

Abstract
Inhibitors of both phosphatidylinositol-3-kinase (PI3-kinase) and MAPK/ERK (mitogen-activate protein kinase/extracellular signal-related kinase) activation inhibit NMDA receptor-dependent long-term potentiation (LTP). PI3-kinase inhibitors also block activation of ERK by NMDA receptor stimulation, suggesting that PI3-kinase inhibitors block LTP because PI3-kinase is an essential upstream regulator of ERK activation. To examine this hypothesis, we investigated the effects of PI3-kinase inhibitors on ERK activation and LTP induction in the CA1 region of mouse hippocampal slices. Consistent with the notion that ERK activation by NMDA receptor stimulation is PI3-kinase dependent, the PI3-kinase inhibitor wortmannin partially inhibited ERK2 activation induced by bath application of NMDA and strongly suppressed ERK2 activation by high-frequency synaptic stimulation. PI3-kinase and MEK (MAP kinase kinase) inhibitors had very different effects on LTP, however. Both types of inhibitors suppressed LTP induced by theta-frequency trains of synaptic stimulation, but only PI3-kinase inhibitors suppressed the induction of LTP by high-frequency stimulation or low-frequency stimulation paired with postsynaptic depolarization. Concentrations of PI3-kinase inhibitors that inhibited LTP when present during high-frequency stimulation had no effect on potentiated synapses when applied after high-frequency stimulation, suggesting that PI3-kinase is specifically involved in the induction of LTP. Finally, we found that LTP induced by theta-frequency stimulation was MEK inhibitor insensitive but still PI3-kinase dependent in hippocampal slices from PSD-95 (postsynaptic density-95) mutant mice. Together, our results indicate that the role of PI3-kinase in LTP is not limited to its role as an upstream regulator of MAPK signaling but also includes signaling through ERK-independent pathways that regulate LTP induction.
Keywords: long-term potentiation, hippocampus, phosphatidylinositol 3-kinase, extracellular signal-related kinase II, NMDA receptor, β-adrenergic receptor
Introduction
The small GTPase Ras as well as Ras regulators and effectors are associated with NMDA-type glutamate receptors in the postsynaptic density of excitatory synapses (Chen et al., 1998; Kim et al., 1998; Hisatsune et al., 1999; Husi et al., 2000). Although the role of Ras in NMDA receptor-mediated signaling has not been well characterized, several findings indicate that Ras signaling pathways have an important role in NMDA receptor-dependent forms of synaptic plasticity, such as long-term potentiation (LTP). For instance, mice with mutations affecting H-Ras (Manabe et al., 2000) or the Ras GTPase-activating proteins NF1 (neurofibromin) (Costa et al., 2002) and SynGAP (a synaptic Ras-GTPase activating protein) (Komiyama et al., 2002) have alterations in hippocampal LTP. Moreover, pharmacological inhibition of the Ras effectors phosphatidylinositol 3-kinase (PI3-kinase) (Kelly and Lynch, 2000; Lin et al., 2001; Raymond et al., 2002; Sanna et al., 2002) and the p44/42 MAPK (mitogen-activated protein kinase) pathway (for review, see Sweatt, 2001; Adams and Sweatt, 2002) disrupts LTP.
Although Ras-activated signaling pathways are clearly involved in LTP, the molecular details of how these pathways contribute to an enhancement of synaptic strength remain unclear and, in some cases, controversial. For example, whereas the enhancement of LTP in H-Ras mutant mice suggests that Ras activation normally suppresses LTP induction (Manabe et al., 2000), the inhibition of LTP in hippocampal pyramidal cells expressing a dominant-negative form of Ras (Zhu et al., 2002) suggests just the opposite, i.e., Ras activation is required for LTP induction. The role of PI3-kinase in LTP is also poorly understood. Activation of the MAPK ERK2 (extracellular signal-related kinase 2) by NMDA receptor stimulation is completely dependent on PI3-kinase activity in cultured neurons (Chandler et al., 2001; Perkinton et al., 2002), suggesting that PI3-kinase inhibitors suppress LTP because PI3-kinase activity is required for NMDA receptor-mediated ERK activation. However, in non-neuronal cells, the requirement for PI3-kinase activity in ERK activation is very dependent on experimental conditions (Duckworth and Cantley, 1997; Wennström and Downward, 1999; Moelling et al., 2002), and, in some cases, PI3-kinase activation can inhibit, rather than facilitate, ERK activation (Rommel et al., 1999; Zimmermann and Moelling, 1999). These complex interactions between PI3-kinase and ERK signaling pathways have not been examined in neurons. Thus, it is not clear whether PI3-kinase inhibitors suppress LTP solely because PI3-kinase activation is required for NMDA receptor-dependent ERK activation or because LTP is also dependent on PI3-kinase signaling through ERK-independent mechanisms.
In the experiments described here, we examined the role of PI3-kinase and ERK in LTP at excitatory synapses in the CA1 region of the mouse hippocampus. Consistent with the notion that PI3-kinase links NMDA receptors to the ERK pathway, PI3-kinase inhibitors significantly reduced both NMDA and high-frequency stimulation (HFS)-induced increases in ERK2 phosphorylation. We found, however, that PI3-kinase inhibitors suppress LTP under conditions in which blocking ERK activation with MEK (MAP kinase kinase) inhibitors has no effect. Thus, although PI3-kinase contributes to NMDA receptor-mediated ERK activation, our results demonstrate that the induction of LTP is also dependent on PI3-kinase signaling through ERK-independent pathways.
Materials and Methods
Slice preparation and electrophysiology. Standard techniques were used to prepare slices (400 μm thick) from the hippocampus of halothane-anesthetized C57BL/6 mice (male, 5–8 weeks old). Slices were maintained in interface-type chambers (Fine Science Tools, Foster City, CA) that were continuously perfused (at 2–3 ml/min) with a warm (30°C), oxygenated (95% O2–5% CO2) artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 4.4 KCl, 25 Na2HCO3, 1 NaH2PO4, 1.2 MgSO4, 2 CaCl2, and 10 glucose. In our initial experiments, field EPSPs (fEPSPs) were recorded in slices maintained in an interface-type recording chamber, whereas in later experiments, extracellular recordings were done using slices completely submerged in ACSF. The type of recording configuration, which is noted in the figure legends, had no apparent affect on our observations.
Standard extracellular recording techniques were used to record fEPSPs in the CA1 region of hippocampal slices (for review, see Thomas et al., 1998; Watabe et al., 2000). Presynaptic fiber stimulation pulses were delivered at 0.02 Hz using a stimulation strength that evoked fEPSPs that were approximately half of the maximal fEPSP amplitude that could be evoked by strong intensity stimulation. LTP was induced using short-duration trains of theta pulse stimulation (TPS) (150 pulses of presynaptic stimulation delivered at 5 Hz), long-duration trains of TPS (900 pulses) delivered in the presence of the β-adrenergic receptor agonist isoproterenol (ISO) (1 μm), or two trains of HFS (100 pulses of 100 Hz stimulation; intertrain interval of 10 sec). The average of fEPSP slopes recorded between 40 and 45 min after TPS or between 55 and 60 min after HFS were used for statistical comparisons.
