

Abstract
The Na+/K+ ATPase asymmetrically distributes sodium and potassium ions across the plasma membrane to generate and maintain the membrane potential in many cell types. Although these pumps have been hypothesized to be involved in various human neurological disorders, including seizures and neurodegeneration, direct genetic evidence has been lacking. Here, we describe novel mutations in the Drosophila gene encoding the α (catalytic) subunit of the Na+/K+ ATPase that lead to behavioral abnormalities, reduced life span, and severe neuronal hyperexcitability. These phenotypes parallel the occurrence of extensive, age-dependent neurodegeneration. We have also discovered that the ATPalpha transcripts undergo alternative splicing that substantially increases the diversity of potential proteins. Our data show that maintenance of neuronal viability is dependent on normal sodium pump activity and establishDrosophila as a useful model for investigating the role of the pump in human neurodegenerative and seizure disorders.
Keywords: neuropathology, paralysis, hyperexcitability, excitotoxicity, alternative splicing, ATPalpha, Na/K ATPase, neurodegeneration
Introduction
Despite the biological and medical significance of neurodegeneration, the cellular and molecular mechanisms that are responsible are still poorly understood. Isolation and characterization of a collection of neurodegeneration mutants should be valuable in dissecting these mechanisms. We developed a screen for identifying neurodegeneration mutants inDrosophila on the basis of the finding that dysfunction in neuronal signaling is often associated with neurodegeneration (Palladino et al., 2002). In Drosophila, mutations in genes required for normal electrical signaling have been readily identified among those with conditional paralytic phenotypes (Loughney et al., 1989; Atkinson et al., 1991; Pallanck et al., 1995; Titus et al., 1997;Littleton et al., 1998). In our previous analysis of a large collection of temperature-sensitive (TS) paralytic mutants, we reported the identification of 15 mutations, defining at least 9 genes, that cause extensive neurodegeneration (Palladino et al., 2002). Two of those mutations caused dominant paralysis and mapped to the same chromosome location. We demonstrate here by genetic and molecular analyses that these mutations are alleles of the ATPalpha gene, which encodes the Na+/K+ ATPase α subunit. At the electrophysiological level, the mutations cause neuronal hyperexcitability and seizure-like activity. Most strikingly, these mutants also have shortened life spans and extensive, progressive spongiform neurodegeneration.
Na+/K+ATPases (sodium pumps) asymmetrically distribute Na+ and K+ions across the plasma membrane of cells. These ion gradients determine the resting potential of cells and drive important secondary processes. Many sodium pump isozymes are highly conserved evolutionarily and widely expressed in animal tissues (for review, see Blanco and Mercer, 1998; Mobasheri et al., 2000). Sodium pumps have at least two essential subunits, α and β. The α subunit of the Na+/K+ ATPase is a large protein (>110 kDa) with multiple transmembrane domains and contains ATP-dependent catalytic activity. The β subunit has a single transmembrane domain and is thought to be involved in pump maturation, membrane localization, and functional regulation of Na+/K+ATPases, as well as to perform additional functions as a cell adhesion molecule (Geering, 1991; Hasler et al., 1998).
Since the discovery of the sodium pumps, an overwhelming body of evidence has demonstrated the fundamental role of this protein in maintaining normal neuronal functions. However, detailed mutational analysis of the in vivo consequences of impaired pump activity has been limited. In Drosophila, mutations that reduce expression of the α subunit but do not alter its structure are associated with a mild bang-sensitive paralytic phenotype (Schubiger et al., 1994). In nematodes, a link between pharyngeal function and sodium pump activity was revealed by Na+/K+ ATPase α loss-of-function eat-6 mutations (Davis et al., 1995; Shima et al., 1998). In mice, targeted knock-outs of the adhesion molecule of glia (AMOG) gene, which encodes the ATPaseβ2 subunit, exhibit progressive motor uncoordination and paralysis of extremities and die within 3 weeks after birth. At this age, the mice were reported to manifest degeneration of astrocyte endfeet and enhanced apoptotic death of photoreceptor cells (Magyar et al., 1994; Molthagen et al., 1996). Although it has been proposed that these phenotypes might arise as a consequence of reduced pump activity and consequent osmotic imbalance, no difference in pump activity was detectable in the mutants. Moreover, interpretation of the mutant phenotypes is complicated by the additional roles of the β2 subunit as a recognition molecule that mediates neuron–astrocyte interactions among other proposed functions. A human family has been reported in which siblings died from neonatal seizures associated with spongiform encephalopathy and low Na+/K+ ATPase activity. An impairment of pump activity was hypothesized to be the primary defect; however, the etiology, including whether it had a genetic basis or what gene was affected if so, remains unknown (Renkawek et al., 1992).
The mammalian Na+/K+ ATPase α gene family contains at least three paralogous genes (ATP1A1–3). Genetic analysis of mouse ATP1A1 and ATP1A2 genes has been performed, and cardiovascular phenotypes have been characterized for each (James et al., 1999). The ATP1A3 gene shows expression predominantly in the mammalian brain and is a candidate for human neurological illnesses, but mutations in this gene have yet to be described.
Analysis of the mutants reported here provides direct genetic evidence that perturbations in the Na+/K+ ATPase α subunit result in severe defects in behavior, neuronal excitability, and maintenance of neuronal viability. Because defects in sodium pump activity have been indirectly implicated in various neural disorders in humans, including epilepsy and spongiform encephalopathies, these mutants should provide a valuable model for elucidating the mechanistic details of these disorders and developing possible therapies.
Materials and Methods
Fly strains and mutagenesis
Fly stocks were maintained on standard cornmeal–molasses agar media at 22–28°C. The DTS1 mutation was obtained in an ethylmethane sulfonate mutagenesis of Canton S in a screen for dominant temperature-sensitive (DTS) paralytic mutations.DTS2 was obtained similarly; however, cn bwanimals were mutagenized. H64 mutations were isolated by Dr. Adelaide Carpenter (Cambridge, UK) from an ethylnitrosurea mutagenesis of a roe pp stock. Revertants of DTS1 andDTS2 were isolated after gamma irradiation of these chromosomes and screening of F1 offspring for loss of the temperature-sensitive paralytic phenotype. Throughout the paper, wild type and control refer to Canton S, unless specified otherwise.
Behavioral assays
Assays of bang-sensitive paralysis and temperature-sensitive paralysis were performed as described previously (Ganetzky and Wu, 1982; Wu et al., 1978).
Genetic and cytological mapping
The conditional paralytic phenotypes of DTS1,DTS2, and H64 were mapped recombinationally relative to the dominant markers Gl, Sb, andH. All three mutations mapped very close and to the left ofH at ∼3–70.0 on the genetic map corresponding to 92D–93B on the cytological map. DTS1, DTS2, and H64 have normal cytology. Of the revertants generated,DTS2R3 andDTS1R1 are also cytologically normal.DTS2R1,DTS2R2, andDTS1R2 are all associated with inversions that include a breakpoint in the 93A5–93B1,2 interval.
Life span analysis
Life spans were generated by collecting newly eclosed animals, placing them at low density, 10–20 animals per vial, with males and females kept in separate vials at 28°C. The animals were passaged daily onto fresh food to minimize moisture, and microbe-related lethality and deaths were noted. Life spans were generated by calculating the percentage of survivorship daily and plotting this as a function of time in days. Animals removed for histological analysis and incidental deaths were subtracted from the population. Viability comparisons used time in days to 50% survival of the population and were analyzed using Student's t test.
Molecular analysis ofATPalpha transcript
Exon six analysis. Standard reverse transcription (RT) reactions were performed with a gene-specific primer directed to exon 9 (TTAATAGTAGGTCTCCTGCTCC-OH), M-MLV Reverse Transcriptase (Promega, Madison, WI), and 10 μg whole RNA isolated from embryos or adults using a modified LiCl/urea preparation (Auffray and Rougeon, 1980). Standard PCR reactions were performed as follows with primers directed toward exons 4 (TCAACACCGACGACATCAACTTCC-OH) and 9 (GGTTGCGGCGCAAGTAGAAACGACG-OH): 94°C denaturing (45 sec); 57°C annealing (45 sec); and 72°C extension (2 min), for 40 cycles. Products were cloned using the TOPO T/A Cloning Kit and One-ShotEscherichia coli (Invitrogen, San Diego, CA). Mini-plasmid preparations of transformants were analyzed by restriction digestion to determine which exon 6 isoform was present.DraIII/EcoRI (New England Biolabs, Beverly, MA) double digests were diagnostic for exon 6b, andBsmBI (New England Biolabs) and BstYI (New England Biolabs) were diagnostic for exon 6c and 6d, respectively. Clones assayed as negative for 6b, 6c, and 6d were sequenced to verify that they contained exon 6a. In total 4, 72, 36, and 11 clones of exons 6a, 6b, 6c, and 6d were identified, respectively.
Wild-type semiquantitative RT-PCR. Semiquantitative RT-PCR was performed on adult whole RNA as published previously (Palladino et al., 2000), with minor modifications. Primers directed to exon 0 (AAATAACATGGCGTTAAGGTCGG-OH) and 12 (CAACGCGAATCGGTTCTAGTGCTGAA-OH) sequences were used separately in combination with a reverse primer directed toward exon 3 (ACCCATTCGGGCGTCTGCTTGGGTGG-OH). Standard RT-PCR reactions were performed except that RP49 primers were added to the reaction at cycle 5, and samples were taken every other cycle from 16 and 28 and resolved on an agarose gel stained with ethidium bromide. Quantification of gel florescence was performed on cycle 20 products using NIH image software (n = 3). The RT-PCR products were cloned, as above, and representatives of each size clone were sequenced directly to document the splicing events.
