

Abstract
Under both pathological and experimental conditions, area CA3 of the adult or juvenile hippocampus generates periodic population discharges known as interictal bursts. Whereas the ionic and synaptic basis of individual bursts has been comprehensively studied experimentally and computationally, the pacemaker mechanisms underlying interictal rhythmicity remain conjectural. We showed previously that rhythmic population discharges resembling interictal bursts can be induced in hippocampal slices from first postnatal week mice, in Mg2+-free solution with GABAAreceptor-mediated inhibition blocked. Here we show that these neonatal bursts occurred with high temporal precision and that their frequency and regularity were greatly reduced by the bradycardic agent ZD-7288 when applied at concentrations and durations that selectively block the hyperpolarization-activated, cationic currentIh. Augmenting Ihby elevating intracellular cAMP dramatically increased burst frequency in a protein kinase A-independent manner. Burst amplitudes were strongly correlated with the preceding, but not the following, interburst intervals. The experimentally observed distribution of interburst intervals was modeled by assuming that a burst was triggered whenever the instantaneous rate of spontaneous EPSPs (sEPSPs) exceeded a threshold and that the mean sEPSP rate was minimal immediately after a burst and then relaxed exponentially to a steady-state level. The effect of blocking Ih in any given slice could be modeled by decreasing only the steady-state sEPSP rate, suggesting that the instantaneous rate of sEPSPs is governed by the level of Ih activation and raising the novel possibility that interburst intervals reflected the slow activation kinetics of Ih in the neonatal CA3.
Keywords: CA3, interictal, pacemaker, neonatal, mouse, Ih, cAMP
Introduction
Area CA3 of the adult and juvenile hippocampus is a well studied generator of synchronous neuronal discharges. Under various experimental conditions, both in vivo and in vitro, it generates paroxysmal population bursts that recur at regular intervals of (depending on preparation) 2–20 sec (Ayala et al., 1973; Lebovitz, 1974; Schwartzkroin and Prince, 1978; Hablitz, 1984; Korn et al., 1987; Jensen and Yaari, 1988;Arvanov et al., 1995; Merlin et al., 1995; Avoli et al., 1996; de Curtis and Avanzini, 2001). These events resemble pathological “interictal spikes” observed in recordings from epileptic foci in human patients or from excised epileptic tissue from such patients (Cohen et al., 2002) and are therefore called interictal bursts (IBs). IBs can be recorded also in adjacent limbic regions but, in almost all cases, are found to propagate to these other regions from their site of origin in CA3 (Bragdon et al., 1992; Stoop and Pralong, 2000). Whether IBs trigger ictal epileptic episodes or, conversely, suppress them is debated (Bragdon et al., 1992; Barbarosie and Avoli, 1997; de Curtis and Avanzini, 2001); either way, IBs are closely linked with epileptogenesis, and understanding their mechanism of generation is crucial for understanding and treating epilepsy.
The intervals between IBs are highly regular: in vivo, the mean coefficient of variation of interburst intervals can be <0.1 (Lebovitz, 1979), a precision comparable with that of the normal human heartbeat (Van Hoogenhuyze et al., 1991). Whereas the ionic and synaptic mechanisms generating individual epileptiform bursts in the hippocampus have been thoroughly studied and modeled (Traub and Miles, 1991), the pacemaker mechanisms underlying the precise timing of IBs remain conjectural. In a seminal study in vivo, Lebovitz (1979) suggested that each IB generates a transient postburst period of suppression during which the system is refractory to the generation of another burst. More recent in vitro studies propose that the refractory period is attributable to synaptic depression (Staley et al., 1998). An alternative to pacing by recovery from suppression is pacing by a slow buildup of excitation, which in the heart, and in a variety of other regularly bursting neuronal networks, is mediated by the hyperpolarization-activated excitatory currentIh (Soltesz et al., 1991) (for review, see Pape, 1996; Bal and McCormick, 1997; Luthi et al., 1998; Dickson et al., 2000). However, although Ih is expressed in the hippocampus by both pyramidal cells (Maccaferri et al., 1993) and inhibitory interneurons (Maccaferri and McBain, 1996;Strata et al., 1997), it has never been implicated in interictal rhythmogenesis.
We demonstrated recently (Wells et al., 2000) that, when GABAA receptors (GABAARs) are blocked in slices of neonatal mouse hippocampus bathed in Mg2+-free artificial CSF (ACSF), highly rhythmic population bursts occur as early as the day of birth. Here we show that the frequency of these neonatal interictal bursts (nIBs) is strongly modulated by pharmacological manipulations that are known to affect the level of Ih activation, suggesting that Ih plays a major role in the timing of nIBs.
Materials and Methods
Slice preparation and solutions. Horizontal brain slices, 500 μm thick, including hippocampus were prepared from neonatal mice of postnatal days 1–6 (P1–P6) (P0 being the first 24 hr after birth). Except for the plane of section, procedures were as described previously (Wells et al., 2000). Slices chosen for experiments (one to two per animal, bisected along the midline to yield two hemislices each) were perpendicular to the long axis of the hippocampus or nearly so (see Fig. 1). After dissection, slices were submerged in a holding chamber with recirculated, oxygenated ACSF at room temperature. For recording, slices were transferred to a submersion chamber and continuously superfused, using a push–pull configuration of peristaltic pumps, with 2.5–3 ml/min ACSF at room temperature, saturated with a mixture of 95% O2–5% CO2. ACSF for dissection and for the holding chamber was composed of the following (in mm): 126 NaCl, 3 KCl, 1.2 NaH2PO4, 2.0 CaCl2, 1.3 MgSO4, 26 NaHCO3, and 20 dextrose. In the recording chamber, slices were initially superfused with Mg2+-free ACSF, which was identical in composition except that equimolar CaCl2 was substituted for MgSO4, for a final Ca2+ concentration of 3.3 mm, to maintain the total divalent ion concentration. To induce paroxysmal discharges (nIBs) (see Fig.2A), 5 μm of the GABAAR antagonist gabazine was then added to the bath. Mg2+-free ACSF with 5 μm gabazine will be referred to as “control ACSF.”
Fig. 1.

A horizontal hippocampal slice from a P4 mouse, as visualized during the experiment. CA1, CA3, and dentate gyrus (DG) are indicated; typical recording position in CA3 stratum radiatum is indicated by the asterisk. Arrows denote lateral (L) and rostral (R) directions.
Fig. 2.

Neonatal IBs in area CA3 are highly regular. A, A 500-sec-long extracellular record from a P3 slice bathed in control ACSF (Mg2+-free, with 5 μm gabazine), illustrating the high regularity of nIBs.B, A segment expanded from the record inA, illustrating that intercluster intervals (two-sided arrows) but not intracluster intervals were included in the analysis in this study. C, The IBI histogram for the experiment ofA shows two well separated peaks; the peak below 5 sec consists of intracluster intervals, which were excluded in the calculation of the CIH (solid line). CVIBI for this experiment was 0.12. D, Superimposed CIHs from all 22 slices recorded in control ACSF. Note the tight clustering and steep slope of 16 CIHs on the left side of the plot, illustrating the pacemaker-like character of nIBs. E, The mean IBI of all slices showed no change with postnatal age. F, In contrast, the CVIBI increased significantly between P1 and P6 (note the regression line).
Drugs. SR-95531 (gabazine), 3-isobutyl-1-methylxanthine (IBMX), and staurosporine were purchased from Sigma (St. Louis, MO). Forskolin, dideoxyforskolin, and ZD-7288 were purchased from Tocris (Ballwin, MO). Drugs were prepared as stock solutions in water or DMSO, as required, at (typically) 1000-fold final concentration, divided into aliquots, and stored at −20°C. During experiments, thawed aliquots were diluted directly into control ACSF.
Electrophysiological recordings. Extracellular field potentials were recorded using thick-walled glass micropipettes broken to a final outside diameter of ∼5 μm under microscopic control and filled with 0.9% NaCl. Differential DC signals (tissue vs bath) were low-pass filtered at 1 kHz, amplified 1000× (Intronix Technologies, Bolton, Ontario, Canada), digitized at 1000 samples/sec, and streamed to disk, using custom software written (by A. Agmon) in the LabView environment (National Instruments, Austin, TX).
Data analysis. Because the raw data were highly oversampled, data records were smoothed and decimated offline by replacing successive blocks of data points with their average; the size of the averaged block (32 points) was chosen empirically to provide optimal noise reduction with minimum loss of signal amplitude and was kept the same for all analyzed records. Effects of drugs were quantified as a ratio over control conditions and are reported as geometric means ± geometric SEMs, together with the number of slices (n) and the number of animals (N) tested in each condition. To quantify drug effects on nIB frequency (see Figs. 3, 4), the number of nIBs in a 500 sec window, starting at least 1000 sec after drug arrival and spanning the period of maximal drug effect, was divided by the number of nIBs in a 500 sec window immediately preceding drug arrival. To calculate nIB amplitudes and interburst intervals (IBIs), nIBs were logged, time-stamped, and measured by custom software written in LabView (A. Agmon); each logged event was examined visually and confirmed by the user, who also checked the record to verify that no nIBs were missed by the program. The amplitude of an nIB was defined as its peak-to-peak voltage difference, and the IBI was defined as the interval between the negative peaks of two adjacent events. When bursts occurred in clusters (see Fig. 2A–C and the first paragraph of Results), each cluster was regarded as a single nIB with amplitude equal to that of the first event in the cluster (which was always the largest), and IBIs were measured from the negative peak of the last event in one cluster to the negative peak of the first event in the next cluster (see Fig. 2B, arrows). Cumulative IBI histograms (CIHs) were calculated using MathCad (MathSoft, Cambridge, MA). All slices with nIBs in control ACSF of at least 200 μV in amplitude and 20 mHz (1 every 50 sec) in frequency were included in the analysis.
