

Abstract
Parkinson's disease (PD) is characterized by a progressive degeneration of the nigrostriatal dopaminergic pathway resulting in movement disorders. Although its etiology remains unknown, PD may be the final outcome of interactions among multiple factors, including exposure to environmental toxins and the occurrence of inflammation in the brain. In this study, using primary mesencephalic cultures, we observed that nontoxic or minimally toxic concentrations of the pesticide rotenone (0.5 nm) and the inflammogen lipopolysaccharide (LPS) (0.5 ng/ml) synergistically induced dopaminergic neurodegeneration. The synergistic neurotoxicity of rotenone and LPS was observed when the two agents were applied either simultaneously or in tandem. Mechanistically, microglial NADPH oxidase-mediated generation of reactive oxygen species appeared to be a key contributor to the synergistic dopaminergic neurotoxicity. This conclusion was based on the following observations. First, inhibition of NADPH oxidase or scavenging of free radicals afforded significant neuroprotection. Second, rotenone and LPS synergistically stimulated the NADPH oxidase-mediated release of the superoxide free radical. Third and most importantly, rotenone and LPS failed to induce the synergistic neurotoxicity as well as the production of superoxide in cultures from NADPH oxidase-deficient animals. This is the first demonstration that low concentrations of a pesticide and an inflammogen work in synergy to induce a selective degeneration of dopaminergic neurons. Findings from this study may be highly relevant to the elucidation of the multifactorial etiology of PD and the discovery of effective therapeutic agents for the treatment of the disease.
Keywords: pesticide, inflammation, microglia, NADPH oxidase, Parkinson's disease, synergistic neurotoxicity
Introduction
The cause of sporadic Parkinson's disease (PD), which accounts for >90% of the incidence of PD, remains unknown. Increasing evidence indicates that PD may represent the final outcome of a complex set of interactions, over decades of time, among factors that include genetic predisposition, innate characteristics of the nigrostriatal dopaminergic system of the brain, and exposure to environmental toxins (Olanow and Tatton, 1999; Kidd, 2000).
Among the various environmental factors suspected to play a role in the pathogenesis of PD, exposure to agrochemicals has been most intensely investigated (Gorell et al., 1998; Ritz and Yu, 2000; Herishanu et al., 2001; Priyadarshi et al., 2001). A number of groups have reported that administration of herbicides such as rotenone and paraquat induces parkinsonism in rodents (Betarbet et al., 2000; Thiruchelvam et al., 2000). Mechanistically, the degeneration of nigral dopaminergic neurons induced by rotenone may not be solely the result of an impairment of neuronal mitochondrial complex I activity but may also involve the participation of the resident immune cells of the brain, microglia (Gao et al., 2002a).
Microglial activation is a hallmark of the pathogenesis of a number of neurodegenerative diseases, including PD and Alzheimer's disease (McGeer et al., 1988; Hauss-Wegrzyniak et al., 1998; Gonzalez-Scarano and Baltuch, 1999; Liu and Hong, 2003). Historically, postmortem analysis of the nigra of PD patients has frequently detected markers for microglial activation, accumulation of proinflammatory cytokines, and footprints for oxidative stress (McGeer et al., 1988). Under physiological conditions, microglia perform the role of immune surveillance (Kreutzberg, 1996). In response to immunological challenges such as invading pathogens and neuronal injuries, microglia readily become activated. Activated microglia produce a wide array of proinflammatory and cytotoxic factors, including cytokines, free radicals, and eicosanoids, which work in concert to induce neurodegeneration (Liu et al., 2002). Within the spectrum of neurotoxic factors produced by activated microglia, reactive oxygen species (ROS) appear to be a key effector of dopaminergic neurodegeneration attributable to the known vulnerability of dopaminergic neurons to oxidative stress as a consequence of reduced antioxidant capacity, high content of iron and dopamine, and possible defect in mitochondrial function (Jenner and Olanow, 1998). Superoxide free radical, released by activated microglia through the multi-subunit and membrane-bound NADPH oxidase, together with other microglia-originated factors, such as nitric oxide (NO), tumor necrosis factor-α (TNFα), and interleukin-1-β (IL-1β), contribute significantly to dopaminergic neurodegeneration (Liu et al., 2000; Gao et al., 2002b). In fact, cerebral inflammation sustained by microglial activation triggered by the inflammogen lipopolysaccharide (LPS) reproduces the delayed and progressive degenerative feature of nigral dopaminergic neurons in PD (Gao et al., 2002b).
In this study, we investigated the effect of exposure to a pesticide (i.e., rotenone) and an inflammogen (i.e., LPS) on the degeneration of dopaminergic neurons using primary mesencephalic neuron–glia cultures as a chronic in vitro model of PD. We report here that rotenone and LPS, at concentrations that were nontoxic or minimally toxic when applied alone, worked in synergy to induce the degeneration of dopaminergic neurons. Results obtained from studies using cultures from NADPH oxidase-null and wild-type mice demonstrated that the release of oxygen free radicals from rotenone and LPS-stimulated microglia appeared to be a key contributor to the dopaminergic neurotoxicity.
Materials and Methods
Animals. NADPH oxidase-deficient (gp91phox−/−) and wild-type C57BL/6J (gp91phox+/+) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Breeding of the mice was performed to achieve timed pregnancy with the accuracy of ±0.5 d. Timed-pregnant Fisher F344 rats were obtained from Charles River Laboratories (Raleigh, NC). Housing and breeding of the animals were performed in strict accordance with the National Institutes of Health guidelines.
Primary mesencephalic neuron–glia cultures. Neuron–glia cultures were prepared from the ventral mesencephalic tissues of embryonic day 13–14 rats or day 12–13 mice, as described previously (Liu et al., 2000; Gao et al., 2002a). Briefly, dissociated cells were seeded at 1 × 105/well and 5 × 105/well to poly-d-lysine-coated 96-well and 24-well plates, respectively. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air, in minimal essential medium (MEM) containing 10% fetal bovine serum (FBS), 10% horse serum (HS), 1 gm/l glucose, 2 mml-glutamine, 1 mm sodium pyruvate, 100 μm nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. Seven-day-old cultures were used for treatment. At the time of treatment, immunocytochemical analysis indicated that the rat neuron–glia cultures were made up of 11% microglia, 48% astrocytes, 41% neurons, and 1% tyrosine hydroxylase-immunoreactive (TH-IR) neurons. The composition of the neuron–glia cultures of NADPH oxidase-deficient mice was very similar to that of the wild-type mice in that there were 12% microglia, 48% astrocytes, 40% neurons, and 1% TH-IR neurons.
Primary rat mesencephalic neuron-enriched cultures.Neuron-enriched cultures were prepared from the ventral mesencephalic tissues of embryonic day 13–14 rats as described previously (Gao et al., 2002a). Briefly, dissociated cells were seeded at 1 × 105/well and 5 × 105/well to poly-d-lysine-coated 96-well and 24-well plates, respectively. Glial proliferation was suppressed by the inclusion of cytosine β-d-arabinocide (5–10 μm). Used for treatment were 7-d-old cultures, which were composed of 91% neurons, 9% astrocytes, and <0.1% microglia.
