

Abstract
The retina adapts to the temporal contrast of the light inputs. One component of contrast adaptation is intrinsic to retinal ganglion cells: temporal contrast affects the variance of the synaptic inputs to ganglion cells, which alters the gain of spike generation. Here we show that slow Na+ inactivation is sufficient to produce the observed variance adaptation. Slow inactivation caused the Na+ current available for spike generation to depend on the past history of activity, both action potentials and subthreshold voltage variations. Recovery from slow inactivation required several hundred milliseconds. Increased current variance caused the threshold for spike generation to increase, presumably because of the decrease in available Na+current. Simulations indicated that slow Na+inactivation could account for the observed decrease in excitability. This suggests a simple picture of how ganglion cells contribute to contrast adaptation: (1) increasing contrast causes an increase in input current variance that raises the spike rate, and (2) the increased spike rate reduces the available Na+current through slow inactivation, which feeds back to reduce excitability. Cells throughout the nervous system face similar problems of accommodating a large range of input signals; furthermore, the Na+ currents of many cells exhibit slow inactivation. Thus, adaptation mediated by feedback modulation of the Na+ current through slow inactivation could serve as a general mechanism to control excitability in spiking neurons.
Keywords: contrast adaptation, slow Na+inactivation, modulation of Na+ current, retinal ganglion cell, models for spike generation, adaptation, spike-frequency adaptation, retinal signal processing
Introduction
Sensory signals are highly variable, and these variations occur on a wide range of time scales. To encode these signals efficiently, sensory systems adapt, trading sensitivity to slowly changing aspects of the input for increased sensitivity to rapid changes. Adaptation permits sensory neurons to use their limited dynamic range effectively. Visual neurons adapt to the mean light intensity (for review, see Walraven et al., 1990) and to the amplitude of the variations about the mean (i.e., the spatial and temporal contrast) (for review, see Shapley, 1997; Meister and Berry, 1999). Although the properties of mean and contrast adaptation have been studied in detail, we have an incomplete understanding of the mechanisms responsible. Here we investigate the contribution of Na+ currents in retinal ganglion cells to temporal contrast adaptation.
Changes in temporal contrast cause adaptation in cells throughout early visual pathways, including those in retina (Shapley and Victor, 1978;Sakai et al., 1995; Smirnakis et al., 1997; Chander and Chichilnisky, 2001) and cortex (Albrecht et al., 1984; Ohzawa et al., 1985;Sanchez-Vives et al., 2000a). Contrast adaptation in the retina includes contributions from the retinal circuitry (Sakai et al., 1995;Rieke, 2001) and intrinsic properties of ganglion cells (Kim and Rieke, 2001b). Retinal cells have a high gain for light increments and decrements about a steady mean, causing saturation at the onset of a high contrast stimulus (Burkhardt et al., 1998). Contrast adaptation permits recovery from such saturation by reducing gain in the maintained presence of time-varying lights.
Changes in temporal contrast alter the variance and in some cases the mean of synaptic inputs to visual neurons. The increase in variance reduces the gain of spike generation in retinal ganglion cells (Kim and Rieke, 2001b). Thus, a component of contrast adaptation is produced by a variance-induced change in the input–output relationship of a ganglion cell. Ca2+-activated K+ currents shape the input–output relationship of many cells through effects such as spike-frequency adaptation (for review, see Sah and Davies, 2000). However, neither K+ nor Ca2+currents contribute substantially to variance adaptation in ganglion cells; instead, variance adaptation is generated by properties of the Na+ currents (Kim and Rieke, 2001b).
We investigated the mechanisms permitting ganglion cells to adapt to the input current variance. These experiments led to three main conclusions: (1) slow inactivation reduced the Na+ current available for spike generation after both subthreshold depolarizations and spikes; recovery from slow inactivation required several hundred milliseconds; (2) slow inactivation caused the fraction of the Na+ current available for spike generation to depend on the variance of the input currents to the cell; and (3) simulations showed that action potentials rather than subthreshold voltages dominated the extent of slow Na+inactivation. These simulations reproduced the experimentally observed variance adaptation. Thus, the mechanism for variance adaptation was simple: increased current variance caused a higher firing rate, which in turn reduced the available Na+ current and increased spike threshold.
Some of this work has been published previously in abstract form (Kim and Rieke, 2001a).
Materials and Methods
Experimental procedures
Dissection. All experiments used retinas from larval tiger salamanders (Ambystoma tigrinum) from Charles Sullivan (Nashville, TN). Animal procedures followed protocols approved by the Administrative Panel on Laboratory Animal Care at the University of Washington (Seattle, WA). Procedures for preparing and dissociating the retina to obtain isolated cells have been described previously (Kim and Rieke, 2001b). Cells were continuously superfused during recording with a HEPES Ringer's solution containing (in mm): 136 NaCl, 2 KCl, 1.5 CaCl2, 10 glucose, 2 NaHCO3, 1.6 MgCl2, and 3 HEPES; pH was adjusted to 7.4 with NaOH, and osmolarity was 270–275 mOsm. Experiments were performed at 20–22°C.
Ganglion and amacrine cells were distinguished from other cell types by their ability to generate Na+ spikes. Data were collected only from cells that were able to generate repetitive action potentials with a width of < 2 msec and a minimum amplitude of ≥60 mV above the resting voltage. In two experiments we labeled the ganglion cells by retrograde transport of rhodamine dextran through the optic nerve (Lukasiewicz and Werblin, 1988). After dissociation, >95% (178 of 185) of the cells with morphology similar to that of the recorded cells were ganglion cells. We did not attempt to distinguish between ganglion cell types.
Patch recording procedure. Voltage and current responses of ganglion cells were measured using perforated-patch or whole-cell recordings. Similar results were obtained in each case. Patch pipettes were filled with a solution containing (in mm):115 Cs-aspartate, 20 CsCl, 10 HEPES, 1 N-methylglucamine (NMG) EGTA, and 0.2 CaCl2; pH was adjusted to 7.2 with NMG-OH, and osmolarity was 260–265 mOsm. For whole-cell recordings, 1 mm ATP and 0.2 mm GTP were added to the internal solution. For perforated-patch recordings, the pipette tip was filled with an amphotericin-free solution, and the pipettes were back-filled with internal solution with an additional 1 mg/ml solubilized amphotericin-B (Sigma, St. Louis, MO). Filled pipettes had resistances of 3–5 MΩ, and the series resistance during recording was 10–20 MΩ. Ganglion cells had resistances between 1 and 4 GΩ and capacitances between 10 and 20 pF.
Na+ current inactivation and recovery were measured in voltage-clamp recordings. To isolate Na+ currents, K+ and Ca2+currents were suppressed by replacing K+with Cs+ in both internal and external solutions and adding 0.1 mmCd2+ to the external solution. Block of the Ca2+ current was confirmed in some cells by adding 100 nm TTX to the external solution; this eliminated all inward current in response to depolarizing voltage steps. To improve the voltage clamp, the Na+ current was reduced by replacing up to 100 mm of external Na+with NMG+. Voltages have been corrected for junction potentials, which were <10 mV.
Adaptation of the spike response of a cell to changes in the variance of the injected current was measured in current-clamp recordings. Random noise with a Gaussian distribution (bandwidth, 0–50 Hz) was injected into the cell, and the variance of the distribution was changed periodically without changing the bandwidth. The mean holding current was between 0 and 10 pA, resulting in a firing rate of 2–6 Hz. Under these conditions, the mean voltage was between −60 and −55 mV.
Data analysis
We quantified adaptation of spike generation to the variance of the injected current using a static nonlinearity model (Brenner et al., 2000; Chichilnisky, 2001; Kim and Rieke, 2001b). This model separates a time-independent (or static) nonlinearity in the response of the cell from true adaptive changes in sensitivity. The model describes the transformation of injected current into the probability of spiking of the cell. Adaptation caused the current-to-spikes transformation to change with the variance of the injected current. The model does not attempt to account for the dynamics of adaptation, but instead describes the steady-state effect of adaptation on the current-to-spikes transformation.
