

Abstract
We examined the cytoskeletal dynamics that characterize neurite sprouting after axonal injury to cortical neurons maintained in culture for several weeks and compared these with initial neurite development. Cultured neocortical neurons, derived from embryonic day 18 rats, were examined at 3 d in vitro (DIV) and at various time points after axotomy at 21 DIV. The postinjury neuritic response was highly dynamic, progressing through an initial phase of retraction, followed by substantial axonal sprouting within 4–6 hr. Postinjury sprouts were motile and slender with expanded growth cone-like end structures. Microtubule markers were localized to sprout shafts and the proximal regions of putative growth cones and filamentous actin was distributed throughout growth cones, whereas neurofilament proteins were restricted to sprout shafts. A similar distribution of cytoskeletal proteins was present in developing neurites at 3 DIV. Exposure of developing and mature, injured cultures to the microtubule stabilizing agent taxol (10 μg/ml) caused growth inhibition, process distension, the transformation of growth cones into bulbous structures, and abnormal neurite directionality. Microtubule and neurofilament segregation occurred after taxol exposure in developing neurites and postinjury sprouts. Exposure to the microtubule destabilizing agent nocodazole (100 μg/ml) resulted in substantial morphological alteration of developing neurons and inhibited neurite growth and postinjury axonal sprouting. Our results indicate that the axons of cortical neurons have an intrinsic ability to sprout after transection, and similar cytoskeletal dynamics underlie neurite development and postinjury axonal sprouting.
Keywords: Alzheimer's disease, axonal sprouting, axonal transection, βIII-tubulin, brain trauma, cortical neuron culture, cytoskeleton, growth cone, microtubules, neurite development, posttraumatic epilepsy, neurofilament, tau, taxol
Introduction
The axonal cytoskeleton, particularly microfilaments and microtubules, is necessary for axonal outgrowth, elongation, branching, and pathfinding (for review, seeTanaka and Sabry, 1995; Brandt, 1998; Gallo and Letourneau, 2000; Korey and Van Vactor, 2000; Suter and Forscher, 2000). The complex interactions between the polymerization states of these cytoskeletal structures provides the dynamic driving force underlying axon elongation and growth cone motility (Kalil et al., 2000). In contrast, neurofilaments, the third main polymer constituent of the axonal cytoskeleton, may act in axonal radial growth and provide maturing neurons with structural support (Hoffman et al., 1984, 1985, 1987;Julien and Grosveld, 1991; Nixon and Sihag, 1991; Lee and Cleveland, 1996) but are not necessary for outgrowth. This is highlighted by the ability of developing axons lacking neurofilaments to grow, albeit more slowly (Walker et al., 2001), and neurofilament deficient animals to develop relatively normally (Zhu et al., 1997; Levavasseur et al., 1999).
The axonal cytoskeleton also plays an important role in the neuronal response to trauma (Maxwell et al., 1997). Nonpenetrative head trauma frequently causes cytoskeletal disruption leading to delayed axotomy (Povlishock and Christman, 1995; Povlishock and Jenkins, 1995; Maxwell et al., 1997). Several studies have demonstrated a reduction in the levels of microtubules and accumulations of neurofilaments in areas of axonal disruption (Jafari et al., 1997, 1998; Maxwell and Graham, 1997; Adlard et al., 2000; Dickson et al., 2000; King et al., 2001).
Traditionally, there has been significant pessimism regarding the potential for regeneration in the mature CNS, attributed primarily to the inhibitory nature of CNS parenchyma (Berry et al., 1994; Fawcett, 1997; Bandtlow and Schwab, 2000; Goldberg and Barres, 2000; Qiu et al., 2000). However, several reports suggest that central neurons respond to injury with a complex sequence of morphological, biochemical, and gene expression alterations, directed toward sprouting, regeneration, and synaptogenesis (Christman et al., 1997;Deller and Frotscher, 1997; Pastor et al., 2000; Vickers et al., 2000;King et al., 2001). Attempted regeneration of mature neurons is associated with substantial cytoskeletal reorganization (Christman et al., 1997; Dickson et al., 2000; King et al., 2001), resulting in the elaboration of fine protrusions (sprouts) into and across lesion sites (McHale et al., 1995; Christman et al., 1997; Deller and Frotscher, 1997; McKinney et al., 1997; King et al., 2001). However, aberrant axonal sprouting has been implicated in the development of posttraumatic epilepsy after human brain trauma (Larner, 1995; McKinney et al., 1997) and the pathogenesis of Alzheimer's disease (Masliah et al., 1991, 1992; Vickers et al., 2000; Arendt, 2001).
We used a novel in vitro model of CNS trauma, involving transection of axonal bundles (Dickson et al., 2000), to characterize the morphological and cytoskeletal alterations underlying the postinjury sprouting response of neurons cultured for several weeks to relative maturity. We compared the sprouting response with the cytoskeletal dynamics underlying initial neurite development of cultured cortical neurons. Finally, we investigated the consequences of microtubule stabilization and destabilization, using taxol and nocodazole, respectively, on initial neurite growth and postinjury axonal sprouting.
Materials and Methods
Cell culture. Primary dissociated cortical neuron cultures were prepared from rat embryos at 18 d of gestation using standard techniques (Banker and Goslin, 1998) and grown in Neurobasal media to selectively promote the growth of neuronal cells (Brewer et al., 1993; Brewer, 1995, 1997). Pregnant Hooded Wistar rats were killed by carbon dioxide exposure. Embryos were rapidly removed using sterile techniques and placed on ice, and, after removal of the skull and meninges, the neocortical hemispheres were dissected out and placed in 10 mm HEPES buffer (in 0.01m PBS), prewarmed to 37°C. Cortical tissue was dissociated by enzymatic digestion (0.025% trypsin) and gentle agitation at 37°C for 20 min. Enzymatic digestion was halted by rinsing the tissue in fresh 10 mm HEPES buffer, and dissociation was completed by gentle trituration using a 1 ml pipette.
Before plating, cell viability was assessed using trypan blue vital dye exclusion. Cells were plated, at a density of 4.5 × 105 cells per coverslip, onto circular 19-mm-diameter glass coverslips (Marienfeld, Lauda-Köigshofen, Germany), immersed in an initial plating media, consisting of Neurobasal media, 10% fetal bovine serum, 2% B-27 supplement (all from Invitrogen, Grand Island, NY), 0.5 mml-glutamine, 25 μm glutamate, and 1 ml/l gentamicin. Coverslips had been previously nitric acid etched and preincubated in 1 mg/ml l-lysine (Sigma, St. Louis, MO) in borate buffer, pH 7.4, overnight, before being placed into individual wells of 12-well microplates (Iwaki, Tokyo, Japan), each containing 2 ml of initial plating media.
Cultures were maintained in a 37°C humidified atmosphere of 5% CO2 for up to 22 d. After 1 d in vitro (DIV), the initial plating media was removed and replaced with a “subsequent,” serum-free growth media containing Neurobasal media, 2% B-27 supplement, 0.5 mml-glutamine, and 3 ml/l gentamicin. One-half of the volume of culture media was replenished every 3–4 d with fresh subsequent growth media.
Axonal injury. Axonal injury was performed as described byDickson et al. (2000). At 20 DIV, coverslips were transferred to individual 35 mm sterile plastic Petri dishes (Iwaki) and allowed to reacclimatize to the incubator conditions. At 21 DIV, individual axonal bundles, interconnecting discrete neuronal aggregates, were transected under microscope guidance using a 12 cm Barkan goniotomy curved-blade diamond knife. Thick axonal bundles, adherent to the underlying substrate, were selected for transection. The knife blade was carefully pressed on the appropriate axonal bundles to completely transect the axons and form a cell-free lesion between two previously interconnected neuronal aggregates. Cuts were made equidistant between neuronal aggregates to produce lesions ∼50–150 μm wide. Several (10–15) injuries were made per coverslip. Cultures were removed from the incubator for no longer than 5 min (because longer durations compromised neuronal integrity). Cultures were reincubated for 4, 14, or 24 hr after injury.
Taxol and nocodazole administration. Taxol powder (Amersham Biosciences, San Francisco, CA) was reconstituted in sterile 0.01 m PBS to achieve a concentration of 1 mg/ml and added to the subsequent media of cultures at a concentration of 10 μg/ml. Nocodozole (Sigma) was reconstituted in sterile 12% DMSO–0.01 m PBS and added to cultures at a final concentration of 100 μg/ml. Vehicle-treated cultures (10 μl/ml PBS for taxol studies or 100 μl/ml 12% DMSO–PBS for nocodozole studies) were processed concurrently to serve as controls. To determine the effect of microtubule stabilization (using taxol) on neurite development, cultures were incubated in taxol for 48 hr, between 3 and 5 DIV. To determine the effect of taxol on postinjury axonal sprouting, taxol was administered to the subsequent media of cultures immediately after localized axotomy at 21 DIV. Cultures were reincubated for 4, 14, or 24 hr after injury. Studies into whether the effects of taxol could be reversed were performed using 1 μg/ml taxol (because higher concentrations caused substantial cell death with prolonged exposure). To investigate the permanency of taxol-induced microtubule stabilization, taxol was washed out of developing cultures at 5 DIV, and cultures were maintained as normal for up to 21 DIV and compared with vehicle-treated cultures, as well as cultures that were constantly exposed to taxol. For postinjury studies, taxol was washed out of mature, injured cultures 4 hr after injury, and cultures were allowed to recover for an additional 20 hr before fixation. To assay the effect of microtubule destabilization on developing and mature, injured cultures, cultures were exposed to nocodazole for 48 hr between 3 and 5 DIV or for 4 hr immediately after axonal transection injury at 21 DIV.
Immunohistochemistry. Cultured neurons were fixed in 4% paraformaldehyde at 37°C for 30 min. Indirect double-immunofluorescent labeling was used to visualize the distribution of specific cytoskeletal components in developing neurites, as well as injured and non-injured mature axons. Briefly, cultured monolayers were incubated in combinations of mouse and rabbit primary antibodies (18–20 hr) (Table 1), followed by incubation in secondary antibodies (1 hr). Mouse and rabbit primary antibodies were visualized with goat anti-mouse AlexaFluor 488 and goat anti-rabbit AlexaFluor 594 secondary antibodies (dilution 1:1000; Molecular Probes, Eugene, OR), respectively. To label for filamentous actin (F-actin), cultures were incubated with AlexaFluor 488 phalloidin (dilution 1:200; Molecular Probes), for 30 min after immunohistochemistry. After washing, coverslips were air dried and mounted onto glass slides using Permafluor mounting media (Immunotech, Marseille, France). All antibodies were diluted in diluent (0.3% Triton X-100 in 0.01 m PBS). Controls, omitting the primary antibodies, were processed concurrently and lacked immunoreactivity for the cellular components under investigation.