Primary microglia-enriched cultures. Microglia were prepared from the whole brains of 1-d-old rats or NADPH oxidase-deficient or wild-type mice, as described previously (B. Liu et al., 2001). Immunocytochemical analysis indicated that the cultures were 95–98% pure for microglia. Cells were seeded at 1 × 105/well in 96-well plates and used for treatment the following day.
Assessment of neurotoxicity. For treatment, cultures were switched to treatment medium consisting of MEM, 2% FBS, 2% HS, 2 mml-glutamine, 1 mm sodium pyruvate, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were treated with vehicle, LPS (E scherichia coli 0111:B4; Calbiochem, San Diego, CA), and/or rotenone (Calbiochem) as described previously (Gao et al., 2002a,b). Degeneration of dopaminergic neurons in the cultures was analyzed by using several parameters. Uptake of [3H]dopamine (DA) was determined by incubation of cultures for 15 min at 37°C with 1 μm [3H]DA (30 Ci/mmol; NEN, Boston, MA) as described previously (Gao et al., 2002a). Nonspecific uptake was determined in the presence of 10 μm mazindol. The number and the average dendrite length of TH-IR neurons were determined as described previously (Gao et al., 2002a). The specificity of neurotoxicity was analyzed by comparing the uptake capacity of cultures for [3H]DA and [3H]GABA and by double-label immunostaining with the anti-TH antibody and an antibody against the neuron-specific nuclear protein (NeuN) as described previously (Gao et al., 2002a).
Measurement of superoxide release. The release of superoxide was determined by measuring the superoxide dismutase (SOD)-inhibitable reduction of cytochrome c as described previously (Liu et al., 2000; Gao et al., 2002a). To measure the immediate release of superoxide from microglia-enriched, neuron–glia, or neuron-enriched cultures after stimulation, cultures grown in 96-well plates were switched to phenol red-free HBSS (100 μl/well). To each well was added 50 μl of HBSS containing vehicle, rotenone, and/or LPS, followed by 50 μl of ferricytochrome c (100 μm) in HBSS, with and without 600 U/ml SOD. The cultures were then incubated at 37°C for 30 min, and the absorbance at 550 nm was read with a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). To determine the effect of NADPH oxidase inhibitors [diphenylene iodonium (DPI) or apocynin] on superoxide release, microglia were pretreated for 30 min at 37°C with vehicle, DPI, or apocynin in HBSS (100 μl/well) before stimulation with rotenone and/or LPS. To measure the levels of superoxide in mouse neuron–glia cultures at extended time points after treatment, cultures grown in 96-well plates were treated with vehicle, rotenone, and/or LPS in treatment medium containing phenol red-free MEM (150 μl/well). Four days later, to each well was added 50 μl of ferricytochrome c (100 μm) in phenol red-free MEM, with and without 600 U/ml SOD. Thirty minutes after the addition of cytochrome c, the absorbance at 550 nm was read.
Assay of intracellular ROS.5-(and-6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2-DCFDA) (Molecular Probes, Eugene, OR), a chloromethyl derivative of H2-DCFDA, passively diffuses into cells in which it is hydrolyzed by intracellular esterases to liberate 2′-7′-dichlorofluoresein which, during reaction with oxidizing species, yields a highly fluorescent compound 2′,7′-dichlorofluorescein (DCF) that is trapped inside the cell (S.X. Liu et al., 2001). For each measurement, a fresh stock solution of CM-H2-DCFDA (5 mm) was prepared in dimethylsulfoxide. CM-H2-DCFDA, diluted to a final concentration of 1 μm in phenol red-free HBSS containing 2% FBS and 2% HS, was added to cultures and incubated for 30 min at 37°C. After washing two times with warm HBSS, vehicle or stimulators in HBSS were added to cultures. After incubation for 30 min at 37°C, fluorescence intensity was measured at 485 nm for excitation and 530 nm for emission using a SpectraMax Gemini XS fluorescence microplate reader (Molecular Devices).
Nitrite and TNFα assays. The production of NO was determined by measuring the accumulated levels of nitrite in the supernatant with the Griess reagent that had a detection limit of 0.5 μm. The amount of TNFα released into the medium was measured with a rat TNFα enzyme-linked immunosorbent assay kit (detection limit, 5 pg/ml; R & D Systems, Minneapolis, MN).
Statistical analysis. Statistical significance was determined using an ANOVA, followed by the Bonferroni'st test using the StatView program (Abacus Concepts, Berkeley, CA). A value of p < 0.05 was considered statistically significant.
Results
Rotenone and LPS synergistically induce a selective degeneration of dopaminergic neurons
To determine the combined neurotoxicity of rotenone and LPS, a nontoxic or minimally toxic concentration of rotenone (0.5 nm) or LPS (0.5 ng/ml) was selected based on our previous reports (Gao et al., 2002a,b). Primary rat mesencephalic neuron–glia cultures were treated with vehicle, 0.5 nm rotenone, 0.5 ng/ml LPS, or a combination of 0.5 nm rotenone and 0.5 ng/ml LPS. Eight days later, the degeneration of dopaminergic neurons was assessed by determining [3H]DA uptake, counting TH-IR neurons, and measuring TH-IR dendrites. As shown in Figure 1A, treatment for 8 d with 0.5 nm rotenone alone did not cause a significant reduction in DA uptake (5%) or loss of TH-IR neurons (2%) compared with vehicle-treated control cultures, consistent with our previous report (Gao et al., 2000a). Treatment with 0.5 ng/ml LPS alone resulted in a significant reduction in DA uptake (25%; p < 0.05 compared with control cultures) and an insignificant loss of TH-IR neurons (9%), similar to that reported previously (Gao et al., 2002b). In contrast, the reduction in DA uptake in cultures treated with the combination of 0.5 nm rotenone and 0.5 ng/ml LPS reached 60%, which was twice the sum of the reduction induced by 0.5 nm rotenone alone (5%) and by 0.5 ng/ml LPS alone (25%). Similarly, the loss of TH-IR neurons induced by 0.5 nm rotenone and 0.5 ng/ml LPS was 48%, which was four times the sum of that induced by 0.5 nmrotenone alone (2%) and by 0.5 ng/ml LPS alone (9%). In addition, the average length of TH-IR dendrites in cultures treated for 8 d with 0.5 nm rotenone and 0.5 ng/ml LPS was shortened by 60 ± 5% (n = 3) compared with control cultures. Cultures treated with 0.5 nm rotenone or 0.5 ng/ml LPS alone exhibited a 2 ± 2 or 19 ± 4% (n = 3) decrease in the average length of TH-IR dendrites.
Fig. 1.
