, Canada) was dissolved in saline. Bicuculline (Sigma) was dissolved in a drop of 0.1m warm HCl and further diluted in saline. Control experiments were performed by injection of the corresponding vehicle solutions. All drugs were administered by intraperitoneal injection.
Behavioral analysis. Animals were transferred into individual cages the day before the experiments to allow them to acclimate to the new environment. Animals were observed in these cages for 3 hr after drug treatment. Seizures were scored according to a modified Racine scoring system from 0–6: 0, no motor seizures; 1, freezing, staring, mouth or facial movements; 2, head nodding or isolated twitches, rigid posture; 3, tail extension, unilateral–bilateral forelimb clonus; 4, rearing, in which the mice sit in an immobile state on their rear haunches with one or both forepaws extended; 5, clonic seizures with loss of posture, jumping, and falling; 6, tonic seizure with hindlimb extension resulting in death (Racine, 1972). Saline- or vehicle-injected animals did not show any seizure behavior. All data are presented as means ± SEM. Statistical significance for seizure scores was determined with the Mann–Whitney U test unless otherwise indicated. For comparison of mortality, Fisher's exact test was used.
Histochemistry. Brains were removed 3 d after drug treatment and paraffin embedded. Serial 5-μm-thick sections were stained with hematoxylin–eosin. Neuronal injury was evident as the appearance of eosinophilic neurons with pyknotic nuclei (Liu et al., 1999). Neuronal damage in the hippocampus was assessed in five randomly chosen sections from each animal.
FAAH enzyme activity assays. FAAH activity assays were performed by measuring the conversion of 100 μm14C-labeled OEA to oleic acid as described previously (Cravatt et al., 2001). Data were expressed as picomoles of oleic acid per minute per milligram of protein ± SEM. Statistical significance was determined with the unpaired ttest.
Determination of N-acyl ethanolamine levels. N-acyl ethanolamine (NAE) levels in the hippocampus, cortex, and cerebellum of FAAH (+/+) and FAAH (−/−) inbred mice were quantified by isotope dilution liquid chromatography mass spectrometry (LC-MS) as described previously (Di Marzo et al., 2000; Cravatt et al., 2001). Briefly, mice were anesthetized by using CO2–O2 and killed by decapitation. Brain regions were removed within 1 min and immediately homogenized in a 2:1:1 mixture of chloroform/methanol/50 mm Tris, pH 8.0, containing d4-anandamide and d4-OEA as standards (2.5 and 50 pmol per brain region, respectively). The organic layer was removed and dried under N2 gas. The remaining residue was solubilized in methanol and injected onto an Agilent (Palo Alto, CA) 1100 series LC-MS. Levels of endogenous anandamide and OEA were quantified by comparing the mass ion peak heights with those of the corresponding d4standards. PEA levels were quantified by comparing the peak height of PEA with that of d4-OEA [a d4-PEA internal standard could not be used because of overlapping retention times and masses with endogenous OEA (the mass of OEA equals the mass of the sodium adduct of d4-PEA)]. NAE levels were expressed as picomoles per gram of wet tissue ± SEM. Statistical significance was determined with the unpaired t test.
Results
Anandamide exhibits proconvulsant activity in limbic seizure models in FAAH (−/−) mice
The rapid rate at which anandamide is degraded in the brain (Willoughby et al., 1997) has hindered experimental efforts to study the function of this endocannabinoid in vivo. We recently described the generation and initial characterization of a mouse model bearing a targeted disruption of the FAAH gene (FAAH (−/−) mice) (Cravatt et al., 2001). FAAH (−/−) mice are severely impaired in their ability to degrade anandamide and show exaggerated CB1-dependent behavioral responses to this fatty acid amide. Additionally, brain levels of anandamide are increased >10-fold in FAAH (−/−) mice, a neurochemical phenotype that correlates with enhanced CB1-dependent analgesia in these animals. Interestingly, more recent studies have shown that the hydrolytic rates of monoacylglycerols are unaltered in FAAH (−/−) mice (Lichtman et al., 2002), indicating that these animals may provide a means to discriminate the functions of amide (e.g., anandamide) and ester (e.g., 2-AG) classes of endocannabinoids. Collectively, these initial studies demonstrated that FAAH (−/−) mice represent a unique animal model in which to examine the activity of both exogenously applied and endogenously produced anandamide.
The effect of anandamide on chemically induced limbic seizures was compared in FAAH (+/+) and (−/−) mice. As reported previously (Cravatt et al., 2001), FAAH (−/−) mice treated with anandamide at doses ranging from 12.5 to 50 mg/kg (intraperitoneally) quickly entered into an immobile state that was periodically interrupted during the first 15 min after treatment by brief fits of spastic motion. After 15 min, FAAH (−/−) mice adopted a flattened, cataleptic state and remained motionless for up to 3–4 hr unless startled by touch or sound. In contrast, FAAH (+/+) mice showed no overt behavioral responses to anandamide other than a modest degree of noncataleptic hypomotility that dissipated within 20 min after injection. One hour after treatment with anandamide, at a time when the effects of this endocannabinoid were maximal in FAAH (−/−) mice (Cravatt et al., 2001), both FAAH (+/+) and (−/−) mice were administered either bicuculline (4 mg/kg, i.p.) or kainate (15 mg/kg, i.p.), and their seizure responses were recorded using a standard scoring method (see Materials and Methods). Compared with vehicle controls, anandamide-treated FAAH (−/−) mice exhibited dramatically enhanced seizures after treatment with either bicuculline (Fig.1A) or kainate (Fig.1C). In contrast, anandamide did not significantly impact the seizure sensitivity of FAAH (+/+) animals. Anandamide was not found to display anticonvulsant activity at lower doses (6.25 mg/kg, i.p.) in either genotype, even when administered before higher doses of bicuculline (6 mg/kg, i.p.) (Fig. 1B) or kainate (25 mg/kg, i.p.) (Fig. 1D), suggesting that this endocannabinoid does not exhibit a dose-dependent paradoxical effect on seizure threshold, as has been observed previously for opioid agonists such as morphine (Lauretti et al., 1994).