Fig. 3.

Blocking Ih strongly reduced nIB frequency and regularity. A, BlockingIh with the specific blocker ZD-7288 caused a more than threefold reduction in nIB frequency, coupled to a pronounced increase in nIB amplitudes. B, The time course of decrease in nIB frequency for the experiment shown inA, plotted at the same time scale. Data points represent instantaneous frequency (1/previous IBI); solid line is a running average, calculated using a sliding window of five data points. Time 0 designates drug arrival in the recording chamber. C, The rightward shift and decreased slope of the CIH for the experiment ofA illustrates the drug-induced increase in the IBI and the CVIBI, respectively. CIHs were calculated using 1 sec bins. D, CVIBI plotted against mean IBI, with data points from the same slice before (open symbols) and after (filled symbols) adding ZD-7288 connected by lines. Note that, in the great majority of cases, the ZD-7288-induced increase in the mean IBI was coupled to a pronounced increase in the CVIBI.E, A reduction in nIB frequency of a similar magnitude to that induced by ZD-7288 was caused by the less specific (but reversible) Ih blocker Cs+ (2 mm). F, Summary of the percent reduction in nIB frequency attributable to ZD-7288 (see legend for concentrations) and Cs+ (2 mm) in all experiments. A andE are from different P3 animals. Calibration:A, 100 μV, 300 sec; E, 150 μV, 250 sec.
Fig. 4.

Increasing intracellular cAMP strongly accelerated nIB frequency in a PKA-independent manner. A, The adenylyl cyclase activator forskolin (25 μm) caused a threefold increase in nIB frequency and a concomitant decrease in amplitudes within minutes after application; theIh blocker ZD-7288 (20 μm) reversed this increase and further reduced the nIB frequency to 60% of its control value before forskolin application (P1 slice).B, Application of the phosphodiesterase inhibitor IBMX (200 μm) caused a 2.5-fold increase in nIB frequency and a concomitant reduction in amplitudes (P3 slice). C, Preincubation for >1 hr with the broad-spectrum protein kinase inhibitor staurosporine (100 nm) did not prevent forskolin (20 μm) from accelerating nIBs by 1.7-fold (P3 slice).D, In the presence of staurosporine, forskolin (25 μm) still caused a 1.5-fold increase in nIB frequency over the frequency in equimolar concentration of the analog DDF, which does not activate adenylyl cyclase (P5 slice). E, Summary plot of all slices tested in DDF and forskolin without staurosporine; data points not connected by lines are from slices tested only in forskolin. Symbols and lines are coded by drug concentration (legend). F, Summary plot as inE but for slices tested in the presence of 100 nm staurosporine. Calibration: A, 100 μV, 500 sec; B, 200 μV, 400 sec; C, 200 μV, 250 sec; D, 100 μV, 250 sec.
Statistics. Statistical significance (pvalue) was computed numerically using exact permutation methods (Good, 1999); calculations were done in MathCad. Specifically, significance of drug-induced changes was computed using the binomial sign test; significance of differences between population means was computed from 10,000 random permutations of the data, and significance of linear correlations (see Fig. 2E,F) was calculated from the Pitman statistic Σi(i ·Xi) computed for 10,000 random permutations of the data. All reported p values are single-tailed probabilities unless noted otherwise.
Computational modeling of nIBs. All modeling was done using MathCad software; the following description uses MathCad notation. The model assumed that, on average, μ spontaneous Poissonian events (e.g., sEPSPs) occur in any given time epoch (a single time epoch in the model represented 100 msec). The probability that exactlyk events will occur in any given time epoch is given by the following Poisson distribution:
The probability C(k,μ) that kor fewer events will occur is given by the cumulative Poisson distribution as follows:
Assume that the occurrence of M (or more) events within a single time epoch triggers a burst. The unconditional probability B(M,μ) of burst occurrence at any given epoch is, therefore, 1 minus the probability that M − 1 or fewer events will occur, or
A normalized IBI histogram gives the probability of occurrence of an IBI of any given length. Once a burst has occurred, the probability that the next burst will occur after exactly jtime epochs is the product of the (j − 1) probabilities (1 − B(M,μ)) that a burst will not occur at any of the first (j − 1) epochs and the probability B(M,μ) that a burst will occur at the jth epoch:
(In the limit of infinitesimally short epochs, this simply gives the interevent interval histogram of Poissonian events, which is a decaying exponential.)
Now assume that μ itself is not stationary but drops to 0 immediately after a burst and then relaxes exponentially to an asymptotic steady-state level μss:
where j is the index of the current time epoch (counting from the occurrence of the last burst), and τ is the time constant of relaxation expressed in time epochs. The expression for the normalized IBI histogram will now be as follows:
From this vector, the mean IBI and the CVIBI (the coefficient of variation of the IBIs) are easily calculated:
Finally, the CIH is simply the time integral of the normalized IBI histogram, as follows:
To fit experimental CIHs with simulated curves (see Fig. 7), this vector was calculated for 1 ≤ j ≤ 1000 (equivalent to 100 sec) for different values of the three free parameters (M, μss, and τ). In addition to computing CIH (see Fig. 7A), IBI, andCVIBI values (see Fig.6E,F), the assumptions of the model were also used to simulate directly trains of nIBs (see Fig.6A–D), using the MathCad functionrpois(m, λ) which returns a vector ofm random numbers having a Poisson distribution with a mean λ.
Fig. 7.

A change in μss, but not in τ, can account for the effect of ZD-7288 on nIB frequency and regularity. A, CIHs of experimental nIBs from a P6 slice in control ACSF (× symbols) was fitted by a curve (solid line) computed with the parameter values μss = 184 and τ = 4 sec. The effect of 20 μm ZD-7288 (squares) was well fitted (solid line) by reducing μss to 167.5, with only a minor increase in τ (to 4.6 sec). With μssunchanged, a 2.5-fold increase in τ (to 10.5 sec) was required to achieve the same rightward shift in the median IBI, but the resulting curve (dotted line) did not reproduce the pronounced decrease in the slope of the experimental CIH. Experimental CIHs were calculated in 1 sec bins. B, The value of the model parameter μ (lefty-axis) in control ACSF (heavy solid line) and in ZD-7288 (heavy dashed-dotted line), as a function of the time elapsed because the previous burst, for the same parameter values used inA. The relatively small reduction in μssrequired to simulate the effect of the drug caused a dramatic drop in the probability of burst occurrence per 100 msec (righty-axis) in ZD-7288 (light dashed-dotted line) compared with control ACSF (light solid line), because now fluctuations around μ only rarely crossed threshold (compare with Fig.6C,D).
Fig. 6.