Table 1.
Primary antibodies
| Antibody name | Type | Immunoreactivity | Dilution | Source |
|---|---|---|---|---|
| Anti-MAP2 | M | Microtubule-associated protein 2 | 1 :1000 | Chemicon (Temecula, CA) |
| Anti-βIII-tubulin | M | Neuron-specific type III β-tubulin | 1 :10,000 | Promega (Madison, WI) |
| Anti-BrdU | M | Nuclei of proliferating cells that have incorporated BrdU | 1 :1000 | Sigma |
| Anti-NeuN | M | Postmitotic neuronal nuclei | 1 :100 | Chemicon |
| SM1312 | MC | Phosphorylated neurofilaments | 1 :5000 | Sternberger Monoclonals (Lutherville, MD) |
| Anti-tau | R | Phosphorylation-independent tau | 1 :5000 | Dako (Carpinteria, CA) |
| Anti-synaptophysin | R | Presynaptic vesicles | 1 :000 | Boehringer Mannheim (Mannheim, Germany) |
| Anti-NSE | R | Neuron-specific enolase | 1 :500 | Dako |
M, Mouse monoclonal antibody; MC, mouse monoclonal mixture; R, rabbit polyclonal antibody.
Assay of cell proliferation. To determine the approximate birth dates of cortical neurons in vitro, cultures were incubated with 10 μm of the thymadine analog 5-bromo-2′-deoxyuridine (BrdU) (Sigma) for 72 hr at either 7 or 17 DIV. Addition of BrdU (1 or 5 μm) to cultures was also attempted at earlier time points (1–3 DIV) but resulted in substantial cell death by 21 DIV. Cultures underwent a complete media change to remove any unincorporated BrdU and were grown to 21 DIV. After fixation (4% paraformaldehyde), DNA denaturation was performed by incubating cultures in 2 m HCl for 1 hr at 37°C. Dividing cells were visualized by immunolabeling with an antibody against BrdU (Table 1). To confirm the absence of neuronal proliferation in cortical cultures, some cultures were also immunolabeled with an antibody against postmitotic neuronal nuclei (Table 1).
Electron microscopy. For scanning electron microscopy studies, cultures growing on circular glass coverslips were fixed with 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, for 20 min at room temperature, postfixed with 0.1% osmium tetraoxide in phosphate buffer, at room temperature for 15 min, and dehydrated through a graded ethanol series. Specimens were critical-point dried with CO2, mounted onto copper stubs and sputter coated with gold for observation with a Jeol (Peabody, MA) JSM-840 scanning electron microscope. For transmission electron microscopy studies, cortical cultures were fixed in 2.5% glutaraldehyde in 0.1 m phosphate buffer, at 37°C for 10 min. Cultures were postfixed in 1.5% osmium tetroxide in 0.1 m phosphate buffer at room temperature for 30 min, followed by 2% uranyl acetate in 70% ethanol for 5 min. Cultures were then dehydrated through a graded ethanol series and embedded in 100% Epon. Specimens were sectioned at 70 nm, stained with 5% uranyl acetate (10 min) and Reynolds lead citrate (10 min), and examined on a Philips EM-410 transmission electron microscope.
Microscopy and digital imaging. Time-lapse digital imaging was used to investigate the effect of taxol on growth cone motility and morphology, as well as the dynamic response of neurons to localized axotomy. At 1 d before imaging, cultured monolayers, growing on glass coverslips, were transferred to individual 35 mm glass Petri dishes. The Neurobasal media, in which the neurons were growing, was gradually replaced with “imaging buffer” (in mm: 124 NaCl, 5 KCl, 0.2 CaCl2, 1 MgCl2, 30 dextrose, and 25 HEPES, pH 7.3) (Zhang and Benson, 2001) over a time course of 1 hr before imaging, to promote neuronal survival when imaging over several hours. Using differential interference contrast (Nomarski) microscopy, a time series of images were captured from a Leica (Nussloch, Germany) DM IRB inverted microscope, equipped with a heating stage. Digital images of immunofluorescence-labeled specimens and live cell images were captured with a Magnifire digital camera (Optronics, Chelmsford, MA), attached to a G4 Macintosh computer (Apple Computers, Cupertino, CA).
Quantitative analysis. The effect of taxol on neurite development was analyzed by determining the proportion of neurites tipped by a growth cone. A growth cone was defined as a fan-shaped structure at the tip of a neurite, with a width three times greater than its neurite of origin. The effect of taxol treatment on neurite length was also determined. Digital images of immunofluorescence-labeled specimens were captured before treatment and after vehicle or taxol treatment, and the lengths of 100 neurites from each category were measured.
The effect of taxol on postinjury axonal sprouting was determined in terms of sprout number and sprout length, 4 hr after injury. Cultures injured by axonal transection were incubated in taxol or vehicle for 4 hr immediately after injury, followed by paraformaldehyde fixation and immunofluorescence labeling. Digital images of several individual injury sites were captured. To quantitate the effect of taxol on sprout elongation, the length of 100 postinjury axonal sprouts were measured from vehicle- and taxol-treated fellow cultures. Additionally, to determine the effect of taxol on sprout elaboration, the number of postinjury axonal sprouts was calculated from 15 individual cuts from vehicle- and taxol-treated fellow cultures. This value was expressed as the number of sprouts per 100 μm of cut site length.
For quantitation, taxol-related experiments were repeated five times (i.e., on cultures derived from five individual litters), and studies were performed by an investigator blind to the treatment of the cultures under investigation. NIH Image (version 1.61) software was used to capture digital images and perform measurements. Statistical analysis was performed using StatView (version 5.0) software (SAS Institute, Cary, NC). As appropriate, paired t tests or ANOVA was performed on quantitative data. Post hoccomparisons (Fisher's test) were performed after ANOVA.
Results
In vitro cortical neuron development
In the days after plating, dissociated cortical neurons formed aggregates, adhered to the coverslip substrate, and elaborated slender neurites. Analysis of the morphological and cytoskeletal characteristics of developing cortical neurons was performed at 3 DIV. Figure 1 illustrates the localization of specific cytoskeletal components in developing neurites at 3 DIV. The distribution of microtubules within developing neurons was visualized using an antibody with specificity for the neuron-specific βIII-tubulin subunit of microtubules (Lee et al., 1990) and an antibody against the microtubule-associated protein tau. Both βIII-tubulin and tau were localized throughout entire neurons, extending into the tapered extremities of dendrites and splayed extremities of axons, as well as the growth cone remnants (Szebenyi et al., 1998; Kalil et al., 2000), spikes, and small branches located along the lengths of some neurites. Colocalization analysis demonstrated extensive labeling of neurites and growth cones for βIII-tubulin (Fig. 1B) and tau (Fig.1C). Both microtubule markers were more abundant in neurite shafts and the central growth cone domain than in the proximal domain of growth cones. Double labeling for phosphorylated neurofilaments and tau verified the localization of tau throughout developing neurites and growth cones (Fig. 1E). Phosphorylated neurofilaments were, however, only localized in a subset of neurites, and labeling was absent in growth cones (Fig. 1D). βIII-Tubulin was uniformly distributed throughout neurites (Fig. 1F), whereas F-actin, as visualized with AlexaFluor 488 phalloidin, was more confined to growth cones, with punctate labeling in neurite shafts (Fig. 1G).
Fig. 1.
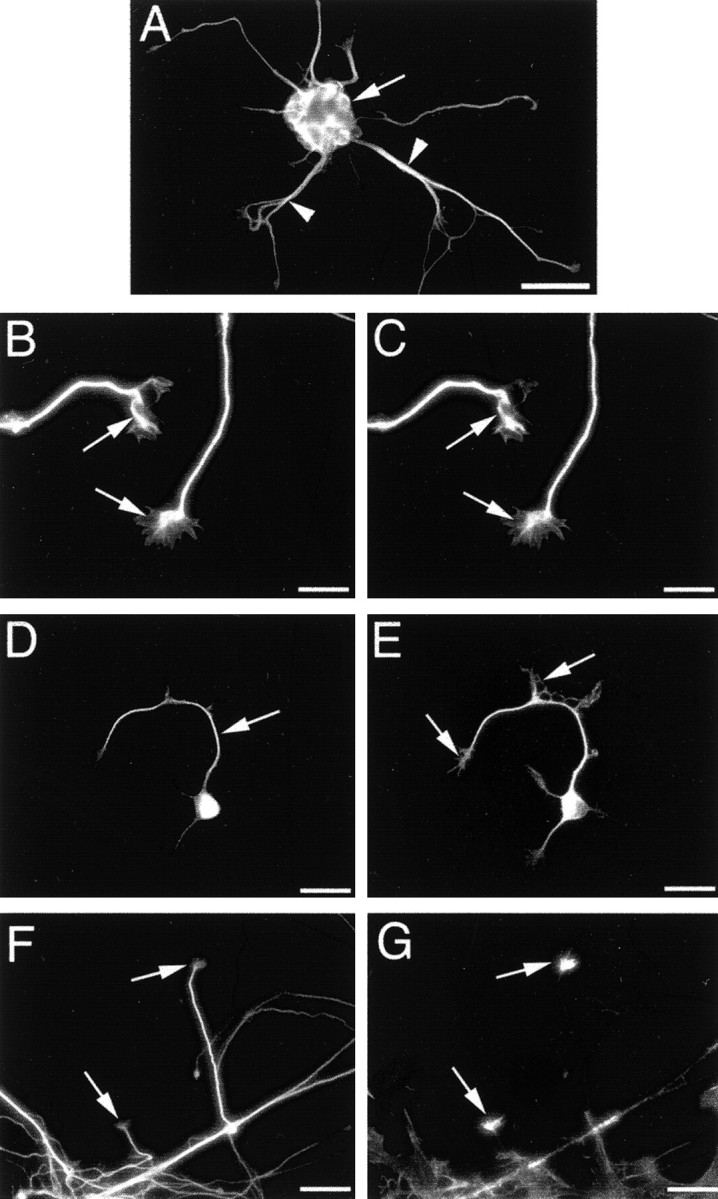
Cultured cortical neurons at 3 DIV.A shows a typical neuronal aggregate (arrow), from which several neurites were elaborated (arrowheads). B andC illustrate neurites double labeled for βIII-tubulin and tau, respectively. Both proteins were distributed throughout neurite shafts and growth cones. D and Eillustrate the same cell double labeled for phosphorylated neurofilaments and tau, respectively. The former were restricted to neurite shafts (arrow in D) and were not localized in growth cones (arrows in E). Figures F andG demonstrate neurites double labeled for βIII-tubulin and F-actin, respectively. F-actin was most abundant in growth cones (arrows in F and G), with punctate distribution along some axons. Scale bars: A, 60 μm;B, C, 15 μm; D,E, 30 μm, F, G, 10 μm.
