

Abstract
Multiple trains of high-frequency synaptic stimulation evoke long-term potentiation (LTP) of synaptic transmission in hippocampal area CA1, which has been correlated with hippocampal long-term memory and requires the activation of cAMP-dependent protein kinase (PKA). To assess whether postsynaptic PKA is necessary for the expression of LTP, we made prolonged whole-cell voltage-clamp recordings from CA1 pyramidal neurons in mouse hippocampal slices during postsynaptic infusion of cell-impermeant modulators of PKA. Repeated stimulation (four 100 Hz trains at 5 min intervals) of the Schaffer collateral pathway increased synaptically evoked EPSCs for up to 2 hr. The postsynaptic infusion of either a cell-permeant PKA inhibitor (Rp-cAMPS) or a cell-impermeant PKA inhibitor (PKI6–22) did not alter post-tetanic peak potentiation, but it caused significant decay of EPSCs to pretetanization amplitudes within 1.5 hr. In contrast, postsynaptic infusion of PKI6–22 did not alter a more modest, decaying form of LTP evoked by a single 100 Hz train. Paired-pulse facilitation was unchanged during most of the duration of LTP, suggesting that postsynaptic mechanisms, including PKA activation, are involved in the expression of LTP induced by multitrain stimulation. The postsynaptic infusion of a constitutively active isoform of the PKA catalytic subunit (Cα) into CA1 pyramidal neurons increased EPSC sizes to elicit long-lasting synaptic facilitation. Thus, mimicking the activation of PKA in postsynaptic CA1 pyramidal neurons is sufficient for inducing persistent synaptic facilitation. Activation of apostsynaptic PKA is necessary for the expression of LTP in CA1 pyramidal neurons and is sufficient for initiating persistent synaptic facilitation.
Keywords: synaptic plasticity, hippocampus, LTP, cAMP-dependent protein kinase, catalytic subunit, PKI, whole-cell recording, postsynaptic, pyramidal neurons, protein kinases
Introduction
Protein kinases critically modulate synaptic plasticity in the mammalian hippocampus (Malinow et al., 1989;Nguyen and Kandel, 1996; Kameyama et al., 1998; Lee et al., 2000; for review, see Meffert et al., 1991; Soderling and Derkach, 2000). Hippocampal long-term potentiation (LTP) is an enhancement of synaptic transmission that may be important for regulating neural information storage (Bliss and Lømo, 1973; Bliss and Collingridge, 1993; Martin et al., 2000; Abraham et al., 2002). One particular protein kinase that is critical for hippocampus-dependent long-term memory and for long-lasting LTP in the Schaffer collateral pathway is cAMP-dependent protein kinase (PKA) (Frey et al., 1993; Abel et al., 1997). Pharmacological and genetic inhibition of PKA blocks both long-lasting LTP in area CA1 of hippocampal slices and the formation of long-term memory (Frey et al., 1993; Huang and Kandel, 1994; Blitzer et al., 1995; Impey et al., 1996; Abel et al., 1997; Bernabeu et al., 1997; Nguyen and Kandel, 1997; Otmakhova et al., 2000). PKA activation by a cell-permeant cAMP agonist (Frey et al., 1993) or activation of adenylate cyclase (AC) by a cell-permeant compound, forskolin (FSK) (Chavez-Noriega and Stevens, 1992), elicits synaptic facilitation that occludes LTP induced by tetanization (Frey et al., 1993; Huang and Kandel, 1994).
PKA is activated after high-frequency stimulation that elicits synaptic potentiation (Roberson and Sweatt, 1996), but the exact cellular locus of PKA activation necessary for the expression of long-lasting LTP is unclear. The postsynaptic injection of a cell-impermeant inhibitor of either AC or PKA only slightly reduced the magnitude of LTP without erasing it (Otmakhova et al., 2000). Because these recordings were followed for only 40 min after tetanization (Otmakhova et al., 2000), it remains unclear whether postsynaptic PKA is necessary and sufficient for establishing long-lasting LTP (1–2 hr). The injection of Sp-cAMPS (a cAMP agonist that activates PKA) into postsynaptic CA1 pyramidal neurons elicits mild synaptic facilitation (Blitzer et al., 1995), whereas postsynaptic injection of Rp-cAMPS, a PKA inhibitor, blocks long-lasting LTP in the Schaffer collateral pathway (Blitzer et al., 1995). However, both of these compounds are cell-permeant; thus, a presynaptic site of action for PKA cannot be excluded in experiments using an intracellular injection of these compounds. Indeed, Sp-cAMPS can recruit presynaptic release sites in hippocampal neurons (Bolshakov et al., 1997; Carroll et al., 1998; Ma et al., 1999). Also, Rp-cAMPS and some cAMP analogs may affect other postsynaptic molecular targets in addition to PKA (Otmakhov and Lisman, 2002).