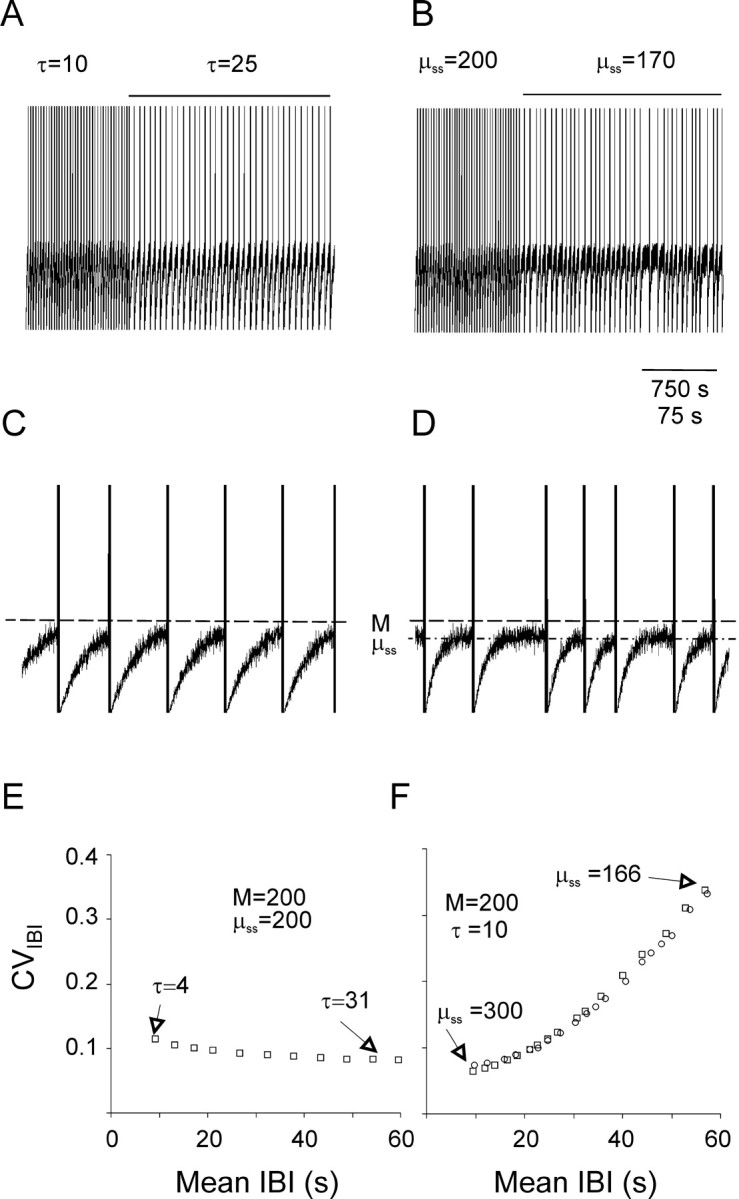
A three-parameter computational model simulated the effect of ZD-7288 on nIB frequency and regularity.A–D, Simulated sequences of nIBs are plotted at a slow (A, B) and a 10-fold faster (C, D) time base. Simulated nIBs were generated by assuming that the mean instantaneous sEPSP rate (μ) is reset to 0 after each burst and then relaxes exponentially to an asymptote (μss) with a time constant τ. Fluctuations around μ were generated by a Poissonian random-number generator; each time a fluctuation crossed the thresholdM (dashed line in C andD), a burst was assumed to be triggered. InA and B, the initial part of the trace was computed using the values M = μss = 200 and τ = 10 sec; during the time indicated by the horizontal lines above the trace, either τ (A, C) or μss(B, D) were changed to the values indicated (μss is also indicated by the dashed-dotted line in D). The traces represent the value of μ, except that the occurrence of a burst is indicated by a vertical line of arbitrary height. Note that increasing τ (A,C) reduced the frequency of bursts without affecting their regularity, whereas reducing μss (B,D) affected both the frequency and the regularity of the bursts. E, The simulated CVIBI versus the simulated mean IBI, computed using the indicated values ofM and μss while varying the value of τ as indicated. Note that changing τ had very little effect on the CVIBI. F, The simulated CVIBIversus the simulated mean IBI, computed using the indicated values ofM and τ while varying μss as indicated (squares). Note that changing μss affected in parallel both the mean IBI and the CVIBI. Changing Mfrom 140 to 238 while keeping μss = 200 generated data points (circles) that fell along the same curve generated by changing μss while keeping Mconstant.
Results
We monitored epileptiform population activity in area CA3 of neonatal mice, by recording extracellular field potentials from stratum radiatum in horizontal hippocampal brain slices (Fig. 1, asterisk denotes a typical recording location). As reported previously (Wells et al., 2000), superfusing the slice with Mg2+-free ACSF with 5 μm of the GABAAR antagonist gabazine (control ACSF) resulted in the appearance of large-amplitude, rhythmic population discharges (Fig.2A), which will be referred to here as nIBs. An initial exposure to Mg2+-free ACSF alone for 20–30 min, before addition of gabazine, seemed to be required to “prime” the burst mechanism, because slices superfused directly with control ACSF often failed to burst regularly. Individual nIBs consisted of a triphasic extracellular potential (Fig. 2B), with an early, large-amplitude negative “spike,” ∼0.5 sec in duration, followed by a slower and smaller positive “wave,” and ending with an even smaller negative undershoot. In many slices, some of the nIBs occurred in doublets or triplets or (rarely) in clusters of four to five events (Fig. 2B illustrates a sequence of two doublets, followed by two single bursts). Intracluster intervals (typically <5 sec) were always much shorter than intercluster intervals and formed a clearly separable peak in the IBI histogram (Fig. 2C). Unlike intercluster intervals, intracluster intervals were not affected by our experimental manipulations (data not shown) and were likely to be under the control of a separate mechanism. Intracluster intervals were therefore excluded from analysis, and each cluster was treated as a single, prolonged event for the sake of determining IBIs (Fig. 2B, arrows demonstrate the definition of IBIs in this study) (see Materials and Methods).
Neonatal IBs exhibited pacemaker-like regularity
The bursting characteristics of each slice were quantified by calculating the mean IBI, the coefficient of variation of the IBIs (CVIBI, i.e., the SD/mean of the IBIs), and the CIH (the normalized integral of the IBI histogram). The CIH of the slice illustrated in Figure 2A is superimposed on the IBI histogram of the same slice in Figure 2C. CIHs of all of the slices in our sample recorded in control ACSF are shown superimposed in Figure 2D. In control ACSF, nIBs occurred at highly regular intervals of 11–32 sec, with the CVIBI averaging 0.20 ± 0.02 (n = 22; N = 17). Moreover, as illustrated in Figure 2D, the majority of our slices (73%) had CIHs tightly clustered at the left end of the range, with IBIs for this group averaging 14.2 ± 0.4 sec (n = 16) and with a mean CVIBI of 0.18 ± 0.02. The mean IBI did not change significantly with age over the developmental period of our study (Fig. 2E) (p = 0.30; two-tailed), but the CVIBI showed a small but significant increase (Fig. 2F) (p < 0.05; two-tailed), indicating some loss of precision with maturation. Indeed, interictal activity in slices from juvenile animals was considerably less regular than in neonatal slices (our unpublished observations).
Blocking Ih strongly reduced nIB frequency and regularity
The high regularity of nIBs prompted us to look for an underlying pacemaker mechanism. A hypothesis proposed over two decades ago (Lebovitz, 1979) suggests that each IB is followed by a postburst refractory period, during which generation of additional bursts is suppressed. This suppression was postulated to be caused by a slowly decaying inhibitory conductance activated by the burst itself; hence, this mechanism was named “autorhythmicity.” If indeed an inhibitory conductance activated by the burst was responsible for pacing nIBs, then blocking it should cause a pronounced acceleration of the rhythm. However, blocking any of the known inhibitory conductances had little or no effect on nIB frequency (our unpublished observations) (Staley et al., 1998).
An alternative to a slowly decaying inhibition is a slow buildup of excitation, for example by a slow inward “pacemaker” current. If such a current is involved in the timing of nIBs, then blocking it should decrease nIB frequency. A current implicated in pacing the heart, as well as various neuronal oscillators in the CNS, is the hyperpolarization-activated cationic currentIh (Pape, 1996). We therefore tested the effect of ZD-7288, which, until very recently (Chevaleyre and Castillo, 2002), was considered a highly selective blocker ofIh (Harris and Constanti, 1995;Gasparini and DiFrancesco, 1997; Satoh and Yamada, 2000), on nIB frequency. In the experiment illustrated in Figure3A, 20 μm ZD-7288 caused a threefold decrease in nIB frequency, with the mean IBI increasing from 12.0 to 35.9 sec. The time course of this effect of ZD-7288 is plotted in Figure 3B, in which the instantaneous burst rate (1/previous IBI) and its running average are plotted against time after drug arrival in the recording chamber. As illustrated by the graph, the reduction in bursting rate developed over several minutes, and the maximal effect was reached within ∼1000 sec from the moment of drug arrival. In a total of six slices in which the time course of drug action was examined in detail, on average, ∼1200 sec superfusion with 20 μmZD-7288 was required for maximal reduction in nIB frequency (range, 700–1650 sec). The time to maximal effect of ZD-7288 on nIB frequency was consistent with the time to maximal block ofIh by ZD-7288, reported to be 10–20 min in studies using 20–100 μm of the drug (Harris and Constanti, 1995; Maccaferri and McBain, 1996; Gasparini and DiFrancesco, 1997; Chevaleyre and Castillo, 2002).
The CIHs of the experiment in Figure 3A, before and after exposure to ZD-7288, are shown in Figure 3C. In addition to the pronounced reduction in the frequency of nIBs, evident from the rightward shift of the CIH, ZD-7288 also markedly reduced their temporal precision, as evident from the pronounced decrease in the slope of the CIH (Fig. 3C). In this slice, the CVIBI increased from 0.11 to 0.28. In Figure3D, the CVIBI is plotted against mean IBI for all slices tested in 10–20 μm ZD-7288, with data points corresponding to the same slice before and after addition of drug connected by lines. The concomitant increase in both mean IBI and CVIBI is clearly evident from the general upward-and-rightward direction of the connecting lines. On average, application of ZD-7288 (10–20 μm) decreased the rate of nIBs to approximately one-half of the control frequency (0.54 ± 0.03; n = 22; N= 17; p < 10−6) and increased the CVIBI by 1.52 ± 0.14-fold over control ACSF (p < 0.001).
CsCl (2 mm), another (although less specific) blocker ofIh (Magee, 1998), reduced nIB frequency in neonatal slices to 0.45 ± 0.10 of control (n = N = 3) (Fig. 3E). Unlike ZD-7288, the effect of Cs+ was readily reversed after washout. Figure 3F summarizes the effects of 10–20 μm ZD-7288 (circles and triangles, respectively) and Cs+ on nIB frequency in all slices tested.