Mutant RT-PCR analysis. Using a polymorphic SacI restriction enzyme site in constitutive exon 4, we assayed expression from the mutant and wild-type chromosomes in ATPalphamutants. Canton S, DTS1, DTS2, and theATPalpha revertant chromosomes contained the SacI restriction enzyme recognition sequence. TheTM6(ATPalpha+) chromosome lacks theSacI site. RT-PCR was performed (as above) on various wild-type and mutant genotypes. Twenty-fold SacI overdigestion of these reaction products was resolved on an agarose gel stained with ethidium bromide. This procedure revealed complete digestion of wild- type samples and partial digestion of DTS1, DTS2, H64, DTS2R3, and DTS1R1 samples; no digestion was detected with DTS2R2, DTS1R2, and DTS2R1 samples (n > 4, each genotype).
Electrophysiology
Extracellular thoracic recordings (ETRs) were performed using procedures similar to those described for recording electroretinograms (Hotta and Benzer, 1969; Pak et al., 1969). Briefly, flies were anesthetized with CO2, and their wings and anterior legs were surgically removed; then the flies were immobilized in plasticine and allowed to recover for 15 min. A temperature-controlled stage was used with a temperature probe inserted into the plasticine adjacent to the animal. Glass recording and reference electrodes filled with 3 m KCl were placed in the thorax and head, respectively. The recording electrode was positioned just below the dorsal cuticle into the vicinity of the dorsal longitudinal flight muscles (DLMs). The activity of these muscles is driven by input from DLM motor neurons and provides an assay for neural activity in the flight motor pathway. Traces were amplified using an Axopatch 1-D amplifier in current-clamp mode (clamping at zero) and recorded using Clampex 6.0.3 software (Axon Instruments, Foster City, CA). Current traces were filtered at 1 kHz, and consecutive traces are reported from representative animals (n >15 for each genotype).
Histology
Heads or bodies from adult flies of wild-type and mutant flies were dissected and preserved in freshly prepared Carnoy's fixative at room temperature for 4–12 hr, washed in 70% ethanol, and processed into paraffin using standard histological procedures. Heads were embedded to obtain frontal sections, and the bodies were embedded to obtain sagittal sections. Serial, 4 μm sections were obtained, stained with hematoxylin and eosin, and examined under a light microscope (n >40, each genotype). Occurrence of neurodegeneration was indicated by the vacuolar appearance of neural tissues of the brain or ganglia. Young animals were collected within 24 hr of eclosion, aged for 24–48 hr at 28oC, and processed as above. Aged animals were collected within 24 hr of eclosion, aged at 28oC and screened for gross pathology at the age of 50% survival for that population.
Results
DTS1, DTS2, and H64 are dominant conditional paralytic mutants
In our previous examination of TS paralytic mutants (Palladino et al., 2002), we identified two dominant mutations, referred to here as DTS1 and DTS2, that were placed in the same approximate chromosome location by recombination mapping. These mutants also display a similar behavioral phenotype. After exposure to 37–38°C, DTS1/+ and DTS2/+adults become paralyzed within 10–30 sec with complete penetrance. Wild-type animals never become paralyzed from acute exposures to 37–38°C. After a 3 min exposure to the restrictive temperature,DTS1/+ and DTS2/+ flies regain the ability to stand after 1–2 min at the permissive temperature (<30°C) and require another several minutes before they begin to walk. Even without exposure to elevated temperatures, both DTS1/+ andDTS2/+ heterozygotes are somewhat sluggish. DTS1and DTS2 cause embryonic lethality when homozygous.
The H64 mutation, isolated on the basis of its dominant bang-sensitive paralytic phenotype (see Materials and Methods), mapped to the same region as DTS1 and DTS2 and was therefore examined further to determine whether these dominant mutations were all related. H64/+ flies become completely paralyzed for 10–35 sec when subjected to a mechanical shock. After paralysis, up to 5 min of recovery is required before the mutants regain full activity. Although H64 heterozygotes are somewhat sluggish at 20–22°C even without mechanical stimulation, they do not show TS paralysis at 37–38°C. H64, likeDTS1 and DTS2, is lethal when homozygous.
Although DTS1 and DTS2 do not cause bang-sensitive paralysis when maintained at 20–22°C, they do manifest a novel temperature-dependent bang-sensitive phenotype. When the stocks are maintained at 28°C, DTS1/+ andDTS2/+ flies show bang-sensitive paralysis lasting for 5–30 sec when tested at room temperature (20–22°C), even after the flies are allowed to accommodate to the temperature shift for several hours. Thus, all three mutations map to the same chromosome interval, share similar dominant conditional paralytic phenotypes, and are lethal as homozygotes, suggesting the possibility of allelism.
DTS1, DTS2, and H64 areATPalpha alleles
To further test the possibility of allelism of H64,DTS1, and DTS2, complementation tests for recessive lethality were performed in all pair-wise combinations. The results (Table 1) show that all three mutants fail to complement, suggesting that they all share lethal mutations of the same gene.
Table 1.
Viability of existing ATPalpha alleles with the new conditional mutants and their revertants
| DTS1 | DTS1R1 | DTS1R2 | DTS2 | DTS2R1 | DTS2R2 | DTS2R3 | H64 | |
|---|---|---|---|---|---|---|---|---|
| ATPalpha+ | TS, BStd | BS | BS | TS, BStd | BS | mTS, BS | BS | BS |
| DTS1 | ℓ | l | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| DTS1R1 | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| DTS1R2 | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| DTS2 | sℓ, TS | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| DTS2R1 | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| DTS2R2 | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| DTS2R3 | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| H64 | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
| ATPalpha2206 | BS, TS | ℓ | ℓ | dℓ | ℓ | ℓ | ℓ | ℓ |
| ATPAlpha01453 | sℓ, TS | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ | ℓ |
TS, Temperature-sensitive paralysis (38°C); BS, bang-sensitive paralysis; BStd, temperature-dependent bang-sensitive paralysis (28°C); mTS, modified TS; ℓ, early developmental lethal;sℓ, semi-lethal (5 vs expected 25%);dℓ, lethal during pupariation and eclosion. Note: ATPAlpha2206/ATPAlpha01453 flies are not bang-sensitive but have reduced viability (10 vs expected 50% of offspring).
The ATPalpha gene is located in the same chromosome region where recombination mapping placed H64, DTS1, andDTS2. Moreover, ATPalpha2206 is a recessive hypomorphic allele of ATPalpha and confers a bang-sensitive paralytic phenotype because of a P element insertion (Schubiger et al., 1994). These observations raised the possibility that H64, DTS1, and DTS2 were dominant mutations of ATPalpha. Complementation tests withATPalpha2206 orATPalpha01453, a recessive lethal allele also associated with a P element insertion (Feng et al., 1997), support the conclusion that H64, DTS1, andDTS2 are dominant mutations of ATPalpha (Table1).
This conclusion is further supported by the isolation and characterization of revertants of DTS1 and DTS2. We took advantage of the fact that DTS1 and DTS2behave as dominant gain-of-function mutations and therefore should be revertible by second-site mutations within the gene that eliminates its function. Thus, we screened for γ-ray-induced revertants ofDTS1 and DTS2 that were not paralyzed at the restrictive temperature. Two revertants of DTS1(DTS1R1–2) and three revertants ofDTS2 (DTS2R1–3) were recovered. DTS1R2,DTS2R1, andDTS2R2 were all associated with cytologically visible breakpoints in the 93A–B region. The other two revertants appeared cytologically normal. Molecular analysis of these revertants (see below) revealed that all of them contained lesions within ATPalpha, confirming that the originalDTS1 and DTS2 mutations, and by inferenceH64 as well, are alleles of ATPalpha. The revertants were all found to contain lesions consistent with loss-of-function mutations, and all butDTS2R2 appear to be null alleles ofATPalpha. We find no significant difference in phenotype between the heterozygous null revertants and H64heterozygotes, suggesting that the H64 mutation is a loss-of-function allele, probably a null, and that phenotypes arise inH64 and the revertants because of haploinsufficiency ofATPalpha. These data are consistent with the observation that a large deficiency, Df(3R)r-1G6, which removesATPalpha, causes a dominant bang-sensitive phenotype (Lebovitz et al., 1989).
DTS1 and DTS2 cause neuronal hyperexcitability
Activity of the Na+/K+ ATPase is required to maintain ionic gradients and therefore the resting potential across the neuronal cell membrane. Reduction of Na+/K+ ATPase activity with the inhibitor ouabain in vertebrates induces seizure discharges (Bignami and Palladini, 1966). Therefore, we examined the dominant ATPalpha mutations to determine whether they caused any gross defects in neuronal activity. For this analysis, we performed ETRs by placing the recording electrode just beneath the dorsal cuticle to monitor electrical activity from the DLMs. Because the activity of these muscles is driven by motor neurons in the thoracic ganglion, the recorded activity provides a measure of neuronal activity in the flight motor pathway. We discovered a striking temperature-dependent bursting phenotype in DTS1 (Fig. 1), indicating neuronal hyperexcitability in the flight motor pathway. This interpretation is supported by the observation of a similar, but somewhat more severe, phenotype for seizurets2(seits2), a K+channel mutation known to cause neuronal hyperexcitability (Elkins and Ganetzky, 1990; Titus et al., 1997). Similar results (data not shown) were obtained for DTS2. The bursting activity seen inDTS1 and DTS2 is consistent with defects in Na+/K+ ATPase activity that could result in more depolarized membrane resting potentials.