Fig. 1.

The effects of anandamide on bicuculline- and kainate-induced seizures in FAAH (+/+) and (−/−) mice. Treatment with anandamide (6.25–50 mg/kg, i.p.) 1 hr before administration of either bicuculline (A; 4 mg/kg, i.p.) or kainate (C; 15 mg/kg, i.p.) significantly enhanced the severity of seizures in FAAH (−/−) mice (filled bars) but not in FAAH (+/+) mice (open bars); n = 4 mice per group for 6.25 and 12.5 mg/kg anandamide, and n = 6–14 mice per group for 25 and 50 mg/kg anandamide. Pretreatment with a low dose of anandamide (6.25 mg/kg, i.p.) did not affect the seizure responses to high doses of bicuculline (B; 6 mg/kg) or kainate (D; 25 mg/kg) in FAAH (+/+) or (−/−) mice;n = 6–14 mice per group. For A–D, seizure scores are presented as mean ± SEM. All data were compared with the Mann–Whitney U test. ★p < 0.05 for FAAH (+/+) versus FAAH (−/−) mice receiving the same treatment. *p < 0.05 and ***p < 0.001 for FAAH (−/−) mice under different treatment conditions.
In the bicuculline model, the proconvulsant activity of anandamide (25 mg/kg, i.p.) in FAAH (−/−) mice was blocked by pretreatment with the CB1 antagonist SR141716A (1 mg/kg, i.p.) (Fig.2A), indicating that CB1 receptors may mediate the effects of anandamide in these animals. Consistent with a proconvulsant role for CB1 receptors in the bicuculline model, the CB1 agonist (R)-(+)-WIN 55,212–2, but not its inactive enantiomer (S)-(−)-WIN 55,212–3, was found to enhance the severity of bicuculline-induced seizures in both FAAH (+/+) and (−/−) mice (Fig. 2B). Although the administration of SR141716A alone did not affect bicuculline-induced seizures in either FAAH (+/+) or (−/−) mice (Fig. 2A), pretreatment with this CB1 antagonist did significantly increase the severity of kainate-induced seizures in both FAAH (+/+) and (−/−) mice (Fig. 2C). This proconvulsant activity of SR141716A was not dependent on CB1 receptors, because the agent also augmented kainate-induced seizures to an equivalent magnitude in CB1 (−/−) mice (Fig.2C). A two-way ANOVA supported this observation, as reflected by a significant increase in seizure magnitude by SR141716A compared with vehicle (F(1,76) = 30;p < 0.001) and the effect of SR141716A being independent of genotype. Collectively, these findings suggest that SR141716A augments kainate-induced seizures through a non-CB1 receptor mechanism of action. On this note, recent studies of arterial relaxation in CB1 (−/−) mice have also uncovered a CB1-independent activity for SR141716A (Bukoski et al., 2002), indicating that this compound may not be completely selective for the CB1 receptor in vivo. Regardless, the proconvulsant effects of SR141716A precluded its further use in the kainate-induced seizure model.
Fig. 2.
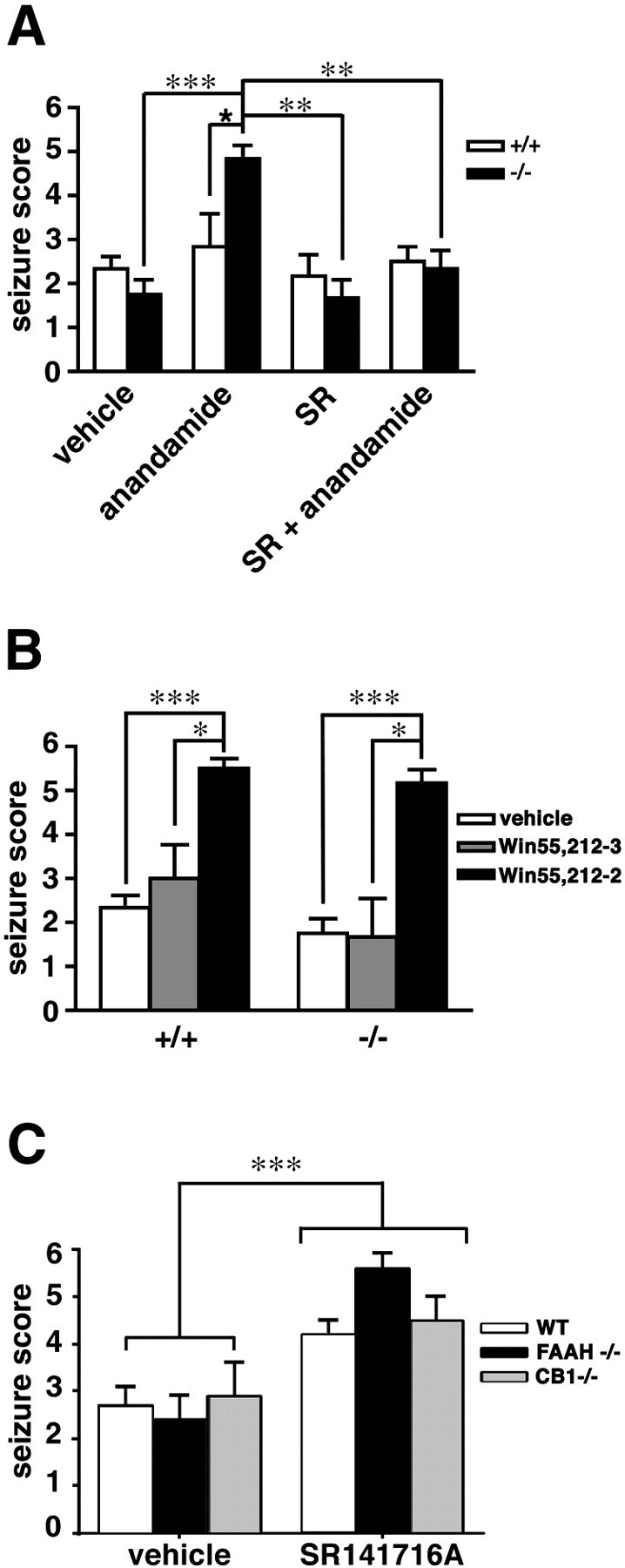
The effects of CB1 receptor agonists and antagonists on chemically induced seizures in FAAH (+/+) and (−/−) mice. A, FAAH (+/+) and (−/−) mice were treated with the CB1 receptor antagonist SR141716A (1 mg/kg, i.p.) or vehicle 10 min before administration of anandamide (25 mg/kg, i.p.) or vehicle. Seizures were induced with (bicuculline, 4 mg/kg, i.p.) 60 min after anandamide injection. SR141716A blocked the effects of anandamide on seizures in FAAH (−/−) mice but did not affect seizure responses in vehicle-treated animals of either genotype (SR group). n = 6–12 mice per group. ★p < 0.05 for FAAH (+/+) versus FAAH (−/−) mice receiving the same treatment. **p < 0.01 and ***p < 0.001 for FAAH (−/−) mice under different treatment conditions (Mann–Whitney U test).B, Treatment with the CB1 agonist (R)-(+)-WIN 55,212–2, but not its inactive enantiomer (S)-(−)-WIN 55,212–3 (15 mg/kg, i.p.), 1 hr before administration of bicuculline (4 mg/kg) significantly enhanced the severity of seizures in both FAAH (+/+) and (−/−) mice. n = 6–12 