Although generally considered a highly selective blocker ofIh in various systems (BoSmith et al., 1993; Briggs et al., 1994; Harris et al., 1994; Satoh and Yamada, 2000), ZD-7288 was very recently reported to depress synaptic transmission in the hippocampus through an unknown mechanism, apparently independent of its effect onIh (Chevaleyre and Castillo, 2002). The time course of synaptic depression was very slow compared with the effect on Ih examined in the same study, with no obvious depression occurring until at least 30 min of exposure to drug and with maximal effect requiring at least 60 min of exposure to 50 μm ZD-7288; lower concentrations of drug (10 μm) caused only a slight depression after a similar exposure time. Thus, it seemed highly unlikely that these nonspecific effects of ZD-7288 could have contributed to the pronounced reduction in nIB frequency after 20 min of exposure to 20 μm ZD-7288 in our experiments. Also, the pronounced increase in nIB amplitudes induced by ZD-7288 in our experiments seemed inconsistent with synaptic depression. Nevertheless, we tested for any synaptic depression under the conditions of our experiments by quantifying the effect of ZD-7288 on field EPSPs (fEPSPs) evoked in CA1 by stimulation of the CA3 to CA1 pathway, the same pathway tested by Chevaleyre and Castillo (2002). These control experiments were done on slices from P5–P29 mice, because, in slices from younger animals, the evoked fEPSP was too small and labile for reliable analysis. Seven slices in which the evoked fEPSP was stable (to ±20% of control amplitude) for at least 30 min before drug application were selected for analysis. In these slices, the evoked response remained virtually unchanged after 60 min of superfusion with 20–25 μm ZD-7288 (1.01 ± 0.14 of control amplitude; n = 7; N = 6; data not shown). We conclude that the typical concentration of ZD-7288 used in our study did not cause any appreciable synaptic depression within the time course of the experiments. However, very high concentrations (500 μm) of ZD-7288 caused drastic reduction in the frequency of nIBs (to 0.21 ± 0.05 of control; n = 5; N = 3; p < 0.05) (Fig.3F, squares) and, in contrast to the lower concentrations, also strongly depressed nIB amplitudes (data not shown), suggesting that high concentrations of ZD-7288 may indeed cause synaptic depression.
Increasing intracellular cAMP strongly increased nIB frequency
Ih is strongly modulated by cyclic nucleotides, which bind directly to the cytoplasmic domain of the channel (Pape, 1996; Santoro and Tibbs, 1999; Wainger et al., 2001); we therefore tested whether modulating intracellular cAMP affected nIB frequency. In the experiment illustrated in Figure4A, the adenylyl cyclase activator forskolin (25 μm) accelerated nIBs more than threefold and markedly reduced their amplitude; both effects were reversed by 20 μm ZD-7288. Overall, forskolin (10–25 μm) caused a 2.0 ± 0.5-fold increase in nIB frequency (n =N = 3). Forskolin (5–25 μm) still increased nIB frequency in the presence of ZD-7288 (15 μm), causing a 2.1- to 3-fold increase over the frequency in ZD-7288 alone (n = N = 2; data not shown); this could have represented the effect of elevated intracellular cAMP concentration on residualIh channels, because 15 μm ZD-7288 is expected to block only ∼50% of the channels in hippocampal pyramidal neurons (Gasparini and DiFrancesco, 1997).
Because forskolin also adds to excitability by blocking K+ channels, we tested the effects of the analog dideoxyforskolin (DDF), which also blocks K+ channels but does not activate adenylyl cyclase (Hoshi et al., 1988). DDF (5–25 μm) caused a modest (1.23 ± 0.11-fold) increase in nIB frequency compared with control ACSF; however, replacing DDF by equimolar amounts of forskolin caused an additional increase (compared with the frequency in DDF) of 1.47 ± 0.09-fold (n = 11; N = 9;p < 0.001), as summarized in Figure4E. We also tested the effect on nIB frequency of IBMX, a nonselective inhibitor of the enzymatic degradation of cAMP by phosphodiesterase (Fredholm et al., 1976). Application of 200 μm IBMX caused, on average, a 1.87 ± 0.25-fold increase in nIB frequency (n = 6;N = 3; p < 0.02) (Fig.4B). Because both forskolin and IBMX are expected to increase the intracellular concentration of cAMP, we conclude that intracellular cAMP has a strong positive modulatory effect on nIB frequency.
cAMP effects on nIB frequency were protein kinase A independent
cAMP could potentially modulate nIB frequency by mechanisms independent of Ih, for example by reducing the slow IAHP (Dunwiddie et al., 1992; Pedarzani and Storm, 1995a), by increasing synaptic efficacy (Chavez-Noriega and Stevens, 1992; Boulanger and Poo, 1999;Castro-Alamancos and Calcagnotto, 1999), or by opening gap junctions (Burghardt et al., 1995; Banoub et al., 1996; Paulson et al., 2000; van Rijen et al., 2000; Carystinos et al., 2001). All f these effects, however, are thought to be mediated by protein kinase A (PKA), whereas the effect of cAMP on Ih in CA1 and elsewhere is attributed to direct gating of the channel by cAMP (DiFrancesco and Tortora, 1991; Pedarzani and Storm, 1995b; Wainger et al., 2001). To test whether PKA-mediated effects could have contributed to the observed effect of forskolin, we superfused the slices with the broad-spectrum protein kinase blocker staurosporine (100 nm) for 30–60 min before addition of forskolin. Preincubation in staurosporine caused a variable reduction in nIB amplitude but no change in frequency (data not shown; n= 7; N = 4; p = 0.47; two-tailed). Preincubation in staurosporine did not affect the large increase in nIB frequency seen with 10–25 μm forskolin (Fig.4C) (nIB frequency increased by 2.1 ± 0.2-fold;n = 5; N = 4; p < 0.05). In the presence of staurosporine, 25 μmDDF caused a small, 1.13 ± 0.10-fold increase in nIB frequency, but replacing DDF by equimolar amounts of forskolin (Fig.4D) caused an additional increase in frequency (compared with DDF) of 1.50 ± 0.03-fold (n = 5;N = 4; p < 0.05), which was not significantly different from the effect of forskolin without staurosporine in the bath (p = 0.80; two-tailed). The effects of DDF and forskolin in the presence of staurosporine are summarized in Figure 4F. We conclude that the strong acceleration of nIBs by forskolin and IBMX was not dependent on PKA but was consistent with a direct action of cAMP on the Ih channel. This does not exclude the possibility of PKA-mediated effects onIh (Mellor et al., 2002; Vargas and Lucero, 2002), because these indirect effects could have been occluded by the direct effects of cAMP.
[To verify that our batch of staurosporine was effective, we tested it on the cAMP-mediated presynaptic enhancement of the mossy fibers to CA3 pathway, which is PKA dependent (Weisskopf et al., 1994; Lonart et al., 1998). In two slices from two P13 mice, a 20 min application of 25 μm forskolin caused a 1.6- to 2.4-fold increase in the amplitude of the postsynaptic (but not the presynaptic) component of the field potential evoked in CA3 by a 50–80 μA stimulus in the hilus of the dentate gyrus. In a second slice from each animal, preincubation with 100 nm staurosporine for 30–45 min totally blocked this forskolin-induced potentiation (data not shown), providing evidence that our batch of staurosporine effectively blocked PKA.]
Burst amplitudes were strongly correlated with the preceding but not the following IBI
As is evident from Figures 3 and 4, drugs that reduced the frequency of nIBs also increased their amplitude, whereas drugs that accelerated the bursts reduced their amplitude. The change in amplitude could have been a result of the change in frequency; conversely, the change in frequency could have been secondary to the change in amplitude. To determine whether the primary effect of ZD-7288 was on the frequency of nIBs or on their amplitudes, we examined the correlation between event amplitudes and both the preceding and the following IBIs. If the primary effect of the drug was to increase nIB amplitude, and the increased amplitude in turn slowed down the rhythm, one would expect that nIB amplitudes would be correlated with the following, but not with the preceding, IBIs. As evident from Figure5, the opposite was the case: nIB amplitudes were strongly dependent on the IBIs preceding the events (Fig. 5A,C) and not at all on the IBIs following them (Fig. 5B,D). In slices with IBIs not exceeding 30 sec, the relationship between the amplitudes of nIBs and the preceding IBIs was well described by linear regression (Fig. 5A); the mean coefficient of determination (r2) of the linear regression line was 0.64 ± 0.04 in control and 0.78 ± 0.03 in ZD-7288 (n = 25; N = 20). When nIB amplitudes from the same slices were plotted against the following IBIs (Fig.5B), no such correlation was observed (r2 = 0.04 ± 0.01 for both control and ZD-7288 conditions). In slices with IBIs longer than 30 sec, nIB amplitudes seemed to reach a ceiling at IBIs of 30–40 sec, and the relationship between nIBs and preceding IBIs was better fit by a decaying exponential (Fig. 5C); again, no such relationship was observed with the following IBIs (Fig. 5D). This analysis strongly suggested that ZD-7288 directly affected the frequency of the nIBs and that the change in amplitude was a secondary effect caused by the slowing of the rhythm and not vice versa. In most slices (e.g., the two illustrated in Fig.5A,C), data points from nIBs in control solution were distributed along or close to the same line fitted to data points collected in the presence of ZD-7288, suggesting that the drug affected only the timing of the events and did not disturb the relationship between timing and amplitude.