Microtubule stabilization and destabilization during development
To determine the effect of microtubule stabilization on neuronal development, developing cultures were exposed to taxol (10 μg/ml) and examined for morphological and cytoskeletal alterations. Taxol was administered to cultures immediately before imaging, and a series of digital images were captured at 1 min intervals using differential interference contrast microscopy. Initially, characteristically splayed growth cones existed at the tips of neurites; however, in the presence of taxol, the neurites rapidly formed bulbous end structures, many of which lacked the filopodial protrusions characteristic of growth cones (Fig. 2A).
Fig. 2.
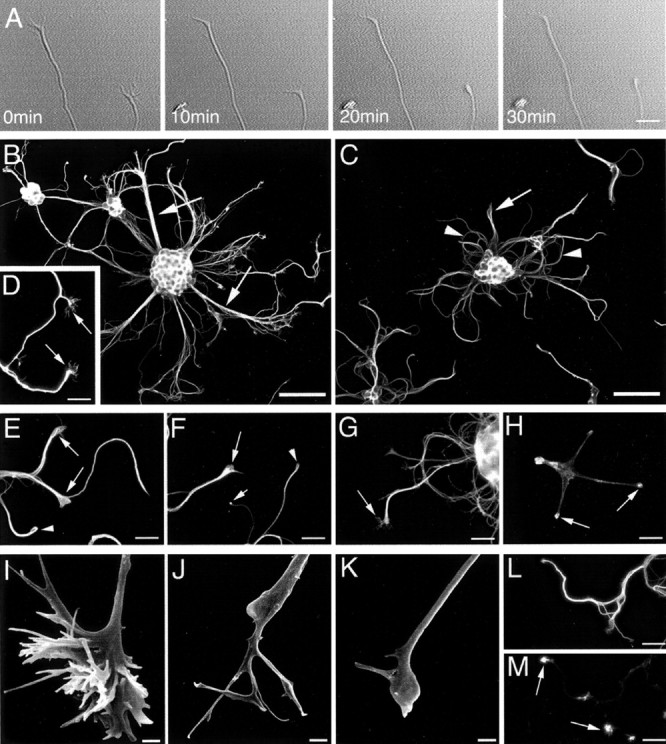
Effect of taxol exposure (10 μg/ml) on neurite morphology and cytoarchitecture. A shows a selection of images from a time-lapse series. Times shown on each image indicate minutes after taxol administration. In the presence of taxol, the originally splayed tips of neurites take on a bulbous morphology (arrows). Immunofluorescence labeling for βIII-tubulin demonstrated that neurites elaborated from vehicle-treated cultures (3–5 DIV) were slender and extended radially, often in defined bundles (arrows inB), whereas neurites from taxol-treated cultures (3–5 DIV) were distended, particularly in their distal domains (arrow inC) and often demonstrated a curled morphology (arrowheads in C). D, βIII-Tubulin labeling of vehicle-treated cultures demonstrating examples of growth cones (exhibiting defined filopodia) located at the distal tips of developing axons (arrows). In the distal tips of taxol-treated developing neurites, βIII-tubulin was localized to club-like (arrows) and loop-like (arrowhead) structures (E). Phosphorylated neurofilaments accumulated into club-like (long arrow), ball-like (short arrow), or loop-like (arrowhead) structures (F). Tau was localized throughout distended neurites and in the filopodial-like protrusions emanating from their distal tips (arrow in G), whereas F-actin was most abundant in neurite tips, forming bulb-like accumulations (H). Scanning EM images of growth cones from vehicle-treated (I) and taxol-treated (J, K) neurons (3–5 DIV). Vehicle-treated growth cones had an elaborate three-dimensional structure (H), whereas taxol-treated growth cones were bulbous, with few remaining filopodia (I, J). Double-labeling analysis demonstrated that some neurites regained growth cones after taxol washout (M, F-actin labeling) and underwent more neurite growth than cultures continually exposed to taxol; however, neurites often remained distended (L, βIII-tubulin). Scale bars: A, D–H, L–M, 20 μm; B, C, 60 μm; I–K, 1 μm.
Developing cultures, incubated in taxol between 3 and 5 DIV, demonstrated substantially altered morphology compared with vehicle-treated controls. Vehicle-treated neuronal aggregates elaborated numerous slender neurites, which extended radially and often in defined bundles. These neurite bundles splayed out distally into individual neurites, many of which were tipped by a growth cone (Fig.2B,I). In contrast, the neurites of taxol-treated neurons remained localized around their neuronal aggregates, often curling and looping back toward the aggregates from which they originated (Fig. 2C). Compared with vehicle-treated neurites (Fig. 2D), taxol-treated neurites were thickened and often swollen into bulbous structures at their distal tips (Fig.2E–H,J,K). Furthermore, some neurites retained aberrant growth cones with filopodial-like protrusions distally, whereas others completely lacked protuberances at their distal tips. Neuron-specific βIII-tubulin was localized to club- and loop-like structures in the distal tips of taxol-treated neurites (Fig. 2E), whereas phosphorylated neurofilaments (Fig. 2F) and tau (Fig.2G) were evenly distributed throughout the distended neurites. Additionally, accumulations of phosphorylated neurofilaments into ring- or ball-like structures were present in some neurite tips after taxol treatment (Fig. 2F). F-actin was most abundant in neurite tips. Figure 2H illustrates an example of a phalloidin-labeled neuron in which tight ball-like structures of F-actin have formed within neurite tips and filopodia have been completely eliminated. F-actin, βIII-tubulin, and tau were distributed throughout the filopodial-like structures emanating from the swollen tips of some taxol-treated neurites; presumably these structures correspond to growth cones, in which normal morphology has been retarded by microtubule stabilization. Taxol washout demonstrated that the effects of taxol were long lasting, with stunted neuronal morphology persistent at 21 DIV. However, the effects of taxol were partially reversible because increased neurite growth was observed in cultures from which taxol was washed out compared with cultures grown in the presence of taxol (cell death was high in the latter cultures). Moreover, some of the neurites from which taxol was removed regained their growth cones.
Quantitative analysis demonstrated that taxol significantly (p < 0.05) reduced the proportion of neurites tipped by a growth cone from 53.2 ± 7.8% (mean ± SE throughout) in vehicle-treated controls to 18.2 ± 3.7% after taxol treatment (Fig. 3A). Additionally, taxol inhibited neurite elongation between 3 and 5 DIV (Fig. 3B). At 3 DIV, mean neurite length for untreated cultures was 64.0 ± 3.9 μm. By 5 DIV, vehicle-treated neurites had elongated significantly (p < 0.05) to 90.9 ± 2.1 μm; however, taxol-treated neurites did not extend significantly (p > 0.05) from pretreatment and were significantly shorter (p < 0.05) than vehicle-treated controls, with a mean neurite length of 59.8 ± 1.4 μm.
Fig. 3.
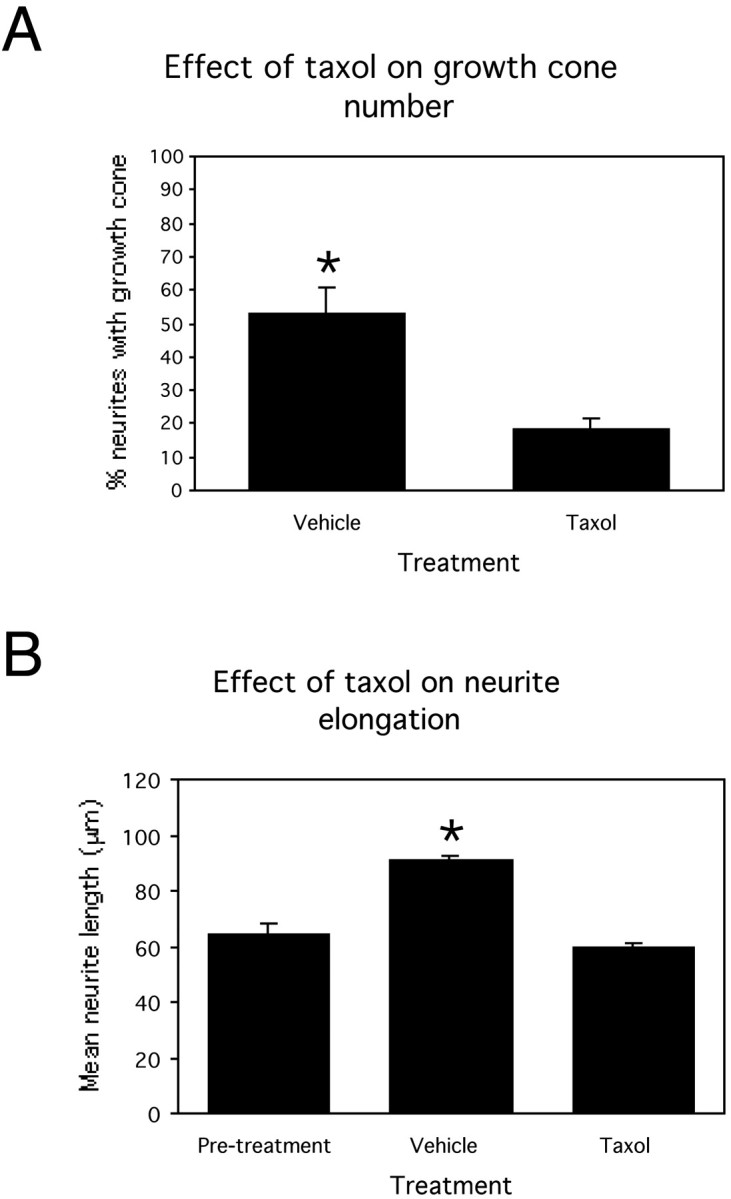
Bar graphs showing the effect of exposing developing cortical cultures to vehicle or taxol for 48 hr between 3 and 5 DIV. A, Proportion of neurites tipped by a growth cone after 48 hr vehicle or taxol treatment. B, Mean neurite length before treatment and after vehicle or taxol treatment. *p < 0.05. Error bars are SEMs.