mice per group. *p < 0.05 and ***p < 0.001 for WIN 55,212–2-treated versus WIN 55,212–3-treated and vehicle-treated mice of the same genotype, respectively (Mann–WhitneyU test). C, SR141716A augmented kainate-induced seizures in wild-type (WT), FAAH (−/−), and CB1 (−/−) mice. Subjects from each genotype were treated with either vehicle or SR141716A (1 mg/kg, i.p.) 70 min before kainic acid (15 mg/kg, i.p.). Data are depicted as means ± SEM (n = 8–14 FAAH (−/−) and CB1 (−/−) mice; 20–22 wild-type mice). ***p < 0.001 for SR141716A-treated versus vehicle-treated animals (ANOVA).
Unable to assess the CB1 dependence of the effects of anandamide on kainate-induced seizures with SR141716A, we sought indirect evidence for the involvement of CB1 receptors by testing the activity of two structurally related, non-CB1-binding NAEs: OEA and PEA. Neither OEA nor PEA (50 mg/kg, i.p.) affected kainate-induced seizures in FAAH (+/+) or (−/−) mice (data not shown). The structural specificity of the proconvulsant activity of anandamide is consistent with this NAE acting at CB1 receptors as opposed to other potential sites of action in the CNS such as gap junctions (Guan et al., 1997), which exhibit less discrimination among endogenous NAEs. Collectively, these results demonstrate that anandamide exhibits proconvulsant activity in two models of limbic seizures. These effects are tightly regulated by FAAH and appear to be mediated by CB1 receptors.
The proconvulsant effects of anandamide result in hippocampal neuronal death in FAAH (−/−) mice
Anandamide and its precursor N-arachidonoyl phosphatidylethanolamine are produced in a calcium-dependent manner in response to brain injury and/or intense neuronal excitation (Hansen et al., 2001a,b), suggesting that these compounds may serve a neuroprotective function in vivo (Hansen et al., 1998). In support of this premise, CB1 agonists reduce excitotoxic cell death in hippocampal neuronal cultures (Shen and Thayer, 1998), apparently through the presynaptic inhibition of glutamate release. Additionally, both anandamide and 2-AG reduce ischemic damage in cortical neurons cultured under hypoxic conditions (Sinor et al., 2000). Most recently, anandamide was found to reduce neuronal damage in vivo in response to oubain-induced excitotoxicity (van der Stelt et al., 2001). Nonetheless, attempts to block the neuroprotective effects of anandamide with CB1 antagonists have produced mixed results (Sinor et al., 2000; van der Stelt et al., 2001), suggesting that at least a portion of the neuroprotection afforded by this endocannabinoid may be attributable to the action of its metabolite, arachidonic acid, which has also been shown to inhibit excitotoxicity (Lauritzen et al., 2000).
If anandamide exhibited a neuroprotective effect in vivo in response to excitotoxic stimuli, then one might anticipate that the proconvulsant effects of this NAE in FAAH (−/−) mice would result in minimal hippocampal neuronal damage. However, in FAAH (−/−) mice, anandamide-dependent seizures induced by bicuculline and kainate resulted in substantial hippocampal neuronal death in the CA1 and CA3 regions, respectively (Fig. 3). In the bicuculline model, the neurotoxic effects of anandamide were blocked by pretreatment with CB1 antagonist SR141716A (1 mg/kg, i.p.) (Fig. 3A). A more detailed examination did not uncover a qualitative neuroprotective effect for anandamide, because FAAH (−/−) mice that displayed similar seizure scores after treatment with either a high dose of kainate (25 mg/kg, i.p.) or a low dose of kainate (15 mg/kg) plus anandamide (50 mg/kg) showed similar degrees of hippocampal damage (Fig. 3B). Collectively, these results do not support a role for anandamide as an endogenous substance that protects against excitotoxic neuronal death in the hippocampus.