Fig. 1.

Electrophysiological dysfunction inATPalpha mutants. Thoracic recordings from the dorsal flight muscles were performed on wild-type,ATPalphaDTS1, andseizurets2 animals. Recordings were taken starting at 20°C followed by an increase in temperature to 37°C and then returned to 20°C. At elevated temperatures, continuous spiking activity is observed inATPalphaDTS1 andseits2 mutants but not in wild type (n >15 animals per genotype). Eachtrace represents 5 sec. The ordinate is in millivolts.
DTS1 and DTS2 cause shortened life spans and neurodegeneration
In the course of maintaining stocks of DTS1 andDTS2, we discovered that these mutants display a marked age-dependent decrement in locomotor activity. In comparison with age-matched wild-type flies, the mutants become quite sedentary, with a premature loss of both walking activity and flight ability.
To assess the consequence of these age-dependent deficits in a more quantitative manner, we measured the life spans of the mutants. As shown in Figure 2A,DTS1 and to a lesser extent DTS2 andATPalpha2206 are short-lived, whereasH64 has an essentially normal life span. Comparisons of the time required to reach 50% survival for populations of each genotype demonstrate a significant reduction in life span for DTS1,DTS2, and ATPalpha2206 relative to wild type (Fig. 2B).
Fig. 2.

Reduced life span in ATPalphamutants. Survival as a function of age was determined for populations of flies of various genotypes at 28oC.A, Survival curves for four independent populations ofATPalphaDTS1(gray circle),ATPalphaDTS2(gray square),ATPalpha2206(gray triangle),ATPalphaH64 (gray diamond), and ATPalpha+genotypes (black symbols) are shown. Control strains are Ubx/±(UBX), TM6,Tb (TM6)/±, and Canton S (C.S.).B, The time required to reach 50% survivorship for each population was used to compare the life spans ofATPalpha mutants with the controls. Life span is significantly reduced inATPalphaDTS1,ATPalphaDTS2 animals (17 and 27 d) and moderately reduced inATPalpha2206 (36 d) versus controls (41–45 d) (p < 0.001; all comparisons with Canton S). In contrast, the life span ofATPalphaH64/+ flies does not differ significantly from Canton S (p > 0.5).
The premature loss of motor activity exhibited by both dominant TS mutations and the reduction in life span are consistent with the phenotypes manifested by other mutants in Drosophila known to be associated with neurodegeneration (Buchanan and Benzer, 1993;Kretzschmar et al., 1997; Min and Benzer, 1997, 1999). We performed a histological analysis of DTS1/+ and DTS2/+ to examine neurodegeneration in these mutants. Wild-type and mutant adults were aged to ∼50% survival on their respective life span curves and then examined histologically. Serial frontal sections revealed extensive neuropathology in the brains of all ATPalphamutants examined (Fig. 3). InDTS2/+ and DTS1/+ animals, neurodegeneration is evident as the appearance of vacuolar structures distributed widely throughout the central brain and optic regions (Fig.3A,B). Similar vacuolar neuropathology has been observed in several other characterized neurodegeneration mutants identified in Drosophila and is a typical manifestation of neurodegeneration in both flies and mammals (Buchanan and Benzer, 1993; Kretzschmar et al., 1997; Min and Benzer, 1997; Palladino et al., 2002). This phenotype was never observed in wild-type animals, which only rarely contained small vacuolar structures (Fig. 3G). Additionally, many DTS2/+and DTS1/+ animals showed a more extreme phenotype manifested as clusters of large holes that were highly localized in the ventral lateral region of the central brain (Fig.3C,D). H64 andATPalpha2206 were also found to undergo neurodegeneration. In contrast with that seen in the dominant TSATPalpha alleles, the neurodegeneration in H64heterozygotes and ATPalpha2206 homozygotes was less severe, especially inATPalpha2206, and appeared as sporadically localized vacuolar pathology throughout the brain. Another conditional mutant with a profound bursting physiological defect,seits2, was examined for neurodegeneration. In contrast to the massive degeneration seen inDTS1 and DTS2,seits2 shows only sporadic individual large vacuolar structures that were uncommon in age-matched control animals. Histological examination of a number of individuals of each genotype (n >50 for each genotype) demonstrated that the neuropathology observed in DTS1 and DTS2 was 100% penetrant, and the distinctive patterns of neurodegeneration were reproducible for each of the mutants.
Fig. 3.
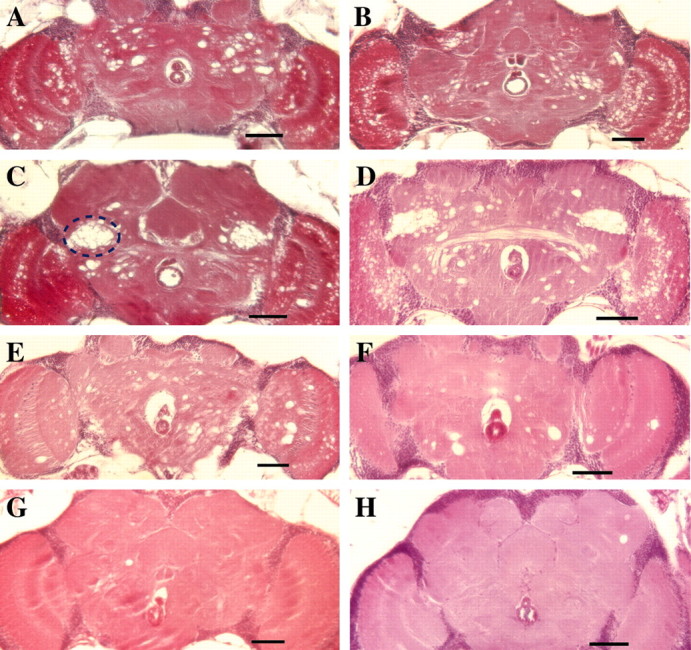
Neurodegeneration in the CNS ofATPalpha mutants. Shown are frontal sections of brains from aged Drosophila. Brains ofATPalphaDTS2/+(A, C)ATPalphaDTS1/± (B, D) animals demonstrate neurodegeneration marked by extensive vacuolar pathology throughout the central brain and optic lobes. The pathology seen in A and B is typical of these genotypes. In addition, for bothATPalphaDTS2/+(C) andATPalphaDTS1/± (D) a number of individuals also demonstrated a distinct region located symmetrically on either side of the brain (dashed outline in C) with more extreme focal neuropathology. The large hole just below the center of the brain in all panels is the esophagus. Neurodegeneration is also observed inATPalphaH64/± (E) andATPalpha2206/ATPalpha2206(F) animals. However, the pathology is less than that observed inATPalphaDTS1/± andATPalphaDTS2/+, particularly forATPalpha2206/ATPalpha2206. Brains from seits2 flies also showed evidence of neuropathology (H). Although this degeneration was much less common and extensive than that seen in ATPalphaDTS1/± andATPalphaDTS2/+, it was more prevalent than that observed in controls. Small vacuolar neuropathology was observed only rarely in aged wild-type flies (G). Tissues from all genotypes examined were from animals aged approximately to the midpoints of their respective survival curves. Scale bar, 50 μm.
Similar histological analysis of young individuals of the same genotypes revealed little or no evidence of neurodegeneration in any of the ATPalpha mutants (Fig. 4). Thus, neurodegeneration in these mutant animals appears to be age dependent and not the result of developmental defects, paralleling the neuropathological onset of many progressive human degenerative conditions.
Fig. 4.

Onset of neurodegeneration inATPalphaDTS1/± andATPalphaDTS2/+flies is age dependent. Frontal sections of brains from young adultATPalphaDTS2/+(A) andATPalphaDTS1/± (B) animals (day 2–3 after eclosion) show little or no overt pathology and do not differ noticeably from wild-type controls. Scale bar, 50 μm.
Similar to vertebrates, a significant proportion of the CNS inDrosophila is found within the thoracic cavity. Sagittal sections of the thoracic ganglion were examined for pathology in young and aged animals. In accord with the results found in the brain, the thoracic ganglion also undergoes age-dependent neurodegeneration inATPalphaDTS1 andATPalphaDTS2 (Fig.5). Thus, the dominant alleles ofATPalpha undergo extensive neurodegeneration that is widely distributed throughout their CNS.
Fig. 5.