Preliminary investigations were performed to determine the effect of microtubule destabilization on neurite development using nocodazole. Nocodazole exposure for 48 hr between 3 and 5 DIV resulted in profound morphological alteration compared with control cultures (Fig.4A). Neurite growth was clearly stunted, neuronal cell bodies and neurites were often bordered by sections of lamellipodial-like membrane, which were not observed in vehicle-treated cultures by this stage of development (Fig.4B), whereas many growth cones lacked obvious lamellipodia (Fig. 4C).
Fig. 4.
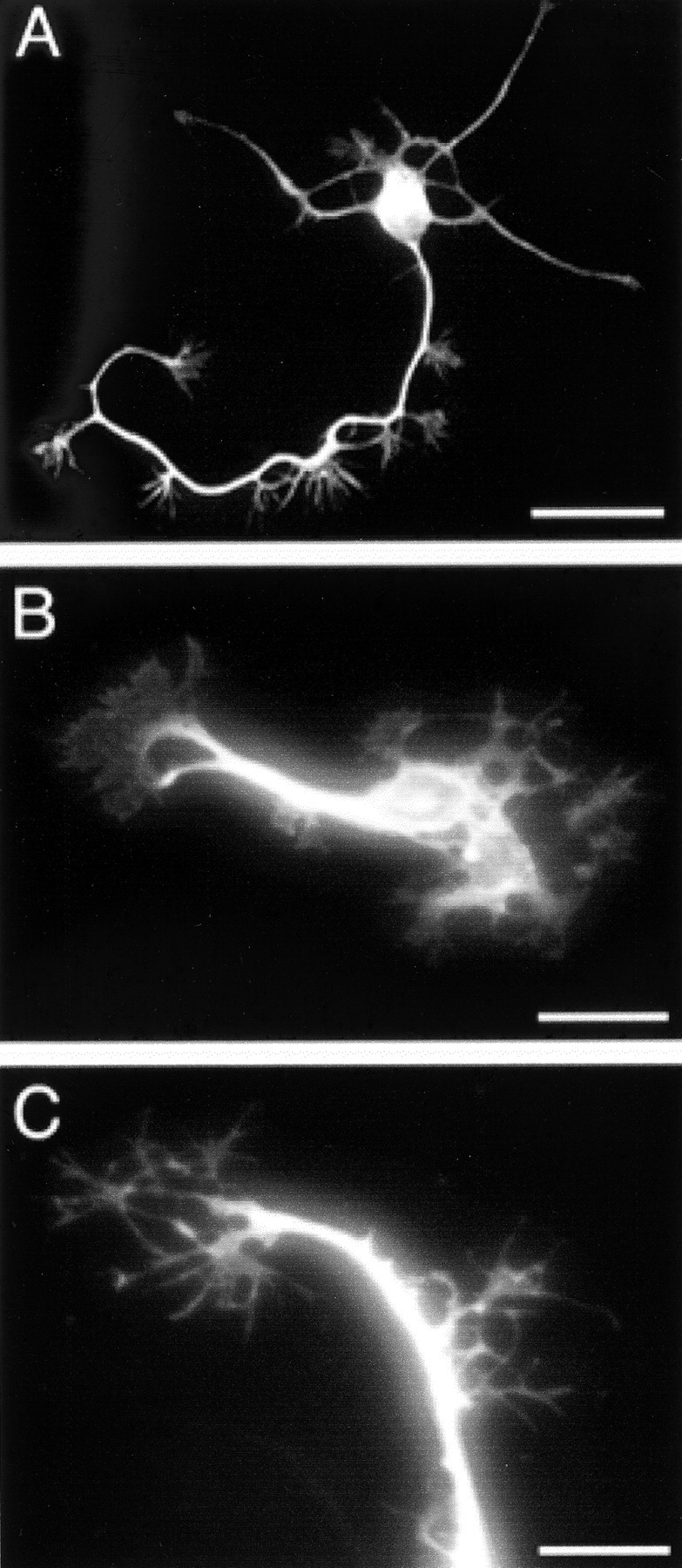
Effect of nocodazole (100 μg/ml) on neuronal morphology in developing cortical cultures. Vehicle-treated neurons (3–5 DIV, 1.2%DMSO–PBS) had defined cell bodies, axons were slender and tipped by growth cones, and future axonal branch points were indicated by growth cone “remnants” (A). Nocodozole-treated cultures had vastly altered morphology: neurites were stunted and often existed as thick protrusions of the cell, cell bodies were often bordered by a lamellipodial-like fringe (B), and large axonal growth cones often lacked lamellipodia (C). Scale bars, A, 20 μm; B, C, 10 μm.
Response of mature cortical neurons to localized axonal transection
Relatively mature (21 DIV) cultures were characterized by large, multicellular aggregates, from which elaborate networks and bundles of neuronal processes extended, forming interconnections with other aggregates. Even by this stage of development, some growth cones (with similar morphology and cytoskeletal constitution to developmental growth cones) were present in more sparsely populated regions of the cultured monolayers. The nuclei of neurons cultured for 21 d lacked immunoreactivity for BrdU (applied at either 7 or 17 DIV) (Fig.5A–D) but were immunoreactive for a marker of NeuN (neuronal-specific postmitotic nuclear protein) (Fig. 5E,F). Additionally aggregated neurons were immunoreactive for other markers of mature neurons, such as MAP2 (microtubule-associated protein 2) and neurofilament triplet proteins. Electron microscopy combined with immunohistochemistry (for synaptophysin immunoreactivity) confirmed the presence of substantial synaptic contacts throughout the cultures (Fig.5G–I); however, axons in axonal bundles were not myelinated (Fig. 5J). Collectively, these results demonstrate that cortical neurons cultured for 21 d were postmitotic, were at least 14 d old, and were relatively mature.
Fig. 5.
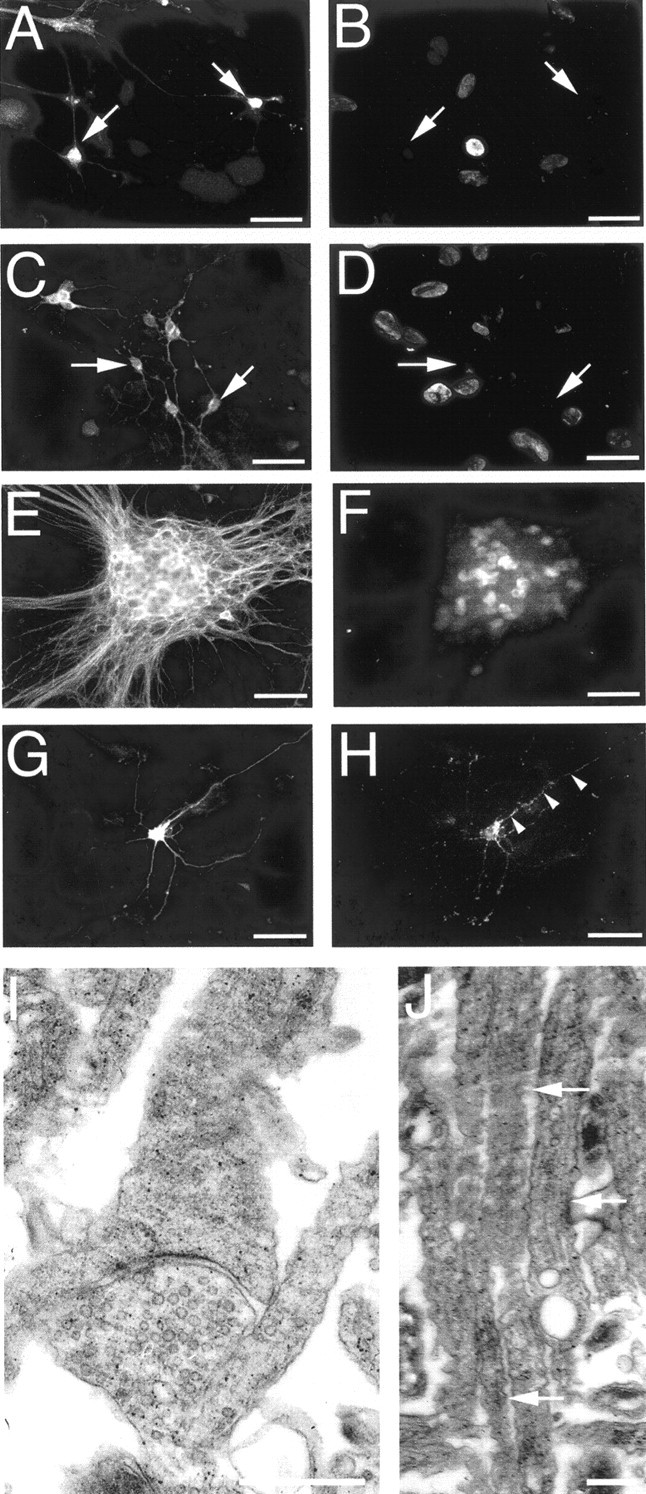
Confirmation of cortical neuron maturity. BrdU was administered to cultures at 7 (A, B) or 17 (C, D) DIV, for cell proliferation assays, and cultures were grown to 21 DIV. Neuron-specific enolase (NSE) immunopositive neurons (arrows in A andC) lacked BrdU-immunoreactive nuclei (B,D) in double-labeling studies, indicating that they were not proliferating. Double labeling for tau (E) and postmitotic neuronal nuclei (F) at 21 DIV demonstrated that aggregated neurons were not dividing. NSE-labeled neuron (G) in apposition with a profile of synaptophysin-immunoreactive puncta (arrows depict examples inH), demonstrating that neurons had abundant synaptic connections. The presence of synapses was confirmed with electron microscopy (I). J, Electron microscopy demonstrating that axons within axonal bundles were not myelinated (arrows depict examples). Scale bars:A–H, 30 μm; I, 2 μm;J, 1 μm.