Fig. 5.

Amplitudes of nIBs were strongly correlated with the preceding but not the following interburst intervals. Illustrated data were recorded before (circles) and after (triangles) adding ZD-7288 to a P2 slice (A, B) and a P4 slice (C, D). In A andB, data points in control and in ZD-7288 were fitted separately with linear regression lines, illustrating a strong correlation in A(r2 = 0.80 andr2 = 0.87, respectively) but lack of correlation in B(r2 = 5 × 10−5 and r2= 0.03, respectively). Note that the two regression lines inA nearly coincide. In C, data points in ZD-7288 were fitted by a decaying exponential with an asymptote of 0.6 mV and a time constant of 12.5 sec; note that data points in control ACSF fall along nearly the same curve
A simple computational model simulated the effect of ZD-7288 on nIB frequency and regularity
To gain insight into the mechanism by whichIh modulates nIB rhythm, we constructed a network-level mathematical model of the pacemaker driving nIBs, which generated computed IBI distributions closely resembling experimental CIHs (for mathematical details of the model, see Materials and Methods). Our model formally resembled a model recently described by Staley et al. (2001) but differed in some of its underlying assumptions and in using Poissonian rather than binomial statistics. The model assumed that excitatory synapses in CA3 are spontaneously active at a rate that fluctuates with a Poissonian distribution around a mean instantaneous rate μ (Fatt and Katz, 1952; Rotshenker and Rahamimoff, 1970; Isaacson and Walmsley, 1995). It further assumed that μ falls to 0 immediately after an nIB and then recovers exponentially, with a time constant τ, to a steady-state level μss (Staley et al., 1998, 2001). Finally, it assumed that a burst is triggered whenever the actual instantaneous rate of sEPSPs in the network exceeds a threshold M (Prida and Sanchez-Andres, 1999). A Monte-Carlo implementation of the model (based on a random number generator) was used to generate simulated trains of nIBs (Fig.6A–D), and a computational implementation was used to calculate mean IBI, CVIBI, and CIH values of modeled nIBs (Fig.6E,F) and to fit them to experimental CIHs (Fig. 7A). It should be noted that the model made no assumptions about the underlying cellular constituents. For example, the parameter μ could be interpreted as the aggregate rate of sEPSPs in the network or it could refer to the rate of sEPSPs in a subset of cells functioning as a pacemaker “kernel.”
Although the model depended on three free parameters (M, μss, and τ), within a relatively wide range of values, the simulated CIHs were sensitive mostly to the difference between M and μss and not to their absolute values (Fig. 6F); we therefore effectively reduced the number of free parameters to two by fixing M at the arbitrary value M = 200 and varied the other two parameters (τ and μss) independently. Varying these two parameters had very different effects on the behavior of the model. When τ was increased, the frequency of simulated nIBs was reduced, but little change in their regularity was evident (Fig.6A,C), as corroborated by calculating the mean IBI and the CVIBI for a range of values of τ; the computed CVIBI values slightly decreased as the computed mean IBIs increased (Fig.6E). In contrast, decreasing μss also reduced the frequency of simulated nIBs but, at the same time, markedly reduced their regularity (Fig.6B,D). When CVIBI values were plotted against corresponding IBIs computed for a range of values of μss, it was evident that the CVIBI values increased in parallel with the mean IBI (Fig. 6F, squares). When, instead of decreasing μss while keeping the threshold M constant, M was increased while keeping μss constant, the computed data points (Fig. 6F, circles) fell along the same line as the data points computed by varying μss while holding M constant.
The effect of blocking Ih was well modeled by a reduction in the steady-state rate of sEPSPs but not by an increase in its time constant of recovery
As evident from a comparison of Figure 3D with Figure6, E and F, a reduction in μss or an increase in M, but not a change in τ, could reproduce the experimental effect of blockingIh, which was an increase in both the mean IBI and in the CVIBI. The model would therefore be consistent with our data if blockingIh increased the differenceM − μss, but not if it changed τ. To test this directly, we fitted computed CIHs to experimentally determined ones, by keeping M = 200 and varying independently both μss and τ. Figure7A illustrates experimental CIHs from a P6 slice, recorded before and after addition of ZD-7288. The superimposed solid lines were the best fit computed from our model. In control ACSF, the best fit was achieved with the parameter values μss = 184 and τ = 4.0 sec. In ZD-7288, the best fit was achieved with parameter values μss = 167.5 and τ = 4.6 sec, which represents a doubling of the difference M − μss but only a 15% increase in the time constant τ. Increasing τ alone (by 2.5-fold), without changing μss, shifted the computed control CIH to the right but did not change its slope (dotted line), corresponding to an increase in the median IBI with little or no change in CVIBI, as demonstrated in Figure6E. On average, ZD-7288 application had no effect on the best-fit value of τ (in control, τ = 6.7 ± 0.6 sec,n = 19, N = 15; in ZD-7288, τ increased by 1.04 ± 0.05-fold and was not significantly greater than control; p = 0.23). In contrast, the best fit for the mean difference M − μssnearly tripled, from 10.7 ± 3.0 in control to 28.8 ± 2 in ZD-7288 (p < 10−6).
In the example of Figure 7A, a reduction in μss from 184 to 167.5 sEPSPs/100 msec, which represents a fractional reduction by <9%, caused >2.5-fold increase in the mean IBI and >1.5-fold increase in the CVIBI. To understand why a relatively small change in the steady-state rate of synaptic activity could cause a dramatic decrease in both the frequency and the regularity of nIBs, it is instructive to examine how the probability for the occurrence of a burst (B(M,μ) in the model; see Materials and Methods) was influenced by the difference M − μss. The drug-induced decrease in μss is illustrated in Figure 7B, which plots μ (heavy solid line for control, heavy dashed-dotted line for ZD-7288) as a function of time elapsed from the previous burst, for the same values of the model parameters used to fit the experimental data in Figure 7A. Although the reduction in μss induced by ZD-7288 was modest, the maximal probability for a burst to occur decreased from 0.127/100 msec in control ACSF (Fig. 7B, light solid line) to only 0.0088/100 msec in the presence of ZD-7288 (light dash-dotted line), which is a 14-fold decrease. The result of this large reduction in burst probability was that, by the time 20 sec have elapsed from the previous burst, a burst had occurred with near certainty (99.9% probability) in control ACSF but with only 20% probability in the presence of ZD-7288, as seen from the CIHs in Figure 7A.
Discussion
Our results indicate that the frequency and the temporal precision of spontaneous interictal bursts in the neonatal CA3 are positively modulated by the hyperpolarization-activated, cyclic nucleotide-sensitive cationic currentIh. This conclusion is based on the pronounced decrease in nIB frequency and regularity whenIh was blocked by either Cs+ or ZD-7288 (Fig. 3), a specific bradycardic agent whose only known effect, at the concentrations and exposure times we used, is to block Ih(Harris and Constanti, 1995; Gasparini and DiFrancesco, 1997; Satoh and Yamada, 2000). This conclusion was further supported by the pronounced increase in nIB frequency induced by manipulations that are expected to elevate intracellular cAMP levels (Fig. 4). The cAMP-mediated effect was not affected by blocking PKA with staurosporine, consistent with a direct action of cAMP on the Ihchannel (Pedarzani and Storm, 1995b; Wainger et al., 2001). To our knowledge, this is the first report implicatingIh in the timing of interictal epileptiform bursts. The strong acceleration of interictal epileptiform bursts by cAMP is also demonstrated here, to our knowledge, for the first time (for modest effects in adult rats, see Boulton et al., 1993) and may need to be considered when prescribing to epilepsy-prone patients drugs that may potentially affect cAMP levels.
Blocking Ih can be modeled by either an increase in the burst threshold or a decrease in the rate of spontaneous synaptic release
How does Ih modulate burst frequency? To gain insight into this question, we developed a simple computational model based on the assumption that a burst is triggered whenever the instantaneous rate of spontaneous EPSPs exceeds a threshold M; a similar mechanism appears to determine the timing of “giant depolarizing potentials,” another form of oscillatory activity in the neonatal hippocampus (Prida and Sanchez-Andres, 1999). The rate of sEPSPs, in turn, was assumed to be a Poissonian variable, with a mean μ that dropped to zero immediately after a burst and then recovered to an asymptotic steady-state value μss with a time constant τ. [A formally similar model described by Staley et al. (2001) uses binomial, rather than Poissonian, statistics and is based on somewhat different assumptions.] Once an appropriate set of parameters (M, μss, and τ) was found to generate a cumulative IBI histogram that fit the experimental record from a given slice, the effect of blockingIh in the same slice could be satisfactorily reproduced by increasing the differenceM − μss, by either increasing the threshold M or decreasing the steady-state sEPSP rate μss but not by increasing the time constant τ. This allows two different interpretations for the role ofIh in the timing of nIBs.