Fig. 3.
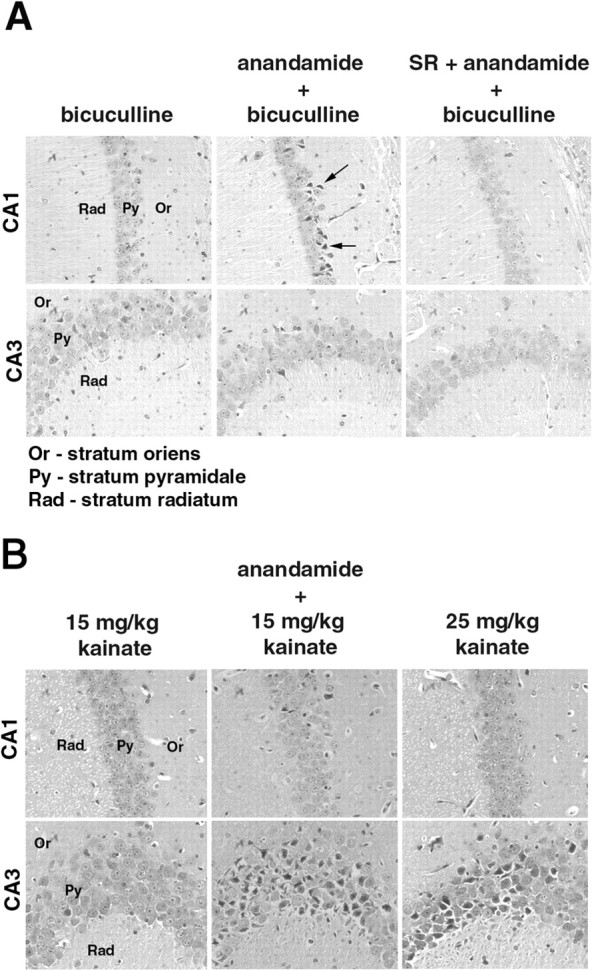
The effects of anandamide on hippocampal neurotoxicity associated with bicuculline- and kainate-induced seizures in FAAH (−/−) mice. Brains from FAAH (−/−) mice (n = 5–7 for each treatment group) were removed 3 d after drug treatment, and hematoxylin–eosin-stained slices through the hippocampal region were examined for neuronal cell death. Subthreshold doses of bicuculline (4 mg/kg) and kainate (15 mg/kg) treatment that did not result in clonic–tonic seizures were not found to induce neuronal cell death in the hippocampal region (A, B, left panels). Administration of anandamide (50 mg/kg, i.p.) 60 min before treatment with bicuculline or kainate resulted in severe seizures (Fig. 1) that were accompanied by prominent hippocampal neuronal death. In the bicuculline-treated group, five of the seven surviving animals exhibiting seizure scores of 4–5 displayed pyramidal cell death mainly in the CA1 region of the hippocampus (A, middle panels). Arrows point to examples of injured neurons, which appear as dark, eosinophilic cells with shrunken nuclei. Administration of SR147161A (SR) (3 mg/kg, i.p.) 10 min before treatment with anandamide blocked the neurotoxic (A, right panels) effects of this endocannabinoid. B, In the kainate model, three of the five surviving FAAH (−/−) mice pretreated with anandamide that exhibited seizure scores of 4–5 showed substantial neuronal damage mainly in the CA3 region (B, middle panels). A similar degree of neuronal damage was observed in a separate group of FAAH (−/−) mice that exhibited strong seizure responses (scores of 4–5) to a high dose of kainate (25 mg/kg, i.p.) (B, right panels).
Increased sensitivity to kainate-induced seizures in FAAH (−/−) mice
FAAH (+/+) and (−/−) mice were compared for their seizure responses to increasing doses of kainate and bicuculline. No genotype differences were observed in the severity of seizures induced by lower doses of either agent (Fig. 4); however, at the highest dose of kainate tested (30 mg/kg), all FAAH (−/−) mice responded with intense clonic–tonic seizures that resulted in death within 25 min (Fig. 4A,B). In contrast, the majority of the FAAH (+/+) mice survived treatment with a high dose of kainate, despite displaying relatively intense seizures that lasted for up to 60 min (Fig.4A,B). FAAH (+/+) and (−/−) mice exhibited similar seizure responses to low or high doses of bicuculline (Fig. 4C).
Fig. 4.
