

Abstract
During neuroinflammation, activated microglial cells migrate toward dying neurons, where they exacerbate local cell damage. The signaling molecules that trigger microglial cell migration are poorly understood. In this paper, we show that pathological overstimulation of neurons by glutamate plus carbachol dramatically increases the production of the endocannabinoid 2-arachidonylglycerol (2-AG) but only slightly increases the production of anandamide and does not affect the production of two putative endocannabinoids, homo-γ-linolenylethanolamide and docosatetraenylethanolamide. We further show that pathological stimulation of microglial cells with ATP also increases the production of 2-AG without affecting the amount of other endocannabinoids. Using a Boyden chamber assay, we provide evidence that 2-AG triggers microglial cell migration. This effect of 2-AG occurs through CB2 and abnormal-cannabidiol-sensitive receptors, with subsequent activation of the extracellular signal-regulated kinase 1/2 signal transduction pathway. It is important to note that cannabinol and cannabidiol, two nonpsychotropic ingredients present in the marijuana plant, prevent the 2-AG-induced cell migration by antagonizing the CB2 and abnormal-cannabidiol-sensitive receptors, respectively. Finally, we show that microglial cells express CB2 receptors at the leading edge of lamellipodia, which is consistent with the involvement of microglial cells in cell migration. Our study identifies a cannabinoid signaling system regulating microglial cell migration. Because this signaling system is likely to be involved in recruiting microglial cells toward dying neurons, we propose that cannabinol and cannabidiol are promising nonpsychotropic therapeutics to prevent the recruitment of these cells at neuroinflammatory lesion sites.
Keywords: microglia, migration, cannabinoids, glutamate, purinergic, kinase
Introduction
Marijuana intake produces a large variety of biological effects. These include the well known psychotropic effects, some adverse effects (e.g., memory loss, sedation, and motor impairment), and several beneficial effects (e.g., relief of muscle spasticity, analgesia, and reduction of inflammation) (Consroe et al., 1997; Hall and Solowij, 1998; Hampson and Deadwyler, 2000; Watson et al., 2000; Solowij et al., 2002). Because marijuana produces remarkable beneficial effects, patients with multiple sclerosis, for example, commonly use this plant as a therapeutic agent; however, we still lack essential information on the mechanistic basis of these beneficial effects (Lyman et al., 1989; Meinck et al., 1989;Wirguin et al., 1994; Martyn et al., 1995; Baker et al., 2000,2001).
The marijuana plant, Cannabis sativa, contains >60 cannabinoid compounds, the best known being Δ9-tetrahydrocannabinol (THC), cannabinol (CBN), and cannabidiol (CBD) (for review, see Howlett et al., 2002). Cannabinoid compounds produce their biological effects by acting through at least three cannabinoid receptors (see Table1). These include the cloned cannabinoid CB1 receptors, which are expressed predominately in the CNS (Matsuda et al., 1990), the cloned cannabinoid CB2 receptors, which are expressed predominately by immune cells (Munro et al., 1993), and the abnormal-cannabidiol-sensitive receptors (Járai et al., 1999) (hereafter referred to as abn-CBD receptors). The latter receptors have not been cloned yet, but they have been pinpointed pharmacologically in mice lacking CB1 and CB2 receptors and are also known as anandamide (AEA) receptors (Járai et al., 1999).
Table 1.
Cannabinoid receptor subtypes
| Cannabinoid receptor subtypes | Agonists | Antagonists |
|---|---|---|
| CB1 | THC, AEA, 2-AG | SR141716A |
| CB2 | THC, 2-AG, CBN | SR144528, CBN |
| abn-CBD | AEA, abn-CBD, CBD | O-1918, CBD |
The three subtypes of cannabinoid receptors referred to in this study, with their allied agonists and antagonists. Note that CBN and CBD act as partial agonists at CB2 and abn-CBD receptors, respectively, and therefore also act as antagonists.
The induced psychotropic and adverse effects of marijuana are attributable to THC acting at CB1 receptors expressed by neurons (Mallet and Beninger, 1998; Zimmer et al., 1999; Hampson and Deadwyler, 2000; Huestis et al., 2001), whereas the immunomodulatory effects are attributable to THC and CBN acting at CB2 receptors expressed by immune cells and to CBD acting on abn-CBD receptors (Kaminski, 1998; Klein et al., 1998; Buckley et al., 2000; Malfait et al., 2000). Because CBN and CBD do not have significant intrinsic activity on CB1 receptors (Felder et al., 1995; Showalter et al., 1996), these compounds do not produce psychotropic and adverse side effects (Perez-Reyes et al., 1973), which makes them promising candidates as anti-inflammatory therapeutics. It should be emphasized that plant cannabinoids often act as partial agonists on cannabinoid receptors (Howlett et al., 2002), which suggests that they might antagonize the efficacious effects of certain endocannabinoids (i.e., the endogenous cannabinoid ligands produced by cells) (Piomelli et al., 1998; Stella and Piomelli, 2001).
Whether plant cannabinoids or endocannabinoids affect microglial cells, the immune cells of the CNS, is poorly understood. Microglial cells are macrophage-like cells that originate from myeloid tissue and migrate into the developing CNS (Rezaie and Male, 1999). Once this early wave of microglial cell migration is completed, these cells remain in the CNS and become quiescent, a resting state that lasts as long as the CNS remains healthy. Quiescent microglial cells have a ramified morphology and express plasma membrane receptors, enabling them to survey the CNS and respond to pathological events (Kreutzberg, 1996). For example, microglial cells express purinergic receptors that detect ATP, which is released in high amounts by dying cells (Möller et al., 2000; Kim et al., 2001). Engagement of plasma membrane receptors on quiescent microglial cells initiates a rapid, multiple-step change in phenotype that is referred to as microglial cell activation (Kreutzberg, 1996;Bruce-Keller, 1999; Becher et al., 2000). Initially, quiescent microglial cells retract their ramifications, transforming themselves into amoeboid-like cells with motile protrusions (Stence et al., 2001). They then migrate toward the site of injury, where they release proinflammatory cytokines and cytotoxic agents (Kreutzberg, 1996;Bruce-Keller, 1999; Becher et al., 2000). Thus, as the initial step, microglial cell migration plays a crucial role in the propagation of the neuroinflammatory response; however, the signaling molecules that trigger microglial cell migration are poorly understood. We sought to determine whether plant cannabinoids and/or endocannabinoids regulate microglial cell migration and assessed whether pathological conditions affect endocannabinoid production from neurons and microglial cells.