First, Ih could be increasing neuronal excitability in general, for example by contributing to a more depolarized resting potential, as has been demonstrated in the hippocampus and elsewhere (Akasu et al., 1993; Li et al., 1993;Maccaferri et al., 1993; Travagli and Gillis, 1994; Maccaferri and McBain, 1996; Seutin et al., 2001). According to this interpretation,Ih would be contributing to a reduced threshold M but would have no effect on μ. The postburst depression and recovery of μ would then need to be explained by other mechanisms, e.g., by depletion of synaptic vesicles (caused by high-frequency firing during the burst), followed by their gradual replenishment (Zucker and Regehr, 2002). The major determinant of the time constant τ, according to this hypothesis, would be the time course of synaptic vesicle replenishment, andIh would only be modulating the rhythm, not pacing it. Whereas recovery from synaptic depression has been successfully used to model rhythmic bursting in a variety of systems (Staley et al., 1998, 2001; Tabak et al., 2000; Tsodyks et al., 2000), the role of synaptic vesicle replenishment in pacing interictal bursts is yet to be tested directly by pharmacological or genetic manipulations that accelerate it or slow it down.
An alternative interpretation for the role ofIh in timing nIBs, also consistent with the model, is that instead of decreasing the thresholdM, Ih increases μss, the steady-state rate of sEPSPs, e.g., by directly depolarizing presynaptic terminals (Beaumont and Zucker, 2000;Southan et al., 2000; Mellor et al., 2002). Depolarization of presynaptic terminals will increase calcium influx through voltage-gated calcium channels, enhancing the probability of spontaneous neurotransmitter release (Alger et al., 1996; Frerking et al., 2001; Turecek and Trussell, 2001). This interpretation is consistent with the forskolin-induced increase in miniature GABAergic currents described in immature CA3 neurons (Sciancalepore and Cherubini, 1995) and is attractive because it also explains the postburst modulation in μ: if μ is governed byIh, then its time course between bursts should reflect the time course ofIh, which is expected to be inactivated immediately after the burst (attributable to the strong depolarization during the burst) and then to reactivate slowly (by the postburst hyperpolarization) to a steady-state level. The time constant of recovery τ would therefore be determined by the activation kinetics of Ih, andIh would actually function as the pacemaker for nIBs. This interpretation is consistent with our finding that the effect of ZD-7288 could be simulated by reducing μss and not by increasing τ,because ZD-7288 strongly reduces the steady-state amplitude of theIh current with little or no change in its activation kinetics (Harris and Constanti, 1995; Satoh and Yamada, 2000).
Ih channels with the appropriate properties are expressed in the neonatal CA3
A major constraint on the alternative interpretation above is thatIh needs to be activated with a time constant comparable with τ, which in our data averaged ∼7 sec. A very recent study of Ih in the developing mouse hippocampus (Vasilyev and Barish, 2002) indicates that activation kinetics of Ih in neonatal (P1–P5) CA3 pyramidal neurons are indeed dominated by a slow component with a time constant of 5–10 sec at physiological hyperpolarized potentials. Of the four known subunits from whichIh channels may be assembled, pyramidal cells in the neonatal CA3 express HCN1 and HCN2 mRNA (Bender et al., 2001) and protein (Vasilyev and Barish, 2002). In heterologous expression systems, Ihchannels consisting solely of the HCN2 subunit exhibit much slower kinetics and considerably higher cAMP sensitivity compared with HCN1-only or with mixed HCN1/HCN2 channels (Santoro et al., 2000; Chen et al., 2001; Wainger et al., 2001); it is therefore tempting to speculate that the HCN2 subunit is the major contributor toIh channels in the neonatal CA3. Regardless of molecular composition, however,Ih channels in the neonatal CA3 exhibit activation kinetics consistent with our model and with a possible role in pacing interictal bursts.
Burst amplitudes reflect recovery from synaptic depression
As shown in Figure 5, the amplitude of nIBs was correlated with the preceding, but not with the following, IBI; a similar relationship was demonstrated previously in the mature CA3 (Staley et al., 1998), in the embryonic spinal cord (Streit, 1993; Tabak et al., 2001), in the retina (Grzywacz and Sernagor, 2000), and in networks of dissociated cortical neurons (Opitz et al., 2002), suggesting that this relationship is a common feature of rhythmically bursting synaptic networks (O'Donovan, 1999). Burst intensity (which can be reflected in burst amplitude and/or in burst duration) is thought to be determined by the total number of readily releasable synaptic vesicles at the moment of burst initiation (Staley et al., 1998); this number presumably declines to near zero immediately after the burst, because virtually all synapses are activated during a paroxysmal event, and then gradually increases as vesicles are replenished from the reserve pool (Zucker and Regehr, 2002). In our experiments, burst amplitudes did not recover fully until 30–40 sec after the previous burst (Fig.5C), suggesting that the time constant of vesicle replenishment was considerably longer than the time constant τ, calculated to be ∼7 sec from our experimental data. This lends additional support to the alternative interpretation above, that the activation of Ih, and not recovery from synaptic depression, was the process pacing nIBs in the neonatal CA3.
The mechanisms of pacing by Ih are system specific
Whether Ih acts as a bona fide pacemaker of interictal activity in the neonatal hippocampus or merely modulates the rhythm by affecting overall excitability remains to be determined. Indeed, the common characterization ofIh as a “pacemaker current” (Pape, 1996) may obscure its very different modes of action in different systems, which may oscillate at frequencies orders of magnitude apart. For example, Ih accelerates both the heartbeat (DiFrancesco and Ojeda, 1980) and the much faster oscillations in thalamocortical relay neurons (McCormick and Pape, 1990; Soltesz et al., 1991); it also accelerates nIBs in CA3, which are an order of magnitude (at least) slower than the heartbeat. In contrast, Ih actually slows down spindles in the thalamus, which oscillate at approximately the same frequency as nIBs (Luthi et al., 1998). The recent cloning of HCN channels (Ludwig et al., 1998; Monteggia et al., 2000) will no doubt be followed by the generation of knock-out mice in which the precise role of Ih in pacing oscillations in different systems could be tested directly. Note that an HCN2 knock-out mouse was reported recently by Ludwig et al. (2003).
Footnotes
This work was supported by National Institutes of Health Grant HD33463 (A.A.) and by the Epilepsy Foundation through the generous support of the American Epilepsy Society (J.E.W.). We thank Drs. Arlette Colta, Liset M. de la Prida, James Porter, George Spirou, Richard Warren, William Wonderlin, and members of the West Virginia University Sensory Neuroscience Research Center for helpful comments on this and previous versions of this manuscript. We thank Cary Johnson for excellent technical support.
Correspondence should be addressed to Ariel Agmon, Department of Neurobiology and Anatomy, West Virginia University, Health Science Center Drive, Morgantown, WV 26506-9128. E-mail: aagmon@wvu.edu.