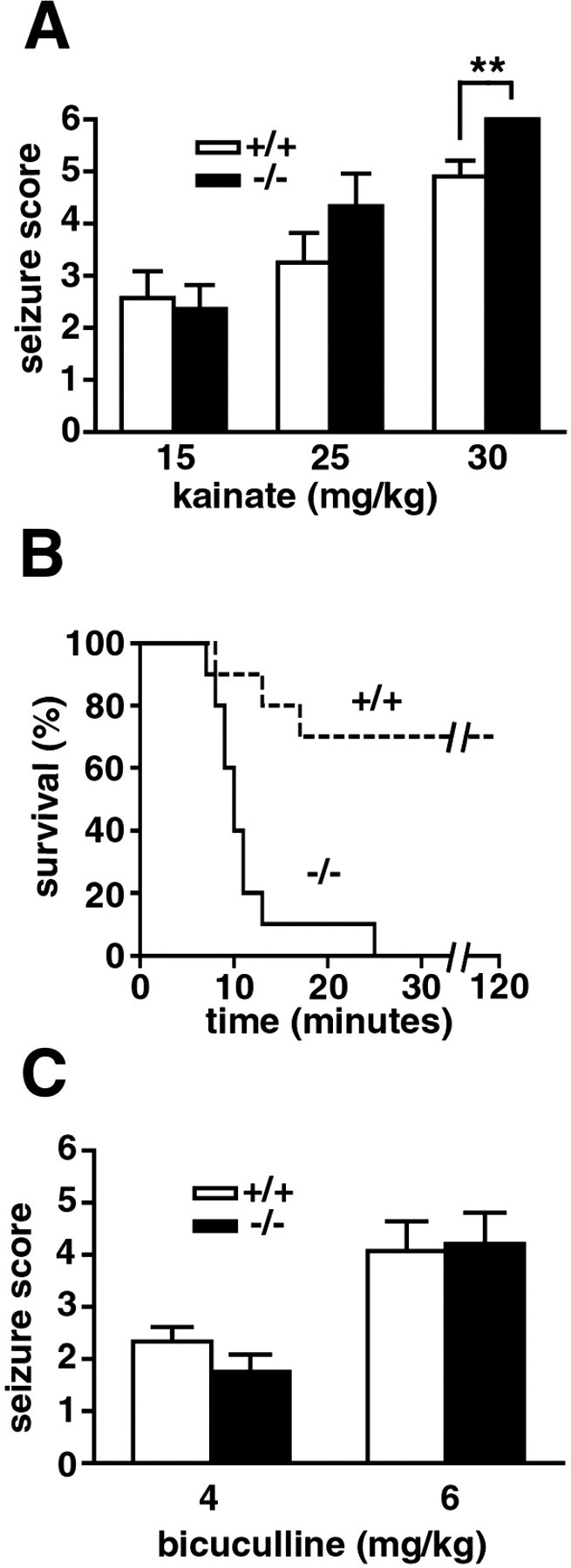
Seizure sensitivities of naive FAAH (+/+) and (−/−) mice. In response to high (30 mg/kg, i.p.), but not low (15 and 25 mg/kg), doses of kainate, FAAH (−/−) mice exhibited more intense seizures (A, filled bars) and reduced survival (B, solid line) compared with FAAH (+/+) mice (A, open bars; B, dashed line). No genotype differences were observed in the severity of seizures induced by low (4 mg/kg, i.p.) and high (6 mg/kg, i.p.) doses of bicuculline (C). **p < 0.01 for FAAH (+/+) versus (−/−) mice receiving the same treatment (Mann–Whitney U test). The survival curve (B) showed a significantly higher mortality for FAAH (−/−) mice than for FAAH (+/+) mice (p < 0.01; Fisher's exact test) after treatment with 30 mg/kg kainate. n = 10 mice per group.
The heightened severity of kainate-induced seizures in FAAH (−/−) mice prompted us to examine whether increased endogenous levels of anandamide were present in the hippocampus of FAAH (−/−) mice that might account for their altered behavior. Hippocampal levels of anandamide, as well as other NAEs, were measured by isotope dilution liquid chromatography mass spectrometry (Di Marzo et al., 2000) and found to be >10-fold higher in FAAH (−/−) mice relative to FAAH (+/+) mice (Table 1), consistent with the virtual absence of fatty acid amide hydrolytic activity in the hippocampus of FAAH (−/−) animals (Table2). Additional brain regions examined, including the cortex and cerebellum, displayed similar increases in endogenous NAE levels in FAAH (−/−) mice, although the absolute levels of anandamide in these regions were slightly lower than those found in the hippocampus (Table 1). These data, in conjunction with the proconvulsant activity observed for anandamide, but not other NAEs, are consistent with an augmented anandamide-based endocannabinoid tone contributing to the heightened seizure sensitivity of FAAH (−/−) mice.
Table 1.
Endogenous levels of NAEs in brain regions of FAAH (+/+) and (−/−) mice
| Brain regions | NAE levels (pmol/gm wet weight), mean ± SEM | |||||
|---|---|---|---|---|---|---|
| Anandamide | OEA | PEA | ||||
| +/+ | −/− | +/+ | −/− | +/+ | −/− | |
| Hippocampus | 3.3 ± 0.8 | 38.0 ± 2.8* | 61.5 ± 13.6 | 1341.8 ± 123.0 | 154.5 ± 41.7 | 2149.50 ± 226.7 |
| Cortex | 1.8 ± 0.1 | 26.0 ± 2.4 | 55.4 ± 7.2 | 1094.7 ± 130.7 | 103.3 ± 27.6 | 1685.33 ± 59.3 |
| Cerebellum | 1.3 ± 0.3 | 29.7 ± 2.1 | 101.0 ± 20.1 | 1669.8 ± 161.8 | 237.2 ± 56.0 | 2401.67 ± 164.9 |
Levels for anandamide, OEA, and PEA were significantly higher in all three brain regions in FAAH (−/−) mice compared with FAAH (+/+) mice (p < 0.001; unpaired t test;n = 3–6 per group). Anandamide levels in FAAH (−/−) hippocampus were higher than in FAAH (−/−) cortex and cerebellum (
p < 0.05; unpaired ttest).
Table 2.
OEA hydrolytic activities in brain regions of FAAH (+/+) and (−/−) mice
| Brain region | FAAH activity (pmol/min/mg protein), mean ± SEM | |
|---|---|---|
| +/+ | −/− | |
| Hippocampus | 57.93 ± 1.84 | 0.38 ± 0.16 |
| Cortex | 45.86 ± 5.09 | 0.98 ± 0.44 |
| Cerebellum | 28.4 ± 1.1 | 0.78 ± 0.11 |
Rates of hydrolysis of OEA were significantly reduced in FAAH (−/−) mice compared with FAAH (+/+) mice in all three brain regions tested (p < 0.001; unpaired t test; n = 3 per group).