Cultured cortical neurons were injured by local neurite transection at 21 DIV. Time-lapse digital imaging demonstrated that neurons respond to transection with a specific, highly dynamic sequence of changes. Immediately after injury, neuronal processes began to retract away from the site of injury (Fig.6A). This retraction response persisted for up to 4–6 hr after injury. Within 2–6 hr after injury, an extensive axonal sprouting response was initiated (Fig.6B).
Fig. 6.
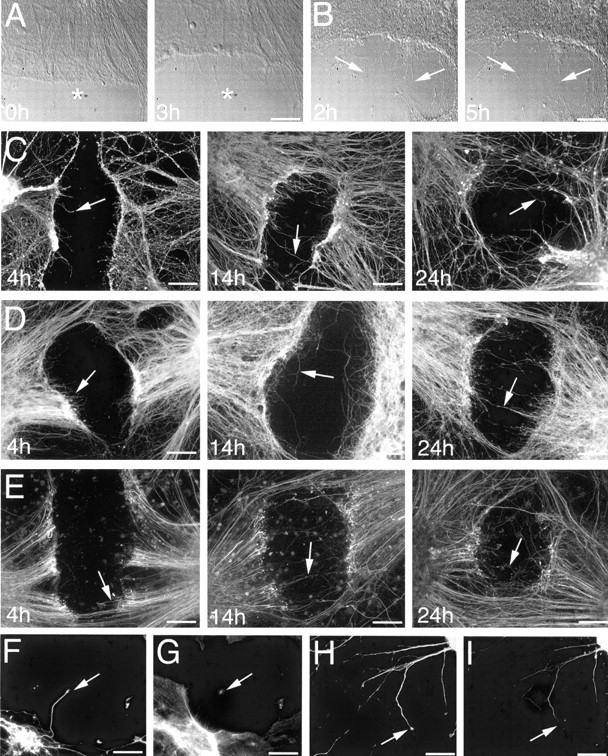
Time course of the posttransection response.A, Time-lapse imaging of a cut site immediately after injury, demonstrating retraction away from the injury site (note retraction in relation to the asterisk in each image).B, Time-lapse imaging of a cut site imaged from 2 hr after injury, with sprout growth indicated by arrows at each time point. Time course of postinjury axonal sprouting in cultures immunofluorescence labeled for βIII-tubulin (C), tau (D), and phosphorylated neurofilaments (E). Arrows in each image depict examples of sprouts. By 24 hr after injury, sprouting was extensive and several sprouts had crossed the lesion sites. Times shown on each image indicate postinjury interval in hours. Double labeling for βIII-tubulin and F-actin (F, G, respectively), as well as tau and phosphorylated neurofilaments (H, I, respectively), demonstrated that βIII-tubulin and tau were distributed throughout postinjury sprouts and proximal growth cones, whereas F-actin was most abundant in sprout growth cones and phosphorylated neurofilament were restricted to sprout shafts. Scale bars: A, B, 40 μm;C–E, 60 μm; F–I, 30 μm.
Immunofluorescent labeling was used to determine the distribution of cytoskeletal components in postinjury neurite sprouts at 4, 14, and 24 hr after injury. Postinjury analysis demonstrated that the alterations occurring in response to axonal transection were consistent across all cultures at each time point investigated. Double-labeling studies demonstrated that MAP2 labeled processes (dendrites) only extended short distances from the neuronal aggregates and were rarely transected during neuronal injury. βIII-Tubulin (Fig. 6C) and tau (Fig. 6D) were consistently distributed throughout postinjury axonal sprouts at all time points investigated. At 4 hr after injury, numerous axonal sprouts had extended into the injury sites. By 14 hr after injury, sprouts had continued to elongate and were extending toward the opposite side of the injury sited from their origin. At 24 hr after injury, sprouting was extensive, and many sprouts had crossed the injury site. Phosphorylated neurofilaments became distributed in postinjury axonal sprouts at 14 and 24 hr after injury, as opposed to either βIII-tubulin or tau, which were distributed throughout sprouts at all postinjury time points examined (Fig. 6E).
Postinjury axonal sprouts were generally characterized by slender sprout shafts tipped by expanded growth cone-like end structures, although some sprouts tapered distally. Both βIII-tubulin and tau were uniformly distributed throughout sprout shafts and their splayed end structures (Fig. 6B,H, respectively). F-actin was abundant in sprout growth cones (Fig.6G); however, phosphorylated neurofilaments were restricted to the sprout shafts (Fig. 6I). A prominent feature marking the injury site border was the accumulation of phosphorylated neurofilaments into ring- and bulb-like structures as we described previously in in vivo and in vitro models of neuronal injury (King et al., 1997, 2000, 2001; Dickson et al., 2000). Ring- and bulb-like structures lacked immunoreactivity for βIII-tubulin and tau and were apparent at all time points examined, existing either in isolation or as a continuum of the severed axonal tips.
Microtubule stabilization and destabilization after axonal transection
The effect of microtubule stabilization on postinjury axonal sprouting was investigated by exposing cultures, grown for 21 DIV, to taxol (10 μg/ml culture media) immediately after axonal transection. Taxol exposure had a substantial effect on postinjury axonal sprouting. In vehicle-treated control cultures, the degree of sprouting increased as time progressed, resulting in extensive sprout growth across the injury sites by 24 hr after injury (Fig.7A,B). However, postinjury axonal sprouting was comparably limited in taxol-treated cultures (Fig. 7D,E). In addition, taxol exposure resulted in substantial alteration in sprout morphology. Injured, vehicle-treated cultures elaborated sprouts that were generally tipped by expanded growth cone-like structures (Fig. 7C). Conversely, injured taxol-treated cultures elaborated sprouts tipped by bulbous end structures (Fig.7F). Taxol washout at least partially reversed the effects of microtubule stabilization on sprout growth inhibition, with increased sprouting observed in cultures in which taxol had been removed compared with those continually exposed to taxol after injury at 21 DIV. Additionally, some sprouts regained growth cone-like structures at their tips after taxol washout (Fig. 7G).
Fig. 7.
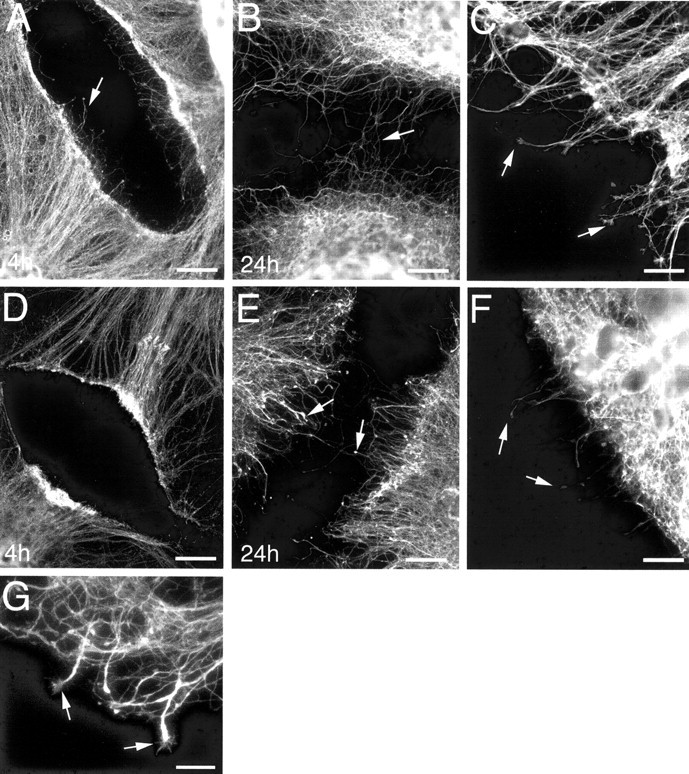
Immunofluorescence labeling for tau, demonstrating the effect of taxol exposure on postinjury axonal sprouting. Times shown on each image indicate hours of vehicle or taxol exposure after injury. Vehicle-treated cultures demonstrated an extensive and progressive sprouting response after axonal transection, with several sprouts crossing the lesion site by 24 hr after injury (A, B). Postinjury sprouts elaborated from vehicle-treated cultures were typically slender with expanded growth cone-like distal domains (arrows in C). Limited postinjury axonal sprouting was demonstrated in taxol-treated cultures up to 24 hr after injury (D, E), and bulb-like structures formed at the tips of sprouts (arrows inF). Arrows in A, B,D, and E depict examples of postinjury sprouts. C and F were captured at 4 and 14 hr after injury, respectively. Taxol washout, after 4 hr of postinjury taxol exposure, resulted in increased sprout growth across injury sites, and some sprouts gained growth cone-like structures at their tips (arrows in G). Scale bars: A,B, D, E, 60 μm;C, F, 20 μm; G, 10 μm.
Quantitative analysis demonstrated that taxol substantially influenced sprout outgrowth and elongation. Sprout outgrowth was significantly (p < 0.05) inhibited by taxol, with vehicle-treated cultures elaborating an average of 14.7 ± 1.6 sprouts per 100 μm of cut site length compared with taxol-treated cultures, which had elaborated on average only 6.6 ± 1.5 sprouts per 100 μm of cut site length, 4 hr after injury (Fig.8A). Similarly, statistical analysis indicated a significant (p< 0.05) difference between mean sprout length in injured, taxol-treated (21.6 ± 0.8 μm) and vehicle-treated (42.6 ± 0.5 μm) cultures at 4 hr after injury, demonstrating that taxol profoundly inhibited sprout elongation (Fig. 8B). Cell death analysis, using the vital dye propridium iodide, demonstrated the localization of dead and dying cells along injury site boarders. Taxol exposure did not affect the level of cell death up to 24 hr after axotomy. However increased cell death was observed when cultures were exposed to taxol for >72 hr.
Fig. 8.