The static nonlinearity model predicts the spike probability as a function of time by passing the injected current through a linear filter and applying a static nonlinearity to the filter output. The linear filter estimates the time dependence of the relationship between injected current and spike probability. The static nonlinearity captures the abrupt increase in firing probability associated with the membrane voltage crossing threshold for spike generation. The linear filter was estimated by calculating the average current waveform preceding a spike. The static nonlinearity was estimated by comparing the output of this filter (the filter convolved with the injected current) with the measured spiking probability.
Variance adaptation in experiments and simulations (see below) was analyzed using this model. The linear filter and static nonlinearity were calculated from several minutes of measured or simulated responses to random injected currents. This procedure was repeated for currents of several variances. The first 2 sec of data after a change in variance were discarded to allow variance adaptation to reach a steady state. As reported previously (Kim and Rieke, 2001b), adaptation to changes in the variance of the injected current could be captured by a change in the linear filter. Thus, the response of the cell was described as a variance-dependent linear filter followed by a variance-independent static nonlinearity. Changes in the amplitude of the linear filter were used to quantify the extent of adaptation.
Computer simulation of ganglion cells
As described in Results, Na+currents in ganglion cells underwent both fast (Hodgkin and Huxley, 1952) and slow (Crill, 1996; Fleidervish et al., 1996; Mickus et al., 1999) inactivation. We used a computer simulation to test whether the measured properties of the Na+ current contributed to variance adaptation. The simulation provided a relatively simple description of the cell while capturing activation and inactivation of the Na+ current in detail. Other conductances, particularly K+ and Ca2+conductances, are known to shape the spike response of a cell (Hodgkin and Huxley, 1952; Fohlmeister and Miller, 1997). However, adaptation of ganglion cells to changes in current variance was not altered by K+-, Ca2+-, or Ca2+-activated currents (Kim and Rieke, 2001b); to minimize the number of parameters in the model, these currents were not simulated separately. We begin by describing the simulated Na+ current and then describe how this current was incorporated in a spiking model for the ganglion cell.
Na+ current simulation. The Na+ current was simulated using the Hodgkin–Huxley formalism with the addition of two slow inactivation variables: s1 ands2. Thes1 variable had both slow onset and recovery. Because of its slow kinetics, changes ins1 were dominated by subthreshold voltages. We refer to this form of inactivation as spike independent. The s2 variable describes slow inactivation that was entered after a spike and recovered slowly. We refer to this form of inactivation as spike dependent.
In the Hodgkin–Huxley formalism, the activation variable mand inactivation variable h are independent. We assume the same is true for s1 ands2. Although the assumption of independent inactivation gates is not likely to hold (Hille, 2001), this simulation captured the measured properties of the Na+ current and thus allowed us to explore the role of slow inactivation in variance adaptation. Furthermore, the relative simplicity afforded by this assumption permitted us to eliminate all free parameters in the simulation.
The m, h, s1, and s2 variables describe the fraction of gating particles in the Na+ channel that are in the permissive state. The Na+current is given by the probability that all gating particles are in the permissive state (i.e., that the channel is open and not inactive):
| Equation 1 |
where V is the membrane voltage,ENa is the Na+ equilibrium potential, andGNa is the maximal Na+ conductance.
The activation and inactivation variables were described by first-order kinetics (Fig. 1) (e.g., the spike-independent slow inactivation particles1 obeyed):
| Equation 2 |
where βs1 and αs1 are rate constants describing the entry into and recovery from slow inactivation. With first-order kinetics, the s1 variable exponentially approaches its steady-state value at a given voltage. The rate constants, αs1 and βs1, were calculated from measurements of the steady-state value,s , and the exponential time constant τs1:
| Equation 3 |
and
| Equation 4 |
Spike-dependent slow inactivation, described bys2, was entered only after a spike. Thus at voltages below spike threshold, the recovery rate constant αs2 was simply the inverse of the recovery time constant:
| Equation 5 |
The rate constants for the h and mvariables were from Hodgkin and Huxley (1952). The activation variable,m, was described by the rate constants:
| Equation 6 |
and
| Equation 7 |
where V is in millivolts and αm and βm are in msec−1. The fast-inactivation variableh was described by:
| Equation 8 |
and
| Equation 9 |
The spike-independent slow inactivation variables1 was described by:
| Equation 10 |
and
| Equation 11 |
The spike-dependent slow inactivation variables2 was described by:
| Equation 12 |
Onset of spike-dependent slow inactivation was simulated by multiplying s2 by a factor of 0.77 after each Na+ spike (see Results).
Fig. 1.

Schematic of ganglion cell simulation. The simulation describes the transformation between the input current and membrane voltage of a cell. The cell was simulated as a single compartment with leak, capacitive, cellular noise, and Na+ currents (see Eq. 13). If an Na+ spike was generated, the simulation was switched to voltage clamp and forced to repolarize along the voltage trajectory of an experimentally measured action potential. The Na+ current parameters were updated continuously during this voltage trajectory. After the voltage returned to rest, the simulation was switched back into current clamp, and the voltage was controlled again by the currents.
Note that the rate constants controlling the slow inactivation variables s1 ands2 are several orders of magnitude smaller than those for the fast inactivation variable h.
Model for spike generation. The Na+ current described above formed the basis of the simulation describing how input currents were converted to membrane voltage (Fig. 1). The input to the simulation was the injected current Istim, and the output was the membrane voltage V, including action potentials. In addition to the Na+ current, the simulation included leak and capacitive currents and a Gaussian current noise Inoise (bandwidth, 0–50 Hz) to simulate cellular noise. These currents were summed to obtain an equation describing the dynamics of the membrane voltage:
| Equation 13 |
Here Cm is the membrane capacitance, Gleak is the leak conductance, and Eleak is the reversal potential for the leak conductance. The Na+ current was calculated from Equation1. The ganglion cell was simulated as a single isopotential compartment, because ganglion cells are believed to be electrotonically compact (Taylor et al., 1996), and our isolation procedure removed all but one or two short (< 40 μm) processes.
Equation 13 determines how the voltage in the simulated cell depends on injected current and is able to generate the rising phase of an action potential. It cannot, however, produce action potentials with a reasonable shape, because it lacks the voltage-dependent K+ conductance that rapidly repolarizes the cell. Thus, to reproduce the voltage changes produced by action potentials, we forced the voltage in the simulation to follow the trajectory of an experimentally measured action potential when the membrane voltage indicated that an action potential was being generated (i.e., whenever a trigger voltage θ was reached). During the action potential, the activation and inactivation variables of the Na+ current were updated continuously in response to the voltage. This allowed spikes to alter the state of the Na+ current without simulating all conductances shaping the action potential. After return to rest, the simulation was switched back to current clamp, and the currents were again free to perturb the membrane voltage.
Computer simulations were performed in Igor Pro (Wavemetrics, Lake Oswego, OR) using a temporal integration scheme described by MacGregor (1987). This integration scheme allows for relatively large step sizes without instability in the integration. Step size for integration was ≤0.1 msec; smaller step sizes gave no noticeable increase in accuracy. The measured action potential used during the voltage-clamp periods was interpolated to the temporal resolution of the integration step size. Values of the parameters in the simulation are given in Table1. θ was chosen to be −15 mV so that only Na+ spikes could cause the switch to voltage clamp. The simulation had no free parameters: the rate constants describing the m and h gates were taken from Hodgkin and Huxley (1952), and all other parameters are the average value from experimental cells (n ≥ 4) (Table1). In Results we discuss how alterations in the simulation parameters affected adaptation.