Materials and Methods
Cells in culture. Newborn mouse neopallia microglial cells in primary cultures were prepared as described previously (Walter et al., 2002). BV-2 cells, a mouse microglial cell line, were grown in DMEM supplemented with FBS (3%), penicillin (100 U), and streptomycin (100 μg/ml) and passaged every 3–4 d for a maximum of 30 passages. Because microglial cells and BV-2 cells were routinely grown with FBS, cells were recovered in a defined cell culture medium ∼18 hr before experiments. Specifically, we recovered them at 200,000 cells/ml MEM/Cellgro (Mediatech, Herndon, VA) (MEM supplemented with 10 mm HEPES, 5 mmNaHCO3, 100 U penicillin, 100 μg/ml streptomycin, 2 mml-glutamine, and 10% Cellgro) (Walter et al., 2002). Newborn mouse neopallia neurons in primary cultures were prepared in B27-supplemented Neurobasal as described by Stella and Piomelli (2001).
Reverse transcription-PCR. To perform reverse transcription (RT), we used superscript first-strand synthesis (Invitrogen, Grand Island, NY). The following mouse cannabinoid receptor primers were used: CB1 forward, 5′-TCT GGC CTA TAA GAG GAT CGT CAC-3′; CB1 reverse, 5′-CAG CAG GCA GAG CAT ACT ACA GAA-3′; CB2 forward, 5′-GGG TCC TCT CAG CAT TGA TTT CT-3′; and CB2 reverse, 5′-GTT AAC AAG GCA CAG CAT GGA AC-3′. Amplification was performed by 35 cycles of the following: 94°C for 30 sec, 63°C for CB1 primers and 57°C for CB2 primers for 30 sec, and 72°C for 2 min. Amplification cycles were followed by 72°C for 10 min. RT-PCR products were separated on 1.2% agarose gels, sequenced, and validated by comparison with the National Center for Biotechnology Information database. Absence of RT-PCR product in the “no RT” reaction confirmed the absence of genomic DNA in the samples.
Immunocytochemistry. To visualize actin filaments cells were fixed with paraformaldehyde and stained as follows: polyclonal rabbit anti-C-terminal CB1 (1:500) (Hájos et al., 2000), polyclonal rabbit anti-C-terminal CB2 (1:500; the antigen was constructed in our laboratory and the antibody was generated by R & R Research and Development, Stanwood, CA), polyclonal rat anti-MAC1 (1:100; Serotec, Oxford, UK), monoclonal mouse-anti-hemagglutinin 11 (HA11) (1:500; Covance Research Products, Princeton, NJ), or with Texas Red-conjugated phalloidin (1:40; Molecular Probes, Eugene, OR). Secondary IgG antibodies were conjugated with Texas Red (1:100) or FITC (1:150; Jackson ImmunoResearch, West Grove, PA). Human embryonic kidney (HEK)293 cells were transfected with 1 μg of construct for hemagglutinin epitope-tagged rCB2 (HA-rCB2/pcDNA3) using 10 μl of Superfect (Qiagen, Hilden, Germany). Images were acquired with a Leica (Nussloch, Germany) TCS SP/NT confocal microscope (Keck Center, University of Washington, Seattle, WA). Nonspecific staining was determined customarily with parallel immunostaining experiments performed in the presence of the appropriate immunizing antigen and using equal gain settings during acquisition and analysis.
Cell migration. Cannabinoid compounds were dissolved at 1000× in DMSO in silane-treated glass vials and added to the lower wells of the Boyden chambers with silane-treated pipette tips. BV-2 cells (5 × 104 in 50 μl of MEM plus 10% Cellgro) were added to the upper chamber and allowed to migrate through polycarbonate filters (pore size, 10 μm) for 3 hr at 37°C (humidified atmosphere of 95% air and 5% CO2). Cells that did not migrate and stayed on the upper surface of the filter were wiped off, whereas cells that had migrated to the lower surface were stained with the DIF-Quick stain kit (IMEB, Inc., San Marcos, CA) and manually counted in random fields at 32× magnification by three scorers that were blind to experimental conditions.
Chemical ionization gas chromatography/mass spectrometry analysis of endocannabinoids. Endocannabinoids were analyzed in two compartments: (1) cells plus adjacent media and (2) distant media. We decided to keep cells covered with their adjacent media to prevent air-induced stress.
Microglial cells, BV-2 cells, and neurons plated in 100 mm culture dishes were placed on a shaking water bath kept at 37°C, keeping them in their defined cell culture media (12.5 ml). Stimulation was initiated by directly adding agents prepared in 1.5 ml of fresh culture media (total volume per dish, 14 ml). To stop the stimulation, we placed dishes on ice and replaced 9 ml of distant media with 5 ml of ice-cold methanol, which fixed the cells plus adjacent media. In some experiments, we fixed the removed 9 ml of distant media by adding it to 9 ml of ice-cold methanol. Lipids from either cells plus adjacent media or distant media were then extracted with chloroform, and endocannabinoids were purified by HPLC and analyzed by chemical ionization gas chromatography/mass spectrometry (CI-GC/MS) as described previously (Stella and Piomelli, 2001; Walter et al., 2002).
Protein content was quantified by using sister cultures (i.e., from the same preparation) grown in 35 mm culture dishes. To avoid a contamination from the proteins present in Cellgro (e.g., 1 mg/ml albumin) and B27-supplemented Neurobasal, the entire medium was removed and the cells were rinsed once with PBS. Cells were then lysed with Triton X-100 (0.1%) and sonicated, and protein content was quantified with a Bio-Rad (Hercules, CA) Dc Protein Assay.
MAP kinase phosphorylation. BV-2 cells in 100 mm dishes were incubated and stimulated as described above for the CI-GC/MS analysis. We replaced the media with ice-cold lysis buffer to stop the stimulation. We used Western blotting with monoclonal antibodies that recognize the dual-phosphorylation state of extracellular signal-regulated kinase 1/2 (ERK1/2) on Thr202 and Tyr204 [phospho-p44/42 MAP kinase (MAPK); Cell Signaling Technology, Beverly, MA] to detect activation of MAPK, and polyclonal antibodies against extracellularly regulated kinase 2 (ERK2; Santa Cruz Biotechnology, Santa Cruz, CA) to determine total ERK2, as described previously (Wade et al., 2001). Samples were scanned and analyzed with the NIH Image analysis program.
Results
It is known that CB1 receptors are widespread in healthy brain, being expressed by many neurons, some astrocytes, and possibly resting microglial cells; however, CB2 receptors are absent in these cells (Tsou et al., 1998; Waksman et al., 1999; Rodríguez et al., 2001). Because the expression of cannabinoid receptor isoform changes as a result of cell activation, as illustrated with macrophage-like cells (Lee et al., 2001), we hypothesized that activated microglial cells express additional cannabinoid receptor isoforms. To test this hypothesis, we took advantage of the fact that microglial cells are activated when transferred into primary culture (Becher and Antel, 1996), and we used RT-PCR to assess whether these cells contain mRNA encoding for CB1 and/or CB2 receptors. Both cultured mouse microglial cells and BV-2 cells contain CB1 and CB2 receptor mRNA (Fig.1a,b). Using an antibody that recognizes the C-terminal domain of CB1 receptors (Hájos et al., 2000), we saw that CB1 receptors were localized primarily in the intracellular compartment of activated microglial cells rather than on their plasma membrane (Fig. 1c,d). To analyze CB2 receptor localization, we generated a rabbit polyclonal antibody specific for the C-terminal domain of CB2 receptors. This antibody labeled HEK293 cells transfected with rat CB2 receptors, whereas no staining was observed when the antibody was preabsorbed with the immunizing peptide or in nontransfected cells (Fig. 1e,f). Using this CB2-specific antibody we found that, unlike CB1 receptors, CB2 receptors were expressed heterogeneously throughout cells, with especially high density at the leading edges of lamellipodia (Fig.1g) and microspikes (Fig. 1h), two cellular protrusions that contain polymerized actin filaments and mediate cell migration (Mitchison and Cramer, 1996; Watanabe and Mitchison, 2002).