Discussion
Depending on the brain region and neural circuits under examination, in vitro electrophysiological studies have shown that the activity of cannabinoids can be either inhibitory (Kreitzer and Regehr, 2001a; Pistis et al., 2002) or disinhibitory (Kreitzer and Regehr, 2001b; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). These apparently contradictory properties of cannabinoids can be reconciled when one considers that both their inhibitory and excitatory effects derive from an ability to suppress neurotransmitter release from presynaptic terminals (Wilson and Nicoll, 2002). If the cannabinoid-sensitive synapse is glutamatergic, the net result is a suppression of excitation, or inhibition; if the cannabinoid-sensitive synapse is GABAergic, the net result is a suppression of inhibition, or excitation. Because the synthesis and release of endocannabinoids appears to occur in an activity-dependent manner from the postsynaptic cell, these lipid messengers may influence neuronal cross-talk by a novel retrograde mode of action (Kreitzer and Regehr, 2001b; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001).
The context-dependent effects that cannabinoids exhibit on neural circuits, in combination with the broad distribution of the CB1 receptor in the CNS, make it difficult to predict the net impact of CB1 activation on complex pathological events such as seizure and neurotoxicity. Additionally, evaluating the function of endocannabinoids is further challenged by the rapid rate at which the compounds are catabolizedin vivo (Willoughby et al., 1997). Here, we attempted to address these issues by evaluating seizure sensitivity and excitotoxicity in mice lacking the protein FAAH (Cravatt et al., 2001), the principal enzyme responsible for degrading the endocannabinoid anandamide. FAAH (−/−) mice exhibit greatly reduced rates of anandamide hydrolysis, and, as a consequence, anandamide produces an array of intense, CB1-dependent behavioral effects in these animals, including hypomotility, hypothermia, analgesia, and catalepsy (Cravatt et al., 2001). The robust cannabinoid activity of anandamide in FAAH (−/−) mice, which results in the appearance of a sedated state, might be anticipated to reflect a widespread inhibition of neuronal activity in the CNS that could provide protection from seizure and excitotoxicity. In general support of this notion, previous studies described an anticonvulsant activity for exocannabinoids and endocannabinoids in mice in the maximal electroshock model (Karler et al., 1974; Wallace et al., 2001, 2002), suggesting that these agents might be useful clinically as anti-epileptics. However, we found here that the endocannabinoid anandamide promotes rather than retards seizures induced by bicuculline and kainate in FAAH (−/−) mice. Additionally, untreated FAAH (−/−) mice exhibited enhanced seizure responses to high doses of kainate that correlated with greatly elevated endogenous levels of anandamide in the hippocampus and other brain regions. Collectively, these data uncovered a previously unappreciated proconvulsant activity for anandamide in two models of limbic seizures.
There are several possible explanations for the apparent discrepancy between the results of the current study and those reported previously. First, anandamide may augment seizure sensitivity in FAAH (−/−) mice through non-CB1 mechanisms. Arguing against this possibility, however, the proconvulsant effects of anandamide in the bicuculline model were blocked by the CB1receptor antagonist SR141716A. Additionally, the failure of structurally related NAEs, such as PEA and OEA, to affect seizure threshold suggests that the proconvulsant activity of anandamide displays a degree of chemical specificity consistent with targeting the CB1 receptor. Finally, the CB1 agonist WIN 55,212–2, but not its inactive enantiomer, also increased the severity of bicuculline-induced seizures. A second potential explanation is that FAAH (−/−) mice, in response to increased steady-state brain levels of anandamide, as well as other endogenous fatty acid amides and possibly their metabolites, exhibit compensatory changes in nervous system function that make them responsive to anandamide in a manner unrepresentative of wild-type animals. However, this hypothesis is not supported by previous studies, which have shown that FAAH (+/+) and (−/−) mice exhibit functionally equivalent CB1 receptor systems. For example, the EC50 values for THC in the tetrad test are equivalent in FAAH (+/+) and (−/−) mice (Cravatt et al., 2001), and the brains from these animals display similar CB1receptor densities (Lichtman et al., 2002). Instead, we speculate that the apparently contradictory effects of cannabinoids on chemically induced and maximal electroshock seizures may derive from the fact that these models promote hyperexcitability in different brain regions. The seizures caused by kainate and bicuculline are mostly limbic in origin (Ben-Ari and Cossart, 2000), and therefore the distribution of the CB1 receptor in brain regions such as the hippocampus may be of primary importance for defining the effects of cannabinoids in these models. With this consideration in mind, the proconvulsant activity of anandamide observed herein is consistent within vitro electrophysiological studies describing a disinhibitory function for endocannabinoids in the hippocampus (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). In contrast, seizures resulting from maximal electroshock reflect a more broad state of hyperexcitability in the CNS (Follesa et al., 1994), and therefore the anticonvulsant activity of cannabinoids in this model may derive from an inhibitory activity of these agents in brain regions outside the hippocampus. Thus, the current studies do not necessarily argue against a role for cannabinoids as anti-epileptics but rather highlight that certain forms of epilepsy, especially ones in which limbic seizures are a major component (temporal lobe epilepsy) (Ben-Ari and Cossart, 2000), may be exacerbated rather than ameliorated by the action of endocannabinoids. Finally, it is important to emphasize that our studies primarily pertain to the fatty acid amide subclass of endocannabinoids (e.g., anandamide), and the effects of ester-based endocannabinoids, such as 2-AG, which are catabolized by enzymes other than FAAH in vivo (Dinh et al., 2002; Lichtman et al., 2002), may be different.