Table 1.
Parameters in computer simulation
| Description | Symbol | Value | Range |
|---|---|---|---|
| Maximal Na+conductance | GNa | 100 nS | 50 to 130 nS |
| Membrane capacitance | Cm | 15 pF | 10 to 20 pF |
| Leak conductance | Gleak | 0.5 nS | 0.2 to 0.9 nS |
| Leak reversal potential | Eleak | −56 mV | −60 to −55 mV |
| Duration of action potential | TAP | 1.5 msec | 1.3 to 2.5 msec |
| Fractional decrease ins2 after spike | s | 0.23 | 0.13 to 0.34 |
| Variance ofInoise | ςnoise | 4 pA2 | 3 to 8 pA2 |
| Equilibrium potential for Na+ | ENa | 35 mV | N/A |
| Trigger voltage for voltage-clamp switch | θ | −15 mV | N/A |
GNa was determined from the maximum amplitude of the Na+ current. Cm was the average measured capacitance. Gleak was measured from the conductance during white noise current injection of 16 pA2 variance. TAP was the average time between the spike waveform exceeding the voltage θ and returning to rest. Inoise was determined from the current fluctuations during voltage clamp at −60 mV.
Results
Spike generation in retinal ganglion cells adapts to the variance of the current injected into the cell (Kim and Rieke, 2001b). The experiments and simulations described below indicate that slow inactivation of the Na+ current can account for most of this adaptation. First, we show that increasing the injected current variance lowered the gain and increased the threshold of spike generation. Second, we show that increasing the current variance reduced the magnitude of the Na+current through slow Na+ inactivation. Third, we simulate the measured properties of the Na+ current and show that slow Na+ inactivation can account for much of the change in gain associated with variance adaptation. Together these results provide a simple mechanistic description for variance adaptation: increased variance decreases excitability by decreasing the available Na+ current.
Effects of variance on spike generation and Na+ current
Variance changes gain and threshold of spike generation
Variance adaptation was measured by injecting Gaussian current fluctuations of two variances into a current-clamped ganglion cell and recording the spike responses. We used the static nonlinearity model (see Materials and Methods) to determine how the current variance affected the gain and kinetics of spike generation. The model characterizes spike generation at a single current variance by passing the injected current through a linear filter (Fig.2A) and applying a static nonlinearity (Fig. 2B). The linear filter estimates the time dependence of the relationship between injected current and spike probability. The static nonlinearity captures the thresholding and rectification of spike generation. Linear filters and static nonlinearities were compared for several variances.
Fig. 2.

Effects of variance on current-to-spikes transformation. The static nonlinearity model was used to characterize the transformation between injected current and the firing rate. Gaussian current fluctuations (bandwidth, 0–50 Hz) with variances of 16 and 144 pA2 were injected into a ganglion cell. The model parameters were calculated from 100 sec of recording.A, Linear filters for the two injected current variances. B, Static nonlinearities for the two current variances. The nonlinearity describes the relationship between the output of the filters (A) and the measured firing rate. C, Histograms of local voltage maxima for each current variance. Maxima were defined as data points with amplitudes larger than those of surrounding points, including both action potential peaks (> 0 mV) and subthreshold maxima (less than −20 mV). Bin size was 0.76 and 1.19 mV for low and high variance. Theinset plots the cumulative distribution of subthreshold maxima. The change in threshold was estimated from the voltages at which the cumulative distributions reached 0.99.
The linear filter and static nonlinearity at each variance are unique up to a single scale factor (Brenner et al., 2000; Chichilnisky, 2001;Kim and Rieke, 2001b). Thus, scaling the y-axis of the filter in Figure 2A and the x-axis of the static nonlinearity in Figure 2B by the same factor does not change the prediction of the model, because the effect of changing the filter amplitude is offset by the change in the static nonlinearity. Although adaptation could affect the shape of both the filter and static nonlinearity, we found that the static nonlinearities could be made to overlap with an appropriate choice of scale factor (Fig. 2B). This allowed spike generation to be described as a variance-dependent linear filter followed by a variance-independent static nonlinearity. As a consequence, changes in excitability (e.g., because of changes in threshold for spike generation) were captured by a change in the filter amplitude.
Changes in variance altered both the amplitude and the time-to-peak of the linear filter, as shown in Figure 2A. The decrease in time-to-peak of the linear filter with increasing variance appeared to be a general property of refractoriness (see Fig. 8). The reduction in the filter amplitude meant that the change in spike probability for a given current change was smaller at high variance than low variance. Similar changes in the linear filter were seen in all 10 cells tested; on average, a ninefold change in variance decreased the filter amplitude by 35 ± 4% (mean ± SEM).
Fig. 8.

Comparison of variance adaptation in real and simulated ganglion cells. Several minutes of Gaussian current fluctuations (bandwidth, 0–50 Hz) of 16 and 144 pA2variance were injected into experimental and simulated cells. Adaptation in the computer simulated spike train was analyzed using the static nonlinear model and treated identically to that of an experimental cell. A, Adaptation in an experimental cell. Left, Linear filters. Right, Filters with the time axes normalized to the time-to-peak of each filter. B, Simulation lacking slow Na+ inactivation. C, Simulation with slow Na+ inactivation. Insets show that the static nonlinearities overlapped in all cases. Simulation parameters are given in Table 1 and Equations 6-12.
The decrease in filter amplitude could be produced by an increased threshold for spike generation. With an increased threshold, a larger current would be required to generate a spike, thereby lowering the sensitivity of spike generation to the injected current. To test for such an effect, we measured spike threshold during periods of high and low current variance.
The change in threshold with variance was estimated from the largest depolarizations that failed to lead to a spike. Figure 2Cshows a histogram of the local voltage maxima, identified as 1 msec time windows in which the measured voltage exceeded that in the adjacent 1 msec windows (adjacent sampling points). Subthreshold voltage fluctuations form the large, broad peak in the histogram centered near −50 mV. Action potentials create the small peak centered near +5 mV. The histogram is 0 in the voltage range from −30 to −10 mV, because the voltage of the cell reaches these values only during an action potential, and action potentials have local maxima only at their peak.
Subthreshold voltage maxima extended to larger depolarizations when the current variance was increased. We quantified this shift using the cumulative distributions of subthreshold maxima (Fig. 2C,inset). In principle, threshold could be determined from the largest subthreshold maxima, the point at which the cumulative distribution reaches 1.0. This definition is impractical with finite data because it relies on a single measurement, the single largest subthreshold point observed. Instead, we estimated relative thresholds for the two variances from the voltages that exceeded 99% of the local maxima. This provided a reliable estimate of shifts in the largest subthreshold voltages. In this cell, threshold was 1.8 mV higher during high variance than during low variance. The mean increase in threshold was 1.6 ± 0.3 mV (mean ± SEM; nine cells) for a ninefold increase in injected current variance. Ignoring local maxima for 100 msec after each action potential had little effect on the apparent threshold change, indicating that it was not caused by the absolute refractory period. The subthreshold voltage changes produced by moderate contrast light inputs are ∼ 10 mV in amplitude. Hence a change in threshold of 1–2 mV represents a substantial change in sensitivity.
Increased variance reduces the available Na+ current
We found previously that the Na+current was necessary for variance adaptation in ganglion cells, whereas K+ or Ca2+ currents were not (Kim and Rieke, 2001b). The central role of the Na+current in adaptation and its impact on the threshold for spike generation suggested that adaptation might be caused by changes in the amount of available (i.e., not inactive) Na+ current and a resulting increase in threshold. Consistent with this idea, we found that the Na+ current was influenced by the magnitude of previous voltage fluctuations.