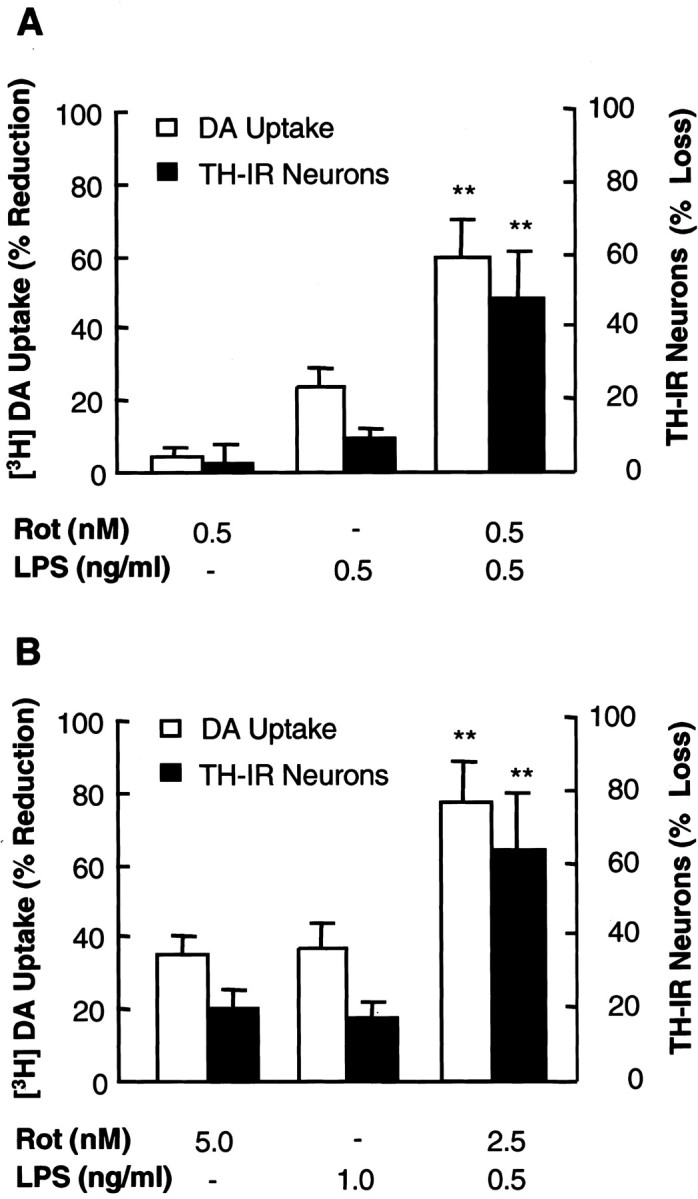
Rotenone and LPS induce synergistic degeneration of dopaminergic neurons. Primary rat neuron–glia cultures were treated with vehicle, indicated concentrations of rotenone or LPS alone, or combinations of indicated concentrations of rotenone and LPS. Eight days later, neurotoxicity was determined by [3H]DA uptake assay and quantification of TH-IR neurons after immunostaining of dopaminergic neurons with an anti-TH antibody. The results are the mean ± SEM of three experiments performed in triplicate.Rot, Rotenone. **p < 0.005 compared with the cultures treated with either rotenone or LPS alone.
Because the dopaminergic neurotoxicity of either rotenone or LPS alone exhibited a positive correlation with the concentrations applied (Gao 2002a,b), an alternative measure of the synergistic neurotoxicity was to determine whether the sum of neurotoxicity induced by each individual agent at a fixed concentration would equal that induced by the combination of both agents at one-half the concentration of individual agent. Therefore, rat neuron–glia cultures were treated with vehicle, 5 nm rotenone, 1 ng/ml LPS, or a combination of 2.5 nm rotenone and 0.5 ng/ml LPS, and neurotoxicity was assessed 8 d later. As shown in Figure 1B, neurotoxicity induced by a combination of 2.5 nmrotenone and 0.5 ng/ml LPS was twice the sum of neurotoxicity induced by 5 nm rotenone alone and that by 1 ng/ml LPS alone. Together, these results indicated that rotenone and LPS acted synergistically to induce the degeneration of dopaminergic neurons.
The specificity of rotenone and LPS-induced synergistic neurotoxicity was determined by measurement of DA and GABA uptake, quantification of TH-IR and NeuN-IR neurons, and morphological analysis after double-label immunostaining for TH and NeuN. As shown in Figure2A, treatment for 8 d with a combination of 0.5 nm rotenone and 0.5 ng/ml LPS did not significantly affect GABA uptake when compared with the marked decrease in DA uptake. Similarly, no significant loss of NeuN-IR neurons was observed compared with the dramatic loss of TH-IR neurons (Fig. 2B). Double-label immunostaining confirmed that the destruction of TH-IR neurons was most prominent, whereas the number and integrity of NeuN-IR neurons were not significantly affected (Fig.3). Compared with the healthy TH-IR neurons in the control cultures, those in the rotenone- and LPS-treated cultures were less numerous, and the remaining TH-IR neurons had markedly shorter and less elaborate dendrites (Fig. 3). These data demonstrated that the synergistic neurotoxicity of rotenone and LPS was preferential to dopaminergic neurons.
Fig. 2.

Rotenone and LPS-induced neurotoxicity is preferential to dopaminergic neurons. Rat neuron–glia cultures were treated with vehicle, 0.5 nm rotenone, and/or 0.5 nm rotenone. Eight days later, cultures were subjected to [3H]DA or [3H]GABA uptake assay (A) or immunostaining with an anti-TH or anti-NeuN antibody followed by quantification of the immunostained neurons (B). The results are the mean ± SEM of three experiments performed in triplicate. R + L, Rotenone plus LPS. **p < 0.005 compared with the control cultures.
Fig. 3.
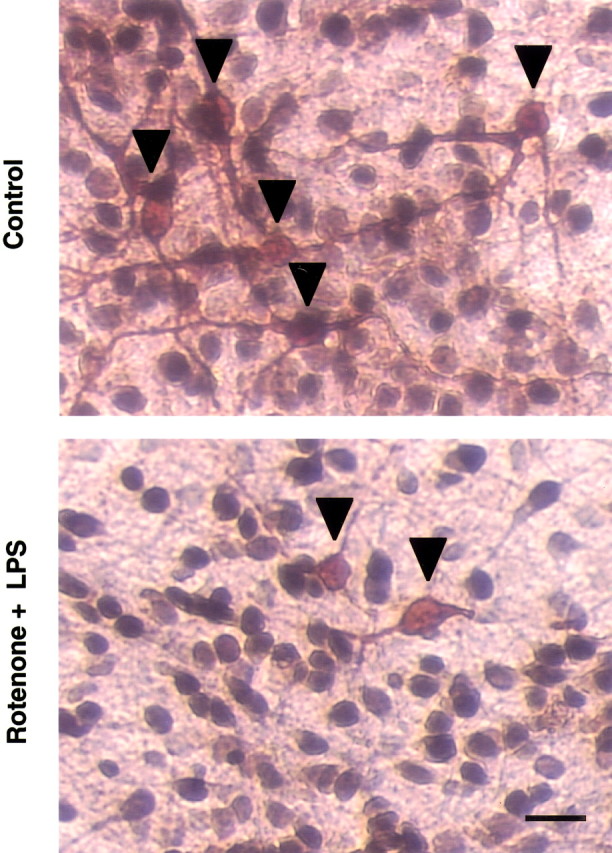
Double-label immunocytochemical analysis for TH- and NeuN-IR neurons. Rat neuron–glia cultures treated for 8 d with vehicle (Control) or 0.5 nmrotenone and 0.5 ng/ml LPS were double immunostained with anti-TH and anti-NeuN antibodies. The images are from one experiment that is representative of three separate experiments. Scale bar, 50 μm.Arrowheads, TH-IR neurons.