The dramatic increase in endogenous levels of anandamide that accompanies excitotoxicity and neurodegeneration (Hansen et al., 2001b) has led to the suggestion that this endocannabinoid may display a neuroprotective function in vivo. Consistent with this notion, anandamide has been shown to reduce neuronal damage caused by hypoxia in cerebral cortical neuron cultures (Sinor et al., 2000) and oubain in rats in vivo (van der Stelt et al., 2001). However, in these studies, the neuroprotective effects of anandamide were not eliminated by SR141716A, suggesting either a non-CB1 site of action for this fatty acid amide or a possible neuroprotective function for its major metabolite arachidonic acid (Lauritzen et al., 2000). If anandamide itself possessed a neuroprotective function, a property that could conceivably be independent of its effects on seizure, we postulated that this activity should be augmented in FAAH (−/−) mice in which the effects of this endocannabinoid are greatly exaggerated. However, anandamide was not found to display a neuroprotective activity in FAAH (−/−) mice in response to either kainite- or bicuculline-induced seizures. Instead, in both of these seizures models, anandamide promoted hippocampal neuronal damage to a degree commensurate with its proconvulsant activity. In the bicuculline model, in which the effects of SR141716A could be experimentally examined, both the proconvulsant and neurotoxic activities of anandamide were blocked by pretreatment with this CB1 receptor antagonist. Interestingly, a recent study examining the role of CB1receptors in excitoxicity elicited by NMDA in the rat cortex also uncovered a neuroprotective function for SR141716A (Hansen et al., 2002). Collectively, these findings suggest that anandamide may promote rather than inhibit neuronal damage caused by excitoxic stimuliin vivo. However, for neuronal damage induced by other stimuli, such as cerebral ischemia, endocannabinoids may display neuroprotective effects, as is suggested by the increased severity of stroke in CB1 (−/−) mice (Parmentier-Batteur et al., 2002).
In summary, we found that, in two models of limbic seizures, the endocannabinoid anandamide exhibits FAAH-regulated proconvulsant and neurotoxic activity. These pharmacological findings, coupled with the enhanced kainate sensitivity of FAAH (−/−) mice, suggest that both exogenously applied and endogenously produced anandamide can promote neuronal hyperexcitability in the hippocampus. Accordingly, these experiments provide important evidence that the disinhibitory activity of endocannabinoids observed in vitroin electrophysiological studies of hippocampal slices also extends to the manner in which these lipids signal in vivo. Nonetheless, it is important to stress that anandamide and related endocannabinoids are not likely to produce a general disinhibition of neuronal activity in the hippocampus or, for that matter, other brain regions. Indeed, a growing body of evidence indicates that, even within the same brain region, endocannabinoids can exhibit either excitatory or inhibitory activity, depending on the neural circuits under examination (Kreitzer and Regehr, 2001b; Wilson and Nicoll, 2002). Regardless, the studies described herein emphasize the central role that FAAH plays in regulating the magnitude of the effects exerted by anandamide in vivo and therefore further promote this enzyme as a target for medicinal chemistry efforts aimed at manipulating endogenous cannabinoid tone for basic research and/or therapeutic purposes.
Footnotes
This work was supported by National Institute of Drug Abuse of the National Institutes of Health Grants DA13173 and DA15197, Allergan, Inc., the Helen L. Dorris Institute for the Study of Neurological and Psychiatric Disorders of Children and Adolescents, and the Skaggs Institute for Chemical Biology. We thank Tamas Bartfai, Floyd Bloom, and the entire Cravatt group for helpful discussions and critical analysis of the work described in this manuscript.
Correspondence should be addressed to Dr. Benjamin F. Cravatt, 10550 North Torrey Pines Road, La Jolla, CA 92037. E-mail:cravatt@scripps.edu.
References
- 1.Adams IB, Martin BR. Cannabis: pharmacology and toxicology in animals and humans. Addiction. 1996;91:1585–1684. [PubMed] [Google Scholar]
- 2.Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- 3.Bukoski RD, Batkai S, Jarai Z, Wang Y, Offertaler L, Jackson WF, Kunos G. CB(1) receptor antagonist SR141716A inhibits Ca(2+)-induced relaxation in CB(1) receptor-deficient mice. Hypertension. 2002;39:251–257. doi: 10.1161/hy0202.102702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 5.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 7.Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, Zimmer A, Martin BR. Levels, metabolism, and pharmacological activity of anandamide in CB1 cannabinoid receptor knockout mice: evidence for non-CB1, non-CB2 receptor-mediated actions of anandamide in mouse brain. J Neurochem. 2000;75:2434–2444. doi: 10.1046/j.1471-4159.2000.0752434.x. [DOI] [PubMed] [Google Scholar]
- 8.Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–825. doi: 10.1038/35071088. [DOI] [PubMed] [Google Scholar]
- 9.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egertova M, Giang DK, Cravatt BF, Elphick MR. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc R Soc Lond B Biol Sci. 1998;265:2081–2085. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Follesa P, Gale K, Mocchetti I. Regional and temporal pattern of expression of nerve growth factor and basic fibroblast growth factor mRNA in rat brain following electroconvulsive shock. Exp Neurol. 1994;127:37–44. doi: 10.1006/exnr.1994.1077. [DOI] [PubMed] [Google Scholar]