Ganglion cells were voltage-clamped at −60 mV and subjected to voltage fluctuations of two variances under conditions that isolated Na+ currents (see Materials and Methods). After the voltage fluctuations, the Na+current was allowed to recover from fast inactivation by holding at −60 mV for 20 msec. The Na+ current was then measured by stepping the voltage to 0 mV (Fig.3A). Figure 3Bshows that the Na+ current depended on the voltage variance. In this cell, the smaller voltage fluctuations reduced the peak Na+ current by 4% and the larger fluctuations by 17%. A similar dependence of the Na+ current on previous voltage variance was seen in each of 17 cells studied.
Fig. 3.

Na+ current is sensitive to preceding voltage fluctuations. A, Voltage-clamp stimulus. The Na+ current was measured in response to a brief test pulse to 0 mV after 2 sec of Gaussian voltage fluctuations (bandwidth, 0–50 Hz). A 20 msec recovery period at a holding voltage of −60 mV was imposed between the noise and test pulse. B, Average Na+ currents after noise with variances of 0, 25, and 100 mV2 are shown. Responses from four pulses after uncorrelated noise stimuli were averaged together to prevent the specific immediate history of the noise from affecting the Na+ current.
We studied the recovery kinetics of the Na+ current by varying the time between an inactivating stimulus and the test pulse used to measure the peak Na+ current. The Na+ current recovered slowly after inactivation by a voltage step or voltage noise. Figure4B shows Na+ currents measured at different times after inactivation caused by voltage noise (Fig. 4A). In all 12 ganglion cells studied, the Na+current continued to recover from inactivation for times well beyond the 10 msec expected from Hodgkin–Huxley kinetics. The time course of recovery of the Na+ current is shown in Figure 4C, which plots the ratio of the Na+ currents with and without inactivation against the recovery time. Full recovery required 1–4 sec.
Fig. 4.

Time course of recovery from noise and voltage steps. A, Voltage-clamp stimulus used to measure slow inactivation produced by noise. The Na+ current was measured by stepping the voltage to 0 mV at various times (trec) after a period of voltage noise (100 mV2 variance; bandwidth, 0–50 Hz).B, Na+ currents in response to the test pulse. From smallest to largest, traces correspond totrec of 10, 100, and 900 msec.C, Time course of recovery of peak Na+ current after inactivation by noise. The Na+ current at each trecwas normalized to that for a trec of 900 msec. This ratio is plotted against the recovery time.D, Voltage-clamp stimulus used to measure slow inactivation produced by voltage steps. The Na+current was measured at various times after a 500 msec depolarization from −80 to −20 mV. E, Na+ currents produced by the test pulse in D. From smallest to largest, traces correspond to trec of 20, 110, and 1100 msec. F, Time course of recovery of peak Na+ current after inactivation by voltage step. Na+ currents were normalized to that for atrec of 3 sec.
Recovery of the Na+ current from inactivation was similar when inactivation was produced by voltage steps rather than a fluctuating voltage (Fig. 4D). Figure 4E shows the Na+currents measured at different times after inactivation caused by a voltage step. Figure 4F shows the time course of recovery. Again the Na+ current recovered much more slowly than expected from Hodgkin–Huxley kinetics. Thus, both noise and step stimuli caused Na+channels to enter an inactive state from which they recovered slowly.
Properties of slow Na+ inactivation
Recovery of the Na+ current from fast inactivation after an action potential typically requires a few milliseconds (Hodgkin and Huxley, 1952; Hille, 2001). As shown above, the ganglion cell Na+ current exhibited slow inactivation with a recovery time constant several orders of magnitude longer. There are two distinct functional components of this slow inactivation. The first decreased the Na+ current after depolarizations above spike threshold; recovery from this spike-dependent inactivation had a time constant of a few hundred milliseconds. The second component of slow inactivation had a slower onset and recovery; this spike-independent component was controlled by subthreshold voltage fluctuations.
Spike-dependent slow Na+ inactivation
The Na+ current recovered slowly after brief voltage-clamp pulses exceeding spike threshold (Fig.5A). We refer to the large Na+ currents in response to these voltage pulses as Na+ spikes. The Na+ current declined exponentially in amplitude in response to a series of pulses (Fig. 5B). This form of slow inactivation reflected the all-or-none properties of spike generation; no slow inactivation occurred when the pulses were too small to produce an Na+ spike, and increasing the length or amplitude of the depolarizing pulse did not produce additional inactivation. We therefore refer to this component of slow inactivation as spike dependent.
Fig. 5.

Spike-dependent slow inactivation.A, Decline in Na+ current in response to a series of voltage pulses. The cell was repeatedly stepped from −50 to 0 mV for 5 msec to generate Na+ spikes. The interval between pulses was 20 msec. B, Peak Na+ current amplitude as a function of pulse number. Peak Na+ currents were normalized by that produced by the first pulse. Normalized Na+ currents are shown for interpulse intervals of 20 and 125 msec. Solid lines are single exponential fits to the data.C, Steady-state peak Na+ current as a function of interpulse interval measured from fits in B.Solid lines are single exponential fits to the data used to estimate the recovery rate constant. D, Fractional reduction in peak Na+ current produced by each pulse calculated from the fits in B. E, Rate constant αs2 for recovery from inactivation. The rate constant was calculated from the inverse of the time constant of the fits in C. Different symbols plot measurements from four cells. The solid line is an exponential fit to αs2 for all four cells.
Spike-dependent slow inactivation was characterized by a first-order kinetic model. Some fraction of thes2-gating variable is permissive, while the remainder, 1 − s2, is slowly inactivated and nonpermissive. A fraction of the non-inactive Na+ current enters the slow-inactive state after a spike; recovery from slow inactivation occurs with a voltage-dependent rate constant, αs2. We varied the duration and holding voltage between pulses to determine the recovery rate constant, αs2, and the change ins2 after a pulse.
The time course of recovery from spike-dependent slow Na+ inactivation was measured from the steady-state Na+ current for pulse series like that in Figure 5A. The steady-state amplitude was reached when recovery from inactivation between pulses balanced inactivation produced by each pulse. Lengthening the interval between pulses increased the steady-state amplitude of the Na+ current. Figure 5C plots the steady-state amplitude against the interpulse interval for two holding voltages. The recovery rate constant, αs2, was estimated from the time constant of the exponential fits in Figure 5C. This rate constant is plotted against the recovery voltage in Figure 5E. All four cells studied showed a decrease in the rate of recovery from spike-dependent slow Na+ inactivation with increasing voltage.
The amount of inactivation produced by each pulse was determined by correcting the reduction in Na+ current between successive pulses for recovery during the intervening interval. Figure 5D shows the fractional reduction in peak Na+ current for different interpulse intervals. Fitting these data with exponentials and extrapolating to an interpulse interval of 0 (i.e., no recovery) estimated the fractional reduction in available Na+ current immediately after a pulse, s . The average fractional reduction in Na+current produced by each pulse was s = 0.23 ± 0.05 (mean ± SEM; four cells). Thus, after each pulse the Na+ current was reduced to 77% of its initial value.
Spike-independent slow Na+ inactivation
The spike-dependent slow Na+inactivation described above did not fully explain the behavior of the Na+ current. Subthreshold changes in voltage also caused slow inactivation; this spike-independent component of inactivation had both slow onset and recovery.
Spike-independent slow inactivation was described as a first-order process analogous to Hodgkin–Huxley fast inactivation. In this case, a single voltage-dependent time constant describes the approach to steady state after a change in voltage. The onset and recovery rate constants βs1 and αs1 were determined from the time constant and steady-state values of slow inactivation (see Eq. 2, Materials and Methods). To reduce errors in measurement, we used voltage-clamp stimuli that produced large changes in steady-state inactivation. For depolarized voltages at which steady-state inactivation was large, we measured the rate of onset of inactivation following a step from a hyperpolarized voltage (Fig.6A). For hyperpolarized voltages at which steady-state inactivation was small, we measured the time constant for recovery from a step producing strong inactivation (Fig. 6D). Combining these measurements provided estimates of the voltage-dependent rate constants βs1 and αs1.