Effect of tandem exposure to rotenone and LPS on dopaminergic neurotoxicity
In addition to simultaneously exposing the mesencephalic neuron–glia cultures to both rotenone and LPS, we also examined the effect of sequential addition of rotenone and LPS on dopaminergic neurons using the following two treatment paradigms. In the first scheme, cultures were first treated with 0.5 nm rotenone, and, 4 d later, LPS was added to a final concentration of 0.5 ng/ml. Conversely, cultures were first treated with LPS (0.5 ng/ml), and rotenone (0.5 nm) was added 4 d later. Eight days after the initial treatment, neurotoxicity was determined by measuring DA uptake and counting TH-IR neurons. For comparison, cultures were treated with either rotenone (0.5 nm) or LPS (0.5 ng/ml) for the entire period (8 d) or only for the last 4 d. Neurotoxicity induced by exposure to rotenone for 8 d and LPS for the last 4 d was more than twice the sum of that induced by exposure to rotenone alone for 8 d and that induced by exposure to LPS alone for the last 4 d. Conversely, neurotoxicity induced by exposure to LPS for 8 d and rotenone for the last 4 d was nearly twice the sum of that induced by rotenone alone for the last 4 d and that by LPS for 8 d (Fig.4A).
Fig. 4.
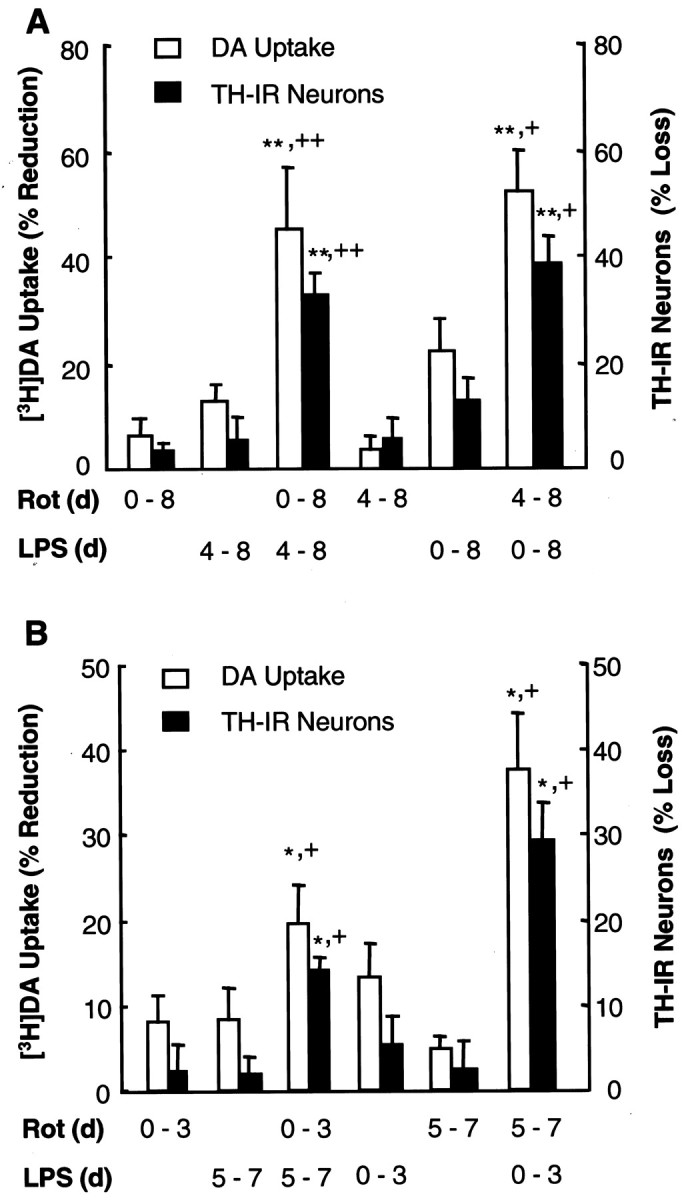
Effect of tandem addition of rotenone and LPS on dopaminergic neurodegeneration. A, Tandem addition without change of medium in between. Rat neuron–glia cultures were treated for the indicated periods with rotenone (0.5 nm) and/or LPS (0.5 ng/ml) in the following manner: rotenone or LPS for the entire 8 d (0–8) or only the last 4 d (4–8), rotenone for 8 d (0–8) plus LPS for the last 4 d (4–8), or LPS for 8 d (0–8) plus rotenone for the last 4 d (4–8). Afterward, neurotoxicity was determined by DA uptake and quantification of TH-IR neurons. **p < 0.005 compared with the cultures treated with rotenone alone;+p < 0.05 and++p < 0.005 compared with the cultures treated with LPS alone. B, Tandem addition with change of medium. Rat neuron–glia cultures were treated for the indicated periods of time with vehicle, 0.5 nm rotenone, and/or 0.5 ng/ml LPS in sequential orders and with medium changes in between as follows: rotenone or LPS alone for the first 3 d only (0–3) or the last 3 d only (5–7), rotenone for the first 3 d (0–3) followed by resting for 1 d and then LPS for 3 d (5–7), LPS for the first 3 d (0–3) followed by resting for 1 d and then rotenone for 3 d (5–7). Afterward, neurotoxicity was determined by DA uptake and quantification of TH-IR neurons. The results are the mean ± SEM of two experiments performed in triplicate. *p < 0.05 compared with the cultures treated with rotenone alone;+p < 0.05 compared with the cultures treated with LPS alone. Rot, Rotenone.
In the second treatment scheme, cultures were first treated with rotenone (0.5 nm). Three days later, the spent medium was changed to fresh medium. On day 5, cultures were treated with LPS (0.5 ng/ml). On day 7, neurotoxicity was assessed. Conversely, cultures were first treated for 3 d with LPS (0.5 ng/ml), allowed to rest for 1 d in fresh medium, and then treated with rotenone (0.5 nm) for 3 d. As shown in Figure 4B, neurotoxicity induced by exposure first to LPS then to rotenone was twice the sum of that induced by exposure for the same length of time to either agent alone. Conversely, the neurotoxicity induced by exposure first to rotenone and then to LPS was slightly greater than the sum of that induced by either agent alone (Fig.4B). These results indicated that exposure of cultures to rotenone and LPS in tandem resulted in additive to synergistic neurotoxicity.