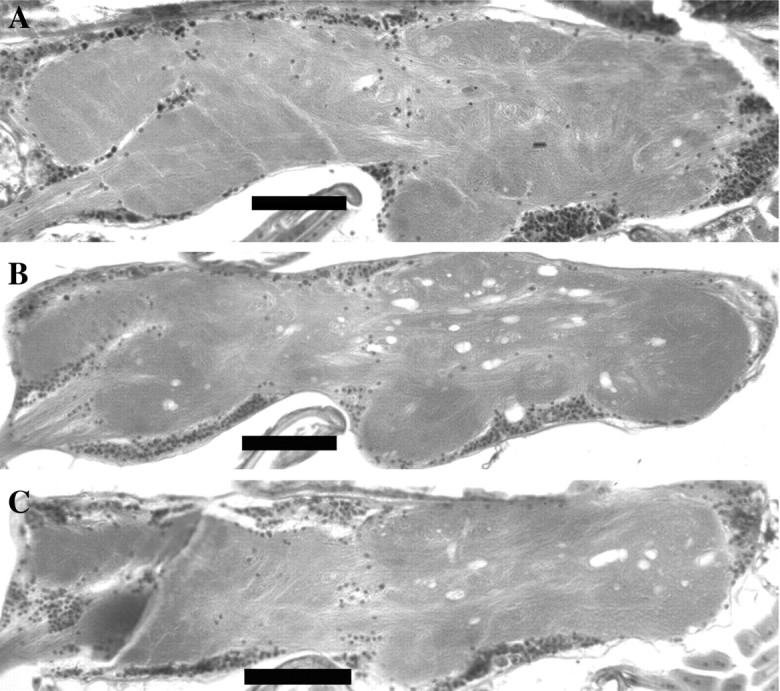
Widespread neurodegeneration throughout the ventral ganglia ofATPalphaDTS1/± andATPalphaDTS2/+flies. Sagittal sections of the thoracic ganglia from aged wild-type (A),ATPalphaDTS1/± (B), andATPalphaDTS2/+(C) flies. Vacuolar pathology is evident in the ganglia of ATPalpha mutants that was never observed in wild-type controls. Tissues from all genotypes examined were from animals aged approximately to the midpoints of their respective survival curves. Scale bar, 50 μm.
DTS2, DTS1, and their revertants have molecular lesions in ATPalpha
To confirm directly the identity of DTS1 andDTS2 as mutations of ATPalpha, we performed molecular analysis of these mutants and their revertants. The revertants, DTS1R2,DTS2R1, andDTS2R2, were all associated with cytologically visible breakpoints in the 93A–B interval and interpreted as mutations causing complete loss of activity of the gene altered by the original DTS1 and DTS2 mutations. Using many pairs of primers directed to the ATPalpha genomic locus, we performed PCR experiments to determine whether the cytological lesions in these revertants fell within theATPalpha gene and to delimit their location on the molecular map. We found that three of these revertants,DTS1R2,DTS2R1, andDTS2R2, had an identifiable molecular lesion that disrupted ATPalpha and would be expectedto abolish its activity (Fig.6A). The remaining two revertants, DTS1R1 andDTS2R3, were not associated with gross physical disruption of the ATPalpha gene by cytological or PCR analysis. However, direct sequence analysis of genomic DNA revealed that DTS1R1 is associated with a 4 bp deletion (ATPalphaΔ2713–16) resulting in a frameshift mutation in the coding region of ATPalphapredicted to cause premature truncation of the protein product resulting in ATPalphaΔ905-C, if in fact any protein is made. Sequence analysis ofDTS2R3 revealed the presence of two point mutations resulting in predicted E to A (39) and L to F (346) substitutions in the ATPalpha protein. Thus, all five revertants ofDTS1 and DTS2 have molecular defects consistent with loss-of-function mutations capable of reverting dominant TS mutations in the ATPalpha gene.
Fig. 6.

Molecular lesions associated withATPalpha mutants. A, Genomic organization of the 25 kb ATPalpha locus. Exons are shown asnumbered boxes, introns are shown aslines, and predicted promoters are shown as bent arrows. Exons shown in black are constitutive, whereas those shown in gray are alternatively spliced. Alternative exons predicted to contain coding sequences are shown aslight gray (0, 15, 1, 6a–d). Sequences of newly identifiedATPalpha exons have been assigned GenBank accession numbers AY174097 and AY174098. Introns are approximately to scale, whereas exons are enlarged. Exon numbering is according to the NCBI database except for exons 6a/b/c/d and 14 and 15, which include newly identified exons. Exons 6b and 6c (formerly predicted exons 6 and 13, respectively in the NCBI database) were identified previously but numbered differently. The location of molecular lesions associated withATPalphaDTS1 andATPalphaDTS2 revertants are shown relative to their location on the genomic map. DTS2R3 contains two point mutations, one each in exons 2 and 4. DTS1R1 has a four-base pair deletion in exon 7. The other revertants have lesions consistent with the location of inversion breakpoints that have been mapped to the regions depicted by dashed lines. Theasterisk indicates the location of the DTS1 and DTS2 mutations shown in C. B, RT-PCR analysis followed by SacI digestion (+) demonstrates a loss of transcripts generated from the mutant homolog in heterozygotes for DTS1R2, DTS2R2 and DTS2R1, the three revertants with cytologically visible inversions breakpoints that disrupt the ATPalphalocus. DTS1R1 appeared to have a reduced level of detectable transcripts generated from the mutant homolog. C, Sequence chromatographic data from genomic PCR products reveals mutations (box) in exon nine ofATPalphaDTS1 (DTS1) andATPalphaDTS2 (DTS2). DTS2 alters a GAC codon to an AAC (D to N), and DTS1 alters a GAG codon to an AAG (E to K).
The effect of these mutations was further verified by generating RT-PCR products from the ATPalpha mRNA isolated fromDTS1, DTS2, H64, and all of the revertants and then digesting these products with the SacI restriction enzyme to distinguish products that derived from mutant allele (SacI sensitive) or wild-type allele (SacI resistant). In each case, the data demonstrated that the mutant chromosomes bearing the primary DTS1, DTS2, andH64 mutations still produced ATPalphatranscripts. However, there was no detectable expression of anATPalpha transcript from theDTS1R2,DTS2R1, orDTS2R2 chromosomes (Fig.6B).
Molecular characterization of the revertants provided a strong indication that the original dominant mutations also resided inATPalpha. We performed direct sequence analysis of genomic DNA to identify the original lesions associated with these mutants (Fig. 6C). Sequence analysis of three control genotypes and the ATPalpha mutants revealed a single base pair change (G to A) in exon 9 of DTS2 that is predicted to cause a D to N substitution affecting residue 981. Remarkably, DTS1 is also associated with a single base pair change (G to A) that results in an apparent E to K substitution of the next residue (982). The mutation inDTS1 destroys a BsmA1 recognition site that was used to confirm the mutation by restriction digestion of genomic PCR products (data not shown). It is extremely unlikely that these changes simply represent silent polymorphisms because they fall within a segment of the protein that is highly conserved overall, and the affected residues in particular are completely invariant in Na+/K+ ATPase α proteins from hydra to humans (Fig.7). In fact, these amino acid residues are even conserved in related H+/K+ATPase α proteins. As expected, these substitutions are still present in the corresponding revertants but are not found in any other control chromosome that we sequenced. These were the only mutations identified that altered the coding potential of the gene inATPalphaDTS1 (DTS1) andATPalphaDTS2 (DTS2) and thus are most likely the cause of the observed phenotypes. Sequencing analysis did not reveal changes that are predicted to change the coding potential of the ATPalpha locus in the H64mutant. In summary, molecular analysis of DTS1 andDTS2 as well as of the revertants derived from these mutants together with the previous genetic complementation data provide definitive evidence that they represent dominant gain-of-function alleles of ATPalpha.
Fig. 7.
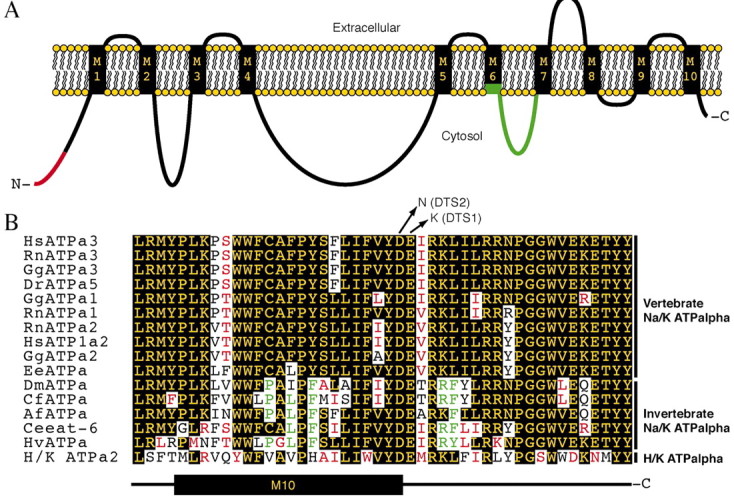
Domain organization of the highly conserved Na+/K+ ATPase proteins.A, Diagram depicting the proposed 10-transmembrane segment model for members of the ATPalpha protein family (Lingrel and Kuntzweiler, 1994). Structural inferences are based on the membrane topology as predicted by Blanco and Mercer (1998). Both the N and C termini are thought to be cytosolic. Boxed regions are predicted transmembrane domain, whereas lines represent the extracellular and cytoplasmic loops. Red depicts the alternative N termini, and green depicts the different coding potentials of alternative exon 6. B, BothATPalphaDTS1 andATPalphaDTS2 mutations cause predicted single amino acid substitutions in the C terminus of the protein. Both of these residues are conserved in all of the Na+/K+ ATPase α subunitproteins shown, which are from representative species throughout the animal kingdom: Hs, Homo sapiens;Rn, Rattus norvegicus; Gg,Gallus gallus; Dr, Danio rerio; Ee, Electrophorus electricus; Dm, Drosophila melanogaster; cf. Ctenocephalides felis, cat flea; As, Artemia franciscana, brine shrimp; Ce, Caenorhabditis elegans;Hv, Hydra vulgaris, hydra. H+/K+ ATPa2 is from Rn and is representative of a related P-type ATPase α family that cotransports H+ and K+. Black shading with yellow lettering represents the consensus among the Na+/K+ ATPase α family (≥50%). Red and black lettering indicateconservative and nonconservative changes from the consensus. Green lettering indicates an invertebrate consensus of ≥80%.