Fig. 6.

Spike-independent slow inactivation.A, Inactivation at depolarized voltages.Top, Test pulses were delivered to measure the Na+ current before and after an inactivating step of variable duration and voltage. A 20 msec recovery period at −80 mV between the inactivating pulse and the second test pulse allowed recovery from fast inactivation. Bottom, The ratio of the Na+ currents produced by the two test pulses is plotted against tinact for two voltages. Measured points were fit with single exponentials (smooth curves) with time constants of 1160 msec for aVinact of −55 mV and 620 msec for aVinact of −35 mV. B, C, Na+ currents in response to test pulses at aVinact of −55 mV (B) and a Vinact of −35 mV (C). From smallest to largest, solid lines correspond to trec of 1600, 250, and 40 msec. The dotted line is Na+ current produced byV1. D, Inactivation at hyperpolarized voltages. Top, Two test pulses were delivered to measure the Na+ current before and after inactivation produced by a 500 msec step to −20 mV followed by a variable duration and voltage recovery period. Bottom, The ratio of the peak Na+ currents produced by the two test pulses is plotted against trec at two voltages. The measured points were fit by a product of two exponentials (smooth curves) with time constants of 130 and 770 msec for a Vrec of −95 mV and time constants of 100 and 890 msec for a Vrec of −75 mV. E, F, Na+ currents in response to test pulses at a Vrec of −95 mV (E) and at a Vrec of −75 mV (F). From smallest to largest,solid lines correspond totrec of 20, 170, and 1380 msec. Thedotted line is the Na+ current produced by V1.
Figure 6A shows the procedure used to measure the time constant and steady-state value of inactivation at depolarized voltages (greater than or equal to −60 mV). A test pulse was delivered to measure the Na+ current in the absence of slow inactivation. This was followed by 1 sec at −80 mV to allow recovery from spike-dependent slow inactivation produced by the test pulse. An inactivating pulse of variable duration was then delivered at one of several voltages, followed by a second test pulse to measure the available Na+ current. Figure 6,B and C, shows the Na+ currents for several inactivation periods at −55 and −35 mV. Dashed lines show the Na+ currents before inactivation. The step to −55 mV, well below spike threshold, produced substantial inactivation, provided the inactivation period was >100 msec.
The effect of slow inactivation on the available Na+ current was measured from the ratio of the Na+ currents in response to the first and second test pulses. Figure 6A, bottom, plots this ratio against the inactivation period for inactivation voltages of −55 and −35 mV. The ratio measured at −35 mV does not extrapolate back to 1 for an inactivation duration of 0 msec, because the voltage step exceeded spike threshold and thus produced a large Na+ current and spike-dependent slow inactivation; this reduced the available Na+ current ∼20% (see above). The time constant with which spike-independent slow inactivation approached a steady-state value was estimated from single exponential fits to the Na+ current ratio at each inactivation voltage (Fig. 6A, smooth curves). The steady-state value of spike-independent slow inactivation was estimated by extrapolating the fit to infinite time and subtracting the contribution of spike-dependent slow inactivation.
Figure 6D shows the procedure used to measure the time constant and steady-state value of inactivation at hyperpolarized (less than or equal to −60 mV) voltages. After a brief voltage step to measure the initial Na+ current, a 500 msec period was imposed to allow recovery from spike-dependent slow inactivation. The voltage was then increased to −20 mV for 500 msec, causing both spike-dependent and spike-independent slow inactivation. The cell was allowed to recover at one of several voltages for a variable time. Finally, the Na+ current was remeasured. Figure 6, E and F, shows Na+ currents for several recovery times. The ratio of the Na+ currents before and after inactivation is plotted as a function of the recovery time (Fig.6D, bottom). A double exponential was fit to the Na+ current ratio at each inactivation voltage. The faster time constant (100–400 msec) was similar to that measured for recovery from spike-dependent slow inactivation. The slower time constant, 500–1500 msec, described recovery from spike-independent slow inactivation. The extrapolated value of the fit at infinite recovery time gave the steady-state value.
Experiments such as that in Figure 6 characterized the first-order kinetic model of spike-independent slow inactivation. The onset and recovery rate constants, βs1 and αs1, were calculated from the time constant and steady-state value of the spike-independent inactivation using Equations 3 and 4. Values for αs1 and βs1 are plotted against voltage in Figure7A,B. These measurements were fit (smooth curves) using Equations 10 and 11 describing thes1 variable to estimate the voltage dependence of each rate constant. Fits were forced to go to zero at large positive voltages for αs1 and large negative voltages for βs1.
Fig. 7.

Rate constants describing spike-independent slow inactivation. A, The recovery rate constant, αs1, plotted versus voltage. The solid line is an exponential fit calculated from Equation 10.B, The entry rate constant, βs1, plotted versus voltage. The solid line is a saturating exponential fit calculated from Equation 1. αs1 and βs1 were determined from the steady-state value and the slow time constant from the fits in Figure 6 using Equations 3 and 4. Different symbols correspond to measurements from different cells.
Recovery from spike-independent slow inactivation was approximately three times slower than recovery from spike-dependent slow inactivation. For example, at −50 mV, αs1 ≈ 0.6 − 1 sec−1, whereas αs2 ≈ 2 − 4 sec−1. This substantial difference in recovery kinetics suggests that the Na+current may have two distinct mechanistic components of slow inactivation (see Discussion).
Slow Na+ inactivation can account for variance adaptation
The experiments described above indicate that slow inactivation causes the amount of available Na+ current to depend on the past history of voltage changes. The simulations described below show that slow Na+inactivation can account for the ability of ganglion cells to adapt to the variance of their input current. Spike-dependent slow inactivation made the dominant contribution to variance adaptation in the simulation. Thus increasing variance caused a higher spike rate in the simulation, which in turn reduced the available Na+ current and decreased excitability.
Motivation for simulation
We found previously that Na+ currents but not K+ or Ca2+ currents were required for variance adaptation (Kim and Rieke, 2001b). Thus to simplify the simulation, we incorporated the Na+ current properties described above without explicit simulation of K+ and Ca2+currents (see Materials and Methods). By itself, such a simulation will not generate action potentials, because K+and other currents are required to shape the action potential and repolarize the cell after an Na+ spike. Instead, spikes were generated by forcing the voltage of the simulated ganglion cell through the trajectory of an experimentally measured action potential once the Na+ current had generated the rising phase. Once the voltage returned to rest, the simulated cellular currents were free to influence the voltage. The criterion level at which the simulation switched modes was −15 mV, well beyond the experimental threshold for action potential generation.
Table 1 gives the parameters of the simulation, and Equations 6-12give the rate constants describing the Na+current. As a test of the Na+ current description, we compared the measured and predicted reduction in Na+ current produced by the voltage fluctuations of Figure 3A. The simulation predicted a reduction in Na+ current by a factor of 0.22, within the range observed experimentally. The simulation showed no change in Na+ current without slow Na+ inactivation. From this we conclude that the simulation captured the key features of slow Na+ inactivation during voltage fluctuations.
Slow inactivation is necessary for variance adaptation in simulation
The contribution of slow inactivation to adaptation in the simulation was determined using the static nonlinearity model (Fig. 2) (see Materials and Methods). Experimentally, increasing the current variance decreased the amplitude and time-to-peak of the linear filter (Fig. 2A). Slow inactivation of the Na+ current could explain the changes in filter amplitude.
Figure 8 compares variance adaptation in real and simulated ganglion cells. Figure 8A shows the linear filters and static nonlinearities for a real ganglion cell. Figure 8, B and C, shows linear filters and static nonlinearities for the simulated ganglion cell with (Fig.8C) and without (Fig. 8B) slow Na+ inactivation. In all cases the static nonlinearities overlapped (insets), and hence the effects of variance on the simulated responses were restricted to changes in the linear filter.