Superoxide released from microglia is a key contributor to the synergistic neurotoxicity of rotenone and LPS
Multiple approaches were taken to investigate the potential underlying mechanism of action responsible for the rotenone and LPS-induced synergistic neurotoxicity. Because LPS-induced neurotoxicity appears to be exclusively mediated by glia, especially microglia, and rotenone-elicited neurodegeneration, at least in vitro, is markedly enhanced by microglia (Bronstein et al., 1995;Araki et al., 2001; Gao et al., 2002a; Liu et al., 2002), the role of microglia was first examined. For this purpose, neuron–glia and neuron-enriched cultures, both prepared from rat ventral mesencephalon, were compared for their susceptibility to neurotoxicity induced by rotenone and LPS. As shown in Figure 5, synergistic dopaminergic neurotoxicity was observed only in neuron–glia cultures treated for 8 d with 0.5 nm rotenone and 0.5 ng/ml LPS but not in neuron-enriched cultures treated in the same manner. Therefore, the presence of glial cells was a prerequisite for rotenone- and LPS-induced synergistic neurotoxicity.
Fig. 5.

Induction of synergistic neurotoxicity by rotenone and LPS depends on the presence of glial cells. Rat neuron–glia or neuron-enriched cultures were treated with vehicle, 0.5 nmrotenone, and/or 0.5 ng/ml LPS. Eight days later, neurotoxicity was determined by [3H]DA uptake assay. The results are the mean ± SEM of three experiments performed in triplicate. **p < 0.005 compared with the cultures treated with either rotenone or LPS alone. N/G, Neuron–glia cultures; N/N, neuron-enriched cultures.
Microglia are known to induce neurodegeneration by the release of a variety of neurotoxic factors, including cytokines such as TNFα and free radicals such as NO and ROS (Liu and Hong, 2003). Hence, the effect of rotenone and LPS on the production of key neurotoxic factors by microglia in the neuron–glia cultures was determined. First, analysis of the accumulation of nitrite, an indicator of NO production, indicated that the levels of nitrite detected in cultures after treatment for 8 d with 0.5 nm rotenone and 0.5 ng/ml LPS were 0.9 ± 0.4 μm (n = 3), similar to that observed in the cultures treated for 8 d with 0.5 ng/ml LPS alone (0.8 ± 0.6 μm;n = 3) (Gao et al., 2002b). The levels of nitrite in vehicle-treated cultures were 0.3 ± 0.4 μm (n = 3). In contrast, the levels of nitrite in cultures treated with 0.5 nmrotenone were below the detection limit of the assay (0.5 μm), an observation consistent with our previous report (Gao et al., 2002a). Therefore, the minute quantities of nitrite produced in cultures treated with rotenone and LPS were probably attributable to the stimulation of glial cells by LPS but not rotenone. Second, the levels of TNFα in cultures treated with 0.5 nm rotenone and 0.5 ng/ml LPS were below the detection limit of the assay (5 pg/ml). Similarly, the amount of TNFα in cultures treated with either agent alone was nondetectable, consistent with our previous reports (Gao et al., 2002a,b). Third, in sharp contrast to the lack of synergy in stimulating the production of NO and TNFα, rotenone (0.5 nm) and LPS (0.5 ng/ml) synergistically stimulated the release of superoxide free radical measured as SOD-inhibitable reduction of cytochromec (Fig. 6A). Although no significant release of superoxide was detected in cultures stimulated with 0.5 nm rotenone, a twofold increase was observed in cultures stimulated with 0.5 ng/ml LPS alone, and a fourfold increase was detected in cultures stimulated with both 0.5 nm rotenone and 0.5 ng/ml LPS (Fig.6A). The fact that rotenone and LPS failed to increase the production of superoxide in neuron-enriched cultures may underlie their inability to induce neurotoxic effect. In addition to the measurement of superoxide release, the levels of intracellular ROS were determined after stimulation with rotenone and LPS. In neuron-enriched cultures, no significant increase in the levels of intracellular ROS was detected. In neuron–glia cultures, however, LPS and rotenone synergistically induced an increase in the levels of intracellular ROS (Fig. 6A).
Fig. 6.

Role of ROS in the induction of synergistic neurotoxicity by rotenone and LPS. A, Measurement of release of superoxide and levels of intracellular ROS. To measure superoxide release, rat neuron–glia or neuron-enriched cultures were stimulated with vehicle, 0.5 nm rotenone, and/or 0.5 ng/ml LPS. The release of superoxide was measured by the SOD-inhibitable reduction of cytochrome c. The levels of intracellular ROS were detected with CM-H2-DCFDA. The relative fluorescence intensities in the control cultures were 300–450 and 450–600 arbitrary units per well for N/N andN/G, respectively. The results are expressed as the percentage of control and are the mean ± SEM of two to three experiments performed in triplicate. *p < 0.05 and **p < 0.005 compared with the control cultures, respectively; ++p < 0.05 compared with the cultures treated with either rotenone or LPS alone.B, Measurement of the release of superoxide from microglia. Microglia-enriched cultures were pretreated for 30 min with 5 μm DPI or 0.25 mm apocynin before stimulation with vehicle, 0.5 nm rotenone, and/or 0.5 ng/ml LPS. Superoxide released from microglia was determined by measuring the SOD-inhibitable reduction of cytochrome c. The results are the mean ± SEM of three experiments performed in triplicate. *p < 0.05 compared with the vehicle control;+p < 0.05 compared with the rotenone and LPS-treated cultures. C, Effect of apocynin,l-NAME, or SOD–catalase on the rotenone and LPS-induced synergistic neurotoxicity. Rat neuron–glia cultures were pretreated for 30 min with vehicle, 0.25 mm apocynin, or 1 mml-NAME before treatment with 0.5 nm rotenone and 0.5 ng/ml LPS. SOD–catalase (100 and 150 U/ml, respectively) were added at the same time with rotenone and LPS. Seven days later, neurotoxicity was determined by [3H]DA uptake assay. The results are the mean ± SEM of three experiments performed in triplicate. **p < 0.005 compared with the vehicle control;+p < 0.05 compared with the rotenone and LPS-treated cultures. N/N, Neuron-enriched cultures;N/G, neuron–glia cultures; C, control;R, rotenone; L, LPS; D, DPI; A, apocynin; S/C, SOD–catalase;L-N, l-NAME.