Fig. 1.
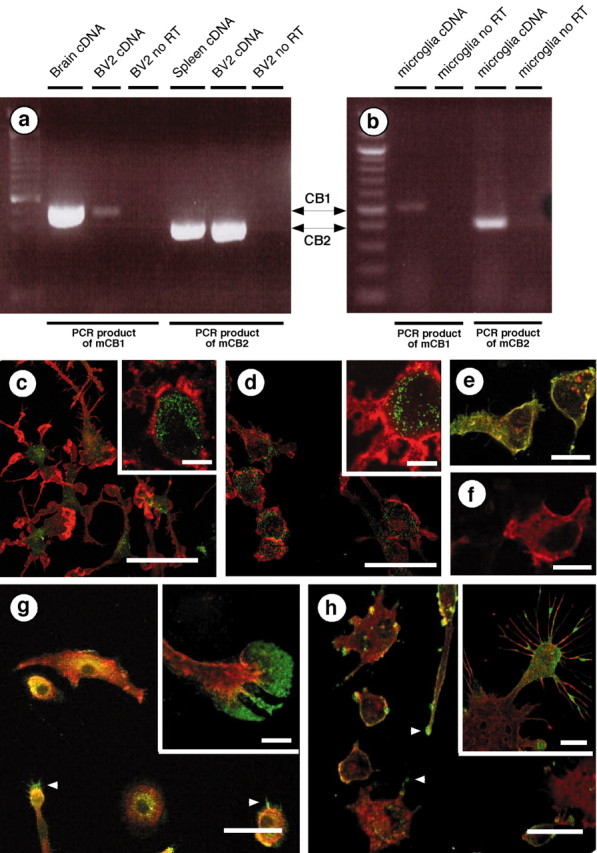
Microglial cells express cannabinoid CB1 and CB2 receptors. a, b, RT-PCR was performed with primers that recognize either mouse CB1 (mCB1) or mouse CB2 (mCB2) mRNA. We used reverse-transcribed total RNA from mouse brain and spleen (i.e., positive controls), BV-2 cells, and mouse microglial cells. RT-PCR products were of the appropriate size (502 bp for CB1 and 401 bp for CB2) and sequence. c–h, Immunofluorescent confocal microscopy. Mouse microglial cells (c) and BV-2 cells (d) were stained with an antibody directed against the CB1 C terminus (green) and phalloidin to label actin (red). Scale bars, 50 μm. Insets, Higher magnifications of the CB1 receptors (green) located in the intracellular compartment (MAC1, a plasma membrane macrophage marker, is in red). Scale bars, 10 μm. e, HEK293 cells transiently transfected with rat CB2 receptors tagged with HA11 (red) and stained with antibodies directed against CB2 C terminus (green). Colocalization isyellow. f, Similar immunostaining as ine, but performed in the presence of the immunizing antigen (i.e., the last 42 aa of the mouse CB2 receptor C terminus). Scale bars, 35 μm. Mouse microglial cells (g) and BV-2 cells (h) stained with antibodies directed against the CB2 receptor C terminus (green) and phalloidin (red).Arrowheads indicate CB2 receptors located at the lamellipodia tip. Scale bars, 50 μm. Insets, Higher magnification showing CB2 receptors at the leading edges of lamellipodia (g) and on microspikes (h). Scale bars, 10 μm.
Cell migration is triggered by chemoattractants that act through seven transmembrane/Gi/o-coupled receptors, some of which accumulate at the leading edges of lamellipodia (Servant et al., 1999). Because it is known that THC triggers the migration of macrophages (Schwartzfarb et al., 1974) [possibly by acting through Gi/o-coupled CB2 receptors (Derocq et al., 2000)], and because we found that activated microglial cells express CB2 receptors at the leading edges of their motile protrusions (Fig.1), we sought to determine whether cannabinoids affect microglial cell migration. To address this possibility, we used the Boyden chamber assay (Wilkinson, 1998). BV-2 cells were added to the upper chamber, and their migration through a filter toward a lower chamber containing cannabinoids was quantified. We found that THC had no significant effect on basal BV-2 cell migration at concentrations of <3 μm (Fig. 2a), whereas CBD slightly increased migration with an EC50 of 250 nm. Next, we tested the effect of abnormal-cannabidiol, a synthetic agonist that acts as a full agonist on abn-CBD receptors (Járai et al., 1999). Abnormal-cannabidiol significantly increased cell migration with an EC50 of 600 nm (Fig.2c). It is important to note that abnormal-cannabidiol and THC acted synergistically when both were added at low nanomolar concentrations (Fig. 2d), which suggests that microglial cell migration is regulated by abn-CBD receptors and cannabinoid receptors (possibly CB2 receptors) acting in a cooperative/synergistic manner.
Fig. 2.

Cannabinoids increase microglial cell migration.a–d, In the lower chamber of the Boyden chamber assay, we added THC (a), CBD (b), abn-CBD (c), and THC plus abn-CBD (d) (at a 1:1 molar ratio). BV-2 cell migration toward these ligands was quantified, and results are expressed as a percentage of basal BV-2 cell migration (i.e., vehicle = 0.1% DMSO; dashed line) measured in individual experiments. *p < 0.05; **p < 0.01; significantly different from basal BV-2 cell migration (ANOVA followed by Dunnett's post hoc test). Values are mean ± SEM of 9–45 independent quantifications of migration (i.e., 3–15 separate experiments performed in triplicate).
AEA and 2-arachidonylglycerol (2-AG), two endocannabinoids produced by neurons (Di Marzo et al., 1994; Stella et al., 1997; Stella and Piomelli, 2001), increased BV-2 cell migration in a concentration-dependent manner (Fig.3a,c), whereas arachidonic acid, a result of the hydrolysis of these lipids, had no effect (data not shown). Two putative endocannabinoids, homo-γ-linolenylethanolamide (HEA) and docosatetraenylethanolamide (DEA) (Felder et al., 1993; Hanus et al., 1993; Pertwee et al., 1994), also increased BV-2 cell migration in a concentration-dependent manner (Fig. 3e,f), whereas palmitylethanolamide (PEA), which is a congener of AEA that does not act on CB1 or CB2 receptors (Griffin et al., 2000), had no effect (Fig. 3d). It is noteworthy that AEA, 2-AG, HEA, and DEA at 1 μmappear to act through the same receptors and/or signal transduction pathway, because their effects were not additive (data not shown).