Alternative splicing generates structurally diverseATPalpha proteins
In the course of our molecular analysis of ATPalpha, we uncovered previously undescribed exons for this gene and a complex pattern of alternative splicing that resulted in previously unsuspected complexity in the protein isoforms encoded by ATPalpha. Using primers directed toward exons 4 and 9 for RT-PCR reactions, we identified four exons that appeared to be mutually exclusive and named them 6a, 6b [formerly exon 6, National Center for Biotechnology Information (NCBI) database], 6c (formerly 13), and 6d (Fig.8). All four of the alternative exons are identical in length (94 bp) and have similar coding potentials (Fig.8). In previous studies of ATPalpha inDrosophila, including functional assays, cDNAs that were examined contained the exon corresponding to 6b (Lebovitz et al., 1989;Sun et al., 1998, 2001). Among 123 ATPalpha cDNA subclones (exon 4–9), each was found to contain one and only one member from the set of exons 6a, 6b, 6c, and 6d, indicating that these exons are used as mutually exclusive alternative cassettes.
Fig. 8.

Exon 6 alternative splicing ofATPalpha in Drosophila. A, Genomic organization of the region encompassing exon 5 to exon 7, showing the location of the exon 6 cluster (colored boxes). Amino acid sequence alignments (C) and sequence analysis of cDNA clones suggest that exons 6a, 6b, 6c, and 6d represent mutually exclusive versions of an alternatively spliced cassette. B, Alignment of the nucleotide sequences of the exon 6 cluster demonstrates their sequence similarity and identical size. Metazoan splice-site consensus is shown on the consensus line (y, pyrimidine;R, purine). C, Alignment of the predicted amino acid sequences encoded by the exon 6 cluster. For comparison, the sequences of the corresponding segments from the nematode Eat-6 protein (eat6), the consensus for vertebrate Na+/K+ ATPase α proteins (v-cons.), and the rat H+/K+ ATPa2 protein sequence are included. Absence of shading indicates identity throughout the Na+/K+ATPase family. Yellow and blue boxesindicate differences from the vertebrate consensus that are conservative and nonconservative changes, respectively (dot indicates lack of a strong consensus). Red lettering indicates residues that differ between rat H+/K+ ATPase α and the vertebrate consensus.
Each alternative exon 6 encodes part of the M6 transmembrane segment and the entire M6–M7 intracellular domain of the ATPalpha protein (Fig. 7A). Evolutionary comparisons of this region show that it is highly conserved between worms and humans and that all four alternative exons encode most of these conserved residues (Fig.8C); however, there are intriguing variations among the three isoforms as well. In comparison with α subunits found in other species, exon 6c is most similar to the corresponding region of theCaenorhabditis elegans protein encoded by eat-6and to the sequence encoded by the vertebrate orthologs (Fig.8C). It is of interest that several of the residues that vary among vertebrate ATPalpha Na+/K+paralogs and between H+/K+ATPalpha sequences are those that vary among exons 6a, 6b, 6c, and 6d. These data suggest the possibility that Drosophila generates functionally as well as structurally diverse ATPalpha proteins through alternative splicing of exon 6.
In addition to alternative splicing of exon 6, our molecular characterization of ATPalpha has uncovered extensive alternative splicing at the 5′ end of this gene. Semiquantitative RT-PCR was performed to investigate the relative abundance and diversity of transcripts generated containing exons 0 and 12. Using forward primers with similar melting temperatures directed either toward exon 0 or 12 and a common reverse primer directed toward constitutive exon 3, relative comparisons of RT-PCR products are reasonable provided the annealing temperature is not limiting for either forward primer, the reaction products are similar in size, and the PCR amplification remains exponential through the cycle in which comparisons are being made. Semiquantitative RT-PCR of theATPalpha gene suggests that transcripts containing exon 12 are more abundant and diverse than those containing exon 0 (Fig.9A,B). Exon 12 products become evident approximately four cycles before those from exon 0, suggesting that these transcripts are ∼12- to 16-fold more abundant in adults. Consistent with this interpretation, fluorescence quantification of cycle 20 reaction products revealed that the ratio of ATPalpha products to RP49 product was 5.6 ± 1.1 and 0.4 ± 0.07 for exon 12 and exon 0 products, respectively (error is SEM; n = 3). Also, where exon zero produces only one visible product by RT-PCR, at least four distinct products are evident when the upstream primer is directed toward exon 12 sequences. Analysis of multiple, independently isolated clones of these products is consistent with these interpretations and identifies the most abundant products from exon 12, as well as rare splice products (Fig. 9B). These analyses have identified a new multi-exon, 14/15, and a potentially new translational start in exon 15. These data suggest that three alternate N termini exist for this protein; two long forms with putative translational initiation sites in exons 0 and 15, and a short form with an initiation site in constitutive exon 2 (Fig. 9C,D).
Fig. 9.
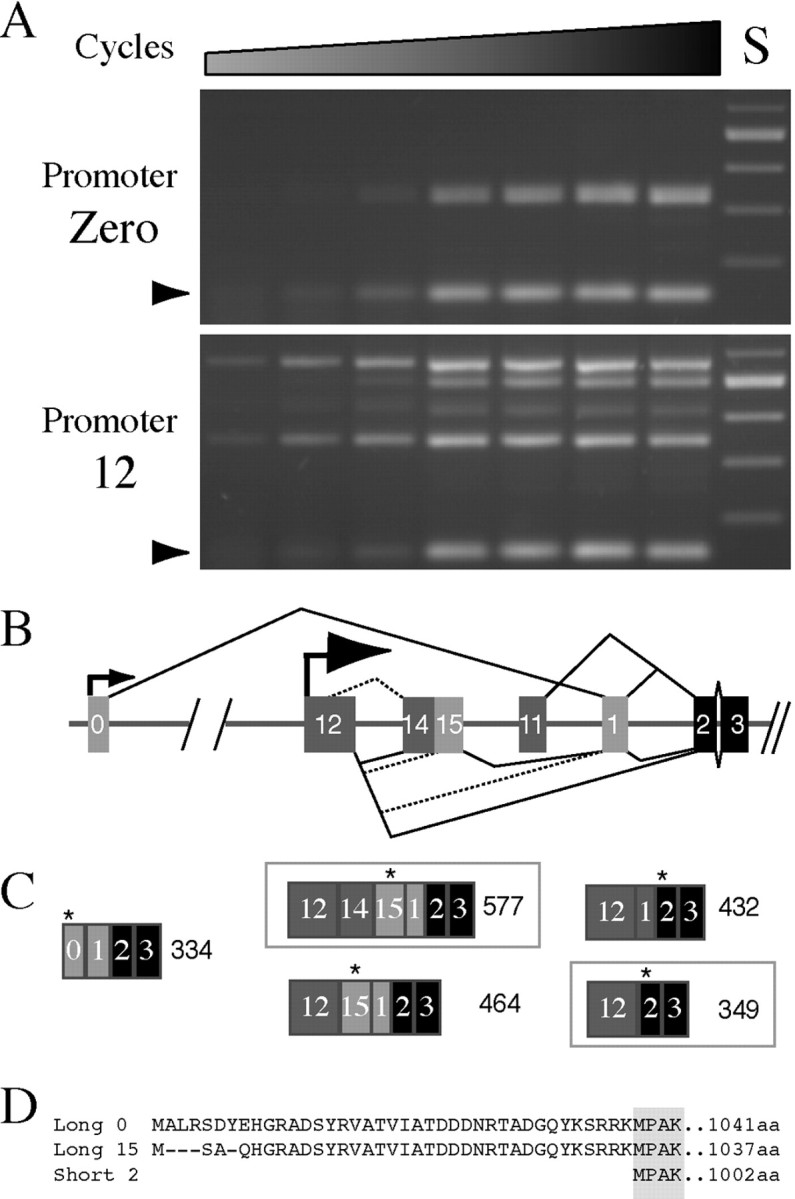
5′ alternative splicing in ATPalphain Drosophila. A, Semiquantitative RT-PCR demonstrates that exon 12 transcripts (bottom panel) are more abundant and diverse than exon 0 transcripts (top panel). Thearrowhead indicates amplification ofRP49. Each lane represents a two-cycle increase in amplification from 16 to 28. Standard (S) is a 100 base pair ladder. B, Molecular analysis of many individually isolated clones has revealed that diverse splice forms are generated from exon 12 that are consistent with the RT-PCR results. We have diagramed all of the apparent splicing events suggested by analysis of these clones.Dashed splicing lines depict less frequent events as determined both by RT-PCR analysis and characterization of cloned products. C, These analyses suggest that exon 0 gives rise to one predominant product (334 bp) and that exon 12 gives rise to many variants, the most abundant of which are highlighted with abox (577 and 349 bp). The first in-frame translational start for each transcript type is indicated by a star.D, The coding potential of the N-terminal ATPalpha isoforms is shown. Black boxes in B andC are constitutive exons, whereas gray boxes depict alternative exons (light gray boxeshave coding potential).