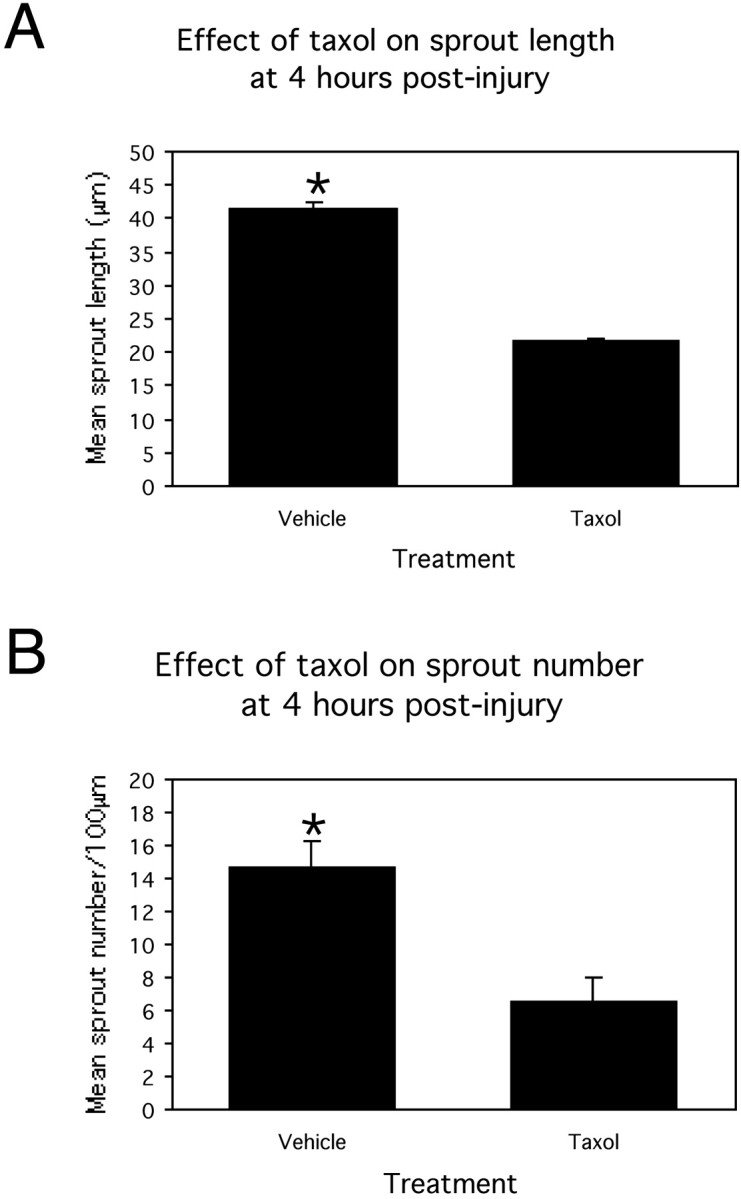
Bar graphs demonstrating the effect of exposing cultures to taxol for 4 hr after axonal transection. A, Mean sprout length. B, Mean number of sprouts per 100 μm of injury site length. *p < 0.05. Error bars are SEMs.
Exposure to the microtubule destabilizing agent nocodazole (100 μg/ml) substantially inhibited postinjury axonal sprouting, with little evidence of sprouting observed by 4 hr after injury (Fig.9).
Fig. 9.
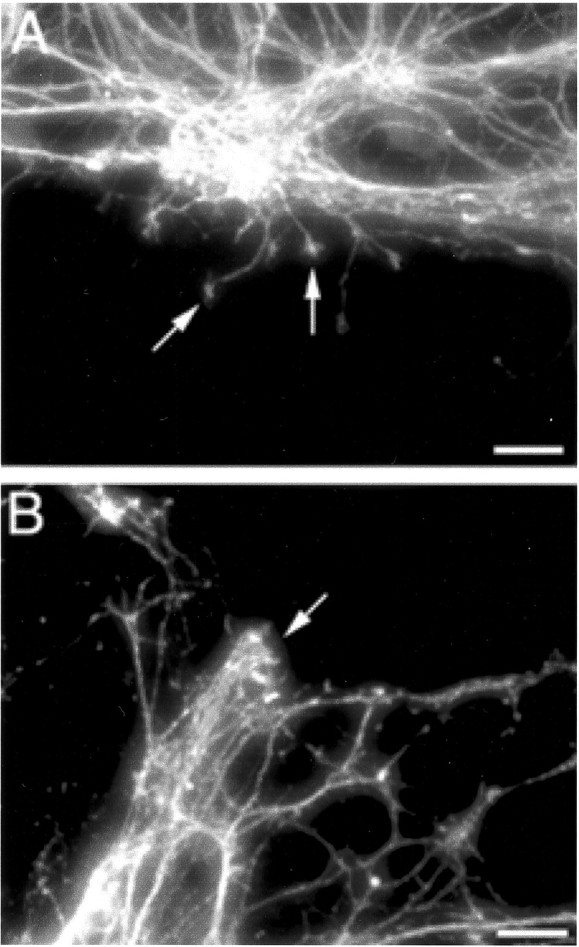
Effect of nocodazole on postinjury axonal sprouting. Nocodazole (100 μg/ml) exposure for 4 hr immediately after axonal transection injuries at 21 DIV markedly inhibited sprouting from transacted axonal bundles (arrow in B) compared with vehicle-treated controls (1.2%DMSO–PBS) in which short sprouts, tipped by growth cones, were elaborated into cut sites (arrows inA).
Discussion
Using live digital imaging and immunofluorescence-labeling techniques, we compared initial neurite development with the sprouting response exhibited by relatively mature neurons after localized axonal transection in vitro. After injury, a substantial regenerative attempt was indicated by the elaboration of sprout-like protuberances into injury sites. The overall morphology, motility, and cytoskeletal composition of axonal sprouts were highly homologous with developing neurites. Exposure to the microtubule stabilizing agent taxol and microtubule destabilizing agent nocodazole inhibited neurite development and postinjury axonal sprouting, demonstrating that similar microtubule dynamics may underlie both processes.
In vitro neuronal development
Both βIII-tubulin and tau were distributed throughout neurite shafts and the central domain of growth cones, with less abundant labeling exhibited in peripheral growth cone regions. F-actin was punctately distributed along some neurites and abundant in developmental growth cones. Conversely, phosphorylated neurofilaments were only localized in a subset of developing neurites (presumably those destined to become axons), and immunoreactivity was absent in growth cones. The distributions of these cytoskeletal components may reflect their relative importance in axonal growth. For instance, microtubules have been demonstrated to be essential in axonal outgrowth (Okabe and Hirokawa, 1990; Walker et al., 2001), whereas axonal growth can proceed in the absence of neurofilaments (Jiang et al., 1996; Zhu et al., 1997; Levavasseur et al., 1999), albeit at a slower rate (Walker et al., 2001).
Microtubule stabilization and destabilization inhibits neurite growth
Early-stage cultures (3 DIV) were exposed to taxol to investigate the effects of microtubule stabilization on neurite development. Taxol, widely used in the treatment of various cancers, promotes the polymerization of tubulin monomers and the formation of stable, nonfunctional microtubules (Gotaskie and Andreassi, 1994). The microtubule network is a particularly motile component of the axonal cytoskeleton within developing neurites (for review, see Brandt, 1998;Kalil et al., 2000). Exposure of cortical neurons to taxol in the current study resulted in neurite distension, cessation of neurite elongation, and a significant reduction in growth cone formation. Similar effects of taxol have been observed in cultured chick sensory neurons (Letourneau and Ressler, 1984; George et al., 1988). We also demonstrated that, after taxol treatment, loops of tubulin immunoreactivity were localized within the deformed end structures that replaced developmental growth cones at neurite tips, with ring- and bulb-like accumulations of phosphorylated neurofilaments often observed within the central core of the microtubule loops. The segregation of microtubules and neurofilaments is unusual because these cytoskeletal components are normally intermixed (Letourneau and Ressler, 1984). The dilated bulbous structures that formed at neurite tips after taxol exposure frequently lacked filopodia and lamellipodia and thus probably lacked the ability to effectively transduce guidance cues, which is a factor likely to account for the curling and looping of neurites around their aggregates of origin.
Abnormalities occurring after taxol exposure are likely to reflect the bundling and proliferation of microtubules within neurites, as well as altered microtubule spacing and interaction with microtubule-associated proteins (Black, 1987). Taxol-promoted microtubule proliferation may exhaust the local pool of tubulin monomers available for use in neurite elongation and presumably causes steric hindrance of axonal transport, which may prevent the delivery of constituents necessary for neurite extension. Indeed, Theiss and Meller (2000) demonstrated that taxol inhibits anterograde axonal transport. Furthermore, we demonstrated that, although some taxol-treated neurites regained growth cones and neurite growth increased after taxol washout, many neurites remained distended, indicating that taxol-induced microtubule stabilization is relatively permanent.
Nocodazole binds to tubulin and causes microtubule depolymerization (Dinter and Berger, 1998). Exposure of developing cortical cultures to nocodozole inhibited neurite growth and caused substantial morphological reorganization, which differed from the morphological aberrations induced by taxol exposure. Nocodazole-exposed neurites developed growth cones that lacked lamellipodia, as reported previously by Gallo (1998), and some neurons were bordered by lamellipodial-like fringes, phenomena that were not observed in taxol-treated cultures.
Axons respond to transection by retraction and sprouting
Using an in vitro model of axonal trauma capable of eliciting a substantial axonal sprouting response, we demonstrated that transection of the axonal bundles and networks interconnecting neuronal aggregates induced a characteristic and highly dynamic response. Time-lapse imaging verified that axons retract away from injury sites, toward their aggregate of origin, immediately after transection. Postinjury axonal retraction has been reported previously after lamprey spinal cord transection (McHale et al., 1995). The retraction response was accompanied by other reactive alterations, including swelling in the distal region of severed axons and the accumulation of neurofilaments into ring- and bulb-like structures, as reported previously (Meller et al., 1993: King et al., 1997, 2000, 2001; Dickson et al., 2000).
Extensive neurite sprouting was initiated by 4–6 hr after injury. Time course immunofluorescence-labeling studies confirmed that, by 4 hr after injury, several fine βIII-tubulin and tau immunoreactive sprouts emanated into injury sites. By 24 hr after injury, several sprouts had traversed the lesion sites. Several of the postinjury sprouts exhibited small growth cone-like structures at their tips, indicating the sprouting response was possibly a guided mechanism and not simple elongation along random pathways.