A change in the time-to-peak of the filter occurred both in the simulations based on models of the Na+current and in simple threshold-crossing models with a refractory period (data not shown). This change was absent when the refractory period was removed from the threshold-crossing model. Similar changes in time-to-peak of the filter have been seen in other threshold-crossing models (Pillow and Simoncelli, 2003). Thus, the change in time-to-peak was attributable to a time-dependent nonlinearity (refractoriness) rather than a variance-dependent change in the behavior of the cell. Refractoriness in the simulation, resulting from the membrane time constant and the repolarization after the action potential, reproduced the change in time-to-peak seen experimentally (Fig. 8B,C). We did not study this effect further because it was an inherent property of any cell with a refractory period.
The change in amplitude of the filter reflected a change in excitability mediated by slow Na+inactivation. To separate changes in amplitude from those in kinetics, the time axes of the linear filters were normalized by the time-to-peak. Figure 8A–C shows these rescaled filters (right). Increasing the variance decreased the filter amplitude in the real cell (Fig. 8A) and in the simulation with slow Na+ inactivation (Fig. 8C) but did not produce a change in amplitude without slow Na+ inactivation (Fig.8B). With slow Na+inactivation, the filter amplitude was reduced by 20–25% for a ninefold change in variance, close to that seen experimentally.
Slow inactivation contributed to adaptation in the simulation by reducing the available Na+ current when the current variance increased. This effect was dominated by spike-dependent slow inactivation. Figure9B shows the simulated voltage responses to injected currents of low and high variance. Spike-independent and spike-dependent slow inactivation are shown in Figure 9C,D. All panels in Figure 9 show behavior of the simulated cell several seconds after a change in variance. After a variance change, inactivation approached its steady-state value with time constants of 0.15 and 1 sec for spike-dependent and spike-independent slow inactivation. Both components of slow inactivation increased with increasing variance.
Fig. 9.
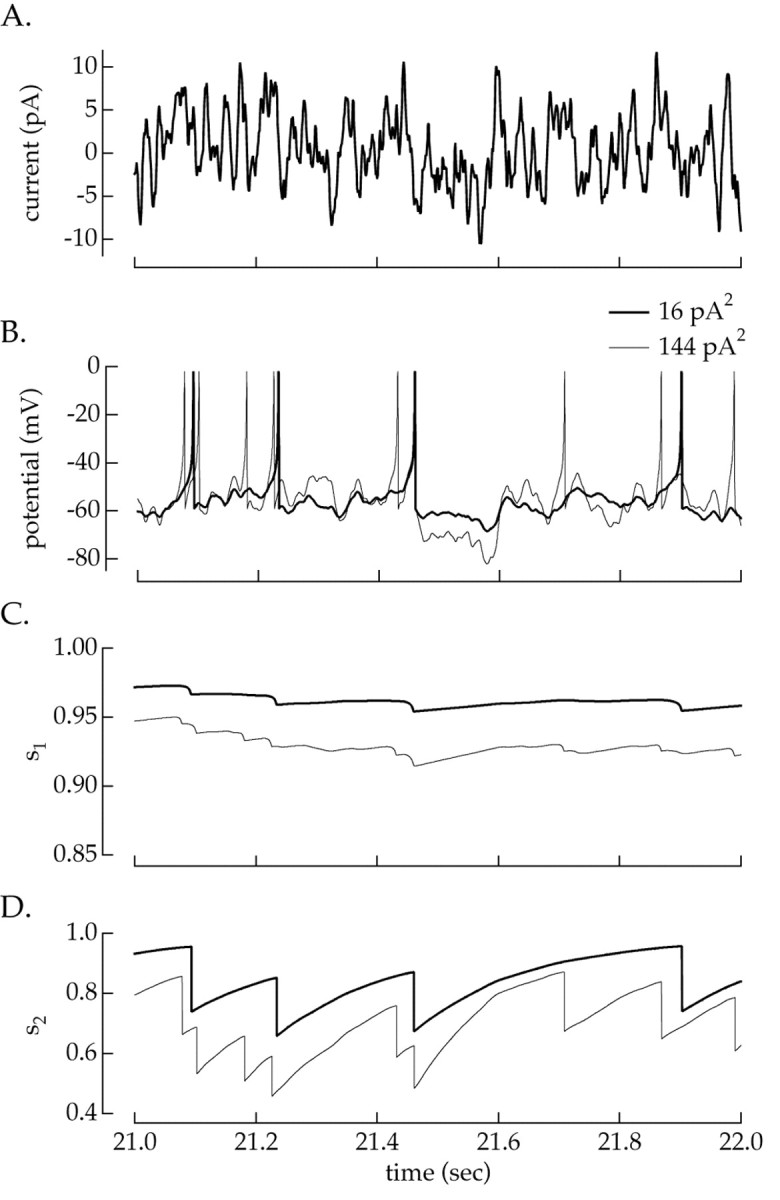
Increase in slow Na+inactivation with variance. Gaussian current fluctuations (0–50 Hz) of 16 and 144 pA2 were injected into a simulated ganglion cell. A, The 16 pA2 variance current fluctuations. The 144 pA2 current fluctuations were identical to that shown except for a scaling factor.B, Voltage output of a simulated ganglion cell.C, Value of the spike-independent slow inactivation variable s1. D, Value of the spike-dependent slow inactivation variables2. Several seconds of current injection at each variance were delivered before time 0, so variables had time to reach steady state.
Spike-dependent slow inactivation increased substantially with variance, as shown in Figure 9D. The greater firing rate at high variance combined with the slow recovery from inactivation causeds2 to depend strongly on variance. However, spike-independent slow inactivation changed little (3–5%) when the variance increased, as shown in Figure 9C. When spike-dependent slow inactivation was removed from the simulation, ∼90% of the variance-induced change in available Na+ current disappeared, whereas removal of spike-independent slow inactivation had little effect. Thus, spike-dependent slow inactivation accounted for the majority of the variance adaptation.
Variance adaptation is robust to changes in simulation parameters
The simulations described above indicate that slow Na+ inactivation can account for variance adaptation. Next we investigated the robustness of adaptation to the parameters of the simulation. As described above, adaptation was measured by the variance-dependent change in amplitude of the linear filters in the static nonlinearity model. We explored parameters that led to spike rates near the physiological range of 2–6 Hz.
Not surprisingly, changes in the parameters controlling the extent of slow-dependent Na+ inactivation affected the extent of adaptation. Varying the rate constant of recovery from spike-dependent slow inactivation, αs2, or the fractional decrease in s2 following a spike, s , between the experimental extremes changed the extent of adaptation by up to 40%. The spike-dependent inactivation produced much larger changes than using the extreme measured values of the rate constants and voltage dependence of spike-independent slow inactivation, αs1 and βs1, which changed the extent of adaptation by <10%. Adaptation was also sensitive to the membrane capacitance and leak conductance, which varied substantially between cells (Table 1). Variations in these parameters likely represent differences between different ganglion cell types as well as variability introduced by the isolation procedure. The resulting cell-to-cell variability in membrane time constant and in spike shape caused up to 50% changes in the extent of adaptation.
Changes in other parameters of the model produced only small effects on the extent of adaptation. The rate constants for the h andm variables of the Na+ current are of particular interest. Using α and β from a recent ganglion cell model (Fohlmeister and Miller, 1997) rather than the Hodgkin–Huxley values had little effect (< 10%) on variance adaptation. Adaptation was also changed minimally by shifts of up to 10 mV in the voltage dependence of the Na+current.