Whole-cell current-clamp recordings were used to examine the effects of various inhibitors on LTP induced by pairing low-frequency presynaptic fiber stimulation with postsynaptic depolarization. In these experiments, slices were bathed in a modified ACSF in which the concentrations of CaCl2 and MgSO4 were increased to 4 mm each, the concentration of KCl was reduced to 2.2 mm, and picrotoxin (100 μm) was added to block GABAA-mediated inhibitory postsynaptic potentials. The CA3 region of the slices was removed to prevent spontaneous bursting. Low-resistance (5–10 MΩ) patch-clamp electrodes filled with a solution containing 122.5 mmCs-gluconate, 17.5 mm CsCl, 10 mmtetraethylammonium (TEA)-Cl, 0.2 mm EGTA, 10 mm HEPES, 2 mm Mg-ATP, and 0.3 mmGTP, pH 7.2, were used to record EPSPs evoked by 0.05 Hz presynaptic fiber stimulation. Current was injected into the cells throughout the experiment to hyperpolarize membrane potentials to between −80 and −90 mV and thus minimize action potential generation by potentiated EPSPs. An additional 50-msec-long, 0.1 nA pulse of hyperpolarizing current was injected 150 msec after each evoked EPSP to monitor input and access resistance. At the start of each experiment, the intensity of presynaptic fiber stimulation was adjusted to evoke EPSPs between 5 and 10 mV in amplitude. After a 10 min period of baseline recording, current injected through the recording electrode was used to depolarize the postsynaptic cell to near 0 mV, and LTP was induced by pairing this tonic depolarization with 100 presynaptic fiber stimulation pulses delivered at 2 Hz. Pairing was done within 20 min of obtaining whole-cell recordings to minimize the “wash-out” of LTP that can occur during whole-cell recordings. Statistical significance of results from electrophysiological experiments was assessed using ANOVAs (followed by Bonferroni t tests) or paired and unpairedt tests (two-tailed).
Western immunoblotting. Hippocampal slices were prepared as described above and allowed to recover for at least 2 hr before an experiment. For each experiment, slices obtained from the same animal were placed into four separate interface type chambers (typically three slices were loaded per chamber) and constantly perfused at 2 ml/min with warm (30°C) ACSF. The slices in one chamber were exposed to ACSF alone and served as untreated controls, whereas the slices in the other three chambers were exposed ACSF containing various combinations of agonists and antagonists. To examine the effects of NMDA receptor activation on ERK2 activation, we first conducted a series of preliminary experiments in which we measured phospho-ERK2 levels in slices exposed to different concentrations of NMDA (1–100 μm) for various periods of time (2.5–10 min). A concentration (20 μm) and incubation time (5 min) of NMDA that reliably induced a robust, but submaximal, increase in phospho-ERK2 levels was then used for all subsequent experiments. Similar preliminary experiments were also performed to find an appropriate dose (1.0 μm) and incubation time (5 min) for activation of ERK2 by the β-adrenergic receptor agonist ISO. After drug treatments, the slices were rapidly frozen on dry ice and stored for up to 1 week at −80°C.
CA1 “mini-slices” containing just the CA1 region of the hippocampus were used to examine changes in ERK activation induced by high-frequency synaptic stimulation. In these experiments, the CA3, dentate gyrus, and subiculuar regions of freshly cut hippocampal slices were removed to produce isolated CA1 regions. After a 2 hr recovery period, we determined the health of each mini-slice by examining postsynaptic responses evoked by low-frequency presynaptic stimulation pulses delivered by a stimulating electrode placed in stratum radiatum. Slices that showed evidence of epileptiform activity (evoked and/or spontaneous bursting) were discarded. Three mini-slices were then each stimulated with two trains of HFS (100 pulses delivered at 100 Hz; intertrain interval of 10 sec) using a stimulation intensity sufficient to evoke a 2–6 mV population spike. These mini-slices were pooled (times after HFS ranged from 2.5 to 5 min) and rapidly frozen on dry ice. CA1 mini-slices that received low-frequency stimulation alone served as controls.
For Western analysis, the slices were homogenized in ice-cold buffer containing 50 mm Tris-HCl, 50 mm NaCl, 10 mm EGTA, 10 mm EDTA, 80 μm sodium molybdate, 5 mm sodium pyrophosphate, 1 mmsodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 0.01% Triton X-100, 4 mm para-nitrophenylphosphate, and Protease Inhibitors Complete (Roche Molecular Biochemicals, Indianapolis, IN). A small aliquot (2 μl) was removed from each sample for protein determination using the Bradford method, and 2× loading buffer was added to the rest of the sample. Samples were boiled for 3 min and then loaded (20–40 μg of protein per lane) onto 12% SDS-PAGE gels. After electrophoresis, proteins were transferred onto either nitrocellulose (Protran; Schleicher and Schuell, Keene, NH) or polyvinylidene difluoride (Immun-Blot; Bio-Rad, Hercules, CA) membranes. Blots were incubated in a blocking buffer consisting of Tris-buffered saline containing 0.05% Tween 20 (TBST) plus either 4% nonfat dry milk or 5% bovine serum albumin for 1 hr and then incubated overnight (at 4°C) with primary antibodies diluted in blocking buffer. After this, the blots were washed three times with TBST and then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (usually at a dilution of 1:2000 in blocking buffer) for 1–2 hr. Proteins were visualized using enhanced chemiluminescence [ECL Western Blotting Analysis System (Amersham, Arlington Heights, IL) or Immun-Star HRP detection kit (Bio-Rad)]. Images of the blots were acquired using a cooled CCD camera-based image acquisition system (Chemi-Doc; Bio-Rad), and densitometric analysis was performed using the Quantity One software package (Bio-Rad). Primary antibodies to phospho-p44/p42 MAPK (Thr202/Tyr204, 1:2000), total p44/p42 MAPK (1:1000), phospho-glutamate receptor type 1 (GluR1) (S845, 1:1000), phospho-Akt (Thr308, 1:500), and total Akt (1:1000) were obtained from Cell Signaling Technology (Beverly, MA). A primary antibody to a neuronal-specific isoform (βIII) of tubulin (1:5000) was obtained from Upstate Biotechnology (Lake Placid, NY). To control for potential variations in loading, the optical density of bands for each protein of interest were first normalized to the optical density values obtained for tubulin bands in each lane. These normalized values were then expressed as a percentage of the levels seen in untreated control slices. In some experiments, signals were normalized to actin levels determined using an anti-actin antibody (10 μg/ml) obtained from Chemicon (Temecula, CA). One-way ANOVAs or, when appropriate, Kruskal–Wallis one-way ANOVA on ranks were used to assess statistical significance of Western blot data. In both cases, Student–Newman–Keuls tests were used for multiple pairwise comparisons.
Results
PI3-kinase inhibitors suppress NMDA receptor-dependent activation of the MAPK pathway
As shown in Figure 1, phospho-ERK2 levels were increased more than threefold in hippocampal slices exposed to 20 μm NMDA for 5 min. Although the increase in phospho-ERK2 levels induced by NMDA was significantly (p < 0.01) smaller in slices pretreated with the PI3-kinase inhibitor wortmannin, this increase was surprisingly robust (more than twofold) and statistically significant compared with basal phospho-ERK2 levels in untreated control slices (p < 0.001). Thus, although PI3-kinase inhibitors completely block NMDA-induced ERK activation in cultured neurons (Chandler et al., 2001; Perkinton et al., 2002), our results suggest that activation of the ERK pathway by NMDA receptor stimulation is only partially PI3-kinase dependent in hippocampal slices from young adult animals.
Fig. 1.

NMDA-induced increases in phosphorylated ERK2 are only partially PI3-kinase dependent. A, Average ± SEM results from nine experiments in which slices from the same animal were either left untreated (open bar) or exposed to 20 μmNMDA (5 min), 200 nm wortmannin (Wort) (≥40 min), or NMDA plus wortmannin. Phospho-ERK2 levels were significantly increased by NMDA in both control and wortmannin-treated slices (*p < 0.01 compared with untreated controls). The increase in phospho-ERK2 levels induced by NMDA in wortmannin-treated slices (250 ± 25.2% of untreated control slices) was significantly less than that induced by NMDA in slices bathed in ACSF (341 ± 30.4% of untreated control slices; #p < 0.05). Wortmannin did not have a significant effect on basal levels of phospho-ERK2. Total levels of ERK2 were unchanged in slices exposed to wortmannin alone, NMDA alone, or wortmannin plus NMDA (levels were 108 ± 5.5, 116 ± 6.4, and 108.3 ± 8.1% of that seen in untreated control slices, respectively). B, Results from individual experiments summarized in A. The plot shows the NMDA-induced increase in phospho-ERK2 levels in slices bathed in ACSF (control) versus ACSF plus 200 nm wortmannin (filled symbols) or 5 μm wortmannin (open symbols). C, Wortmannin inhibits NMDA-induced increases in phospho-Akt in hippocampal slices. Average ± SEM results from nine experiments. In the absence of wortmannin, NMDA (20 μm, 5 min) increased phospho-Akt levels to 214 ± 58% of baseline (*p < 0.05 compared with untreated control). Wortmannin reduced basal levels of phospho-Akt to 15.4 ± 6.2% of control (p < 0.05 compared with untreated controls) and completely blocked NMDA-induced increases in phospho-Akt (phospho-Akt levels were 9.3 ± 2% of control in wortmannin-treated slices exposed to NMDA). D, Representative immunoblots showing the effects of wortmannin on levels of phospho-ERK1/2 and phospho-Akt (Thr308) in untreated (UT) and NMDA treated (N) slices.