The alternative exons in the exon 6 cluster and those in the 5′ region of the gene were discovered in our sequencing efforts to identify the molecular lesions associated withATPalphaDTS1 andATPalphaDTS2. The coding potential of all of the newly described exons, as well as those previously known, appear wild type in these mutants, with the exception of the mutations discovered to affect residues 981 and 982, as described above.
In summary, the DrosophilaNa+/K+ ATPase α gene harbors previously unrecognized complexity, which results in the generation of multiple different protein isoforms through alternative splicing. Because the Drosophila genome contains only one known gene for the Na+/K+ ATPase α subunit and one predicted paralog (CG17923) rather than at least three or four α subunit genes, as appear to be present in most vertebrate genomes, alternative splicing may be a mechanism for generating diversity produced in vertebrates through the use of multiple structural genes. Although the mutants characterized here do not appear to be defective in the generation or use of specific splice variants, a full understanding of the biological functions of the Na+/K+ ATPase in various cell types will ultimately require a more complete elucidation of why Drosophila encodes and deploys such a multitude of Na+/K+ ATPase isoforms.
Discussion
The existence of Na+/K+ATPases was first hypothesized on the basis of the need for neurons to create and maintain resting membrane potentials (Dean, 1941). A substantial body of research over the past 45 years has amply confirmed the biological importance of this enzyme, which uses energy stored in ATP to generate steep ion gradients required to drive many essential secondary processes. Because of its biological importance, it is not surprising that Na+/K+ ATPase function has been implicated in a wide range of diseases: cardiac hypertrophy, hypertension, renal dysfunction, bipolar mood disorder, and spongiform encephalopathies such as those caused by prion diseases, namely Kuru, Crutzfeld-Jakob disease, and Gerstmann-Straussler-Scheinker syndrome (Herrera et al., 1988;Calandriello et al., 1995; el-Mallakh and Wyatt, 1995; Mynett-Johnson et al., 1998; Mobasheri et al., 2000). Nonetheless, direct mutation of Na+/K+ATPase α subunit genes has not previously been identified as the cause of neural disease or other syndromes in humans. Our data demonstrate that dominant and, to a lesser degree, recessive mutations in Na+/K+ATPalpha in Drosophila cause neural dysfunction, leading to seizures and neurodegeneration.
Neurodegenerative diseases and Na+/K+ ATPase alpha
Although a direct connection between mutations inNa+/K+ATPase alpha genes and neurodegeneration has not been established previously, our data are supported by reports suggesting that loss of Na+/K+ ATPase function can cause neuropathology. These studies include investigations of the phenotypic effects of administering Na+/K+ ATPase α inhibitors (Bignami and Palladini, 1966; Lees and Leong, 1994) and of mutations affecting the Na+/K+ ATPase β2 subunit (Magyar et al., 1994). Both perturbations resulted in neuropathological effects, including neurodegeneration. In addition, a family has been reported in which two affected children with neonatal seizures and spongiform encephalopathy also had reduced ATPase activity (Renkawek et al., 1992). However, these studies have not allowed a definitive connection to be made between altered sodium pump activity and phenotypes such as neurodegeneration. Pharmacological agents often have nonspecific effects, which can complicate their interpretation. The β subunits have several known functions in addition to regulation of the catalytic α subunits, so the precise basis of the phenotypes caused by the β2 knock-out mutation in mice is also uncertain. A genetic basis for the affected human family has not been established, and the etiology of the reduced Na+/K+ ATPase levels in the postmortem brain tissues is unknown. We have found that recessive loss-of-function mutations of ATPalpha cause neurodegeneration in Drosophila. These mutations in the catalytic α subunit cause spongiform neuropathology and provide direct genetic evidence that alterations of the sodium pump lead to dramatic neuropathological phenotypes. Additionally, dominant mutations in the same gene cause even more striking neuropathological defects and are associated with physiological seizure activity. These new mutants will be valuable in studies to elucidate the mechanism by which dysfunction of Na+/K+ATPases leads to neurodegeneration and associated disease conditions.
Hyperexcitability and neurodegeneration inATPalpha mutants
The two dominant TS paralytic mutations of ATPalphacause neuronal hyperexcitablity, which may underlie the associated neuropathology in these animals. The physiological defect is present in very young adults, before the occurrence of any overt neurodegeneration. This result supports the conclusion that neural dysfunction, manifested as hyperexcitability, might lead to the observed neuropathology. However, seits2, a mutation in the gene encoding ERG-type K+ channels (Titus et al., 1997; Wang et al., 1997), which also causes extensive bursting activity (Kasbekar et al., 1987), is not associated with the kind of extensive neurodegeneration seen in ATPalpha mutants. These data suggest that hyperexcitability alone is not sufficient to cause neurodegeneration in Drosophila.
The physiological bursting phenotypes such as those seen inseits2 and reported here for dominantATPase α mutants have been observed in several otherDrosophila behavioral mutants. Such mutants are being used to investigate the physiological basis for seizure disorders such as epilepsy (Kuebler et al., 2001). Our results demonstrate thatATPalpha is another gene that can cause physiological seizures when mutated in particular ways. It will be of interest to determine whether mutations of this gene might have similar phenotypic consequences in mammals. In any case, the dominant ATPalphamutants in Drosophila should provide a very useful experimental model for investigating physiological seizures, neurodegeneration, and the connection between them.
Molecular defects in ATPalpha mutants
The two independent, dominant temperature-sensitiveATPalpha alleles that we examined were found to have both reduced life spans and similar histological and electrophysiological defects. Surprisingly, they both have mutations that alter adjacent, highly conserved residues in the C terminus of the encoded protein. The most parsimonious explanation, given the data presented here, is that the mutations identified are responsible for the phenotypes observed in ATPalphaDTS1 andATPalpha DTS2. These mutations may therefore identify key residues that serve important functional roles. Controlled proteolysis and chemical cross-linking experiments have demonstrated that the C terminus (M8–M10 region) of this protein makes intrasubunit contacts with the M1–M2 region as well as intersubunit contacts with the β subunit (Sarvazyan et al., 1995). Thus, the dominant ATPalpha mutations might perturb one or both of these interactions, affecting regulation of the protein and resulting in gain-of-function phenotypes. Scanning mutagenesis of oxygen-containing residues predicted to be cytosolic or at the membrane/cytosol interface has been performed (Arguello et al., 1999a,b). As such, one of the residues in which we identified a lesion, D981 (D995 in sheep ATPase α 1), has already been the subject of investigation. These studies demonstrate that the D995A mutations do not affect cation–enzyme interaction but do appear to impair the maturation of the protein. The dominant phenotypes that we observed inATPalphaDTS1 and ATPalphaDTS2, which are more severe than those caused by null mutations of the same gene, suggest that these mutations cause a gain-of-function or have a dominant-negative effect. Until recently it was thought that an α−β protomer, which is the minimum unit required for function in vitro, was also the in vivo functional unit of the enzyme, making it more difficult to account for a dominant-negative effect. However, more recent data indicate that the protein may exist as a tetraprotomer in vivo (Donnet et al., 2001; Taniguchi et al., 2001). If the presence of even one mutant subunit could affect the activity or processing of the oligomeric complex, a dominant-negative effect could be readily explained. The dominant ATPalpha mutations could result from a classical gain-of-function, such as misregulated pump activity or altered ion selectivity, or via a dominant-negative model; our data do not distinguish between these possibilities. Because the phenotypes resulting from the dominant mutations are similar but more severe than the recessive phenotypes and mutation of the D981 residue has previously been to impair protein localization, the dominant-negative model seems more likely.
Scanning calorimetry studies of wild-type pig kidney Na+/K+ATPases have uncovered three domains of thermal unfolding, one mapping to the β subunit and two to the α subunit (Grinberg et al., 2001). Possibly, the specific mutant residues identified inATPalphaDTS1 andATPalphaDTS2 further destabilize ATPalpha protein, resulting in some thermal unfolding at temperatures that are permissive for wild-type ATPalpha.
Alternative splicing and functional diversity of Na+/K+ ATPase
We have found that the ATPalpha gene inDrosophila contains extensive 5′ alternative splicing and four alternative versions of exon 6 that are spliced in a mutually exclusive manner. These exons have similar coding potential but differ at residues that may be of key importance to the function of the protein. Thus, 5′ alternative splicing and splicing of this cassette may be an important mechanisms for generating functional diversity of ATPase α subunits.