Fig. 3.
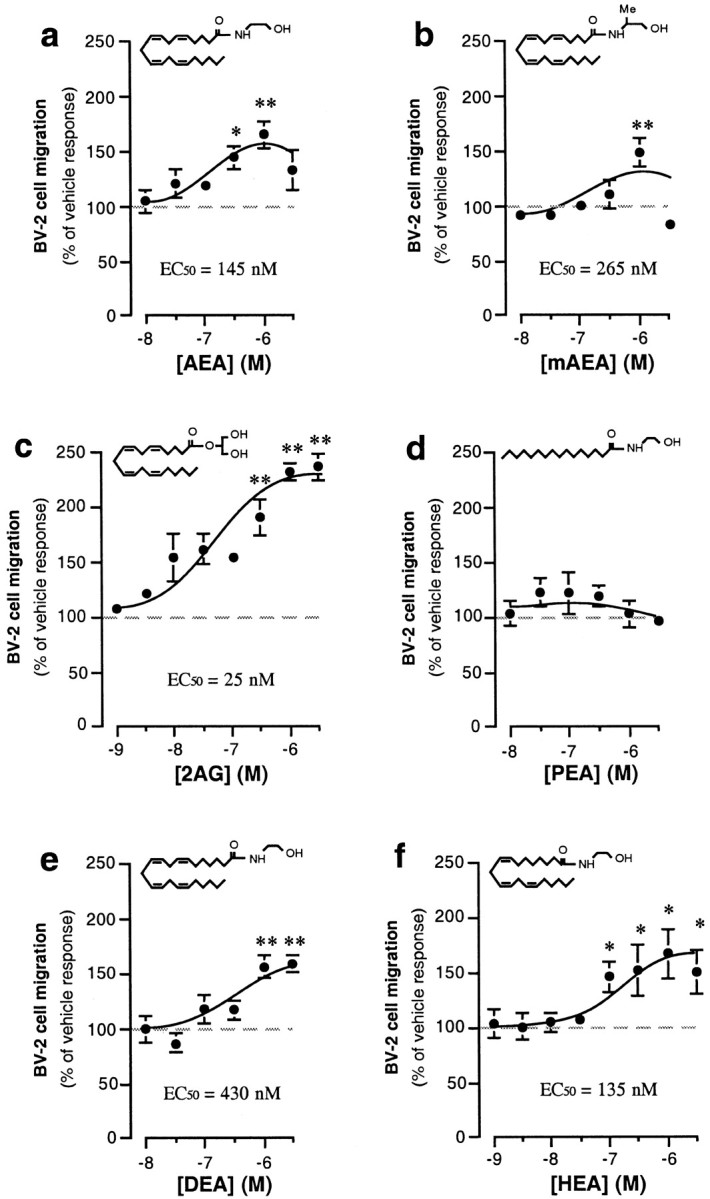
Endocannabinoids increase microglial cell migration. a–f, In the lower chamber of the Boyden chamber assay, we added AEA (a), meth-AEA (mAEA; b), 2-AG (2AG;c), PEA (d), DEA (e), and HEA (f). BV-2 cell migration toward these ligands was quantified, and results are expressed as a percentage of basal BV-2 cell migration (i.e., vehicle = 0.1% DMSO; dashed line) measured in individual experiments. *p < 0.05; **p < 0.01; significantly different from basal BV-2 cell migration (ANOVA followed by Dunnett's post hoc test). Values are mean ± SEM of 9–45 independent quantifications of migration (i.e., 3–15 separate experiments performed in triplicate).
Because 2-AG triggers microglial cell migration with the highest potency and efficacy, we also characterized its effect. Cell motility can be divided into (1) basal random motion, which occurs in the absence of a stimulus, (2) chemokinesis, which is random motion increased by a chemical stimulus, and (3) chemotaxis, which is directed cell migration along a chemical gradient (Wilkinson, 1998). To determine whether 2-AG triggers chemokinesis, chemotaxis, or a combination of both, we performed a checkerboard experiment in which equal concentrations of 2-AG were added to both the upper and the lower chambers, thereby disrupting the chemical gradient, and followed BV-2 cell migration under these conditions. If 2-AG produces chemokinesis, the response should persist in this disrupted gradient. If 2-AG produces chemotaxis, the response should be absent. We found that the 2-AG-triggered microglial cell migration was reduced by 50% when performing the checkerboard experiments, indicating that 2-AG induces both chemokinesis and chemotaxis (n = 3).
Figure 4, a andb, shows that 2-AG-triggered migration was prevented by (1) pertussis toxin pretreatment, which uncouples Gi/o-coupled receptors; (2)N-(1,S)-endo1,3,3-trimethylbicyclo(2,2,1)heptan-2-yl)-5(4-chloro-3-methyl-phenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide (SR144528), a CB2 receptor antagonist (Rinaldi-Carmona et al., 1998); (3) CBN, a CB2 receptor partial agonist (Felder et al., 1995); (4) O-1918, an abn-CBD receptor antagonist (G. Kunos, unpublished observations); and (5) CBD, an abn-CBD receptor partial agonist (Járai et al., 1999). These results indicate that 2-AG triggers microglial cell migration by acting through Gi/o-coupled CB2 and abn-CBD receptors. The 2-AG response did not involve CB1 receptors, becauseN-(piperidiny-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole -3-carboxamide (SR141716A), a CB1 receptor antagonist (Rinaldi-Carmona et al., 1994), did not decrease migration significantly (Fig. 4a,b); the response also was not mimicked by 100 nmmethanandamide (meth-AEA), a CB1/CB2 receptor agonist (Fig.3b). Note that 1 μm methanandamide significantly increased BV-2 cell migration, likely because of its agonist effect on CB2 receptors (Goutopoulos et al., 2001).
Fig. 4.

2-AG increases microglial cell migration by acting through CB2- and CBD-sensitive receptors and stimulating ERK1/2 activity. a, Effects of various agents on basal migration. Values in a and b are means ± SEM of 9–36 independent quantifications of migration (i.e., 3–12 separate experiments performed in triplicate). Agents that were added to the lower chamber are as follows: 30 nmSR141716A (SR1), 30 nm SR144528 (SR2), 300 nm CBN, 300 nm CBD, 1 μm O-1918, or 10 μm PD98059 (PD). To test for the involvement of Gi/o-proteins, BV-2 cells were pretreated with 1 μg/ml pertussis toxin (PTX) for 18 hr.b, Effects of various agents on 2-AG (2AG)-induced migration. Results are expressed as a percentage of the control 2-AG-induced migration determined in each experiment (i.e., migration induced by 1 μm 2-AG added alone to the lower chamber minus corresponding basal migration obtained with the same agent; dashed line; see a). *p < 0.05; **p < 0.01; significantly different from the control 2-AG response (Student'st test). c, Representative Western blot with phospho-ERK1/2 antibodies. d, Quantification of three separate experiments performed in duplicate (n = 6). **p < 0.01; significantly different from basal (ANOVA followed by Dunnett'spost hoc test).