One possible explanation for the modest effect of wortmannin on NMDA-induced ERK activation observed in our experiments is that PI3-kinase activity was not adequately inhibited by the wortmannin concentration and/or incubation time (≥40 min) used in our experiments. To address this possibility, we performed two additional, control experiments. Activation and phosphorylation of the protein kinase Akt is typically PI3-kinase dependent (Toker, 2000), and changes in levels of Akt phosphorylated at S473 or Thr308 can been used as a convenient assay of PI3-kinase activity (Lin et al., 2001; Sanna et al., 2002). We thus used an antibody that specifically recognizes Thr308 phosphorylated Akt to measure levels of activated Akt in slices treated with NMDA and/or wortmannin. As shown in Figure 1, Cand D, phospho-Akt was readily detected in untreated control slices, and levels were increased approximately twofold in NMDA-treated slices. Wortmannin (200 nm, 40 min) strongly reduced basal levels of phospho-Akt and completely prevented the increase in Akt phosphorylation induced by NMDA. This indicates that 200 nm wortmannin effectively inhibits PI3-kinase in hippocampal slices. As an additional control, we also examined whether incubating slices in ACSF containing a 25-fold higher concentration of wortmannin (5 μm) might produce a more robust inhibition of NMDA-induced ERK2 activation. As shown by the open symbols in Figure 1B, increasing the wortmannin concentration to 5 μm did not produce a stronger inhibition of NMDA-induced MAPK activation. In these experiments, phospho-ERK2 levels were increased to 350 ± 36% of control in slices exposed to NMDA alone and were increased to 227 ± 43% of control in slices exposed to NMDA in the presence of 5 μm wortmannin (n = 3).
Although these results indicate that activation of ERK2 by NMDA receptor stimulation is partially PI3-kinase dependent in hippocampal slices from adult animals, bath-applied NMDA will activate both synaptic and extrasynaptic receptors. Because these two pools of NMDA receptors can couple to distinct signaling pathways (Hardingham et al., 2002), we also examined whether PI3-kinase inhibitors suppress activation of ERK2 by patterns of synaptic stimulation that activate synaptic NMDA receptors and induce NMDA receptor-dependent LTP. In addition, we used isolated CA1 mini-slices in these experiments to lessen the possibility that our biochemical results might be confounded by regional differences in the types of signaling pathways that couple NMDA receptors to the ERK pathway. In control experiments (normal ACSF), HFS reliably induced a statistically significant (p < 0.05) increase in levels of phospho-ERK2 with no effect on total ERK2 levels (Fig.2). In contrast, HFS in wortmannin-treated slices failed to induce a statistically significant increase in phospho-ERK2 levels compared with unstimulated control slices (Fig. 2). These results indicate that activation of ERK pathway by LTP-inducing patterns of synaptic stimulation in the hippocampal CA1 region is strongly dependent on PI3-kinase activity.
Fig. 2.

Blocking PI3-kinase with wortmannin inhibits HFS-induced ERK2 activation. A, Western immunoblots showing the effect of HFS delivered in the presence and absence of wortmannin (Wort) on phospho-ERK1/2 and total ERK1/2 levels in CA1 mini-slices. Lane 1, Untreated control (ACSF alone, no HFS); lane 2, HFS in ACSF; lane 3, 200 nm wortmannin alone; lane 4, HFS in wortmannin. Note that, although HFS delivered to slices bathed in normal ACSF induced a robust increase in phospho-ERK2 levels, it had little effect on phospho-ERK2 levels in wortmannin-treated slices.B, C, Average ± SEM results from seven experiments showing basal and stimulated (2.5–5 min after HFS) levels of phospho-ERK2 (B) and total ERK2 (C) in slices bathed in normal ACSF or ACSF containing 200 nm wortmannin. High-frequency stimulation delivered to slices bathed in ACSF increased phospho-ERK2 levels to 182 ± 19% of unstimulated controls (*p < 0.05 compared with unstimulated controls). High-frequency stimulation-induced ERK activation was significantly reduced in wortmannin-treated slices (levels were 125 ± 16% of unstimulated controls; #p < 0.05 compared with HFS in ACSF) and not significantly different from levels of phospho-ERK2 in unstimulated slices bathed in ACSF alone or ACSF plus wortmannin. None of the treatments had any effect on total ERK2 levels.
PI3-kinase inhibitors suppress ERK-dependent forms of LTP
To examine whether PI3-kinase and ERK signaling might have distinct roles in NMDA receptor-dependent forms of synaptic plasticity, we compared the effects of MEK and PI3-kinase inhibitors on LTP induced by different patterns of synaptic stimulation. In the CA1 region of mouse hippocampal slices, the induction of LTP by a short train (150 pulses) of TPS alone or by a longer train (900 pulses) of TPS delivered in the presence of the β-adrenergic receptor agonist ISO is especially sensitive to inhibitors of ERK activation (Winder et al., 1999; Watabe et al., 2000). Thus, to examine whether PI3-kinase is required for the induction and/or maintenance of ERK-dependent forms of LTP, we first investigated whether PI3-kinase inhibitors suppressed the amount of potentiation induced by these TPS protocols.
As shown in Figure 3A, the PI3-kinase inhibitor LY294002 strongly suppressed LTP induced by a 150 pulse train of TPS. In control experiments, fEPSPs were potentiated to 167.9 ± 9.5% of baseline after TPS (n = 6), whereas fEPSPs were just 109.1 ± 5.9% of baseline after TPS in slices continuously bathed in ACSF containing 20 μm LY294002 (n = 6). The inhibition of LTP by LY294002 is most likely not attributable to direct effects on NMDA receptors because previous studies have found that LY294002 and wortmannin do not inhibit NMDA receptor-mediated responses in hippocampal neurons (Shanley et al., 2001; Sanna et al., 2002). Moreover, at this concentration, LY294002 had no obvious effect on basal synaptic transmission (data not shown) or on synaptic transmission during the TPS train (Fig. 3B). LY294002 also had no effect on the number of EPSP-evoked complex spike bursts that were evoked during the TPS train [complex spike bursts were elicited by 77.3 ± 2.7% of the EPSPs during the TPS train in control slices (n = 6) and by 77.2 ± 1.6% of the EPSPs during the TPS train in LY294002-treated slices (n = 6)]. This indicates that LY294002 does not inhibit LTP induction through indirect effects on postsynaptic excitability (Thomas et al., 1998). As described below (see Fig. 9), TPS-induced LTP was also inhibited by wortmannin (200 nm).
Fig. 3.

The PI3-kinase inhibitor LY294002 suppresses TPS-induced LTP. A, A 150 pulse train of TPS delivered at time 0 induced LTP in vehicle control experiments (0.1% DMSO; open symbols; n = 6) but had little lasting effect on synaptic transmission in slices treated with 20 μmLY294002 for at least 40 min before TPS (filled symbols;n = 6). LY294002 was present throughout the experiment. Inset shows fEPSPs recorded during baseline and 45 min after TPS in a control slice (left set of traces) and in a slice bathed in LY294002 (right set of traces). Calibration: 2 mV, 5 msec.B, LY294002 has no effect on synaptic responses evoked during TPS. Note that both the facilitation at the start of TPS and the depression of synaptic transmission at the end of the TPS train are similar in control (open symbols) and LY294002-treated slices (filled symbols). On average, fEPSPs elicited by pulse 2 to pulse 6 of the TPS train facilitated to 147.6 ± 6.1% of baseline in control experiments and were facilitated to 145.8 ± 4.8% of baseline in LY294002-treated slices (not significantly different;p = 0.82). In control experiments, fEPSPs were depressed to 37.1 ± 6.2% of baseline at the end of the TPS train (average of the last 5 stimulation pulses) and were depressed to 45.2 ± 5.8% of baseline in LY294002-treated slices (not significantly different; p = 0.363). Recordings were done using slices maintained in an interface chamber.