The cassette encoded by exon 6 extends from I/V796 to R827 (corresponding to V810 to R845 of sheep ATPase α 1) and is predicted to encode the entire cytosolic loop between M6 and M7 and part of the M6 transmembrane domain. Data have demonstrated that the M5–M6 region contains many residues, S775, E779, D804, and D808, that are critical for cation–ATPase interactions (Swarts et al., 1996; Arguello et al., 1999b). The segment including YTLTSNIPEI in the fifth transmembrane segment is especially important in determining ion selectivity of the pump (Jorgensen et al., 1998). Additionally, ADP binding has been shown to protect the M6–M7 cytoplasmic loop against tryptic digestion (Lutsenko and Kaplan, 1994), and the M5–M6 region appears to interact with protein and not lipid (Lutsenko et al., 1995; Beguin et al., 1998), together suggesting that these regions have mechanistic significance to the protein's function. Cysteine scanning of H+/K+– and Na+/K+–ATPases suggests that the intracellular half of M6 is important for energy transduction between the K+ binding site and the phosphorylation site (Asano et al., 2001; Guennoun and Horisberger, 2002). Together, these and other data demonstrate the central importance of the M5–M6 region to ATPase α function. The existence of multiple, alternatively spliced versions of exon 6 in theDrosophila ATPalpha gene suggests that the sequence differences encoded by these alternative exons could have profound functional consequences on pump kinetics, ion selectivity, or regulatory properties. Previous studies of ATPalpha function inDrosophila have used cDNAs that contained the same exon 6 splice variant (exon 6b). The discovery of multiple exons that generate additional structural diversity for this important region of the protein may reveal previously unsuspected functional diversity as well.
Neurodegenerative mechanisms involve ATPalpha
Our data show that both loss-of-function and dominant, possible gain-of-function mutations of ATPalpha cause neurodegeneration, although the effect of the latter is more severe. Mechanistically, this is interesting because excitotoxic amino acids irreversibly inhibit Na+/K+ ATPase activity, the Na+/K+ ATPase is the single most important consumer of ATP in the brain, and seizure activity initiates energy supply failure attributable to the high metabolic requirements of maintaining cation gradients across the plasma membrane [Beal et al. (1993); Lees (1993); and references therein]. Together these findings implicate Na+/K+ATPase function in a diverse array of neurodegenerative conditions arising secondarily from ischemia, seizures, oxidative defects, and mitochondrial encephalopathies, all of which share an underlying neuronal bioenergetic defect. We propose that normal Na+/K+ ATPase function plays a central role in neuronal maintenance and that neuropathogenesis in our mutants is the result of energetic defects in the CNS. The more severe neuropathology observed inATPalphaDTS1 andATPalphaDTS2 is consistent with this model, given the increased energy requirements associated with excitatory seizures.
We have provided genetic evidence that ATPalpha dysfunction in Drosophila is responsible for neural disorders, namely seizures and neurodegeneration. We believe the Na+/K+ ATPase will prove to be a central maintenance protein in the nervous system and that these mutations will prove to be valuable tools to incisively dissect the relevant pathways leading to these neuropathological conditions. These studies establish Drosophila as a model that can be used to investigate the cellular and physiological mechanisms underlying human diseases associated with genetic and nongenetic factors that perturb activity of the Na+/K+ pump. The molecular characterization that we report, revealing previously unsuspected complexity of regulation of ATPalpha at the transcriptional and post-transcriptional levels, will provide further insights into the biological roles of the sodium pump in both normal and disease conditions.
Footnotes
This work was supported by the Jane Coffin Childs Medical Research Foundation (M.J.P.), National Institutes of Health Grant NS15390 (B.G.), and the Wills Foundation (M.J.P.). This is publication number 3600 from the Laboratory of Genetics. We thank Drs. Tim Fergestad, Julie Simpson, and Jay Hirsh for many helpful suggestions with the work and on this manuscript and Adelaide Carpenter for kindly providing the H64 mutation.
Correspondence should be addressed to Michael J. Palladino, Laboratory of Genetics, University of Wisconsin, Madison, 445 Henry Mall, Madison, WI 53706. E-mail: mjpalladino@facstaff.wisc.edu.
References
- 1.Arguello JM, Whitis J, Cheung MC, Lingrel JB. Functional role of oxygen-containing residues in the fifth transmembrane segment of the Na, K-ATPase alpha subunit. Arch Biochem Biophys. 1999a;364:254–263. doi: 10.1006/abbi.1999.1124. [DOI] [PubMed] [Google Scholar]
- 2.Arguello JM, Whitis J, Lingrel JB. Alanine scanning mutagenesis of oxygen-containing amino acids in the transmembrane region of the Na, K-ATPase. Arch Biochem Biophys. 1999b;367:341–347. doi: 10.1006/abbi.1999.1278. [DOI] [PubMed] [Google Scholar]
- 3.Asano S, Io T, Kimura T, Sakamoto S, Takeguchi N. Alanine-scanning mutagenesis of the sixth transmembrane segment of gastric H+, K+-ATPase alpha-subunit. J Biol Chem. 2001;276:31265–31273. doi: 10.1074/jbc.M103698200. [DOI] [PubMed] [Google Scholar]
- 4.Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- 5.Auffray C, Rougeon F. Purification of mouse immunoglobulin heavy-chain messenger RNAs from total myeloma tumor RNA. Eur J Biochem. 1980;107:303–314. doi: 10.1111/j.1432-1033.1980.tb06030.x. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF, Hyman BT, Koroshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci. 1993;16:125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- 7.Beguin P, Hasler U, Beggah A, Horisberger JD, Geering K. Membrane integration of Na, K-ATPase alpha-subunits and beta-subunit assembly. J Biol Chem. 1998;273:24921–24931. doi: 10.1074/jbc.273.38.24921. [DOI] [PubMed] [Google Scholar]
- 8.Bignami A, Palladini G. Experimentally produced cerebral status spongiosus and continuous pseudorhythmic electroencephalographic discharges with a membrane-ATPase inhibitor in the rat. Nature. 1966;209:413–414. doi: 10.1038/209413a0. [DOI] [PubMed] [Google Scholar]
- 9.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 10.Buchanan RL, Benzer S. Defective glia in the Drosophila brain degeneration mutant drop-dead. Neuron. 1993;10:839–850. doi: 10.1016/0896-6273(93)90200-b. [DOI] [PubMed] [Google Scholar]
- 11.Calandriello L, Curini R, Pennisi EM, Palladini G. Spongy state (status spongiosus) and inhibition of Na, K-ATPase: a pathogenetic theory. Med Hypotheses. 1995;44:173–178. doi: 10.1016/0306-9877(95)90132-9. [DOI] [PubMed] [Google Scholar]
- 12.Davis MW, Somerville D, Lee RY, Lockery S, Avery L, Fambrough DM. Mutations in the Caenorhabditis elegans Na, K-ATPase alpha-subunit gene, eat-6, disrupt excitable cell function. J Neurosci. 1995;15:8408–8418. doi: 10.1523/JNEUROSCI.15-12-08408.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dean RB. Theories of electrolyte equilibrium in muscle. Biol Symp. 1941;3:331–348. [Google Scholar]
- 14.Donnet C, Arystarkhova E, Sweadner KJ. Thermal denaturation of the Na, K-ATPase provides evidence for alpha-alpha oligomeric interaction and gamma subunit association with the C-terminal domain. J Biol Chem. 2001;276:7357–7365. doi: 10.1074/jbc.M009131200. [DOI] [PubMed] [Google Scholar]
- 15.el-Mallakh RS, Wyatt RJ. The Na, K-ATPase hypothesis for bipolar illness. Biol Psychiatry. 1995;37:235–244. doi: 10.1016/0006-3223(94)00201-D. [DOI] [PubMed] [Google Scholar]
- 16.Elkins T, Ganetzky B. Conduction in the giant nerve fiber pathway in temperature-sensitive paralytic mutants of Drosophila. J Neurogenet. 1990;6:207–219. doi: 10.3109/01677069009107111. [DOI] [PubMed] [Google Scholar]
- 17.Feng Y, Huynh L, Takeyasu K, Fambrough DM. The Drosophila Na, K-ATPase alpha-subunit gene: gene structure, promoter function and analysis of a cold-sensitive recessive-lethal mutation. Genes Funct. 1997;1:99–117. doi: 10.1046/j.1365-4624.1997.00006.x. [DOI] [PubMed] [Google Scholar]
- 18.Ganetzky B, Wu CF. Drosophila mutants with opposing effects on nerve excitability: genetic and spatial interactions in repetitive firing. J Neurophysiol. 1982;47:501–514. doi: 10.1152/jn.1982.47.3.501. [DOI] [PubMed] [Google Scholar]
- 19.Geering K. Posttranslational modifications and intracellular transport of sodium pumps: importance of subunit assembly. Soc Gen Physiol Ser. 1991;46:31–43. [PubMed] [Google Scholar]