Analysis of the morphology, cytoskeletal composition, and dynamic properties of postinjury sprouts indicated some notable similarities with developing neurites. Developing neurites and sprouting axons were motile, slender structures. Additionally, a majority of sprouts were tipped by expanded growth cone-like structures, which exhibited immunoreactivity for βIII-tubulin, tau, and F-actin, although they were generally smaller than developmental growth cones. Phosphorylated neurofilaments were restricted to sprout shafts and appeared at later time points than observed for the microtubule markers. In a similar manner, it has been suggested that the sequence of cytoskeletal gene expression occurring during neuronal regenerative attempts recapitulates the developmental pattern (Hoffman and Cleveland, 1988;Lee and Cleveland, 1996). The delay in the appearance of phosphorylated neurofilaments in sprout shafts may indicate they are not required for initial sprout outgrowth. Similarly, neurite outgrowth proceeds in the absence of neurofilaments (Zhu et al., 1997; Levavasseur et al., 1999). Neurofilaments, however, may play an important role in sprout elongation. In this regard, reduced regenerative ability of the peripheral nervous system has been reported in neurofilament-deficient animals (Jiang et al., 1996; Zhu et al., 1997). Furthermore, Meller et al. (1993) speculates that neurofilaments may provide the propulsive force underlying sprout growth.
Microtubule stabilization and destabilization inhibits postinjury sprouting
Administration of taxol immediately after axonal transection at 21 DIV resulted in distinct morphological changes similar to those observed in developing taxol-treated neurites. Taxol exposure substantially inhibited postinjury axonal sprouting, causing a significant reduction in both sprout length and number at 4 hr after injury. Interestingly, postinjury sprouting was not completely inhibited by taxol but rather reduced by approximately one-half. This indicates that either the concentration of, or exposure time to, taxol was insufficient to stabilize all microtubules. Conversely, mechanisms other than microtubule dynamics may be responsible for sprout formation and growth. For instance, the actin and microtubule cytoskeletons may act in a synergistic and coordinated manner to dictate the cytoskeletal response underlying sprout growth, as is the case during neurite development (Letourneau et al., 1987), with disruption of one component affecting the dynamics of the other (Dent and Kalil, 2001). Unlike the limited sprouting observed in taxol-treated cultures, postinjury nococazole exposure completely abolished axonal sprouting.
Our results indicate some of the notable similarities between developing and sprouting axons and verify that the dynamic properties of microtubules are fundamental to the processes of sprout outgrowth and elongation. The effects of taxol on postinjury axonal sprouting were partially reversible, with increased sprouting observed after taxol washout in addition to the formation of growth-cone like structures at the tips of some sprouts.
Taxol has been demonstrated to reduce β-amyloid toxicity in Alzheimer's disease (Michaelis et al., 1998), as well as to inhibit detrimental alterations in intracellular calcium levels that may result in neuronal death (Burke et al., 1994), aberrant changes in tau antigenicity (Mattson, 1992), and excitotoxicity (Furukawa and Mattson, 1995). At low doses, taxol has also been demonstrated to promote recovery of function after spinal cord injury (Perez-Espejo et al., 1996). However, Figueroa-Masot et al. (2001) have reported that taxol potently induces apoptosis in cultured cortical neurons in a dose-dependent and exposure-time-dependent manner. Accordingly, although we recorded relatively little cell death after taxol exposure for 24 hr, cell death was substantially increased after 72 hr of taxol treatment. Using an in vivo model of brain trauma, Adlard et al. (2000) demonstrated that taxol inhibits microtubule loss and neurofilamentous accumulation. However, taxol exposure may also delay the resolution of reactive axonal alterations after injury, which may not be helpful for the adaptive response to trauma (Adlard et al., 2000).
Conclusion
The inability of axons to successfully regenerate after brain trauma has been attributed to several factors within the CNS milieu (Berry et al., 1994; Fawcett, 1997; Bandtlow and Schwab, 2000; Goldberg and Barres, 2000; Qiu et al., 2000), as well as an intrinsic incapacity for regeneration by the damaged neurons (Fawcett, 1997). The current study shows that the axons of relatively mature neurons can respond rapidly to injury by adaptive sprouting, and the cytoskeletal dynamics that underlie this attempt at regeneration are similar to the mechanisms underlying initial neurite development. The microtubule disrupting agents taxol and nocadazole effectively inhibited both initial neurite development and the regenerative sprouting response after injury, indicating the importance of cytoskeletal, particularly microtubule dynamics, in neurite growth and the neuronal response to injury. Although taxol and nocodazole have opposing effects on microtubule structure (polymerization and depolymerization, respectively), both agents essentially act to suppress microtubule dynamics, which, as our studies have demonstrated, are crucial to normal neurite growth and postinjury axonal sprouting. Cytoskeletal disrupting agents, such as taxol, could potentially be useful for preventing the maladaptive regenerative sprouting response that may underlie posttrauma epilepsy (Larner, 1995; McKinney et al., 1997) and Alzheimer's disease (Masliah et al., 1991, 1992; Vickers et al., 2000;Arendt, 2001).
Footnotes
This research was supported by the National Health and Medical Research Council, the Royal Hobart Hospital Research Foundation, and the Tasmanian Masonic Centenary Medical Research Foundation. We thank Sandrine Chopin, Graeme McCormack, Marian Quilty, and Irene Jacobs for their technical assistance in completing this work.
Correspondence should be addressed to Prof. James C. Vickers, Discipline of Pathology, Faculty of Health Sciences, University of Tasmania, 43 Collins Street, Hobart, Tasmania, 7000, Australia, E-mail:james.vickers@utas.edu.au.
References
- 1.Adlard PA, King CE, Vickers JC. The effects of taxol on the central nervous system response to physical injury. Acta Neuropathol (Berl) 2000;100:183–188. doi: 10.1007/s004019900160. [DOI] [PubMed] [Google Scholar]
- 2.Arendt T. Alzheimer's disease as a disorder of mechanisms underlying structural brain self-organization. Neuroscience. 2001;102:723–765. doi: 10.1016/s0306-4522(00)00516-9. [DOI] [PubMed] [Google Scholar]
- 3.Bandtlow CE, Schwab ME. NI-35/250/nogo-a: a neurite growth inhibitor restricting structural plasticity and regeneration of nerve fibers in the adult vertebrate CNS. Glia. 2000;29:175–181. doi: 10.1002/(sici)1098-1136(20000115)29:2<175::aid-glia11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 4.Banker G, Goslin K. Types of nerve cell cultures, their advantages and limitations. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1998. pp. 11–36. [Google Scholar]
- 5.Berry M, Hall S, Shewan D, Cohen J. Axonal growth and its inhibition. Eye. 1994;8:245–254. doi: 10.1038/eye.1994.53. [DOI] [PubMed] [Google Scholar]
- 6.Black MM. Taxol interferes with the interaction of microtubule-associated proteins with microtubules in cultured neurons. J Neurosci. 1987;7:3695–3702. doi: 10.1523/JNEUROSCI.07-11-03695.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandt R. Cytoskeletal mechanisms of axon outgrowth and pathfinding. Cell Tissue Res. 1998;292:181–189. doi: 10.1007/s004410051049. [DOI] [PubMed] [Google Scholar]
- 8.Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- 9.Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- 10.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 11.Burke WJ, Raghu G, Strong R. Taxol protects against calcium-mediated death of differentiated rat pheochromocytoma cells. Life Sci. 1994;55:313–319. doi: 10.1016/0024-3205(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 12.Christman CW, Salvant JB, Jr, Walker SA, Povlishock JT. Characterization of a prolonged regenerative attempt by diffusely injured axons following traumatic brain injury in adult cat: a light and electron microscopic immunocytochemical study. Acta Neuropathol (Berl) 1997;94:329–337. doi: 10.1007/s004010050715. [DOI] [PubMed] [Google Scholar]
- 13.Deller T, Frotscher M. Lesion-induced plasticity of central neurons: sprouting of single fibres in the rat hippocampus after unilateral entorhinal cortex lesion. Prog Neurobiol. 1997;53:687–727. doi: 10.1016/s0301-0082(97)00044-0. [DOI] [PubMed] [Google Scholar]
- 14.Dent W, Kalil K. Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci. 2001;21:9757–9769. doi: 10.1523/JNEUROSCI.21-24-09757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickson TC, Adlard PA, Vickers JC. Sequence of cellular changes following localized axotomy to cortical neurons in glia-free culture. J Neurotrauma. 2000;17:1095–1103. doi: 10.1089/neu.2000.17.1095. [DOI] [PubMed] [Google Scholar]
- 16.Dinter A, Berger EG. Golgi-disturbing agents. Histochem Cell Biol. 1998;109:571–590. doi: 10.1007/s004180050256. [DOI] [PubMed] [Google Scholar]
- 17.Fawcett JW. Astrocytic and neuronal factors affecting axon regeneration in the damaged central nervous system. Cell Tissue Res. 1997;290:371–377. doi: 10.1007/s004410050943. [DOI] [PubMed] [Google Scholar]
- 18.Figueroa-Masot XA, Hetman M, Higgins MJ, Kokot N, Xia Z. Taxol induces apoptosis in cortical neurons by a mechanism independent of Bcl-2 phosphorylation. J Neurosci. 2001;21:4657–4667. doi: 10.1523/JNEUROSCI.21-13-04657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furukawa K, Mattson MP. Cytochalasins protect hippocampal neurons against amyloid beta-peptide toxicity: evidence that actin depolymerization suppresses Ca2+ influx. J Neurochem. 1995;65:1061–1068. doi: 10.1046/j.1471-4159.1995.65031061.x. [DOI] [PubMed] [Google Scholar]