Fig. 9.

TPS-induced LTP is ERK independent but still PI3-kinase dependent in hippocampal slices from PSD-95 mutant mice.A, Both MEK and PI3-kinase inhibitors suppress the induction of LTP by a 150 pulse train of TPS in slices from wild-type animals. In vehicle control experiments (0.1- 0.2% DMSO; open circles), fEPSPs were potentiated to 205 ± 7% of baseline 45 min after TPS (n = 9). The amount of potentiation induced by TPS was significantly reduced (p< 0.001) in slices continuously bathed in ACSF containing either 200 nm wortmannin (Wort; filled circles; fEPSPs were 125 ± 11% of baseline; n = 6) or 20 μmU0126 (filled triangles; fEPSPs were 137 ± 9% of baseline;n = 11). B, The amount of LTP induced by a 150 pulse train of TPS was not reduced by U0126 in slices from PSD-95 mutant mice. In vehicle control experiments, fEPSPs were potentiated to 279 ± 15% of baseline (open circles;n = 14) and were potentiated to 270 ± 12% of baseline (n = 14) in slices continuously bathed in ACSF containing U0126 (triangles). LTP induced by TPS in PSD-95 mutant slices was inhibited by wortmannin. In slices from PSD-95 mutant mice that were continuously bathed in 200 nm wortmannin, fEPSPs were potentiated to 195 ± 13% of baseline (filled circles;n = 7; p < 0.05 compared with control levels of LTP in PSD-95 mutant slices). All experiments were done in an interface-type recording chamber.
Although the induction of LTP by a short, 150-pulse-long train of TPS by itself induces LTP, longer trains of TPS (300–900 pulses) only induce significant levels of potentiation when combined with β-adrenergic receptor activation (Thomas et al., 1996; Winder et al., 1999). Because the induction of LTP by TPS paired with β-adrenergic receptor activation is also ERK dependent (Winder et al., 1999), we examined whether PI3-kinase inhibitors inhibit LTP induced by this pattern of synaptic stimulation as well. As shown in Figure4, in control experiments (n = 15), fEPSPs were potentiated to 189.7 ± 7.9% of baseline 45 min after 900 pulses of TPS delivered at the end of a 10 min application of the β-adrenergic receptor agonist ISO. In contrast, TPS in the presence of ISO induced a significantly smaller potentiation of synaptic transmission in wortmannin-treated slices (fEPSPs were potentiated to 165.7 ± 6.8% of baseline;n = 14; p < 0.05 compared with control). Wortmannin also significantly reduced ISO-induced increases in phospho-ERK2 levels in hippocampal slices (Fig.4B), a finding consistent with a recent report showing that cAMP-induced activation of ERK is PI3-kinase dependent in neurons (Lin et al., 2001). Although both of these findings are consistent with the hypothesis that PI3-kinase activity is required for ERK-dependent forms of plasticity, both TPS-induced LTP (Thomas et al., 1996) and ISO-induced ERK activation are protein kinase A (PKA) dependent (Roberson et al., 1999). Thus, the inhibition of both ISO-induced ERK2 activation and LTP induced by TPS in the presence of ISO might be attributable to nonselective effects of wortmannin on PKA. We thus examined whether 200 nm wortmannin interfered with β-adrenergic receptor activation-induced increases in phosphorylation of the GluR1 subunit of AMPA receptors at a site phosphorylated by PKA (Ser845). As shown in Figure 4C, the increase in phospho-GluR1 levels induced by ISO was not altered in wortmannin-treated slices, suggesting that wortmannin does not appreciably affect PKA under our experimental conditions.
Fig. 4.
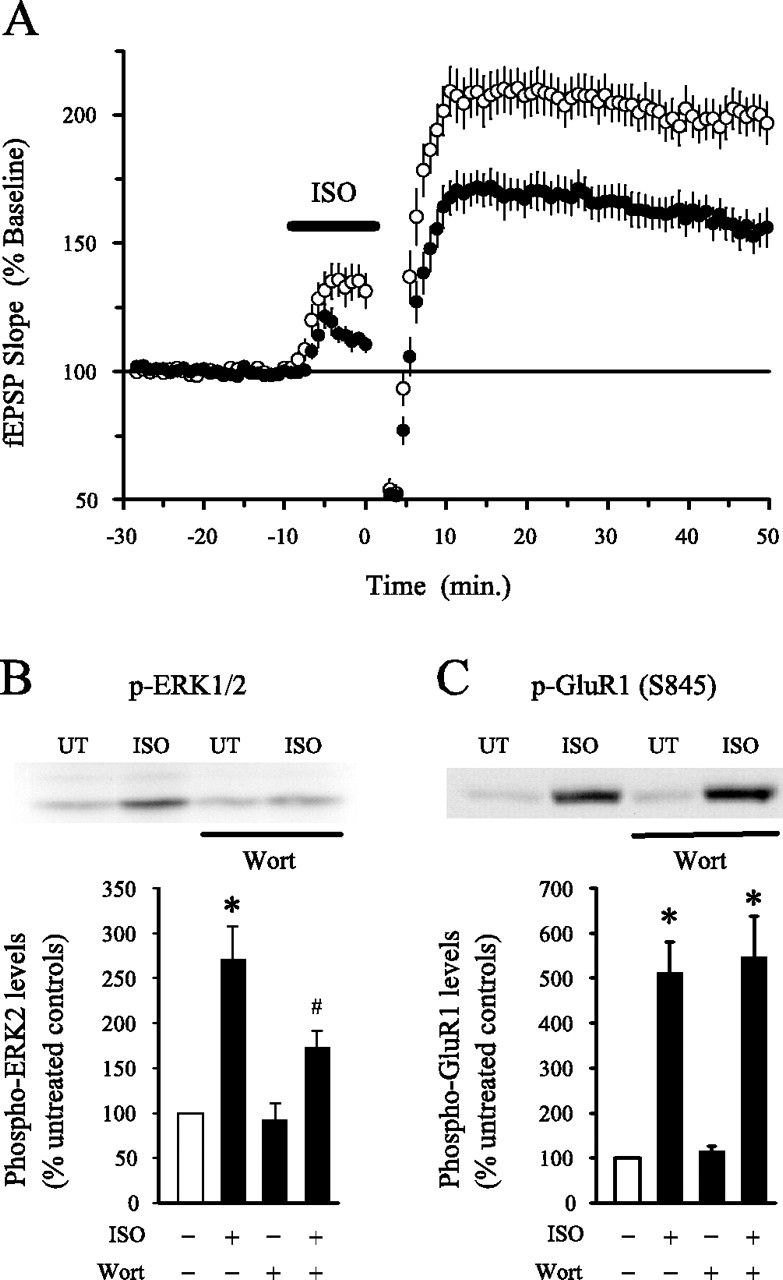
PI3-kinase inhibitors suppress the induction of LTP by long trains of TPS paired with β-adrenergic receptor activation and inhibit ISO-induced ERK2 activation. A, Nine hundred pulses of TPS (delivered at time 0) paired with a 10 min application of 1 μm ISO (indicated by the bar) induced significantly less potentiation in slices continuously bathed in 200 nm wortmannin (filled symbols; n = 14) compared with slices bathed in normal ACSF plus vehicle (0.1% DMSO; open symbols; n = 15). A long train of TPS delivered in the absence of ISO had little lasting effect on synaptic transmission in control and wortmannin-treated slices (data not shown).B, Wortmannin (Wort) inhibits ISO-induced ERK2 activation. Average ± SEM results from five separate experiments in which slices from the same animal were either left untreated (UT; open bar) or exposed to 1 μm ISO (5 min), 200 nm wortmannin (≥40 min), or ISO plus wortmannin. In vehicle control experiments (0.01–0.1% DMSO), phospho-ERK2 levels were increased to 250 ± 40.2% of control in ISO-treated slices (*p < 0.05 compared with untreated controls). A significantly smaller increase in phospho-ERK2 levels was induced by ISO in wortmannin-treated slices (phospho-ERK2 levels were increased to 164.1 ± 17.8% of control; #p < 0.05 compared with levels in slices treated with ISO alone). Total levels of ERK2 were unchanged in slices exposed to wortmannin alone, ISO alone, or wortmannin plus ISO (data not shown). The inset shows a representative immunoblot showing basal and ISO-stimulated levels of phospho-ERK2 in slices bathed in ACSF alone and ACSF plus 200 nm wortmannin. C, Wortmannin has no effect on the increase in phospho-S845 GluR1 levels induced by ISO. In slices bathed in ACSF, a 5 min application of 1.0 μm ISO increased phospho-GluR1 levels to 512 ± 69% of control (n = 5). In wortmannin-treated slices, phospho-GluR1 levels were 547.7 ± 90.4% of control. The inset shows a representative immunoblot showing basal and ISO-stimulated levels of phospho-S845 GluR1 in slices bathed in ACSF and ACSF plus wortmannin.