- 20.Grinberg AV, Gevondyan NM, Grinberg NV, Grinberg VY. The thermal unfolding and domain structure of Na+/K+-exchanging ATPase. A scanning calorimetry study. Eur J Biochem. 2001;268:5027–5036. doi: 10.1046/j.0014-2956.2001.02436.x. [DOI] [PubMed] [Google Scholar]
- 21.Guennoun S, Horisberger JD. Cysteine-scanning mutagenesis study of the sixth transmembrane segment of the Na, K-ATPase alpha subunit. FEBS Lett. 2002;513:277–281. doi: 10.1016/s0014-5793(02)02323-2. [DOI] [PubMed] [Google Scholar]
- 22.Hasler U, Wang X, Crambert G, Beguin P, Jaisser F, Horisberger JD, Geering K. Role of beta-subunit domains in the assembly, stable expression, intracellular routing, and functional properties of Na, K-ATPase. J Biol Chem. 1998;273:30826–30835. doi: 10.1074/jbc.273.46.30826. [DOI] [PubMed] [Google Scholar]
- 23.Herrera VL, Chobanian AV, Ruiz-Opazo N. Isoform-specific modulation of Na+, K+-ATPase alpha-subunit gene expression in hypertension. Science. 1988;241:221–223. doi: 10.1126/science.2838907. [DOI] [PubMed] [Google Scholar]
- 24.Hotta Y, Benzer S. Abnormal electroretinograms in visual mutants of Drosophila. Nature. 1969;222:354–356. doi: 10.1038/222354a0. [DOI] [PubMed] [Google Scholar]
- 25.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na, K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell. 1999;3:555–563. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen PL, Nielsen JM, Rasmussen JH, Pedersen PA. Structure-function relationships of E1–E2 transitions and cation binding in Na, K-pump protein. Biochim Biophys Acta. 1998;1365:65–70. doi: 10.1016/s0005-2728(98)00043-7. [DOI] [PubMed] [Google Scholar]
- 27.Kasbekar DP, Nelson JC, Hall LM. Enhancer of seizure: a new genetic locus in Drosophila melanogaster defined by interactions with temperature-sensitive paralytic mutations. Genetics. 1987;116:423–431. doi: 10.1093/genetics/116.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuebler D, Zhang H, Ren X, Tanouye MA. Genetic suppression of seizure susceptibility in Drosophila. J Neurophysiol. 2001;86:1211–1225. doi: 10.1152/jn.2001.86.3.1211. [DOI] [PubMed] [Google Scholar]
- 30.Lebovitz RM, Takeyasu K, Fambrough DM. Molecular characterization and expression of the (Na+ + K+)-ATPase alpha-subunit in Drosophila melanogaster. EMBO J. 1989;8:193–202. doi: 10.1002/j.1460-2075.1989.tb03364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lees GJ. Contributory mechanisms in the causation of neurodegenerative disorders. Neuroscience. 1993;54:287–322. doi: 10.1016/0306-4522(93)90254-d. [DOI] [PubMed] [Google Scholar]
- 32.Lees GJ, Leong W. Brain lesions induced by specific and non-specific inhibitors of sodium-potassium ATPase. Brain Res. 1994;649:225–233. doi: 10.1016/0006-8993(94)91068-5. [DOI] [PubMed] [Google Scholar]
- 33.Lingrel JB, Kuntzweiler T. Na+, K(+)-ATPase. J Biol Chem. 1994;269:19659–19662. [PubMed] [Google Scholar]
- 34.Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- 35.Loughney K, Kreber R, Ganetzky B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- 36.Lutsenko S, Kaplan JH. Molecular events in close proximity to the membrane associated with the binding of ligands to the Na, K-ATPase. J Biol Chem. 1994;269:4555–4564. [PubMed] [Google Scholar]
- 37.Lutsenko S, Anderko R, Kaplan JH. Membrane disposition of the M5–M6 hairpin of Na+, K(+)-ATPase alpha subunit is ligand dependent. Proc Natl Acad Sci USA. 1995;92:7936–7940. doi: 10.1073/pnas.92.17.7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magyar JP, Bartsch U, Wang ZQ, Howells N, Aguzzi A, Wagner EF, Schachner M. Degeneration of neural cells in the central nervous system of mice deficient in the gene for the adhesion molecule on Glia, the beta 2 subunit of murine Na, K-ATPase. J Cell Biol. 1994;127:835–845. doi: 10.1083/jcb.127.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Min KT, Benzer S. Spongecake and eggroll: two hereditary diseases in Drosophila resemble patterns of human brain degeneration. Curr Biol. 1997;7:885–888. doi: 10.1016/s0960-9822(06)00378-2. [DOI] [PubMed] [Google Scholar]
- 40.Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science. 1999;284:1985–1988. doi: 10.1126/science.284.5422.1985. [DOI] [PubMed] [Google Scholar]
- 41.Mobasheri A, Avila J, Cozar-Castellano I, Brownleader MD, Trevan M, Francis MJ, Lamb JF, Martin-Vasallo P. Na+, K+-ATPase isozyme diversity: comparative biochemistry and physiological implications of novel functional interactions. Biosci Rep. 2000;20:51–91. doi: 10.1023/a:1005580332144. [DOI] [PubMed] [Google Scholar]
- 42.Molthagen M, Schachner M, Bartsch U. Apoptotic cell death of photoreceptor cells in mice deficient for the adhesion molecule on glia (AMOG, the beta 2-subunit of the Na, K-ATPase). J Neurocytol. 1996;25:243–255. doi: 10.1007/BF02284800. [DOI] [PubMed] [Google Scholar]
- 43.Mynett-Johnson L, Murphy V, McCormack J, Shields DC, Claffey E, Manley P, McKeon P. Evidence for an allelic association between bipolar disorder and a Na+, K+ adenosine triphosphatase alpha subunit gene (ATP1A3). Biol Psychiatry. 1998;44:47–51. doi: 10.1016/s0006-3223(97)00343-0. [DOI] [PubMed] [Google Scholar]
- 44.Pak WL, Grossfield J, White NV. Nonphototactic mutants in a study of vision of Drosophila. Nature. 1969;222:351–354. doi: 10.1038/222351a0. [DOI] [PubMed] [Google Scholar]
- 45.Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA. 2000;6:1004–1018. doi: 10.1017/s1355838200000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palladino MJ, Hadley TJ, Ganetzky B. Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila. Genetics. 2002;161:1197–1208. doi: 10.1093/genetics/161.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pallanck L, Ordway RW, Ganetzky B. A Drosophila NSF mutant. Nature. 1995;376:25. doi: 10.1038/376025a0. [DOI] [PubMed] [Google Scholar]
- 48.Renkawek K, Renier WO, de Pont JJ, Vogels OJ, Gabreels FJ. Neonatal status convulsivus, spongiform encephalopathy, and low activity of Na+/K(+)-ATPase in the brain. Epilepsia. 1992;33:58–64. doi: 10.1111/j.1528-1157.1992.tb02283.x. [DOI] [PubMed] [Google Scholar]
- 49.Sarvazyan NA, Modyanov NN, Askari A. Intersubunit and intrasubunit contact regions of Na+/K(+)-ATPase revealed by controlled proteolysis and chemical cross-linking. J Biol Chem. 1995;270:26528–26532. doi: 10.1074/jbc.270.44.26528. [DOI] [PubMed] [Google Scholar]
- 50.Schubiger M, Feng Y, Fambrough DM, Palka J. A mutation of the Drosophila sodium pump alpha subunit gene results in bang-sensitive paralysis. Neuron. 1994;12:373–381. doi: 10.1016/0896-6273(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 51.Shima Y, Tada Y, Furuki M, Hara Y, Ohta H. A missense mutation of the gene for Na+, K(+)-ATPase alpha-subunit causes abnormal feeding behavior in Caenorhabditis elegans. Biochem Biophys Res Commun. 1998;248:778–782. doi: 10.1006/bbrc.1998.8981. [DOI] [PubMed] [Google Scholar]
- 52.Sun B, Wang W, Salvaterra PM. Functional analysis and tissue-specific expression of Drosophila Na+, K+-ATPase subunits. J Neurochem. 1998;71:142–151. doi: 10.1046/j.1471-4159.1998.71010142.x. [DOI] [PubMed] [Google Scholar]
- 53.Sun B, Xu P, Wang W, Salvaterra PM. In vivo modification of Na(+), K(+)-ATPase activity in Drosophila. Comp Biochem Physiol [B] Biochem Mol Biol. 2001;130:521–536. doi: 10.1016/s1096-4959(01)00470-5. [DOI] [PubMed] [Google Scholar]
- 54.Swarts HG, Klaassen CH, de Boer M, Fransen JA, De Pont JJ. Role of negatively charged residues in the fifth and sixth transmembrane domains of the catalytic subunit of gastric H+, K+-ATPase. J Biol Chem. 1996;271:29764–29772. doi: 10.1074/jbc.271.47.29764. [DOI] [PubMed] [Google Scholar]
- 55.Taniguchi K, Kaya S, Abe K, Mardh S. The oligomeric nature of Na/K-transport ATPase. J Biochem (Tokyo) 2001;129:335–342. doi: 10.1093/oxfordjournals.jbchem.a002862. [DOI] [PubMed] [Google Scholar]
- 56.Titus SA, Warmke JW, Ganetzky B. The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J Neurosci. 1997;17:875–881. doi: 10.1523/JNEUROSCI.17-03-00875.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang XJ, Reynolds ER, Deak P, Hall LM. The seizure locus encodes the Drosophila homolog of the HERG potassium channel. J Neurosci. 1997;17:882–890. doi: 10.1523/JNEUROSCI.17-03-00882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu CF, Ganetzky B, Jan LY, Jan YN. A Drosophila mutant with a temperature-sensitive block in nerve conduction. Proc Natl Acad Sci USA. 1978;75:4047–4051. doi: 10.1073/pnas.75.8.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]