The above results show that the specific values of the parameters in the simulation influence the extent of variance adaptation but not its presence. Much of the cell-to-cell variability in adaptation can be explained by the measured variability in parameters controlling adaptation. The robustness of variance adaptation to changes in the parameters of the simulation comes about because adaptation is dominated by spike-dependent slow inactivation. After an action potential, slow inactivation reduces the available Na+ current and decreases excitability for several hundred milliseconds.
Discussion
The experiments described above lead to three conclusions about how retinal ganglion cells adapt to the variance of their input currents: (1) the Na+ currents of retinal ganglion cells exhibit spike-dependent and spike-independent slow inactivation; (2) slow inactivation reduces the available Na+ current when the variance of the input current of the cell increases, leading to a decrease in excitability; and (3) the role of slow inactivation in variance adaptation holds for a wide range of cellular and Na+ current parameters. Below we discuss the role of slow Na+ inactivation in contrast adaptation and more generally in controlling neural excitability.
Contrast adaptation, variance adaptation, and slow Na+ inactivation
Slow Na+ inactivation influences neural excitability directly by controlling the available transient Na+ current (Fleidervish et al., 1996;Colbert et al., 1997; Jung et al., 1997) and indirectly through modulation of persistent Na+ currents (Schwindt and Crill, 1995; Crill, 1996; Taddese and Bean, 2002). Slow inactivation is usually studied by measuring the influence of current or voltage steps on the Na+ current. Increased inactivation decreases the Na+current and decreases excitability.
Retinal ganglion cells also show alterations in excitability caused by slow inactivation. We studied slow Na+inactivation induced by Gaussian current fluctuations because they mimic physiological inputs to ganglion cells more closely than current steps. The variances of fluctuating currents used here approximate those of the synaptic inputs to a ganglion cell for high and low contrast light inputs. Recovery from slow inactivation required several hundred milliseconds. Thus, after a change in the variance of the input currents, the available Na+ current will reach a steady-state level relatively slowly. This gradual approach to steady state will cause the excitability of the cell to reflect the voltage changes occurring in the previous 0.2–0.5 sec.
Slow inactivation caused the available Na+current to depend on the past history of subthreshold and superthreshold voltage changes. Other neurons also exhibit spike-dependent and spike-independent slow Na+ inactivation (Colbert et al., 1997;Mickus et al., 1999). If the Na+ channels enter the slow inactive state only from the open state, one inactivation mechanism could give rise to both spike-dependent and spike-independent slow inactivation (Mickus et al., 1999). However, the substantial difference in the recovery kinetics of slow inactivation in ganglion cells suggests either distinct mechanisms or that recovery from inactivation depends on the past history of voltage changes.
Slow Na+ inactivation can explain the component of contrast adaptation intrinsic to spike generation in ganglion cells (Kim and Rieke, 2001b): increases in temporal contrast increase the variance of the input currents to a ganglion cell, causing a decrease in available Na+ current because of slow inactivation and hence decreasing excitability. The decrease in excitability increases the dynamic range of the cell, permitting a ganglion cell to encode a wider range of input currents.
Slow inactivation reduced the available Na+ current during high variance by 30–40%. This slow inactivation contributes to a fast kinetic component (time constant of < 1 sec) of temporal contrast adaptation; other mechanisms operating in the retinal circuitry operate ∼10 times more slowly (Smirnakis et al., 1997; Chander and Chichilnisky, 2001; Kim and Rieke, 2001b; Rieke, 2001).
Slow Na+ inactivation and sustained Na+ currents
The persistent current is a sustained Na+ current that inactivates exclusively through slow inactivation (Crill, 1996). This current typically activates at voltages 10–15 mV below the transient Na+ current and is smaller by 2–3 orders of magnitude. The persistent current can alter cell excitability by providing an inward current that helps depolarize a cell to spike threshold (Schwindt and Crill, 1995; Colbert et al., 1997; Koizumi et al., 2001). Changes in the availability of the persistent current through slow inactivation can modulate excitability.
Persistent currents have been found in retinal neurons (Koizumi et al., 2001). However, the effects of slow inactivation (described in Results) appear distinct from those produced by modulation of a persistent current. Our simulation had a non-inactivating Na+ current in a narrow voltage range because of the overlap of the activation and fast inactivation gating variables (the window current) (French et al., 1990), and modulation of this current to more closely match the persistent current did not affect the extent of adaptation.
Slow inactivation as a general modulator of excitability
A variety of cellular and network mechanisms allows cells throughout the nervous system to accommodate a wide range of input signals. These mechanisms operate on time scales ranging from tens of milliseconds to minutes. They also are controlled by a variety of statistical properties of the input signals, including the mean and variations about the mean. In several cases these adaptation mechanisms have been linked to functional properties of adaptation (Sanchez-Vives et al., 2000b; Kawai, 2002; Smith et al., 2002).
In motion-sensitive neurons on the fly visual system, the time scale of adaptation itself adapts (Fairhall et al., 2001). Thus the rate of onset of adaptation after an increase in motion variance depends on the duration of the previous period of low motion variance. This provides an interesting challenge for understanding the underlying mechanisms, because scaling of the adaptation time scale is not expected from a simple mechanism characterized by a single time constant. However, scaling of the recovery kinetics of slow Na+ inactivation has been observed (Toib et al., 1998) and could contribute to a rescaling of the adaptation time course. It will be interesting to determine whether this observation applies more generally.
Two observations suggest that adaptation mediated by control of the available Na+ current through slow inactivation may be a general mechanism. First, slow Na+ inactivation is a common, although not universal, property of voltage-activated Na+ currents. In pyramidal cells, for example, Na+ currents in the dendrites show substantially greater slow inactivation than those in the soma (Colbert et al., 1997). Second, variance adaptation mediated by slow inactivation was robust to large changes in many cell properties, including spike shape, firing rate, fast Na+ activation and inactivation, and membrane time constant.
The Na+ currents of many spiking neurons are much larger than the minimum needed to generate action potentials (Crill, 1996). This is also true for retinal ganglion cells. The surplus of Na+ current allows phenomena that constitute a small fraction of the Na+ current, such as persistent (Crill, 1996) and resurgent (Raman and Bean, 2001) currents, to be large enough to modulate excitability. The surplus of Na+ current also allows slow inactivation to reduce excitability in a graded manner. As emphasized above, this allows adaptation to match the input–output relationship of a cell to the dynamic range of its inputs. Slow Na+inactivation also provides a feedback mechanism that shapes the spike output of a cell by lowering excitability during periods of high activity. Such a feedback, operating on a time scale similar to the slow inactivation characterized here, is an essential part of a recent model describing the relationship between light inputs and ganglion cell outputs (Keat et al., 2001).
Footnotes
This work was supported by National Institutes of Health (NIH) Grant EY-11850, the McKnight Foundation, NIH Training Grant GM07108 (K.J.K.), and the University of Washington Graduate School Fund for Excellence and Innovation Special Fellowship (K.J.K.). We thank Helen Brew, Greg Field, Bertil Hille, Randy Powers, Cecilia A. Gold, Bill Catterall, A. P. Sampath, Todd Scheuer, Mike Shadlen, and Bill Zagotta for helpful discussions, and Eric Martinson for excellent technical assistance.
Correspondence should be addressed to Fred Rieke, Department of Physiology and Biophysics, Health Sciences Building, Room G424, 1959 Northeast Pacific Street, University of Washington, Seattle, WA 98195. E-mail: rieke@u.washington.edu.