PI3-kinase inhibitors suppress ERK-independent forms of LTP
Previously, we found that the MEK inhibitor SL327 has no effect on the induction of LTP by low-frequency presynaptic fiber stimulation paired with postsynaptic depolarization, suggesting that the induction of LTP by this stimulation protocol is MAPK independent (Watabe et al., 2000). If PI3-kinase inhibitors block LTP solely because they prevent NMDA receptor-mediated ERK activation, then PI3-kinase inhibitors should have no effect on pairing-induced LTP. We thus examined whether PI3-kinase inhibitors affect pairing-induced LTP to determine whether ERK-independent forms of LTP are also PI3-kinase dependent. To confirm that this stimulation protocol induces an ERK-independent form of potentiation, we first examined whether a different MEK inhibitor, U0126, affects pairing-induced LTP. As shown in Figure5B, the amount of potentiation observed 30 min after pairing postsynaptic depolarization with low-frequency presynaptic fiber stimulation in cells recorded from slices bathed in ACSF containing 20 μm U0126 was not significantly different from the potentiation seen in cells recorded from slices bathed in ACSF plus 0.2% DMSO. To confirm that 20 μm U0126 effectively blocked NMDA receptor-mediated ERK2 activation under our experimental conditions, we examined the effects of U0126 on NMDA-induced increases in ERK2 phosphorylation in hippocampal slices. As shown in Figure5A, U0126 strongly reduced basal levels of phospho-ERK2 and blocked increases in ERK2 phosphorylation induced by a 5 min bath application of 20 μm NMDA. Thus, it seems unlikely that the inability of U0126 to block pairing-induced LTP can be attributed to an insufficient level of MEK inhibition. In contrast to U0126, pairing-induced LTP was strongly suppressed in cells in which the PI3-kinase inhibitor LY294002 was present in the recording electrode solution (Fig. 5B) (p< 0.001 compared with the potentiation induced in control cells). The differential sensitivity of pairing-induced LTP to MEK and PI3-kinase inhibitors indicates that the effects of PI3-kinase inhibitors on LTP are not simply attributable to alterations in ERK signaling. Moreover, because LY294002 was selectively delivered into the postsynaptic CA1 pyramidal cells, our results suggest that postsynaptic PI3-kinase activity is especially important for LTP.
Fig. 5.

Low-frequency presynaptic fiber stimulation paired with postsynaptic depolarization induces an ERK-independent but PI3-kinase inhibitor-sensitive form of LTP. A, The MEK inhibitor U0126 (20 μm) blocks NMDA-induced ERK2 activation. Western immunoblot showing phospho-ERK2 levels (top) and total ERK levels (bottom) in a representative experiment. Lane 1, Untreated control (slices exposed to ACSF alone); lane 2, NMDA (20 μm, 5 min) in ACSF; lane 3, 20 μm U0126 alone (≥40 min); lane 4, U0126 plus NMDA. Note that U0126 strongly reduces basal levels of phospho-ERK2 and blocks the increase in ERK2 phosphorylation induced by NMDA. None of the treatments had an affect on total ERK2 levels. The same results were obtained in three separate experiments. B, Pairing-induced LTP is not blocked by U0126 but is inhibited by LY294002. Presynaptic stimulation pulses paired with postsynaptic depolarization to near 0 mV (at time 0) induced nearly identical levels of potentiation in vehicle control experiments (0.1–0.2% DMSO; open circles; EPSPs were potentiated to 267 ± 13% of baseline; n = 13) and in cells recorded from slices continuously bathed in ACSF containing 20 μm U0126 (triangles; EPSPs were potentiated to 275 ± 13% of baseline; n = 9). Significantly less LTP was induced, however, in cells in which LY294002 (100 μm) was included in the electrode filling solution to block postsynaptic PI3-kinase (filled circles; EPSPs were potentiated to 159 ± 22% of baseline; n = 8; p < 0.001 compared with vehicle control cells). The inset shows EPSPs (average of 3 responses) recorded during baseline (smaller responses) and 30 min after pairing. Calibration: 5 mV, 20 msec.
In agreement with previous reports (Winder et al., 1999; Watabe et al., 2000), we also found that MEK inhibitors do not block HFS-induced LTP in mouse hippocampal slices (Liu et al., 1999). As shown in Figure 6A, the MEK inhibitor SL327 (10 μm) had no effect on the amount of potentiation present 60 min after HFS, although this concentration of SL327 completely blocked HFS-induced increases in levels of phospho-ERK2 in CA1 mini-slices. In contrast, blocking PI3-kinase with either LY294002 (Fig. 6B) or wortmannin (Fig.7A) strongly inhibited HFS-induced LTP. Thus, PI3-kinaseinhibitors not only suppress ERK-dependent forms of LTP but also inhibit LTP induced by patterns of synaptic stimulation (pairing, HFS) that induce an ERK-independent form of LTP. This later finding is difficult to reconcile with the simple notion that PI3-kinase activity is required for LTP only because PI3-kinase is an essential link between NMDA receptors and ERK activation. Instead, these observations indicate that PI3-kinase and ERK signaling have distinct roles in NMDA receptor-dependent LTP.
Fig. 6.