CB2 receptors activate ERK1/2 (Bouaboula et al., 1996), a signal transduction pathway known to regulate cell migration (Klemke et al., 1997). Therefore, we assessed whether the 2-AG response requires this signal transduction pathway. Indeed, 2-AG increased ERK1/2 activity in BV-2 cells (Fig. 4c,d), and an inhibitor of this pathway, PD98059, abolished the 2-AG-triggered BV-2 cell migration (Fig.4a,b).
In the second part of our study, we assessed whether pathological stimuli affect endocannabinoid production, because this could participate in recruiting microglial cells to the lesion site. First, we investigated how the neuronal production of AEA, 2-AG, HEA, and DEA is affected by acute challenge with (1) ionomycin, a calcium ionophore that induces large increases in intracellular calcium, and (2) glutamate plus carbachol, which mimics pathological overstimulation (Stella and Piomelli, 2001). To our knowledge, HEA and DEA production by neurons has never been documented. Acute stimulation of neurons with ionomycin for 2.5 min selectively increased HEA and DEA production (Fig. 5a–d), whereas it had no effect on AEA and 2-AG production, which is consistent with reports showing that only longer applications of ionomycin (e.g., 20 min) increase AEA and 2-AG production (Di Marzo et al., 1994; Stella et al., 1997). Glutamate plus carbachol applied for 2.5 min dramatically increased the production of 2-AG, modestly increased the production of AEA and DEA, and did not affect HEA production (Fig. 5a–d). These results show that the ionomycin-induced increase in intracellular calcium or the pathological stimulation of neurons enhances the production of different endocannabinoids, with 2-AG being the most abundant endocannabinoid produced under pathological conditions.
Fig. 5.

Mouse neurons and microglial cells produce four endocannabinoids. a–d, Mouse neurons were incubated with ionomycin (iono; 5 μm) for 2.5 and 5 min or with glutamate (Glut; 100 μm) and carbachol (Carb; 1 mm) for 2.5 min, lipids were extracted from chloroform from cells plus adjacent media and purified by HPLC; AEA (a), 2-AG (2AG; b), HEA (c), and DEA (d) were then quantified by CI-GC/MS.e–h, BV-2 cells were incubated with ionomycin (5 μm) for increasing periods of time, and endocannabinoids were quantified. prot, Protein. The arrowindicates that EGTA (5 mm) prevents the ionomycin response.i–l, Mouse microglial cells were incubated with ionomycin (5 μm) for 2.5 and 5 min or with ATP (1 mm) for 10 min, and endocannabinoids were quantified. Values are means ± SEM of 6–32 independent endocannabinoid quantifications, each performed on one 100 mm dish of cells. *p < 0.05; **p < 0.01; significantly different from basal amount (see Table 1) (ANOVA followed by Dunnett's post hoc test).
We then determined whether microglial cells produce endocannabinoids, because (1) this has never been addressed directly, (2) it is known that macrophages produce these ligands in a calcium-dependent manner (Wagner et al., 1997; Schmid et al., 2000), and (3) endocannabinoid produced by microglial cells could participate in recruiting more distant microglial cells toward dying neurons. Ionomycin increased 2-AG and DEA production from BV-2 cells, whereas AEA and HEA production was unaffected (Fig. 5a–d). Chelation of extracellular calcium with EGTA prevented this response. We found that under basal conditions mouse microglial cells in culture contained higher amounts of endocannabinoids than neurons in culture (Table2). Like BV-2 cells, ionomycin increased the production of 2-AG and DEA from mouse microglial cells without affecting AEA and HEA (Fig. 5e–h). It is important to note that ATP selectively increased 2-AG production from mouse microglial cells (Fig. 5e–h). These results show that the production of 2-AG and DEA from microglial cells can be increased, although only 2-AG production is increased by a pathological stimulus.
Table 2.
Endocannabinoid contents in primary cultures of mouse neurons and microglial cells under basal conditions
| Endocannabinoid content | ||
|---|---|---|
| Neurons (pmol/mg protein) | Microglial cells (pmol/mg protein) | |
| AEA | 0.7 ± 0.2 | 13.6 ± 4.3 |
| 2-AG | 0.4 ± 0.1 | 8.2 ± 2.0 |
| HEA | 0.2 ± 0.1 | 1.7 ± 0.4 |
| DEA | 0.4 ± 0.1 | 4.1 ± 1.1 |
Mouse neurons and microglial cells cultured in 100 mm dishes contained 4.15 ± 0.2 and 0.55 ± 0.04 mg of protein per dish, respectively (n = 14). Cells were analyzed under basal conditions (i.e., in cell culture media); endocannabinoids were extracted from cells plus adjacent media, purified by HPLC, and quantified by CI-GC/MS. Values are mean ± SEM of 14 independent quantifications of endocannabinoids, each performed on one 100 mm dish of cells.
Finally, we determined whether endocannabinoids produced by microglial cells are released, because a paracrine function for these lipids has been suggested in neuronal signaling (Bisogno et al., 1997; Giuffrida et al., 1999; Kreitzer and Regehr, 2001; Wilson and Nicoll, 2001). To do this, we used BV-2 cells that either were kept under basal condition or were stimulated with ionomycin for 5 min and assessed the relative amount of endocannabinoids present in (1) cells plus adjacent media and (2) distant media. We found that 60% of total AEA and 54% of total HEA was released toward distant media under both basal and ionomycin-stimulated conditions (n = 10 independent endocannabinoid quantifications, each performed on cells contained in one 100 mm dish). Under basal conditions, 2-AG was undetectable in cells plus adjacent media, as well as in distant media, whereas after ionomycin stimulation, 36% of 2-AG was released toward distant media. It is interesting to note that under basal conditions, 59% of DEA was released toward distant media, whereas after ionomycin stimulation, only 34% of DEA was released toward distant media. These results indicate that a portion of endocannabinoids produced by microglial cells is released from cells toward distant media, reinforcing the notion that these lipids serve a paracrine signaling function. Additionally, our results indicate that the relative amount of endocannabinoid that is released toward distant media varies depending on endocannabinoid subtype and whether cells are acutely stimulated.