Previously, we showed that superoxide released from LPS- or rotenone-activated microglia appeared to be a major effector of the dopaminergic neurodegeneration (Gao et al., 2002a,b). To determine the source of extracellular superoxide in rotenone and LPS-stimulated cultures, microglia-enriched cultures were tested for response to both rotenone and LPS. As shown in Figure 6B, significant release of superoxide was detected in microglia stimulated with 0.5 nm rotenone and 0.5 ng/ml LPS, whereas no apparent superoxide release was observed in microglia stimulated with the same concentrations of either agent alone. Furthermore, the rotenone and LPS-stimulated superoxide release was almost completely prevented by preincubation with DPI and apocynin, inhibitors of NADPH oxidase (Fig. 6B).
The relevance of the release of superoxide to the rotenone and LPS-induced synergistic neurotoxicity was also determined. Preincubation of neuron–glia cultures with apocynin (0.5 mm) or coincubation with SOD–catalase (100 and 150 U/ml, respectively) significantly reduced the neurotoxicity induced by rotenone and LPS (Fig. 6C). Preincubation with 1 mmNG-nitro-l-arginine methyl ester (l-NAME), an inhibitor of nitric oxide synthase, did not afford a statistically significant degree of neuroprotection (Fig. 6C). These results demonstrated that stimulation of the production of superoxide in microglia, at least in part, underlie the mechanism of action responsible for the synergistic neurotoxicity of rotenone and LPS.
Microglial NADPH oxidase is the primary source of superoxide production
The observation that both microglial production of superoxide and synergistic neurotoxicity induced by rotenone and LPS were sensitive to modulation by inhibitors of NADPH oxidase prompted us to speculate that NADPH oxidase played a critical role in mediating the synergistic neurotoxicity. To test this hypothesis, we established primary mesencephalic neuron–glia cultures from NADPH oxidase-deficient (PHOX−/−) and wild-type (PHOX+/+) mice. Similar to rat neuron–glia cultures (Fig. 1), synergistic dopaminergic neurotoxicity was observed in neuron–glia cultures from wild-type mice (PHOX+/+) after treatment for 8 d with 0.5 nm rotenone and 0.5 ng/ml LPS (Fig.7A). However, neuron–glia cultures from PHOX−/− mice were nearly completely resistant to neurotoxicity induced by 0.5 nm rotenone and 0.5 ng/ml LPS (Fig.7A). The resistance of PHOX−/− cultures to rotenone and LPS neurotoxicity suggested that production of superoxide was a key event in the induction of neurotoxicity. Indeed, in neuron–glia cultures treated for 4 d with 0.5 nm rotenone and 0.5 ng/ml LPS, significant production of superoxide was observed only in PHOX+/+ but not in PHOX−/− cultures (Fig. 7B). Furthermore, the primary source of superoxide production was microglia, as exemplified by the markedly increased production of superoxide in rotenone and LPS-stimulated microglia from PHOX+/+ mice but not those from PHOX−/− mice (Fig. 7C). These results lend strong support to the notion that ROS production mediated by the microglial NADPH oxidase plays a critical role in the synergistic neurotoxicity induced by rotenone and LPS.
Fig. 7.

Effect of NADPH oxidase deficiency on the induction of neurotoxicity and production of superoxide induced by rotenone and LPS. A, Neurotoxicity. Neuron–glia cultures from wild-type (PHOX+/+) or NADPH oxidase-deficient (PHOX−/−) mice were treated with vehicle, 0.5 nm rotenone, and/or 0.5 ng/ml LPS. Eight days later, neurotoxicity was determined by [3H]DA uptake assay. The results are the mean ± SEM of three experiments performed in triplicate. **p < 0.005 compared with the rotenone or LPS-treated cultures. B, Determination of superoxide production in neuron–glia cultures 4 d after treatment. PHOX+/+ or PHOX−/−neuron–glia cultures were treated with vehicle, 0.5 nmrotenone, and/or 0.5 ng/ml LPS. Four days later, levels of superoxide were measured as SOD-inhibitable cytochrome c reduction. **p < 0.005 compared with the vehicle control;++p < 0.005 compared with the cultures treated with either rotenone or LPS alone. C, Production of superoxide in microglia. PHOX+/+ or PHOX−/− microglia-enriched cultures were stimulated with vehicle, 0.5 nm rotenone, and/or 0.5 ng/ml LPS. Release of superoxide were measured as SOD-inhibitable cytochromec reduction. **p < 0.005 compared with the vehicle control; ++p < 0.005 compared with the cultures treated with either rotenone or LPS alone.C, Control; R, rotenone;L, LPS.
Discussion
Idiopathic PD is an age-related neurodegenerative disease that usually occurs late in life and progresses over many years. The pathogenesis of PD involves a progressive and selective destruction of the nigrostriatal dopaminergic pathway, which is critical for the regulation of body movements. Although the exact cause of PD remains unknown, the late onset and slow progressing nature of the disease suggests the possibility that interactions among a variety of intrinsic and environmental factors may set in motion a cascade of events leading to the eventual destruction of the nigrostriatal dopaminergic pathway.
One of those suspected factors is exposure to agrochemicals, such as pesticides. Results from a number of epidemiological and case-controlled studies appear to indicate a positive correlation between early exposure to pesticides and development of PD (Gorell et al., 1998; Ritz and Yu, 2000; Herishanu et al., 2001). Experimentally, several groups have reported recently that administration of pesticides and/or related agrochemicals results in the degeneration of the nigrostriatal dopaminergic pathway. For example, chronic administration of rotenone has been reported to reproduce parkinsonian features in rats (Betarbet et al., 2000). Alteration of striatal dopaminergic chemistry and induction of behavioral abnormalities have also been reported in rats after exposure to a combination of the herbicide paraquat and the fungicide maneb (Thiruchelvam et al., 2000). The exact mechanism of action responsible for the preferential degeneration of the nigrostriatal dopaminergic pathway by these agrochemicals, however, is not completely clear.
Another intriguing factor that has been increasingly associated with the pathogenesis of PD is inflammation in the brain. Clues to the involvement of inflammation in the pathogenesis of PD have derived from postmortem analyses of PD brains in which microglial activation, elevated levels of proinflammatory mediators such as TNFα, IL-1β, and NO, and evidence of ROS production have been detected in the substantia nigra (McGeer et al., 1988; Cassarino et al., 1997; Hunot et al., 1999; Nagatsu et al., 2000). However, it is not clear whether these inflammatory “footprints” observed in the late stages of the pathogenesis of PD may merely be a reflection of reactive gliosis after the occurrence of the initial neuronal injuries. On the other hand, increasing evidence has suggested that early-life occurrence of inflammation in the brain, either as a result of exposure to infectious agents or brain injury, plays an important role in later-life development of PD (Ravenholt and Foege, 1982; Factor et al., 1988;Mattock et al., 1988; Williams et al., 1991; Casals et al., 1998;Plassman et al., 2000). The likelihood that inflammation plays a key role in the earlier stages of PD is significantly enhanced by the observation that the midbrain region is particularly rich in microglia (Lawson et al., 1990; Kim et al., 2000), which are readily activated by immunological challenges and neuronal injuries (Kreutzberg, 1996). In fact, we demonstrated recently that chronic intranigral infusion of LPS results in a delayed and progressive degeneration of nigral dopaminergic neurons (Gao et al., 2002b). In addition, Ling et al. (2002) has also demonstrated that in utero exposure of developing fetuses to LPS results in a loss of nigral dopaminergic neurons in neonates.