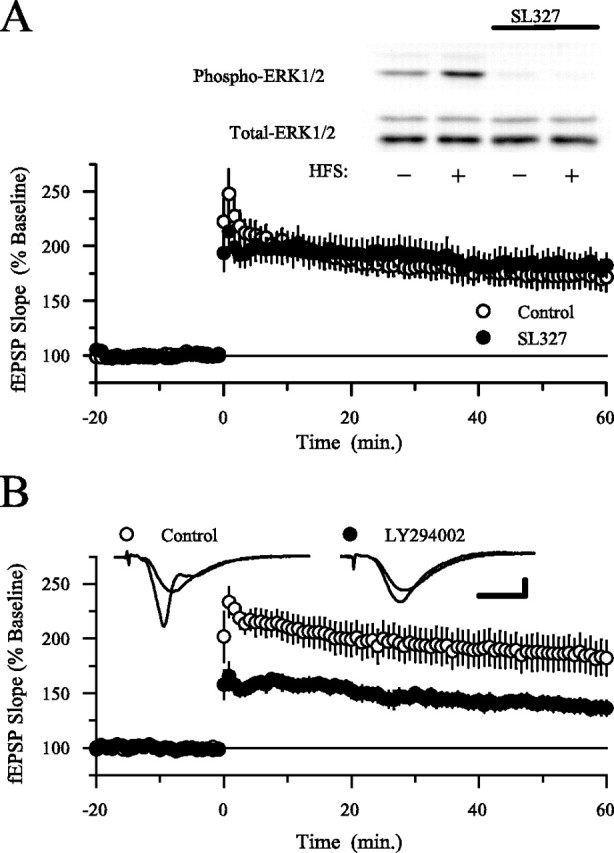
HFS-induced LTP is not blocked by MEK inhibitor SL327 but is suppressed by PI3-kinase inhibitors. A, Concentrations of the MEK inhibitor SL327 that block HFS-induced ERK2 activation have no effect on HFS-induced LTP. In experiments done in interface slice chambers, the amount of potentiation present 60 min after HFS (delivered at time 0) is the same in control and SL327-treated slices. In control experiments (open symbols; 0.1% DMSO), fEPSPs were potentiated to 173 ± 12% of baseline 60 min after HFS (n = 6) and were potentiated to 182 ± 12% of baseline in slices continuously bathed in 10 μm SL327 (filled symbols; n = 6). The inset shows example immunoblots probed with antibodies specific for phospho-ERK1/2 (top) and total ERK1/2 (bottom). Lane 1, Untreated control (ACSF alone); lane 2, HFS in ACSF; lane 3, SL327 alone (≥40 min); lane 4, HFS in SL327. Note that SL327 strongly reduced both basal levels of phospho-ERK2 and the increase in ERK2 phosphorylation induced by HFS. None of the treatments affected total ERK2 levels. The same results were obtained in three separate experiments. B, Inhibiting PI3-kinase activity with LY294002 significantly reduces HFS-induced LTP. In vehicle control experiments (0.1% DMSO; open symbols), fEPSPs were potentiated to 185 ± 16% of baseline 60 min after HFS; n = 6). In contrast, fEPSPs were potentiated to 138 ± 6% of baseline 60 min after HFS in slices continuously bathed in 20 μm LY294002 (filled symbols;n = 6; p < 0.05 compared with control). The inset shows fEPSPs recorded during baseline and 60 min after HFS (larger responses) in a control slice (left set of traces) and in a slice bathed in ACSF containing LY294002 (right set of traces). Calibration: 2 mV, 5 msec. Experiments were done using interface slice recording chambers.
Fig. 7.

PI3-kinase inhibitors suppress the induction but not the maintenance and expression of LTP. A, Wortmannin (Wort) inhibits HFS-induced LTP in submerged slices. Slices were continuously bathed in ACSF containing 0.02% DMSO (vehicle control; open symbols; n = 10) or ACSF containing 200 nm wortmannin (filled symbols; n = 10). One hour after HFS, fEPSPs were potentiated to 174 ± 11% of baseline in control slices but increased to only 132 ± 9% of baseline in wortmannin-treated slices (p < 0.01 compared with control). B, PI3-kinase (PI3K) inhibitors have no effect on established LTP. Wortmannin (200 nm; n = 3) or LY294002 (10 μm; n = 4) were applied starting 30 min after HFS (duration of inhibitors in the bath indicated by the bar). Neither wortmannin nor LY294002 inhibited potentiated synaptic transmission, and the results from all experiments were combined. All experiments were done using a submerged-slice recording chamber.
PI3-kinase inhibitors suppress LTP induction
Because continuous applications of PI3-kinase inhibitors were used in the experiments described above, the inhibition of LTP produced by wortmannin and LY294002 could be attributable to effects on the induction, maintenance, and/or expression of LTP. To examine whether PI3-kinase activity is specifically required for the maintenance and/or expression of LTP, we first induced LTP by delivering HFS in the absence of PI3-kinase inhibitors and then applied either LY294002 (20 μm; n = 4) or wortmannin (200 nm; n = 3) for 30 min starting 30 min after HFS. As shown in Figure 7B, blocking PI3-kinase after LTP induction had no effect on potentiated synaptic transmission. In three additional experiments, we extended the duration of the wortmannin application to at least 60 min and still observed no effect on potentiated synaptic transmission (data not shown). Wortmannin and LY294002 must therefore be present during HFS to inhibit LTP. This suggests that PI3-kinase activity has an important role in the induction of LTP but not its maintenance or expression. In contrast, a recent report found that PI3-kinase is activated for up to 30 min after LTP induction and that PI3-kinase inhibitors applied after HFS produce a reversible inhibition of potentiated EPSPs, suggesting that PI3-kinase activity is specifically required for the expression and not the induction or maintenance of LTP (Sanna et al., 2002). Notably, the concentrations of LY294002 and wortmannin used by Sanna et al. in their experiments (100 and 5 μm, respectively) were much higher than those used in our experiments (20 μm and 200 nm). We thus reexamined the effects of PI3-kinase inhibitors on the maintenance and expression of LTP using these higher inhibitor concentrations. As shown in Figure 8, potentiated synaptic transmission was depressed when either 100 μmLY294002 or 5 μm wortmannin was applied starting 30 min after LTP was induced with HFS. Whereas Sanna et al. (2002) reported that these high concentrations of LY294002 and wortmannin had no effect on unpotentiated synaptic transmission, under our experimental conditions, 100 μm LY294002 and 5 μm wortmannin also depressed transmission at unpotentiated synapses (Fig. 8). Although the reason for this difference is unclear, our results suggest that the inhibition of potentiated synaptic transmission by high concentrations of PI3-kinase inhibitors is not attributable to a selective disruption of mechanisms involved in the expression or maintenance of LTP.
Fig. 8.

High concentrations of PI3-kinase inhibitors depress potentiated synaptic transmission but also inhibit transmission at unpotentiated synapses. A, Potentiated synaptic transmission is inhibited by a high concentration of LY294002 (100 μm) applied for 30 min starting 30 min after LTP induction (n = 5; presence of LY294002 in the recording chamber indicated by the bar). At this concentration, LY294002 also inhibited basal synaptic transmission in a separate series of experiments (open symbols; n = 5).B, A 30 min application of a high concentration of wortmannin (5 μm; Wort) also inhibited both potentiated (filled symbols; n = 5) and basal synaptic transmission (open symbols; n = 4). The bar indicates presence of wortmannin in the recording chamber. All experiments were done using a submerged-slice recoding chamber.
TPS-induced LTP is ERK independent but PI3-kinase dependent in the hippocampus of PSD-95 mutant mice
Recent studies have shown that NMDA receptors form large multiprotein complexes that contain a number of proteins involved in Ras signaling (Husi et al., 2000). PI3-kinase (p85 regulatory subunit) directly binds to the NR2B subunit of NMDA receptors via an interaction mediated by the SH2 (Src homology 2) domain of p85 binding a tyrosine site on NR2B (Hisatsune et al., 1999). This site is phosphorylated by Fyn tyrosine kinase (Hisatsune et al., 1999), which is required for LTP induction (Grant et al., 1992). In contrast to the direct interaction of PI3-kinase with NR2B, MEK and ERK are regulated by SynGAP, a Ras GTPase-activating protein that directly binds PSD-95 (postsynaptic density-95) (Chen et al., 1998; Kim et al., 1998), which in turn binds to NR2 subunits (Kornau et al., 1995). PI3-kinase and ERK are thus linked to the NMDA receptor via distinct protein interactions. Because our pharmacological and biochemical studies indicate that PI3-kinase and MAPK have distinct roles in LTP, we wondered whether alterations in the composition of NMDA receptor complexes could differentially influence the involvement of these signaling pathways in LTP. To begin to address this question, we examined the effects of MEK and PI3-kinase inhibitors on LTP in the hippocampus of mice with a mutation that disrupts PSD-95 (Migaud et al., 1998). As shown in Figure9A, the induction of LTP by 150 pulses of TPS was significantly inhibited by both U0126 and wortmannin in slices from wild-type mice. As expected from previous work (Migaud et al., 1998; Komiyama et al., 2002), TPS induced a larger potentiation of synaptic transmission in slices from PSD-95 mutant mice. Surprisingly, U0126 did not inhibit TPS-induced LTP in slices from PSD-95 mutants (Fig. 9B), suggesting that ERK activation may no longer be required for TPS-induced LTP in the absence of PSD-95. Although the mechanistic basis responsible for this change in ERK dependency is unknown, this aspect of the PSD-95 mutant phenotype provides an additional way to examine the relationship between PI3-kinase and ERK in LTP. Specifically, if a serial pathway with PI3-kinase positioned upstream of ERK activation could account for the role of PI3-kinase in LTP, then the induction of LTP by TPS in slices from PSD-95 mutant mice should also be PI3-kinase independent. We observed, however, that wortmannin still significantly reduced TPS-induced LTP in slices from PSD-95 mutant mice (Fig. 9B). Thus, whereas the absence of PSD-95 dramatically alters the involvement of ERK in TPS-induced LTP, the induction of LTP by this protocol remains partially dependent on PI3-kinase activity. The differential effects of PI3-kinase and MAPK inhibitors on TPS-induced LTP in hippocampal slices from PSD-95 mutant mice thus provides additional evidence that the effects of PI3-kinase inhibitors on LTP cannot be solely attributed to alterations in ERK signaling.