References
- 1.Akasu T, Shoji S, Hasuo H. Inward rectifier and low-threshold calcium currents contribute to the spontaneous firing mechanism in neurons of the rat suprachiasmatic nucleus. Pflügers Arch. 1993;425:109–116. doi: 10.1007/BF00374510. [DOI] [PubMed] [Google Scholar]
- 2.Alger BE, Pitler TA, Wagner JJ, Martin LA, Morishita W, Kirov SA, Lenz RA. Retrograde signalling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J Physiol (Lond) 1996;496:197–209. doi: 10.1113/jphysiol.1996.sp021677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvanov VL, Holmes KH, Keele NB, Shinnick-Gallagher P. The functional role of metabotropic glutamate receptors in epileptiform activity induced by 4-aminopyridine in the rat amygdala slice. Brain Res. 1995;669:140–144. doi: 10.1016/0006-8993(94)01243-b. [DOI] [PubMed] [Google Scholar]
- 4.Avoli M, Barbarosie M, Lucke A, Nagao T, Lopantsev V, Kohling R. Synchronous GABA-mediated potentials and epileptiform discharges in the rat limbic system in vitro. J Neurosci. 1996;16:3912–3924. doi: 10.1523/JNEUROSCI.16-12-03912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayala GF, Dichter M, Gumnit RJ, Matsumoto H, Spencer WA. Genesis of epileptic interictal spikes. New knowledge of cortical feedback systems suggests a neurophysiological explanation of brief paroxysms. Brain Res. 1973;52:1–17. doi: 10.1016/0006-8993(73)90647-1. [DOI] [PubMed] [Google Scholar]
- 6.Bal T, McCormick DA. Synchronized oscillations in the inferior olive are controlled by the hyperpolarization-activated cation current I(h). J Neurophysiol. 1997;77:3145–3156. doi: 10.1152/jn.1997.77.6.3145. [DOI] [PubMed] [Google Scholar]
- 7.Banoub RW, Fernstrom M, Malkinson AM, Ruch RJ. Enhancement of gap junctional intercellular communication by dibutyryl cyclic AMP in lung epithelial cells. Anticancer Res. 1996;16:3715–3719. [PubMed] [Google Scholar]
- 8.Barbarosie M, Avoli M. CA3-driven hippocampal-entorhinal loop controls rather than sustains in vitro limbic seizures. J Neurosci. 1997;17:9308–9314. doi: 10.1523/JNEUROSCI.17-23-09308.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beaumont V, Zucker RS. Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat Neurosci. 2000;3:133–141. doi: 10.1038/72072. [DOI] [PubMed] [Google Scholar]
- 10.Bender RA, Brewster A, Santoro B, Ludwig A, Hofmann F, Biel M, Baram TZ. Differential and age-dependent expression of hyperpolarization-activated, cyclic nucleotide-gated cation channel isoforms 1–4 suggests evolving roles in the developing rat hippocampus. Neuroscience. 2001;106:689–698. doi: 10.1016/s0306-4522(01)00314-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.BoSmith RE, Briggs I, Sturgess NC. Inhibitory actions of ZENECA ZD7288 on whole-cell hyperpolarization activated inward current (If) in guinea-pig dissociated sinoatrial node cells. Br J Pharmacol. 1993;110:343–349. doi: 10.1111/j.1476-5381.1993.tb13815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boulanger L, Poo M. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999;284:1982–1984. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- 13.Boulton CL, McCrohan CR, O'Shaughnessy CT. Cyclic AMP analogues increase excitability and enhance epileptiform activity in rat neocortex in vitro. Eur J Pharmacol. 1993;236:131–136. doi: 10.1016/0014-2999(93)90235-a. [DOI] [PubMed] [Google Scholar]
- 14.Bragdon AC, Kojima H, Wilson WA. Suppression of interictal bursting in hippocampus unleashes seizures in entorhinal cortex: a proepileptic effect of lowering [K+]o and raising [Ca2+]o. Brain Res. 1992;590:128–135. doi: 10.1016/0006-8993(92)91088-v. [DOI] [PubMed] [Google Scholar]
- 15.Briggs I, BoSmith RE, Heapy CG. Effects of Zeneca ZD7288 in comparison with alinidine and UL-FS 49 on guinea pig sinoatrial node and ventricular action potentials. J Cardiovasc Pharmacol. 1994;24:380–387. doi: 10.1097/00005344-199409000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Burghardt RC, Barhoumi R, Sewall TC, Bowen JA. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of connexin43. J Membr Biol. 1995;148:243–253. doi: 10.1007/BF00235042. [DOI] [PubMed] [Google Scholar]
- 17.Carystinos GD, Alaoui-Jamali MA, Phipps J, Yen L, Batist G. Upregulation of gap junctional intercellular communication and connexin 43 expression by cyclic-AMP and all-trans-retinoic acid is associated with glutathione depletion and chemosensitivity in neuroblastoma cells. Cancer Chemother Pharmacol. 2001;47:126–132. doi: 10.1007/s002800000231. [DOI] [PubMed] [Google Scholar]
- 18.Castro-Alamancos MA, Calcagnotto ME. Presynaptic long-term potentiation in corticothalamic synapses. J Neurosci. 1999;19:9090–9097. doi: 10.1523/JNEUROSCI.19-20-09090.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chavez-Noriega LE, Stevens CF. Modulation of synaptic efficacy in field CA1 of the rat hippocampus by forskolin. Brain Res. 1992;574:85–92. doi: 10.1016/0006-8993(92)90803-h. [DOI] [PubMed] [Google Scholar]
- 20.Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chevaleyre V, Castillo PE. Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc Natl Acad Sci USA. 2002;99:9538–9543. doi: 10.1073/pnas.142213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- 23.de Curtis M, Avanzini G. Interictal spikes in focal epileptogenesis. Prog Neurobiol. 2001;63:541–567. doi: 10.1016/s0301-0082(00)00026-5. [DOI] [PubMed] [Google Scholar]
- 24.Dickson CT, Magistretti J, Shalinsky MH, Fransen E, Hasselmo ME, Alonso A. Properties and role of I(h) in the pacing of subthreshold oscillations in entorhinal cortex layer II neurons. J Neurophysiol. 2000;83:2562–2579. doi: 10.1152/jn.2000.83.5.2562. [DOI] [PubMed] [Google Scholar]
- 25.DiFrancesco D, Ojeda C. Properties of the current if in the sino-atrial node of the rabbit compared with those of the current iK, in Purkinje fibres. J Physiol (Lond) 1980;308:353–367. doi: 10.1113/jphysiol.1980.sp013475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- 27.Dunwiddie TV, Taylor M, Heginbotham LR, Proctor WR. Long-term increases in excitability in the CA1 region of rat hippocampus induced by β-adrenergic stimulation: possible mediation by cAMP. J Neurosci. 1992;12:506–517. doi: 10.1523/JNEUROSCI.12-02-00506.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol (Lond) 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- 29.Fredholm BB, Fuxe K, Agnati L. Effect of some phosphodiesterase inhibitors on central dopamine mechanisms. Eur J Pharmacol. 1976;38:31–38. doi: 10.1016/0014-2999(76)90198-9. [DOI] [PubMed] [Google Scholar]
- 30.Frerking M, Schmitz D, Zhou Q, Johansen J, Nicoll RA. Kainate receptors depress excitatory synaptic transmission at CA3→CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci. 2001;21:2958–2966. doi: 10.1523/JNEUROSCI.21-09-02958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gasparini S, DiFrancesco D. Action of the hyperpolarization-activated current (Ih) blocker ZD 7288 in hippocampal CA1 neurons. Pflügers Arch. 1997;435:99–106. doi: 10.1007/s004240050488. [DOI] [PubMed] [Google Scholar]
- 32.Good PI. Resampling methods. Birkhauser; Boston: 1999. [Google Scholar]
- 33.Grzywacz NM, Sernagor E. Spontaneous activity in developing turtle retinal ganglion cells: statistical analysis. Vis Neurosci. 2000;17:229–241. doi: 10.1017/s0952523800172050. [DOI] [PubMed] [Google Scholar]
- 34.Hablitz JJ. Picrotoxin-induced epileptiform activity in hippocampus: role of endogenous versus synaptic factors. J Neurophysiol. 1984;51:1011–1027. doi: 10.1152/jn.1984.51.5.1011. [DOI] [PubMed] [Google Scholar]
- 35.Harris NC, Constanti A. Mechanism of block by ZD 7288 of the hyperpolarization-activated inward rectifying current in guinea pig substantia nigra neurons in vitro. J Neurophysiol. 1995;74:2366–2378. doi: 10.1152/jn.1995.74.6.2366. [DOI] [PubMed] [Google Scholar]
- 36.Harris NC, Libri V, Constanti A. Selective blockade of the hyperpolarization-activated cationic current (Ih) in guinea pig substantia nigra pars compacta neurones by a novel bradycardic agent, Zeneca ZM 227189. Neurosci Lett. 1994;176:221–225. doi: 10.1016/0304-3940(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 37.Hoshi T, Garber SS, Aldrich RW. Effect of forskolin on voltage-gated K+ channels is independent of adenylate cyclase activation. Science. 1988;240:1652–1655. doi: 10.1126/science.2454506. [DOI] [PubMed] [Google Scholar]
- 38.Isaacson JS, Walmsley B. Counting quanta: direct measurements of transmitter release at a central synapse. Neuron. 1995;15:875–884. doi: 10.1016/0896-6273(95)90178-7. [DOI] [PubMed] [Google Scholar]
- 39.Jensen MS, Yaari Y. The relationship between interictal and ictal paroxysms in an in vitro model of focal hippocampal epilepsy. Ann Neurol. 1988;24:591–598. doi: 10.1002/ana.410240502. [DOI] [PubMed] [Google Scholar]
- 40.Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57:325–340. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- 41.Lebovitz RM. Inhibitory phasing of penicillin interictal discharge. Brain Res. 1974;79:301–305. doi: 10.1016/0006-8993(74)90421-1. [DOI] [PubMed] [Google Scholar]
- 42.Lebovitz RM. Autorhythmicity of spontaneous interictal spike discharge at hippocampal penicillin foci. Brain Res. 1979;172:35–55. doi: 10.1016/0006-8993(79)90894-1. [DOI] [PubMed] [Google Scholar]