Despite the intense effort to identify individual agents and/or factors as potential etiological culprits, development of PD may be a result of the interaction of multiple factors, including genetic predisposition, the intrinsic nature of the dopaminergic system, and exposure to environmental toxins (Olanow and Tatton, 1999; Kidd, 2000). In the category of insults from environmental factors, early-life exposures (either simultaneously or in tandem) to multiple agents that are vastly different in identity may contribute significantly to the initiation and/or progression of the disease. In this study, using a chronic (8 d after drug treatment) in vitro model of PD, we provided evidence that the herbicide rotenone and the inflammogen LPS worked in synergy to induce dopaminergic neurotoxicity. Exposure to rotenone (0.5 nm) or LPS (0.5 ng/ml) alone produced little or minimal neurotoxicity (Fig. 1). However, exposure to a combination of the two agents, each at the same concentration as that used individually, resulted in a synergistic dopaminergic neurotoxicity (Fig. 1). Furthermore, enhanced neurotoxicity did not derive only from simultaneous exposure to both agents; tandem exposure also resulted in additive to synergistic dopaminergic neurotoxicity (Fig. 4). These results indicate that exposure to agrochemicals and episodes of inflammation in the brain, occurring either simultaneously or in tandem, may promote, if not trigger, the neurodegenerative processes. Confirmation of these in vitro observations in animal studies and in multifactorial epidemiological studies should shed significant light on the understanding of the etiology of PD.
Analysis of the underlying mechanism of action responsible for the synergistic neurotoxicity of rotenone and LPS indicates that, at the cellular level, the participation of glial cells is indispensable for the induction of the synergistic neurotoxicity (Fig. 5). Because glial cells induce neurodegeneration by the production and release of neurotoxic factors, including cytokines, reactive nitrative species, and ROS, the effect of rotenone and LPS on the release of TNFα, NO, and superoxide was first examined. No detectable release of TNFα was observed in neuron–glia cultures treated with rotenone (0.5 nm) and LPS (0.5 ng/ml) or either agent alone, consistent with previous reports (Gao et al., 2002a,b). Therefore, release of TNFα did not seem to be involved in the synergistic neurotoxicity of rotenone and LPS observed in this study. Production of NO, on the other hand, appeared to be more relevant than TNFα to the observed synergistic neurotoxicity. Although rotenone (0.5 nm) did not appear to elevate NO production (this study; Gao et al. 2002b), modest NO production was detected in neuron–glia cultures treated with 0.5 ng/ml LPS (Gao et al., 2002a) or the same concentration of LPS plus 0.5 nm rotenone (this study). The low micromolar quantities of nitrite (an indicator of NO production) by itself might not be directly toxic to neurons. However, NO could react with other soluble factors such as superoxide free radical to form highly toxic intermediates such as peroxynitrite (Beckman et al., 1993; Xie et al., 2002). Hence, although rotenone at 0.5 nm did not promote the production of NO beyond that induced by 0.5 ng/ml LPS, the involvement of NO in the synergistic neurotoxicity could not be ruled out because of the possibility of formation of peroxynitrite with superoxide that was abundantly produced.
A remarkable finding of this study is that rotenone and LPS act synergistically to induce microglia to release superoxide free radical. Although at higher concentrations, each agent alone is capable of stimulating microglia to release superoxide (Liu et al., 2000; Gao et al., 2002a,b), at lower concentrations, rotenone or LPS is either ineffective or only modestly effective in inducing superoxide generation (Gao et al., 2002a,b). However, when microglia were stimulated with a combination of singularly ineffective or minimally effective concentrations of rotenone and LPS, robust production of superoxide was observed (Figs. 6B, 7C). Mechanistically, the rotenone and LPS-stimulated release of superoxide was mediated through the activity of the membrane-associated and multi-subunit NADPH oxidase, which is the primary enzymatic machinery for the production of superoxide in immune cells of peripheral system and the brain (Babior, 1999). In support of this conclusion were observations that the rotenone and LPS-stimulated superoxide production in microglia was nearly completely prevented by pharmacological inhibition or genetic inactivation of NADPH oxidase (Figs. 6B, 7B,C) and that rotenone and LPS failed to induce neurotoxicity in neuron–glia from NADPH oxidase-deficient mice (Fig. 7A). Hence, NADPH oxidase may be an important therapeutic target for the treatment of PD.
In addition to microglia, it has been reported recently that neurons may also express a functional NADPH oxidase (Noh and Koh, 2000;Tammariello et al., 2000). However, in this study, neuronal NADPH oxidase did not appear to be directly involved in the production of superoxide or the induction of neurodegeneration because rotenone (0.5 nm) and LPS (0.5 ng/ml) neither induced superoxide release (Fig. 6A) nor caused significant neurotoxicity in neuron-enriched cultures (Fig. 5).
Worth noting is the role of intracellular ROS in the induction of synergistic neurotoxicity. At high nanomolar to low micromolar concentrations, rotenone is postulated to work as an inhibitor of mitochondrial complex I and may promote the production of ROS in neurons (Greenamyre et al., 1999). In this study, using CM-H2-DCFDA as a probe, we did not observe significant elevations of intracellular ROS in neuron-enriched cultures stimulated with 0.5 nm rotenone, 0.5 ng/ml LPS, or the combination of both (Fig. 6A). Interestingly, in neuron–glia cultures, although rotenone (0.5 nm) did not induce any changes in fluorescent intensity, LPS (0.5 ng/ml) or LPS (0.5 ng/ml) and rotenone (0.5 nm) did cause a significant increase in fluorescent signal (Fig. 6A). The increased fluorescent signal observed in the neuron–glia cultures may reflect increased ROS generation inside glia, increased levels of intracellular hydrogen peroxide as a consequence of conversion from extracellular superoxide, and/or the intracellular ROS generation in neurons in the presence of superoxide-generating microglia, which were otherwise dormant in the absence of glial cells. Nevertheless, intracellular ROS probably played a role in the synergistic neurotoxicity induced by rotenone and LPS.