References
- 1.Albrecht DG, Farrar SB, Hamilton DB. Spatial contrast adaptation characteristics of neurones recorded in the cat's visual cortex. J Physiol (Lond) 1984;347:713–739. doi: 10.1113/jphysiol.1984.sp015092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner N, Bialek W, de Ruyter van Steveninck R. Adaptive rescaling maximizes information transmission. Neuron. 2000;26:695–702. doi: 10.1016/s0896-6273(00)81205-2. [DOI] [PubMed] [Google Scholar]
- 3.Burkhardt DA, Fahey PK, Sikora M. Responses of ganglion cells to contrast steps in the light-adapted retina of the tiger salamander. Vis Neurosci. 1998;15:219–229. doi: 10.1017/s0952523898152021. [DOI] [PubMed] [Google Scholar]
- 4.Chander D, Chichilnisky EJ. Adaptation to temporal contrast in primate and salamander retina. J Neurosci. 2001;21:9904–9916. doi: 10.1523/JNEUROSCI.21-24-09904.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chichilnisky EJ. A simple white noise analysis of neuronal light responses. Network. 2001;12:199–213. [PubMed] [Google Scholar]
- 6.Colbert CM, Magee JC, Hoffman DA, Johnston D. Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 1997;17:6512–6521. doi: 10.1523/JNEUROSCI.17-17-06512.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- 8.Fairhall AL, Lewen GD, Bialek W, de Ruyter Van Steveninck RR. Efficiency and ambiguity in an adaptive neural code. Nature. 2001;412:787–792. doi: 10.1038/35090500. [DOI] [PubMed] [Google Scholar]
- 9.Fleidervish IA, Friedman A, Gutnick MJ. Slow inactivation of Na+ current and slow cumulative spike adaptation in mouse and guinea-pig neocortical neurones in slices. J Physiol (Lond) 1996;493:83–97. doi: 10.1113/jphysiol.1996.sp021366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fohlmeister JF, Miller RF. Impulse encoding mechanisms of ganglion cells in the tiger salamander retina. J Neurophysiol. 1997;78:1935–1947. doi: 10.1152/jn.1997.78.4.1935. [DOI] [PubMed] [Google Scholar]
- 11.French CR, Sah P, Buckett KJ, Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. J Gen Physiol. 1990;95:1139–1157. doi: 10.1085/jgp.95.6.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hille B. Ion channels of excitable membranes, Ed 3. Sinauer; Sunderland, MA: 2001. [Google Scholar]
- 13.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung HY, Mickus T, Spruston N. Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. J Neurosci. 1997;17:6639–6646. doi: 10.1523/JNEUROSCI.17-17-06639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai F. Ca2+-activated K+ currents regulate odor adaptation by modulating spike encoding of olfactory receptor cells. Biophys J. 2002;82:2005–2015. doi: 10.1016/S0006-3495(02)75549-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keat J, Reinagel P, Reid RC, Meister M. Predicting every spike: a model for the responses of visual neurons. Neuron. 2001;30:646–647. doi: 10.1016/s0896-6273(01)00322-1. [DOI] [PubMed] [Google Scholar]
- 17.Kim KJ, Rieke F. Na+ channels mediate temporal contrast adaptation in the salamander retina (Association for Research in Vision and Ophthalmology abstract). Invest Ophthalmol Vis Sci. 2001a;42:675. [Google Scholar]
- 18.Kim KJ, Rieke F. Temporal contrast adaptation in the input and output signals of salamander retinal ganglion cells. J Neurosci. 2001b;21:287–299. doi: 10.1523/JNEUROSCI.21-01-00287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koizumi A, Watanabe SI, Kaneko A. Persistent Na+ current and Ca2+ current boost graded depolarization of rat retinal amacrine cells in culture. J Neurophysiol. 2001;86:1006–1016. doi: 10.1152/jn.2001.86.2.1006. [DOI] [PubMed] [Google Scholar]
- 20.Lukasiewicz P, Werblin F. A slowly inactivating potassium current truncates spike activity in ganglion cells of the tiger salamander retina. J Neurosci. 1988;8:4470–4481. doi: 10.1523/JNEUROSCI.08-12-04470.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacGregor RJ. Neural and brain modeling. Academic; San Diego: 1987. [Google Scholar]
- 22.Meister M, Berry MJ. The neural code of the retina. Neuron. 1999;22:435–450. doi: 10.1016/s0896-6273(00)80700-x. [DOI] [PubMed] [Google Scholar]
- 23.Mickus T, Jung HY, Spruston N. Properties of slow, cumulative sodium channel inactivation in rat hippocampal CA1 pyramidal neurons. Biophys J. 1999;76:846–860. doi: 10.1016/S0006-3495(99)77248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohzawa I, Sclar G, Freeman RD. Contrast gain control in the cat's visual system. J Neurophysiol. 1985;54:651–667. doi: 10.1152/jn.1985.54.3.651. [DOI] [PubMed] [Google Scholar]
- 25.Pillow JW, Simoncelli EP (2003) Biases in white noise analysis due to non-Poisson spike generation. Neurocomputing, in press.
- 26.Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rieke F. Temporal contrast adaptation in salamander bipolar cells. J Neurosci. 2001;21:9445–9454. doi: 10.1523/JNEUROSCI.21-23-09445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sah P, Davies P. Calcium-activated potassium currents in mammalian neurons. Clin Exp Pharmacol Physiol. 2000;27:657–663. doi: 10.1046/j.1440-1681.2000.03317.x. [DOI] [PubMed] [Google Scholar]
- 29.Sakai HM, Wang JL, Naka K. Contrast gain control in the lower vertebrate retinas. J Gen Physiol. 1995;105:815–835. doi: 10.1085/jgp.105.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Vives MV, Nowak LG, McCormick DA. Membrane mechanisms underlying contrast adaptation in cat area 17 in vivo. J Neurosci. 2000a;20:4267–4285. doi: 10.1523/JNEUROSCI.20-11-04267.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez-Vives MV, Nowak LG, McCormick DA. Cellular mechanisms of long-lasting adaptation in visual cortical neurons in vitro. J Neurosci. 2000b;20:4286–4299. doi: 10.1523/JNEUROSCI.20-11-04286.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwindt PC, Crill WE. Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurosci. 1995;74:2220–2224. doi: 10.1152/jn.1995.74.5.2220. [DOI] [PubMed] [Google Scholar]
- 33.Shapley R. Retinal physiology: adapting to the changing scene. Curr Biol. 1997;7:R421–R423. doi: 10.1016/s0960-9822(06)00207-7. [DOI] [PubMed] [Google Scholar]
- 34.Shapley R, Victor JD. The effect of contrast on the transfer properties of cat retinal ganglion cells. J Physiol (Lond) 1978;285:275–298. doi: 10.1113/jphysiol.1978.sp012571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smirnakis SM, Berry MJ, Warland DK, Bialek W, Meister M. Retinal processing adapts to image contrast and spatial scale. Nature. 1997;386:69–73. doi: 10.1038/386069a0. [DOI] [PubMed] [Google Scholar]
- 36.Smith MR, Nelson AB, Du Lac S. Regulation of firing response gain by calcium-dependent mechanisms in vestibular nucleus neurons. J Neurophysiol. 2002;87:2031–2042. doi: 10.1152/jn.00821.2001. [DOI] [PubMed] [Google Scholar]
- 37.Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- 38.Taylor WR, Mittman S, Copenhagen DR. Passive electrical cable properties and synaptic excitation of tiger salamander retinal ganglion cells. Vis Neurosci. 1996;13:979–990. doi: 10.1017/s0952523800009202. [DOI] [PubMed] [Google Scholar]
- 39.Toib A, Lyakhov V, Marom S. Interaction between duration of activity and time course of recovery from slow inactivation in mammalian brain Na+ channels. J Neurosci. 1998;18:1893–1903. doi: 10.1523/JNEUROSCI.18-05-01893.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walraven J, Enroth-Cugell C, Hood DC, MacLeod DIA, Schnapf JL. The control of visual sensitivity. In: Spillmann L, Werner SJ, editors. Visual perception: the neurophysiological foundations. Academic; San Diego: 1990. pp. 53–101. [Google Scholar]