- 12.Guan X, Cravatt BF, Ehring GR, Hall JE, Boger DL, Lerner RA, Gilula NB. The sleep-inducing lipid oleamide deconvolutes gap junction communication and calcium wave transmission in glial cells. J Cell Biol. 1997;139:1785–1792. doi: 10.1083/jcb.139.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampson RE, Deadwyler SA. Cannabinoids reveal the necessity of hippocampal neural encoding for short-term memory in rats. J Neurosci. 2000;20:8932–8942. doi: 10.1523/JNEUROSCI.20-23-08932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen HS, Lauritzen L, Moesgaard B, Strand AM, Hansen HH. Formation of N-acyl-phosphatidylethanolamines and N-acetylethanolamines: proposed role in neurotoxicity. Biochem Pharmacol. 1998;55:719–725. doi: 10.1016/s0006-2952(97)00396-1. [DOI] [PubMed] [Google Scholar]
- 15.Hansen HH, Ikonomidou C, Bittigau P, Hansen SH, Hansen HS. Accumulation of the anandamide precursor and other N-acylethanolamine phospholipids in infant rat models of in vivo necrotic and apoptotic neuronal death. J Neurochem. 2001a;76:39–46. doi: 10.1046/j.1471-4159.2001.00006.x. [DOI] [PubMed] [Google Scholar]
- 16.Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, Ikonomidou C, Schmid HH, Fernandez-Ruiz J, Hansen HH. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem. 2001b;78:1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x. [DOI] [PubMed] [Google Scholar]
- 17.Hansen HH, Azcoitia I, Pons S, Romero J, Garcia-Segura LM, Ramos JA, Hansen HS, Fernandez-Ruiz J. Blockade of cannabinoid CB1 receptor function protects against in vivo disseminating brain damage following NMDA-induced excitotoxicity. J Neurochem. 2002;82:154–158. doi: 10.1046/j.1471-4159.2002.00961.x. [DOI] [PubMed] [Google Scholar]
- 18.Karler R, Cely W, Turkanis S. Anticonvulsant properties of delta 9-tetrahydrocannabinol and other cannabinoids. Life Sci. 1974;15:931–947. doi: 10.1016/0024-3205(74)90009-5. [DOI] [PubMed] [Google Scholar]
- 19.Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001a;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 21. Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci 21 2001b. RC174(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lauretti GR, Ahmad I, Pleuvry BJ. The activity of opioid analgesics in seizure models utilizing N-methyl-dl-aspartic acid, kainic acid, bicuculline and pentylenetetrazole. Neuropharmacology. 1994;33:155–160. doi: 10.1016/0028-3908(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 23.Lauritzen I, Blondeau N, Heurteaux C, Widmann C, Romey G, Lazdunski M. Polyunsaturated fatty acids are potent neuroprotectors. EMBO J. 2000;19:1784–1793. doi: 10.1093/emboj/19.8.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Bohme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- 25.Lichtman AH, Hawkins EG, Griffin G, Cravatt BF. The pharmacological activity of fatty acid amides is regulated, not mediated by fatty acid amide hydrolase. J Pharmacol Exp Ther. 2002;302:73–79. doi: 10.1124/jpet.302.1.73. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Cao Y, Basbaum AI, Mazarti AM, Sankar R, Wasterlain CG. Resistance to excitotoxin-induced seizures and neuronal death in mice lacking the preprotachykinin A gene. Proc Natl Acad Sci USA. 1999;96:12096–12101. doi: 10.1073/pnas.96.21.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin P, Consroe P. Cannabinoid induced behavioral convulsions in rabbits. Science. 1976;194:965–967. doi: 10.1126/science.982057. [DOI] [PubMed] [Google Scholar]
- 28.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 29.Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 30.Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci. 2002;22:9771–9775. doi: 10.1523/JNEUROSCI.22-22-09771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pistis M, Muntoni AL, Pillolla G, Gessa GL. Cannabinoids inhibit excitatory inputs to neurons in the shell of the nucleus accumbens: an in vivo electrophysiological study. Eur J Neurosci. 2002;15:1795–1802. doi: 10.1046/j.1460-9568.2002.02019.x. [DOI] [PubMed] [Google Scholar]
- 32.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 33.Shen M, Thayer SA. Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol. 1998;54:459–462. doi: 10.1124/mol.54.3.459. [DOI] [PubMed] [Google Scholar]
- 34.Sinor AD, Irvin SM, Greenberg DA. Endocannabinoids protect cerebral cortical neurons from in vitro ischemia in rats. Neurosci Lett. 2000;278:157–160. doi: 10.1016/s0304-3940(99)00922-2. [DOI] [PubMed] [Google Scholar]
- 35.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 36.Thomas E, Cravatt B, Danielson P, Gilula N, Sutcliffe J. Fatty acid amide hydrolase, the degradative enzyme for anandamide and oleamide, has selective distribution in neurons within the rat central nervous system. J Neurosci Res. 1997;50:1047–1052. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1047::AID-JNR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 37.Tsou K, Nogueron M, Muthian S, Sanudo-Pena M, Hillard C, Deutsch D, Walker J. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–140. doi: 10.1016/s0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- 38.van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisogno T, Bar PR, Veldink GA, Vliegenthart JF, Di Marzo V, Nicolay K. Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J Neurosci. 2001;21:8765–8771. doi: 10.1523/JNEUROSCI.21-22-08765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varvel SA, Lichtman AH. Evaluation of CB1 receptor knockout mice in the Morris water maze. J Pharmacol Exp Ther. 2002;301:915–924. doi: 10.1124/jpet.301.3.915. [DOI] [PubMed] [Google Scholar]
- 40.Wallace MJ, Wiley JL, Martin BR, DeLorenzo RJ. Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur J Pharmacol. 2001;428:51–57. doi: 10.1016/s0014-2999(01)01243-2. [DOI] [PubMed] [Google Scholar]
- 41.Wallace MJ, Martin B, DeLorenzo R. Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur J Pharmacol. 2002;452:295–301. doi: 10.1016/s0014-2999(02)02331-2. [DOI] [PubMed] [Google Scholar]
- 42.Willoughby KA, Moore SF, Martin BR, Ellis EF. The biodisposition and metabolism of anandamide in mice. J Pharmacol Exp Ther. 1997;282:243–247. [PubMed] [Google Scholar]
- 43.Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 44.Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- 45.Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]