- 43.Li SJ, Wang Y, Strahlendorf HK, Strahlendorf JC. Serotonin alters an inwardly rectifying current (Ih) in rat cerebellar Purkinje cells under voltage clamp. Brain Res. 1993;617:87–95. doi: 10.1016/0006-8993(93)90617-v. [DOI] [PubMed] [Google Scholar]
- 44.Lonart G, Janz R, Johnson KM, Sudhof TC. Mechanism of action of rab3A in mossy fiber LTP. Neuron. 1998;21:1141–1150. doi: 10.1016/s0896-6273(00)80631-5. [DOI] [PubMed] [Google Scholar]
- 45.Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- 46.Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luthi A, Bal T, McCormick DA. Periodicity of thalamic spindle waves is abolished by ZD7288, a blocker of Ih. J Neurophysiol. 1998;79:3284–3289. doi: 10.1152/jn.1998.79.6.3284. [DOI] [PubMed] [Google Scholar]
- 48.Maccaferri G, McBain CJ. The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurones. J Physiol (Lond) 1996;497:119–130. doi: 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maccaferri G, Mangoni M, Lazzari A, DiFrancesco D. Properties of the hyperpolarization-activated current in rat hippocampal CA1 pyramidal cells. J Neurophysiol. 1993;69:2129–2136. doi: 10.1152/jn.1993.69.6.2129. [DOI] [PubMed] [Google Scholar]
- 50.Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol (Lond) 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mellor J, Nicoll RA, Schmitz D. Mediation of hippocampal mossy fiber long-term potentiation by presynaptic Ih channels. Science. 2002;295:143–147. doi: 10.1126/science.1064285. [DOI] [PubMed] [Google Scholar]
- 53.Merlin LR, Taylor GW, Wong RK. Role of metabotropic glutamate receptor subtypes in the patterning of epileptiform activities in vitro. J Neurophysiol. 1995;74:896–900. doi: 10.1152/jn.1995.74.2.896. [DOI] [PubMed] [Google Scholar]
- 54.Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ. Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res. 2000;81:129–139. doi: 10.1016/s0169-328x(00)00155-8. [DOI] [PubMed] [Google Scholar]
- 55.O'Donovan MJ. The origin of spontaneous activity in developing networks of the vertebrate nervous system. Curr Opin Neurobiol. 1999;9:94–104. doi: 10.1016/s0959-4388(99)80012-9. [DOI] [PubMed] [Google Scholar]
- 56.Opitz T, De Lima AD, Voigt T. Spontaneous development of synchronous oscillatory activity during maturation of cortical networks in vitro. J Neurophysiol. 2002;88:2196–2206. doi: 10.1152/jn.00316.2002. [DOI] [PubMed] [Google Scholar]
- 57.Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 58.Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF, Johnson RG. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci. 2000;113:3037–3049. doi: 10.1242/jcs.113.17.3037. [DOI] [PubMed] [Google Scholar]
- 59.Pedarzani P, Storm JF. Dopamine modulates the slow Ca2+-activated K+ current IAHP via cyclic AMP-dependent protein kinase in hippocampal neurons. J Neurophysiol. 1995a;74:2749–2753. doi: 10.1152/jn.1995.74.6.2749. [DOI] [PubMed] [Google Scholar]
- 60.Pedarzani P, Storm JF. Protein kinase A-independent modulation of ion channels in the brain by cyclic AMP. Proc Natl Acad Sci USA. 1995b;92:11716–11720. doi: 10.1073/pnas.92.25.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prida LM, Sanchez-Andres JV. Nonlinear frequency-dependent synchronization in the developing hippocampus. J Neurophysiol. 1999;82:202–208. doi: 10.1152/jn.1999.82.1.202. [DOI] [PubMed] [Google Scholar]
- 62.Rotshenker S, Rahamimoff R. Neuromuscular synapse: stochastic properties of spontaneous release of transmitter. Science. 1970;170:648–649. doi: 10.1126/science.170.3958.648. [DOI] [PubMed] [Google Scholar]
- 63.Santoro B, Tibbs GR. The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann NY Acad Sci. 1999;868:741–764. doi: 10.1111/j.1749-6632.1999.tb11353.x. [DOI] [PubMed] [Google Scholar]
- 64.Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Satoh TO, Yamada M. A bradycardiac agent ZD7288 blocks the hyperpolarization-activated current (I(h)) in retinal rod photoreceptors. Neuropharmacology. 2000;39:1284–1291. doi: 10.1016/s0028-3908(99)00207-5. [DOI] [PubMed] [Google Scholar]
- 66.Schwartzkroin PA, Prince DA. Cellular and field potential properties of epileptogenic hippocampal slices. Brain Res. 1978;147:117–130. doi: 10.1016/0006-8993(78)90776-x. [DOI] [PubMed] [Google Scholar]
- 67.Sciancalepore M, Cherubini E. Protein kinase A-dependent increase in frequency of miniature GABAergic currents in rat CA3 hippocampal neurons. Neurosci Lett. 1995;187:91–94. doi: 10.1016/0304-3940(95)11348-7. [DOI] [PubMed] [Google Scholar]
- 68.Seutin V, Massotte L, Renette MF, Dresse A. Evidence for a modulatory role of Ih on the firing of a subgroup of midbrain dopamine neurons. NeuroReport. 2001;12:255–258. doi: 10.1097/00001756-200102120-00015. [DOI] [PubMed] [Google Scholar]
- 69.Soltesz I, Lightowler S, Leresche N, Jassik-Gerschenfeld D, Pollard CE, Crunelli V. Two inward currents and the transformation of low-frequency oscillations of rat and cat thalamocortical cells. J Physiol (Lond) 1991;441:175–197. doi: 10.1113/jphysiol.1991.sp018745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Southan AP, Morris NP, Stephens GJ, Robertson B. Hyperpolarization-activated currents in presynaptic terminals of mouse cerebellar basket cells. J Physiol (Lond) 2000;526:91–97. doi: 10.1111/j.1469-7793.2000.t01-1-00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Staley KJ, Longacher M, Bains JS, Yee A. Presynaptic modulation of CA3 network activity. Nat Neurosci. 1998;1:201–209. doi: 10.1038/651. [DOI] [PubMed] [Google Scholar]
- 72.Staley KJ, Bains JS, Yee A, Hellier J, Longacher JM. Statistical model relating CA3 burst probability to recovery from burst-induced depression at recurrent collateral synapses. J Neurophysiol. 2001;86:2736–2747. doi: 10.1152/jn.2001.86.6.2736. [DOI] [PubMed] [Google Scholar]
- 73.Stoop R, Pralong E. Functional connections and epileptic spread between hippocampus, entorhinal cortex and amygdala in a modified horizontal slice preparation of the rat brain. Eur J Neurosci. 2000;12:3651–3663. doi: 10.1046/j.1460-9568.2000.00253.x. [DOI] [PubMed] [Google Scholar]
- 74.Strata F, Atzori M, Molnar M, Ugolini G, Tempia F, Cherubini E. A pacemaker current in dye-coupled hilar interneurons contributes to the generation of giant GABAergic potentials in developing hippocampus. J Neurosci. 1997;17:1435–1446. doi: 10.1523/JNEUROSCI.17-04-01435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Streit J. Regular oscillations of synaptic activity in spinal networks in vitro. J Neurophysiol. 1993;70:871–878. doi: 10.1152/jn.1993.70.3.871. [DOI] [PubMed] [Google Scholar]
- 76.Tabak J, Senn W, O'Donovan MJ, Rinzel J. Modeling of spontaneous activity in developing spinal cord using activity-dependent depression in an excitatory network. J Neurosci. 2000;20:3041–3056. doi: 10.1523/JNEUROSCI.20-08-03041.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tabak J, Rinzel J, O'Donovan MJ. The role of activity-dependent network depression in the expression and self-regulation of spontaneous activity in the developing spinal cord. J Neurosci. 2001;21:8966–8978. doi: 10.1523/JNEUROSCI.21-22-08966.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Traub D, Miles R. Neuronal networks of the hippocampus. Cambridge UP; Cambridge: 1991. [Google Scholar]
- 79.Travagli RA, Gillis RA. Hyperpolarization-activated currents, IH and IKIR, in rat dorsal motor nucleus of the vagus neurons in vitro. J Neurophysiol. 1994;71:1308–1317. doi: 10.1152/jn.1994.71.4.1308. [DOI] [PubMed] [Google Scholar]
- 80. Tsodyks M, Uziel A, Markram H. Synchrony generation in recurrent networks with frequency-dependent synapses. J Neurosci 20 2000. RC50(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Turecek R, Trussell LO. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- 82.Van Hoogenhuyze D, Weinstein N, Martin GJ, Weiss JS, Schaad JW, Sahyouni XN, Fintel D, Remme WJ, Singer DH. Reproducibility and relation to mean heart rate of heart rate variability in normal subjects and in patients with congestive heart failure secondary to coronary artery disease. Am J Cardiol. 1991;68:1668–1676. doi: 10.1016/0002-9149(91)90327-h. [DOI] [PubMed] [Google Scholar]
- 83.van Rijen HV, van Veen TA, Hermans MM, Jongsma HJ. Human connexin40 gap junction channels are modulated by cAMP. Cardiovasc Res. 2000;45:941–951. doi: 10.1016/s0008-6363(99)00373-9. [DOI] [PubMed] [Google Scholar]
- 84.Vargas G, Lucero MT. Modulation by PKA of the hyperpolarization-activated current (Ih) in cultured rat olfactory receptor neurons. J Membr Biol. 2002;188:115–125. doi: 10.1007/s00232-001-0178-y. [DOI] [PubMed] [Google Scholar]
- 85.Vasilyev DV, Barish ME. Postnatal development of the hyperpolarization-activated excitatory current Ih in mouse hippocampal pyramidal neurons. J Neurosci. 2002;22:8992–9004. doi: 10.1523/JNEUROSCI.22-20-08992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- 87.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 88.Wells JE, Porter JT, Agmon A. GABAergic inhibition suppresses paroxysmal network activity in the neonatal rodent hippocampus and neocortex. J Neurosci. 2000;20:8822–8830. doi: 10.1523/JNEUROSCI.20-23-08822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]