Discussion
Many of the signaling molecules in the body (e.g., eicosanoids) are present not as single entities but as large families of structurally related substances. In 1992, when Devane et al. identified AEA as a ligand for CB1 receptors, it seemed reasonable to expect that this lipid was only the first representative of a larger family of endocannabinoids (Devane et al., 1992). Indeed, ensuing studies identified a second endocannabinoid, 2-AG, that acts as a full agonist on CB1 and CB2 receptors (Mechoulam et al., 1995; Sugiura et al., 1995,2000; Stella et al., 1997). In this study, we show that neurons and microglial cells produce four endocannabinoids: the two well known endocannabinoids, AEA and 2-AG, as well as the two new endocannabinoids, HEA and DEA. It is interesting to note that astrocytes in culture also produce these four endocannabinoids (Walter et al., 2002) (L. Walter and N. Stella, unpublished observations). Thus, the endocannabinoid family contains several structurally related lipids that are produced in the CNS by neurons, microglial cells, and astrocytes. It should be emphasized that microglial cells produce ∼20-fold higher amounts of endocannabinoids (expressed in picomoles per nanogram of protein) compared with neurons and astrocytes. On the basis of these data, we propose that, during neuroinflammation, activated microglial cells produce the majority of endocannabinoids that accumulate at the lesion site. A recent study identified yet an additional endocannabinoid in the brain (i.e., noladin) that is an analog of 2-AG that acts on CB1 receptors (Hanus et al., 2001). However, studies that address the cellular source of this lipid, as well as its pathway of biosynthesis, remain to be performed.
The presence of a large family of endocannabinoids leaves open many questions. What are the physiological and pathological stimuli that increase endocannabinoid production? Can different stimuli selectively increase the production of different endocannabinoids? Does each endocannabinoid have distinct biological functions? We show that glutamate plus carbachol stimulation dramatically increases 2-AG production from neurons. Under these conditions, AEA and DEA production is increased only slightly, whereas HEA production is unchanged. It is important to note that HEA and DEA production from neurons is increased by acute application of ionomycin, which shows that the production of these two endocannabinoid candidates is indeed calcium dependent. This result reinforces the notion that different pathways of biosynthesis of endocannabinoids exist in neurons, and that their selective production is driven by specific stimuli (Stella et al., 1997; Giuffrida et al., 1999; Stella and Piomelli, 2001). The same idea holds true for microglial cells and astrocytes. ATP increases the production of 2-AG from microglial cells without affecting the production of AEA, HEA, or DEA. We have shown recently that, in cultured astrocytes, endothelin, a peptide produced by endothelial cells during stroke, increases 2-AG production fivefold and AEA production twofold, whereas it does not affect HEA and DEA production (Walter et al., 2002) (Walter and Stella, unpublished observations). In summary, 2-AG production is dramatically increased by three pathological stimuli that affect neurons, microglial cells, or astrocytes, a finding that corroborates the observed increase in this particular endocannabinoid during neuroinflammation inducedin vivo (Baker et al., 2001; Panikashvili et al., 2001).
We present evidence that 2-AG recruits microglial cells by engaging CB2 and abn-CBD receptors. To our knowledge, this is the first study to report that 2-AG is a full agonist at abn-CBD receptors. We ruled out the involvement of CB1 receptors in the 2-AG-triggered microglial cell migration because this response was not antagonized by 30 nm SR141716A, a concentration that antagonizes CB1 receptors (IC50 = 5 nm) without affecting abn-CBD receptors (IC50 = 600 nm) (Jung et al., 1997; Bukoski et al., 2002). Additionally, we show that THC and abnormal-cannabidiol induce a synergistic increase in microglial cell migration. Although the molecular basis of this synergistic response remains unknown, our results show that engagement of both CB2 and abn-CBD receptors is required to trigger microglial cell migration.
Although both CB1 and CB2 receptors are expressed in activated microglial cells, their cellular location is quite different. This suggests that the functionality of either receptor might be regulated by translocation to the plasma membrane. Changes in the cellular location of cannabinoid receptors that are associated with a change in their functionality have been described with CB2 receptors in transfected cells (Bouaboula et al., 1999). The dynamics of cannabinoid receptor movement between cellular compartments during microglial cell activation, for example, during migration, is a key question. Indeed, studies performed with different G-protein-coupled receptors, namely the complement C5a receptors, propose that these types of receptors accumulate at the leading edges of lamellipodia as a result of plasma membrane accumulation (Servant et al., 1999). Using a CB2 receptor antibody, we show that CB2 receptors are abundant at the leading edges of activated microglial cells, as well as at the microspikes of BV-2 cells. Whether accumulation of CB2 receptors at the leading edges of these protrusions is caused by accumulation of membranes or by a true increase in receptor density is unknown.
Because we found that 2-AG triggers microglial cell migration with high potency and efficacy, we hypothesize that this particular endocannabinoid participates in recruiting microglial cells toward neuroinflammatory lesion sites. The following model can be put forward. First, pathological overstimulation of neurons, which eventually results in their death, increases 2-AG production and recruits surrounding microglial cells. Once these microglial cells arrive at the lesion site, ATP, which is released by dying neurons, acts on the headmost microglial cells, further increasing local 2-AG and further recruiting microglial cells. Such a model is reminiscent of many feedforward mechanisms that lead to local recruitment of immune cells and amplification of inflammation. We also show that CBN and CBD, two nonpsychotropic bioactive compounds of marijuana (Perez-Reyes et al., 1973), may antagonize the 2-AG-induced recruitment of microglial cells. This is in agreement with the fact that nabilone, a synthetic analog of THC, produces minimal palliative effects against multiple sclerosis symptoms, whereas smoking cannabis is reported to be beneficial (Martyn et al., 1995; Consroe et al., 1997). Therefore, our results suggest that bioactive cannabinoids present in the marijuana plant, such as CBN and CBD, are likely to underlie the increased efficacy of cannabis versus nabilone and therefore hold promise as nonpsychotropic therapeutics to treat neuroinflammation.
Footnotes
This work was supported by National Institute on Drug Abuse Grants DA14486 (N.S.) and DA11322 and DA00286 (K.M.), a National Multiple Sclerosis Society grant (N.S.), and National Institute of General Medical Sciences and the Deutsche Forschungs Gemeinschaft fellowship awards (L.W., A.W.). We thank Sanofi Research for providing SR141716A and SR144528, Dr. Thomas Möller (University of Washington) for help with RT-PCR, and Dr. Elisabetta Blasi (University of Perugia, Italy) for BV-2 cells.
Correspondence should be addressed to Nephi Stella, Department of Pharmacology, University of Washington, 1959 Northeast Pacific Street, Seattle, WA 98195-7280. E-mail: nstella@u.washington.edu.