Discussion
Inhibitors of PI3-kinase not only suppress hippocampal LTP (Kelly and Lynch, 2000; Raymond et al., 2002; Sanna et al., 2002) but also inhibit NMDA receptor-mediated ERK activation (Chandler et al., 2001;Perkinton et al., 2002). This suggests that PI3-kinase activity is required for LTP induction because it provides an essential link between NMDA receptors and ERK. In our experiments, wortmannin significantly reduced, but did not block, NMDA-induced ERK activation, suggesting that NMDA receptor-mediated ERK activation is only partially PI3-kinase dependent in the hippocampus of young adult animals. Activation of ERK2 by HFS was strongly suppressed by wortmannin, however, suggesting that PI3-kinase may be particularly important for activation of the ERK pathway by LTP-inducing patterns of synaptic activity. Although these findings are consistent with the hypothesis that PI3-kinase blockers inhibit LTP by antagonizing NMDA receptor-dependent ERK activation, we found that PI3-kinase and MEK inhibitors have distinct effects on LTP. Although blocking ERK activation inhibited LTP induced by two different TPS protocols, it had no effect on LTP induced by HFS or low-frequency synaptic stimulation paired with postsynaptic depolarization. In contrast, PI3-kinase inhibitors suppressed the induction of LTP by all four protocols. In addition, the effects of MEK, but not PI3-kinase, inhibitors on TPS-induced LTP were dramatically altered in PSD-95 mutant mice. The role of PI3-kinase in LTP is thus not limited to its role as an upstream regulator of ERK signaling.
The role of MAPK in the induction of LTP
Although our experiments primarily focused on the role of PI3-kinase in LTP, our findings have important implications regarding the role of ERK in LTP. In our experiments, both pairing and HFS-induced LTP were unaffected by two different MEK inhibitors, indicating that ERK activation is not absolutely required for LTP (Winder et al., 1999; Watabe et al., 2000). In addition, previous studies have shown that ERK activation by itself is not sufficient for LTP induction (Winder et al., 1999; Dudek and Fields, 2001). Thus, ERK activation is not an essential signaling event required for the early phases of NMDA receptor-dependent LTP in the hippocampal CA1 region. This does not mean that ERK activity is not importantly involved in LTP. Indeed, numerous studies have shown that hippocampal LTP is inhibited by MEK inhibitors (English and Sweatt, 1997; Atkins et al., 1998; Coogan et al., 1999; Winder et al., 1999; Watabe et al., 2000;Giovannini et al., 2001). Moreover, as we show here, the induction of LTP by 150 pulse trains of TPS is strongly reduced by MEK inhibitors (Winder et al., 1999; Watabe et al., 2000). Thus, although ERK activation may not be required for the induction of LTP by all patterns of synaptic stimulation, or under all experimental conditions, it clearly represents an important modulator of LTP. In addition to modulating LTP induction, ERK activation is also thought to have an important role in transcriptional events involved in the later, mRNA synthesis and protein synthesis-dependent stages of LTP (Impey et al., 1998; Davis et al., 2000).
Recently, two different models have been proposed to account for the role of ERK signaling in the induction of LTP (Sweatt, 2001; Zhu et al., 2002). In one model (Sweatt, 2001), ERK activation contributes to LTP induction by facilitating activation of synaptic NMDA receptors through a downregulation of dendritic A-type K+ channels (Adams et al., 2000; Watanabe et al., 2002; Yuan et al., 2002). In the second model (Zhu et al., 2002), ERK activation has a more central role in LTP induction and is required for activity-dependent membrane insertion of AMPA receptors. Our observations support the first of these models. First, we found that MEK inhibitors have no effect on pairing-induced LTP in experiments in which postsynaptic K+channels were blocked by the inclusion of Cs+ and TEA in the recording electrode solution. Thus, as would be expected if ERK activation normally contributes to LTP induction by downregulating KAchannels, bypassing ERK and directly inhibiting these channels pharmacologically obviates the need for ERK activation and renders LTP insensitive to the effects of MEK inhibitors. Second, our observation that MEK inhibitors do not block HFS (or pairing-induced LTP) is incompatible with a model in which ERK activation is an essential step in LTP. What is less clear, however, is how the first model can explain the lack of effect of MEK inhibitors on HFS-induced LTP. One possibility is that, under our experimental conditions, the postsynaptic depolarization evoked by HFS is large enough to strongly activate NMDA receptors and induce LTP even in the absence of an ERK-mediated downregulation of KA channels.
The role of PI 3-kinase in LTP
Although LY294002 and wortmannin almost completely blocked LTP induced by a short train of TPS, HFS and presynaptic stimulation paired with postsynaptic depolarization still induced a significant, albeit smaller, potentiation of synaptic transmission in slices pretreated with these inhibitors. One trivial explanation for this incomplete block of LTP is that PI3-kinase activity was not sufficiently inhibited by the concentrations of wortmannin and LY294002 used in our experiments. This seems unlikely, however, because the concentrations of wortmannin (Fig. 1) and LY294002 (data not shown) used in our experiments completely blocked NMDA-induced increases in phospho-Akt levels. Thus, the partial inhibition of LTP by PI3-kinase inhibitors most likely indicates that PI3-kinase activity is not absolutely required for the early stages of LTP up to 1 hr after induction. Instead, like ERK, PI3-kinase may be part of an important modulatory pathway that normally enhances the amount of potentiation induced by some patterns of synaptic stimulation. Alternatively, some patterns of synaptic stimulation may induce two distinct forms of potentiation, one that is dependent on PI3-kinase signaling and another that arises from PI3-kinase-independent mechanisms. The results from our experiments do not distinguish between these two possibilities. In either scenario, however, it seems likely the PI3-kinase activity contributes to the induction of LTP rather than its maintenance or expression because PI3-kinase inhibitors applied after LTP induction do not block LTP.
In contrast to our findings, it has been reported that PI3-kinase inhibitors applied after LTP induction can produce a selective and reversible depression of transmission at potentiated synapses (Sanna et al., 2002), suggesting that the expression of LTP is dependent on persistent PI3-kinase activity. Although we found that potentiated synaptic transmission was depressed by the much higher concentrations of LY294002 and wortmannin used by Sanna et al. (2002), under our experimental conditions, these high concentrations of the inhibitors also depressed transmission at basal, nonpotentiated synapses. The reason for the difference between our observations and those of Sanna et al. (2002) with respect to the effects of high PI3-kinase inhibitor concentrations on basal synaptic transmission is unclear but might be attributable to species differences (rat vs mouse). It is important to note, however, that the lower concentrations of PI3-kinase inhibitors used in our experiments strongly blocked PI3-kinase activity in our slices (as measured by changes in phospho-Akt levels) yet had no effect on transmission at potentiated synapses.