- 20.Gallo G. Involvement of microtubules in the regulation of neuronal growth cones morphologic remodeling. J Neurobiol. 1998;35:121–140. [PubMed] [Google Scholar]
- 21.Gallo G, Letourneau PC. Neurotrophins and the dynamic regulation of the neuronal cytoskeleton. J Neurobiol. 2000;44:159–173. doi: 10.1002/1097-4695(200008)44:2<159::aid-neu6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 22.George EB, Schneider BF, Lasek RJ, Katz MJ. Axonal shortening and the mechanisms of axonal motility. Cell Motil Cytoskeleton. 1988;9:48–59. doi: 10.1002/cm.970090106. [DOI] [PubMed] [Google Scholar]
- 23.Goldberg JL, Barres BA. Nogo in nerve regeneration. Nature. 2000;403:369–370. doi: 10.1038/35000309. [DOI] [PubMed] [Google Scholar]
- 24.Gotaskie GE, Andreassi BF. Paclitaxel: a new antimitotic chemotherapeutic agent. Cancer Pract. 1994;2:27–33. [PubMed] [Google Scholar]
- 25.Hoffman PN, Cleveland DW. Neurofilament and tubulin expression recapitulates the developmental program during axonal regeneration: induction of a specific beta-tubulin isotype. Proc Natl Acad Sci USA. 1988;85:4530–4533. doi: 10.1073/pnas.85.12.4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffman PN, Griffin JW, Price DL. Control of axonal caliber by neurofilament transport. J Cell Biol. 1984;99:705–714. doi: 10.1083/jcb.99.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman PN, Griffin JW, Gold BG, Price DL. Slowing of neurofilament transport and the radial growth of developing nerve fibers. J Neurosci. 1985;5:2920–2929. doi: 10.1523/JNEUROSCI.05-11-02920.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffman PN, Cleveland DW, Griffin JW, Landes PW, Cowan NJ, Price DL. Neurofilament gene expression: a major determinant of axonal caliber. Proc Natl Acad Sci USA. 1987;84:3472–3476. doi: 10.1073/pnas.84.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jafari SS, Maxwell WL, Neilson M, Graham DI. Axonal cytoskeletal changes after non-disruptive axonal injury. J Neurocytol. 1997;26:207–221. doi: 10.1023/a:1018588114648. [DOI] [PubMed] [Google Scholar]
- 30.Jafari SS, Nielson M, Graham DI, Maxwell WL. Axonal cytoskeletal changes after nondisruptive axonal injury. II. Intermediate sized axons. J Neurotrauma. 1998;15:955–966. doi: 10.1089/neu.1998.15.955. [DOI] [PubMed] [Google Scholar]
- 31.Jiang XM, Zhao JX, Ohnishi A, Itakura C, Mizutani M, Yamamoto T, Murai Y, Ikeda M. Regeneration of myelinated fiber after crush injury is retarded in sciatic nerves of mutant Japanese quails deficient in neurofilaments. Acta Neuropathol (Berl) 1996;92:467–472. doi: 10.1007/s004010050548. [DOI] [PubMed] [Google Scholar]
- 32.Julien J-P, Grosveld F. Structure and expression of neurofilament genes. The neuronal cytoskeleton (Burgoyne R, ed), pp 215–232. Wiley; New York: 1991. [Google Scholar]
- 33.Kalil K, Szebenyi G, Dent EW. Common mechanisms underlying growth cone guidance and axon branching. J Neurobiol. 2000;44:145–158. [PubMed] [Google Scholar]
- 34.King CE, Jacobs I, Dickson TC, Vickers JC. Physical damage to rat cortical axons mimics early Alzheimer's neuronal pathology. NeuroReport. 1997;8:1663–1665. [PubMed] [Google Scholar]
- 35.King CE, Dickson TC, Jacobs I, McCormack GH, Riederer BM, Vickers JC. Acute CNS axonal injury models a subtype of dystrophic neurite in Alzheimer's disease. Alzheimers Rep. 2000;3:31–40. [Google Scholar]
- 36.King CE, Canty AJ, Vickers JC. Alterations in neurofilaments associated with reactive brain changes and axonal sprouting following acute physical injury to the rat neocortex. Neuropathol Appl Neurobiol. 2001;27:115–126. doi: 10.1046/j.1365-2990.2001.00317.x. [DOI] [PubMed] [Google Scholar]
- 37.Korey CA, Van Vactor D. From the growth cone surface to the cytoskeleton: one journey, many paths. J Neurobiol. 2000;44:184–193. [PubMed] [Google Scholar]
- 38.Larner AJ. Axonal sprouting and synaptogenesis in temporal lobe epilepsy: possible pathogenetic and therapeutic roles of neurite growth inhibitory factors. Seizure. 1995;4:249–258. doi: 10.1016/s1059-1311(95)80001-8. [DOI] [PubMed] [Google Scholar]
- 39.Lee MK, Cleveland DW. Neuronal intermediate filaments. Annu Rev Neurosci. 1996;19:187–217. doi: 10.1146/annurev.ne.19.030196.001155. [DOI] [PubMed] [Google Scholar]
- 40.Lee MK, Tuttle JB, Rebhun LI, Cleveland DW, Frankfurter A. The expression and posttranslational modification of a neuron-specific beta-tubulin isotype during chick embryogenesis. Cell Motil Cytoskeleton. 1990;17:118–132. doi: 10.1002/cm.970170207. [DOI] [PubMed] [Google Scholar]
- 41.Letourneau PC, Ressler AH. Inhibition of neurite initiation and growth by taxol. J Cell Biol. 1984;98:1355–1362. doi: 10.1083/jcb.98.4.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Letourneau PC, Shattuck TA, Ressler AH. “Pull” and “push” in neurite elongation: observations on the effects of different concentrations of cytochalasin B and taxol. Cell Motil Cytoskeleton. 1987;8:193–209. doi: 10.1002/cm.970080302. [DOI] [PubMed] [Google Scholar]
- 43.Levavasseur F, Zhu Q, Julien JP. No requirement of alpha-internexin for nervous system development and for radial growth of axons. Brain Res Mol Brain Res. 1999;69:104–112. doi: 10.1016/s0169-328x(99)00104-7. [DOI] [PubMed] [Google Scholar]
- 44.Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry R, Baudier J, Saitoh T. Patterns of aberrant sprouting in Alzheimer's disease. Neuron. 1991;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- 45.Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R, Baudier J, Saitoh T. Localization of amyloid precursor protein in GAP43-immunoreactive aberrant sprouting neurites in Alzheimer's disease. Brain Res. 1992;574:312–316. doi: 10.1016/0006-8993(92)90831-s. [DOI] [PubMed] [Google Scholar]
- 46.Mattson MP. Effects of microtubule stabilization and destabilization on tau immunoreactivity in cultured hippocampal neurons. Brain Res. 1992;582:107–118. doi: 10.1016/0006-8993(92)90323-2. [DOI] [PubMed] [Google Scholar]
- 47.Maxwell WL, Graham DI. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J Neurotrauma. 1997;14:603–614. doi: 10.1089/neu.1997.14.603. [DOI] [PubMed] [Google Scholar]
- 48.Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- 49.McHale MK, Hall GF, Cohen MJ. Early cytoskeletal changes following injury of giant spinal axons in the lamprey. J Comp Neurol. 1995;353:25–37. doi: 10.1002/cne.903530105. [DOI] [PubMed] [Google Scholar]
- 50.McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat Med. 1997;3:990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- 51.Meller D, Bellander BM, Schmidt-Kastner R, Ingvar M. Immunohistochemical studies with antibodies to neurofilament proteins on axonal damage in experimental focal lesions in rat. J Neurol Sci. 1993;117:164–174. doi: 10.1016/0022-510x(93)90169-y. [DOI] [PubMed] [Google Scholar]
- 52.Michaelis ML, Ranciat N, Chen Y, Bechtel M, Ragan R, Hepperle M, Liu Y, Georg G. Protection against beta-amyloid toxicity in primary neurons by paclitaxel (Taxol). J Neurochem. 1998;70:1623–1627. doi: 10.1046/j.1471-4159.1998.70041623.x. [DOI] [PubMed] [Google Scholar]
- 53.Nixon RA, Sihag RK. Neurofilament phosphorylation: a new look at regulation and function. Trends Neurosci. 1991;14:501–506. doi: 10.1016/0166-2236(91)90062-y. [DOI] [PubMed] [Google Scholar]
- 54.Okabe S, Hirokawa N. Turnover of fluorescently labelled tubulin and actin in the axon. Nature. 1990;343:479–482. doi: 10.1038/343479a0. [DOI] [PubMed] [Google Scholar]
- 55.Pastor AM, Delgado-Garcia JM, Martinez-Guijarro FJ, Lopez-Garcia C, de La Cruz RR. Response of abducens internuclear neurons to axotomy in the adult cat. J Comp Neurol. 2000;427:370–390. doi: 10.1002/1096-9861(20001120)427:3<370::aid-cne5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 56.Perez-Espejo MA, Haghighi SS, Adelstein EH, Madsen R. The effects of taxol, methylprednisolone, and 4-aminopyridine in compressive spinal cord injury: a qualitative experimental study. Surg Neurol. 1996;46:350–357. doi: 10.1016/s0090-3019(96)00200-5. [DOI] [PubMed] [Google Scholar]
- 57.Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12:555–564. doi: 10.1089/neu.1995.12.555. [DOI] [PubMed] [Google Scholar]
- 58.Povlishock JT, Jenkins LW. Are the pathobiological changes evoked by traumatic brain injury immediate and irreversible? Brain Pathol. 1995;5:415–426. doi: 10.1111/j.1750-3639.1995.tb00620.x. [DOI] [PubMed] [Google Scholar]
- 59.Qiu J, Cai D, Filbin MT. Glial inhibition of nerve regeneration in the mature mammalian CNS. Glia. 2000;29:166–174. [PubMed] [Google Scholar]
- 60.Suter DM, Forscher P. Substrate-cytoskeletal coupling as a mechanism for the regulation of growth cone motility and guidance. J Neurobiol. 2000;44:97–113. [PubMed] [Google Scholar]
- 61.Szebenyi G, Callaway JL, Dent EW, Kalil K. Interstitial branches develop from active regions of the axon demarcated by the primary growth cone during pausing behaviors. J Neurosci. 1998;18:7930–7940. doi: 10.1523/JNEUROSCI.18-19-07930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 63.Theiss C, Meller K. Taxol impairs anterograde axonal transport of microinjected horseradish peroxidase in dorsal root ganglia neurons in vitro. Cell Tissue Res. 2000;299:213–224. doi: 10.1007/s004419900120. [DOI] [PubMed] [Google Scholar]
- 64.Vickers JC, Dickson TC, Adlard PA, Saunders HL, King CE, McCormack G. The cause of neuronal degeneration in Alzheimer's disease. Prog Neurobiol. 2000;60:139–165. doi: 10.1016/s0301-0082(99)00023-4. [DOI] [PubMed] [Google Scholar]
- 65.Walker KL, Yoo HK, Undamatla J, Szaro BG. Loss of neurofilaments alters axonal growth dynamics. J Neurosci. 2001;21:9655–9666. doi: 10.1523/JNEUROSCI.21-24-09655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. J Neurosci. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu Q, Couillard-Despres S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]