References
- 1.Baker D, Pryce G, Croxford LJ, Brown P, Pertwee RG, Huffman JW, Layward L. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature. 2000;404:84–87. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- 2.Baker D, Pryce G, Croxford LJ, Brown P, Pertwee RG, Makriyannis A, Knanolkar A, Layward L, Fezza F, Bisogno T, DiMarzo V. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15:300–302. doi: 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- 3.Becher B, Antel JP. Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia. 1996;18:1–10. doi: 10.1002/(SICI)1098-1136(199609)18:1<1::AID-GLIA1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Becher B, Prat A, Antel JP. Brain-immune connection: immuno-regulatory properties of CNS-resident cells. Glia. 2000;29:293–304. [PubMed] [Google Scholar]
- 5.Bisogno T, Sepe N, Melck D, Maurelli S, De Petrocellis L, Di Marzo V. Biosynthesis, release and degradation of the novel endogenous cannabimimetic metabolite 2-arachidonoylglycerol in mouse neuroblastoma cells. Biochem J. 1997;322:671–677. doi: 10.1042/bj3220671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouaboula M, Poinot-Chazel C, Marchand J, Canat X, Bourrié B, Rinaldi-Carmona M, Calandra B, Le Fur G, Casellas P. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Eur J Biochem. 1996;237:704–711. doi: 10.1111/j.1432-1033.1996.0704p.x. [DOI] [PubMed] [Google Scholar]
- 7.Bouaboula M, Dussossoy D, Casellas P. Regulation of peripheral cannabinoid receptor CB2 phosphorylation by the inverse agonist SR144528. J Biol Chem. 1999;274:20397–20405. doi: 10.1074/jbc.274.29.20397. [DOI] [PubMed] [Google Scholar]
- 8.Bruce-Keller AJ. Microglial-neuronal interactions in synaptic damage and recovery. J Neurosci Res. 1999;58:191–201. doi: 10.1002/(sici)1097-4547(19991001)58:1<191::aid-jnr17>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 9.Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M, Zimmer A. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB2 receptor. Eur J Pharmacol. 2000;396:141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- 10.Bukoski RD, Bátkai S, Járai Z, Wang Y, Offertaler L, Jackson WF, Kunos G. CB1 receptor antagonist SR141617A inhibits Ca2+-induced relaxation in CB1 receptor-deficient mice. Hypotension. 2002;39:251–257. doi: 10.1161/hy0202.102702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Consroe P, Musty R, Rein J, Tillery W, Pertwee R. The perceived effects of smoked cannabis on patients with multiple sclerosis. Eur Neurol. 1997;38:44–48. doi: 10.1159/000112901. [DOI] [PubMed] [Google Scholar]
- 12.Derocq J-M, Jbilo O, Bouaboula M, Ségui M, Clère C, Casellas P. Genomic and functional changes induced by the activation of the peripheral cannabinoid receptor CB2 in the promyelocytic cells HL-60. J Biol Chem. 2000;275:15621–15628. doi: 10.1074/jbc.275.21.15621. [DOI] [PubMed] [Google Scholar]
- 13.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 14.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 15.Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K, Devane WA. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc Natl Acad Sci USA. 1993;90:7656–7660. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- 17.Giuffrida A, Parsons LH, Kerr TM, Rodríguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signalling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- 18.Goutopoulos A, Fan P, Khanolkar AD, Xie X-Q, Lin S, Makriyannis A. Stereochemical selectivity of methanandamides for the CB1 and CB2 cannabinoid receptors and their metabolic stability. Bioorg Med Chem. 2001;9:1673–1684. doi: 10.1016/s0968-0896(01)00088-8. [DOI] [PubMed] [Google Scholar]
- 19.Griffin G, Tao Q, Abood ME. Cloning and pharmacological characterization of the rat CB2 cannabinoid receptor. J Pharmacol Exp Ther. 2000;292:886–894. [PubMed] [Google Scholar]
- 20.Hájos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 21.Hall W, Solowij N. Adverse effects of cannabis. Lancet. 1998;352:1611–1616. doi: 10.1016/S0140-6736(98)05021-1. [DOI] [PubMed] [Google Scholar]
- 22.Hampson RE, Deadwyler SA. Cannabinoids reveal the necessity of hippocampal neural encoding for short-term memory in rats. J Neurosci. 2000;20:8932–8942. doi: 10.1523/JNEUROSCI.20-23-08932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanus L, Gopher A, Almog S, Mechoulam R. Two new unsaturated fatty acid ethanolamides in brain that bind to the cannabinoid receptor. J Med Chem. 1993;36:3032–3034. doi: 10.1021/jm00072a026. [DOI] [PubMed] [Google Scholar]
- 24.Hanus L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, Kustanovich I, Mechoulam R. 2-Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci USA. 2001;98:3662–3665. doi: 10.1073/pnas.061029898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 26.Huestis MA, Gorelick DA, Heishman SJ, Preston KL, Nelson RA, Moolchan ET, Frank RA. Blockade of effects of smoked marijuana by the CBl-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiatry. 2001;58:322–328. doi: 10.1001/archpsyc.58.4.322. [DOI] [PubMed] [Google Scholar]
- 27.Járai Z, Wagner JA, Varga K, Lake KD, Compton DR, Martin BR, Zimmer AM, Bonner TI, Buckley NE, Mezey E, Razdan RK, Zimmer A, Kunos G. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc Natl Acad Sci USA. 1999;96:14136–14141. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung M, Calassi R, Rinaldi-Carmona M, Chardenot P, Le Fur G, Soubrié P, Oury-Donat F. Characterization of CB1 receptors on rat neuronal cell cultures: binding and functional studies using the selective receptor antagonist SR 141716A. J Neurochem. 1997;68:402–409. doi: 10.1046/j.1471-4159.1997.68010402.x. [DOI] [PubMed] [Google Scholar]
- 29.Kaminski NE. Regulation of the cAMP cascade, gene expression and immune function by cannabinoid receptors. J Neuroimmunol. 1998;83:124–132. doi: 10.1016/s0165-5728(97)00228-2. [DOI] [PubMed] [Google Scholar]
- 30.Kim M, Spelta V, Sim J, North AR, Surprenant A. Differential assembly of rat purinergic P2X7 receptor in immune cells of the brain and periphery. J Biol Chem. 2001;276:23262–23267. doi: 10.1074/jbc.M102253200. [DOI] [PubMed] [Google Scholar]
- 31.Klein TW, Friedman H, Specter S. Marijuana, immunity and infection. J Neuroimmunol. 1998;83:102–115. doi: 10.1016/s0165-5728(97)00226-9. [DOI] [PubMed] [Google Scholar]
- 32.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 34.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 35.Lee SF, Newton C, Widen R, Friedman H, Klein TW. Differential expression of cannabinoid CB2 receptor mRNA in mouse immune cell subpopulation and following B cell stimulation. Eur J Pharmacol. 2001;423:235–241. doi: 10.1016/s0014-2999(01)01122-0. [DOI] [PubMed] [Google Scholar]