Significant rises in extracellular levels of reactive nitrative and oxidative species are highly relevant to the induction of dopaminergic neurodegeneration because dopaminergic neurons are known to be particularly vulnerable to oxidative damage attributable to a number of intrinsic characteristics. Among the various types of neurons, dopaminergic neurons have a reduced antioxidant capacity that is exemplified by a lower content of glutathione, an increased accumulation of iron that may participate in the formation of ROS such as hydroxyl radical through Fenton reaction, and a high content of oxidation-prone dopamine and lipids (Jenner and Olanow, 1998;Greenamyre et al., 1999). Oxygen free radicals can trigger chain reactions of lipid peroxidation as well as functional and structural modifications of proteins, enzymes, and nucleic acid macromolecules. In addition, ROS such as superoxide can react with reactive nitrative species such as NO to form more reactive intermediates such as peroxynitrite. Furthermore, it has been proposed that extracellular ROS may promote the mitochondria-mediated generation of intracellular ROS (Hasegawa et al., 1990). In the brain, at least for rodents, the midbrain region, which encompasses the substantia nigra, is particularly rich in microglia (Lawson et al., 1990; Kim et al., 2000). Therefore, the combination of the intrinsic characteristics of dopaminergic neurons and their physical location in a microglia-rich microenvironment renders them especially vulnerable to attacks by free radicals generated both extracellularly and intracellularly.
In summary, this study, for the first time, demonstrated the synergistic dopaminergic neurotoxicity of a pesticide (rotenone) and an inflammogen (LPS). NADPH oxidase-mediated production of superoxide free radical from rotenone and LPS-activated microglia appears to be an important mediator of the synergistic neurotoxicity. Continued exploration of the impact of multiple environmental factors on the degeneration of the nigrostriatal dopaminergic pathway should significantly advance our understanding of the etiology and pathogenesis of Parkinson's disease and our search for effective therapeutic strategies to slow the progression of the disease.
Footnotes
Correspondence should be addressed to Dr. Bin Liu, F1-01, National Institute of Environmental Health Sciences/National Institutes of Health, P.O. Box 12233, Research Triangle Park, NC 27709. E-mail:liu3@niehs.nih.gov.
References
- 1.Araki E, Forster C, Dubinsky JM, Ross ME, Iadecola C. Cyclooxygenase-2 inhibitor NS-398 protects neuronal cultures from lipopolysaccharide-induced neurotoxicity. Stroke. 2001;32:2370–2375. doi: 10.1161/hs1001.096057. [DOI] [PubMed] [Google Scholar]
- 2.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 3.Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 4.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 5.Bronstein DM, Perez-Otano I, Sun V, Mullis Sawin SB, Chan J, Wu GC, Hudson PM, Kong LY, Hong JS, McMillian MK. Glia-dependent neurotoxicity and neuroprotection in mesencephalic cultures. Brain Res. 1995;704:112–116. doi: 10.1016/0006-8993(95)01189-7. [DOI] [PubMed] [Google Scholar]
- 6.Casals J, Elizan TS, Yahr WH. Postencephalitic parkinsonism. J Neural Transm. 1998;105:645–676. doi: 10.1007/s007020050086. [DOI] [PubMed] [Google Scholar]
- 7.Cassarino DS, Fall CP, Swerdlow RH, Smith TS, Halvorsen EM, Miller SW, Parks JP, Parker WD, Jr, Bennett JP., Jr Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson's disease. Biochim Biophys Acta. 1997;1362:77–86. doi: 10.1016/s0925-4439(97)00070-7. [DOI] [PubMed] [Google Scholar]
- 8.Factor SA, Sanchez-Ramos J, Weiner WJ. Trauma as an etiology of parkinsonism: a historical review of the concept. Mov Disord. 1988;3:30–36. doi: 10.1002/mds.870030105. [DOI] [PubMed] [Google Scholar]
- 9.Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002a;22:782–790. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem. 2002b;81:1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 12.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- 13.Greenamyre JT, MacKenzie GM, Peng T-I, Stephans SE. Mitochondrial dysfunction in Parkinson's disease. Biochem Soc Symp. 1999;66:85–97. doi: 10.1042/bss0660085. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa E, Takeshige K, Oishi T, Murai Y, Minakami S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem Biophys Res Commun. 1990;170:1049–1055. doi: 10.1016/0006-291x(90)90498-c. [DOI] [PubMed] [Google Scholar]
- 15.Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer's disease. Brain Res. 1998;780:294–303. doi: 10.1016/s0006-8993(97)01215-8. [DOI] [PubMed] [Google Scholar]
- 16.Herishanu YO, Medvedovski M, Goldsmith JR, Kordysh E. A case-control study of Parkinson's disease in urban population of southern Israel. Can J Neurol Sci. 2001;28:144–147. doi: 10.1017/s0317167100052835. [DOI] [PubMed] [Google Scholar]
- 17.Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;44:S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- 19.Kidd PM. Parkinson's disease as multifactorial oxidative neurodegeneration: implications for integrative management. Altern Med Rev. 2000;5:502–519. [PubMed] [Google Scholar]
- 20.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 22.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 23.Ling Z, Gayle DA, Ma SY, Lipton JW, Tong CW, Hong JS, Carvey PM. In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disord. 2002;17:116–124. doi: 10.1002/mds.10078. [DOI] [PubMed] [Google Scholar]
- 24.Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 25.Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- 26.Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- 27.Liu B, Gao HM, Wang J-Y, Jeohn G-H, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann NY Acad Sci. 2002;962:318–331. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- 28.Liu SX, Athar M, Lippai I, Waldren C, Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc Natl Acad Sci USA. 2001;98:1643–1648. doi: 10.1073/pnas.031482998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattock C, Marmot M, Stern G. Could Parkinson's disease follow intra-uterine influenza? A speculative hypothesis. J Neurol Neurosurg Psychiatry. 1988;51:753–756. doi: 10.1136/jnnp.51.6.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 31.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl. 2000;60:277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 32. Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci 20 2000. RC111(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 34.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 35.Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson's disease: a metaanalysis. Environ Res. 2001;86:122–127. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- 36.Ravenholt RT, Foege WH. 1918 influenza, encephalitis lethargica, parkinsonism. Lancet. 1982;8303:860–864. doi: 10.1016/s0140-6736(82)90820-0. [DOI] [PubMed] [Google Scholar]
- 37.Ritz B, Yu F. Parkinson's disease mortality and pesticide exposure in California 1984–1994. Int J Epidemiol. 2000;29:323–329. doi: 10.1093/ije/29.2.323. [DOI] [PubMed] [Google Scholar]
- 38. Tammariello SP, Quinn MT, Estus S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci 20 2000. RC53(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. J Neurosci. 2000;20:9207–9214. doi: 10.1523/JNEUROSCI.20-24-09207.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams DB, Annegers JF, Kokmen E, O'Brien PC, Kurland LT. Brain injury and neurologic sequelae: a cohort study of dementia, parkinsonism, and amyotrophic lateral sclerosis. Neurology. 1991;41:1554–1557. doi: 10.1212/wnl.41.10.1554. [DOI] [PubMed] [Google Scholar]
- 41.Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, Longo VD. Peroxynitrite mediates neurotoxicity of amyloid β-peptide1–42- and lipopolysaccharide-activated microglia. J Neurosci. 2002;22:3484–3492. doi: 10.1523/JNEUROSCI.22-09-03484.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]