How might PI3-kinase be involved in the induction of LTP? In many cell types, PI3-kinase activates atypical isoforms of protein kinase C (PKC) via activation of phosphoinositide-dependent kinase-1, a protein kinase activated by the phospholipid products generated by PI3-kinase (Chou et al., 1998; Le Good et al., 1998). The atypical PKC isoform PKCζ is activated by LTP-inducing patterns of synaptic stimulation (Sacktor et al., 1993), and inhibitors that selectively block PKCζ inhibit LTP in the hippocampal CA1 region (Ling et al., 2002). Moreover, introduction of activated PKCζ into CA1 pyramidal cells induces a robust potentiation of synaptic transmission that occludes HFS-induced LTP (Ling et al., 2002). Thus, one possibility is that PI3-kinase contributes to the initial activation of PKCζ during LTP induction. PI3-kinase is also involved in the trafficking and insertion of some membrane proteins (Corvera and Czech, 1998; Wu et al., 1998; Rameh and Cantley, 1999; Lhuillier and Dryer, 2002), including AMPA-type glutamate receptors containing GluR1 subunits (Passafaro et al., 2001). Thus another possibility is that PI3-kinase is involved in activity-dependent changes in AMPA receptor trafficking–insertion that are thought to underlie LTP (for review, see Malinow and Malenka, 2002). With regard to this possibility, it is interesting to note that wortmannin completely blocks membrane insertion of GluR1-containing AMPA receptors induced by NMDA receptor activation in cultured neurons (Passafaro et al., 2001) but only partially inhibits LTP in hippocampal slices. Finally, PI3-kinase signaling can regulate organization of the actin cytoskeleton through activation of GTP exchange factors that regulate members of the Rho family of GTPases (Cantrell, 2001; Rodgers and Theibert, 2002). PI3-kinase signaling might thus have an important role in changes in dendritic spine structure induced by activation of synaptic NMDA receptors (for review, see Yuste and Bonhoeffer, 2001). Although the downstream mechanisms underlying the role of PI3-kinase in LTP remain unknown, our results suggest that Ras activation engages a divergent network of multiple downstream signaling events that have distinct regulatory roles in LTP.
Footnotes
This work was supported by a grant from the National Institute of Mental Health (T.J.O.) and the Wellcome Trust (S.G.N.G.). We thank Jane Robinson and Lynsey Forsyth for animal husbandry and genotyping and Rex Parker and Oahn Ho for technical assistance. SL327 was a generous gift from J. M. Trzaskos (DuPont Pharmaceuticals Research Laboratories, Wilmington, DE).
Correspondence should be addressed to Thomas J. O'Dell, Department of Physiology, David Geffen School of Medicine at University of California at Los Angeles, 53-231 Center for Health Science, Box 951751, Los Angeles, CA 90095. E-mail: todell@mednet.ucla.edu.
A. M. Watabe's present address: Division of Neuronal Network, Department of Basic Medical Sciences, The Institute of Medical Science, The University of Tokyo, Tokyo 108-8639, Japan.
References
- 1.Adams JP, Sweatt JD. Molecular psychology: roles for the ERK MAPK kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 2.Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem. 2000;75:2277–2287. doi: 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- 3.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAP kinase cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–621. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 4.Cantrell DA. Phosphoinositide 3-kinase signaling pathways. J Cell Sci. 2001;114:1439–1445. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- 5.Chandler LJ, Sutton G, Dorairaj NR, Norwood D. N-methyl-d-aspartate receptor-mediated biodirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J Biol Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- 6.Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- 7.Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase Cζ by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 8.Coogan AN, O'Leary DM, O'Connor JJ. P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- 9.Corvera S, Czech MP. Direct targets of phosphoinositide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol. 1998;8:442–446. doi: 10.1016/s0962-8924(98)01366-x. [DOI] [PubMed] [Google Scholar]
- 10.Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanisms for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- 11.Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duckworth BC, Cantley LC. Conditional inhibition of the mitogen-activated protein kinase cascade by wortmannin: dependence on signal strength. J Biol Chem. 1997;272:27665–27670. doi: 10.1074/jbc.272.44.27665. [DOI] [PubMed] [Google Scholar]
- 13. Dudek SM, Fields RD. Mitogen-activated protein kinase/extracellular signal-regulated kinase activation in somatodendritic compartments: roles of action potentials, frequency, and mode of calcium entry. J Neurosci 21 2001. RC122 (1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 15.Giovannini MG, Blitzer RD, Wong T, Asoma K, Tsokas P, Morrison JH, Iyengar R, Landau EM. Mitogen-activated protein kinase regulates early phosphorylation and delayed expression of Ca2+/calmodulin-dependent protein kinase II in long-term potentiation. J Neurosci. 2001;21:7053–7062. doi: 10.1523/JNEUROSCI.21-18-07053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant SG, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- 17.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 18.Hisatsune C, Umemori H, Mishina M, Yamamoto T. Phosphorylation-dependent interaction of the N-methyl-D-aspartate receptor ε2 subunit with phosphatidylinositol 3-kinase. Genes Cells. 1999;4:657–666. doi: 10.1046/j.1365-2443.1999.00287.x. [DOI] [PubMed] [Google Scholar]
- 19.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 20.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 21.Kelly A, Lynch MA. Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology. 2000;39:643–651. doi: 10.1016/s0028-3908(99)00169-0. [DOI] [PubMed] [Google Scholar]
- 22.Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family: Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- 23.Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJC, O'Carroll CM, Martin SJ, Morris RGM, O'Dell TJ, Grant SGN. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci. 2002;22:9721–9732. doi: 10.1523/JNEUROSCI.22-22-09721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kornau HC, Scenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 25.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 26.Lhuillier L, Dryer SE. Developmental regulation of neuronal Kca channels by TGFβ1: An essential role for PI3 kinase signaling and membrane insertion. J Neurophysiol. 2002;88:954–964. doi: 10.1152/jn.2002.88.2.954. [DOI] [PubMed] [Google Scholar]
- 27.Lin CH, Yeh SH, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- 28.Ling DSF, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Fukunaga K, Yamamoto H, Nishi K, Miyamoto E. Differential Roles of Ca2+/calmodulin-dependent protein kinase II and mitogen-activated protein kinase activation in hippocampal long-term potentiation. J Neurosci. 1999;19:8292–8299. doi: 10.1523/JNEUROSCI.19-19-08292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 31.Manabe T, Aiba A, Yamada A, Ichise T, Sakagami H, Kondo H, Katsuki M. Regulation of long-term potentiation by H-Ras through NMDA receptor phosphorylation. J Neurosci. 2000;20:2504–2511. doi: 10.1523/JNEUROSCI.20-07-02504.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 33.Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt cross-talk. J Biol Chem. 2002;277:31099–31106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- 34.Passafaro M, Piech V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4:917–925. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- 35.Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signaling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurons. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- 36.Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem. 1999;274:8347–8350. doi: 10.1074/jbc.274.13.8347. [DOI] [PubMed] [Google Scholar]
- 37.Raymond CR, Redman SJ, Crouch MF. The phosphoinositide 3-kinase and p70 S6 kinase regulate long-term potentiation in hippocampal neurons. Neuroscience. 2002;109:531–536. doi: 10.1016/s0306-4522(01)00500-0. [DOI] [PubMed] [Google Scholar]
- 38.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodgers EE, Theibert AB. Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci. 2002;20:187–197. doi: 10.1016/s0736-5748(02)00047-3. [DOI] [PubMed] [Google Scholar]
- 40.Rommel C, Clarke BA, Zimmermann S, Nuñez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- 41.Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E. Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci USA. 1993;90:8342–8346. doi: 10.1073/pnas.90.18.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci 21 2001. RC186 (1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 45.Thomas MJ, Moody T, Makhinson M, O'Dell TJ. Activity-dependent β-adrenergic modulation of low-frequency stimulation-induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 46.Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;18:7118–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57:652–658. [PubMed] [Google Scholar]
- 48.Watabe AM, Zaki PA, O'Dell TJ. Coactivation of β-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci. 2000;20:5924–5931. doi: 10.1523/JNEUROSCI.20-16-05924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wennström S, Downward J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol. 1999;19:4279–4288. doi: 10.1128/mcb.19.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winder DG, Martin KC, Muzzio I, Rohrer D, Chruscinski A, Kobilk B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- 52.Wu RL, Butler DM, Barish ME. Potassium current development and its linkage to membrane expansion during growth of cultured embryonic mouse hippocampal neurons: sensitivity to inhibitors of phosphatidylinositol 3-kinase and other protein kinases. J Neurosci. 1998;18:6261–6268. doi: 10.1523/JNEUROSCI.18-16-06261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- 55.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 56.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]