- 36.Lyman WD, Sonett JR, Brosnan CF, Elkin R, Bornstein MB. Δ9-Tetrahydrocannabinol: a novel treatment for experimental autoimmune encephalomyelitis. J Neuroimmunol. 1989;23:73–81. doi: 10.1016/0165-5728(89)90075-1. [DOI] [PubMed] [Google Scholar]
- 37.Malfait AM, Gallily R, Sumariwalla PF, Malik AS, Andreakos E, Mechoulam R, Feldmann M. The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 2000;97:9561–9566. doi: 10.1073/pnas.160105897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mallet PE, Beninger RJ. The cannabinoid CB1 receptor antagonist SR141716A attenuates the memory impairment produced by Δ9-tetrahydrocannabinol or anandamide. Psychopharmacology. 1998;140:11–19. doi: 10.1007/s002130050733. [DOI] [PubMed] [Google Scholar]
- 39.Martyn CN, Illis LS, Thom J. Nabilone in the treatment of multiple sclerosis. Lancet. 1995;345:579. doi: 10.1016/s0140-6736(95)90485-9. [DOI] [PubMed] [Google Scholar]
- 40.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 41.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 42.Meinck H-M, Schönle PW, Conrad B. Effect of cannabinoids on spasticity and ataxia in multiple sclerosis. J Neurol. 1989;236:120–122. doi: 10.1007/BF00314410. [DOI] [PubMed] [Google Scholar]
- 43.Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 44.Möller T, Kann O, Verkhratsky A, Kettenmann H. Activation of mouse microglial cells affects P2 receptor signaling. Brain Res. 2000;853:49–59. doi: 10.1016/s0006-8993(99)02244-1. [DOI] [PubMed] [Google Scholar]
- 45.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 46.Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 47.Perez-Reyes M, Timmons MC, Davis KH, Wall ME. A comparison of the pharmacological activity in man of intravenously administered Δ9-tetrahydrocannabinol, cannabinol, and cannabidiol. Experientia (Basel) 1973;29:1368–1369. doi: 10.1007/BF01922823. [DOI] [PubMed] [Google Scholar]
- 48.Pertwee R, Griffen G, Hanus L, Mechoulam R. Effects of two fatty acid ethanoloamides on mouse vasa deferentia. Eur J Pharmacol. 1994;259:115–120. doi: 10.1016/0014-2999(94)90499-5. [DOI] [PubMed] [Google Scholar]
- 49.Piomelli D, Beltramo M, Giuffrida A, Stella N. Endogenous cannabinoid signaling. Neurobiol Dis. 1998;5:462–473. doi: 10.1006/nbdi.1998.0221. [DOI] [PubMed] [Google Scholar]
- 50.Rezaie P, Male D. Colonisation of the developing human brain and spinal cord by microglia: a review. Microscopy Res Tech. 1999;45:359–382. doi: 10.1002/(SICI)1097-0029(19990615)45:6<359::AID-JEMT4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 51.Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D, Ferrara P, Soubrié P, Brelière JC, Le Fur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- 52.Rinaldi-Carmona M, Barth F, Millan J, Derocq J-M, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, Portier M, Shire D, Brelière J-C, Le Fur G. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- 53.Rodríguez JJ, Mackie K, Pickel VM. Ultrastructural localization of the CB1 cannabinoid receptor in μ-opioid receptor patches of the rat caudate putamen nucleus. J Neurosci. 2001;21:823–833. doi: 10.1523/JNEUROSCI.21-03-00823.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmid PC, Schwartz KD, Smith CN, Krebsbach RJ, Berdyshev EV, Schmid HHO. A sensitive endocannabinoid assay: the simultaneous analysis of N-acylethanolamines and 2-monoacylglycerols. Chem Phys Lipids. 2000;104:185–191. doi: 10.1016/s0009-3084(99)00124-3. [DOI] [PubMed] [Google Scholar]
- 55.Schwartzfarb L, Needle M, Chavez-Chase M. Dose-related inhibition of leukocyte migration by marijuana and delta-9-tetrahydrocannabinol (THC) in vitro. J Clin Pharmacol. 1974;1:35–41. doi: 10.1002/j.1552-4604.1974.tb02285.x. [DOI] [PubMed] [Google Scholar]
- 56.Servant G, Weiner OD, Neptune ER, Sedat JW, Bourne HR. Dynamics of a chemoattractant receptor in living neutrophils during chemotaxis. Mol Biol Cell. 1999;10:1163–1178. doi: 10.1091/mbc.10.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278:989–999. [PubMed] [Google Scholar]
- 58.Solowij N, Stephens RS, Roffman RA, Babor T, Kadden R, Miller M, Christiansen K, McRee B, Vendetti J. Cognitive functioning of long-term heavy cannabis users seeking treatment. JAMA. 2002;287:1123–1131. doi: 10.1001/jama.287.9.1123. [DOI] [PubMed] [Google Scholar]
- 59.Stella N, Piomelli D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol. 2001;425:189–196. doi: 10.1016/s0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- 60.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 61.Stence N, Waite M, Dailey ME. Dynamics of microglial activation. Glia. 2001;33:256–266. [PubMed] [Google Scholar]
- 62.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 63.Sugiura T, Kondo S, Kishimoto S, Miyashita T, Nakane S, Kodaka T, Suhara Y, Takayama H, Waku K. Evidence that 2-arachidonylglycerol but not N-palmitpylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. J Biol Chem. 2000;275:605–612. doi: 10.1074/jbc.275.1.605. [DOI] [PubMed] [Google Scholar]
- 64.Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 65.Wade CB, Robinson S, Shapiro R, Dorsa DM. Estrogen receptor (ER)a and ERb exhibit unique pharmacological properties when coupled to activation of the mitogen-activated protein kinase pathway. Endocrinology. 2001;142:2336–2342. doi: 10.1210/endo.142.6.8071. [DOI] [PubMed] [Google Scholar]
- 66.Wagner JA, Varga K, Ellis EF, Rzigalinski BA, Martin BR, Kunos G. Activation of peripheral CB1 cannabinoid receptors in haemorrhagic shock. Nature. 1997;390:518–521. doi: 10.1038/37371. [DOI] [PubMed] [Google Scholar]
- 67.Waksman Y, Olson JM, Carlisle SJ, Cabral GY. The central cannabinoid receptor (CB1) mediates inhibition of nitric oxide production by rat microglial cells. J Pharmacol Exp Ther. 1999;288:1357–1366. [PubMed] [Google Scholar]
- 68.Walter L, Franklin A, Witting A, Möller T, Stella N. Astrocytes in culture produce anandamide and other acylethanolamides. J Biol Chem. 2002;277:20869–20876. doi: 10.1074/jbc.M110813200. [DOI] [PubMed] [Google Scholar]
- 69.Watanabe N, Mitchison TJ. Single-molecule speckle analysis of actine filament turnover in lamellipodia. Science. 2002;295:1083–1087. doi: 10.1126/science.1067470. [DOI] [PubMed] [Google Scholar]
- 70.Watson SJ, Benson JA, Joy JE. Marijuana and medicine: assessing the science base. Arch Gen Psychiatry. 2000;57:547–552. doi: 10.1001/archpsyc.57.6.547. [DOI] [PubMed] [Google Scholar]
- 71.Wilkinson PC. Assays of leucocyte locomotion and chemotaxis. J Immunol Methods. 1998;216:139–153. doi: 10.1016/s0022-1759(98)00075-1. [DOI] [PubMed] [Google Scholar]
- 72.Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 73.Wirguin I, Mechoulam R, Breuer A, Schezen E, Weidenfeld J, Brenner T. Suppression of experimental autoimmune encephalomyelitis by cannabinoids. Immunopharmacology. 1994;28:209–214. doi: 10.1016/0162-3109(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 74.Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]