To date, no study has definitively identified postsynaptic CA1 pyramidal neurons as sites of PKA activation critical for long-lasting LTP (1–2 hr duration). In the present study, we asked the following question: Is activation of postsynaptic PKA necessary for establishing long-lasting LTP in CA1 pyramidal neurons? To address this question directly, we used whole-cell patch-clamp recording methods to apply a cell-impermeant, highly specific inhibitor of PKA (PKI6–22 amide), or a catalytic subunit of PKA (to mimic PKA activation), into CA1 pyramidal neurons in mouse hippocampal slices before the induction of LTP by electrical stimulation.
Materials and Methods
Slice preparation. Transverse hippocampal slices from C57BL/6 mice (6–12 weeks of age; Charles River, Montréal, Canada) were prepared as described previously (Nguyen and Kandel, 1997); they were initially incubated in a humidified interface chamber at 28°C. The slices were superfused (1.5 ml/min) with artificial CSF (ACSF) containing (in mm): 124 NaCl, 5 KCl, 10 dextrose, 1.3 MgSO4, 2 CaCl2, 1 NaH2PO4, and 26 NaHCO3, pH 7.35. For whole-cell voltage-clamp recordings, individual slices were transferred to a temperature-controlled submerged chamber (Warner Instruments , Hamden, CT) mounted on an upright microscope (Nikon, Tokyo, Japan) and maintained at 28°C. All data reported were acquired at 28°C; however, we have obtained qualitatively similar results at 32°C (data not shown). Slices were superfused with ACSF bubbled with a gaseous mixture of 95% O2 plus 5% CO2, pH 7.4. The microscope was equipped with infrared differential interference contrast optics for visualization of pyramidal neurons in the stratum pyramidale layer of area CA1. Recordings were obtained from cell bodies ∼50–70 μm beneath the slice surface.
Whole-cell recording and stimulation. Patch pipettes (resistance, 5–6 MΩ) were pulled from 1.5 mm thin-walled borosilicate glass (World Precision Instruments, Sarasota, FL) on a horizontal puller (model P-87; Sutter Instruments, Novato, CA). The pipette solution contained (in mm): 120 potassium gluconate, 25 HEPES, 10 NaCl, 5 MgCl2, 5 K-ATP, 0.3 Na-GTP, 0.2 EGTA, and 2 phosphocreatine, pH 7.35. The initial experiments were performed with cesium-filled electrodes in an attempt to reduce space clamp errors; we found that we could induce robust, long-lasting LTP with this cation (data not shown). However, because cesium increases the activity of mouse PKA catalytic subunits by twofold (Vargas et al., 1999) and elicits prolonged membrane depolarization immediately after tetanic stimulation, we have not used cesium-filled electrodes for our present experiments. The osmolarity of the electrode solution was balanced to 294–298 mOsm with 1 m potassium gluconate. Pipettes were tip-filled with this solution and then back-filled with this same solution plus 10 U/ml creatine phosphokinase and (in some cases) chemical regulators of the cAMP–PKA kinase pathway (see below). A bipolar nickel–chromium stimulating electrode (diameter, 131 μm), placed in the stratum radiatum, was used to evoke whole-cell EPSCs in CA1 pyramidal neurons. The stimulation intensity was adjusted to evoke EPSCs that were 40% of maximal evoked amplitudes (“test intensity”). EPSCs were evoked at a stimulation rate of 0.033 Hz. For clarity, our LTP graphs depict data points at 30 sec intervals and SEs of the means at 2 min intervals. An Axopatch-1D voltage-clamp amplifier (Axon Instruments, Union City, CA) controlled by pClamp-8 software (Axon Instruments) was used to record evoked EPSCs. These were acquired at a holding potential of −90 mV.
In control experiments, 5–10 min after membrane rupture, “baseline” EPSCs were recorded for 20 min at a test stimulation intensity that produced currents of ≈40% maximum. Long-lasting LTP was induced by switching the amplifier to current-clamp mode and delivering four 1 sec, 100 Hz trains at a 5 min intertrain interval (Abel et al., 1997; Nguyen et al., 2000; Woo et al., 2000). In some experiments, only a single 1 sec, 100 Hz train was used to elicit a more modest, decaying form of LTP (Huang and Kandel, 1994). For experiments using PKA blockers, the delay between patch rupture and baseline recording was 10–15 min, to allow 30–35 min of intracellular drug infusion. The recording period for experiments using PKA catalytic subunit α (Cα) began right after patch rupture (1–3 min). In each protocol, individual baseline EPSC amplitudes were normalized against the mean of the baseline EPSC amplitudes. LTP was plotted as the percentage increase in EPSC amplitude relative to this average pretetanization EPSC amplitude (% initial in all figures).
Control LTP experiments (no PKA modulators in the patch pipette) were rejected if the post-tetanization increase in the EPSC amplitude decayed to pretetanization values within 15 min. These LTP failures constituted 18% of the total cells stimulated with the multiburst LTP induction protocol. Potentiated EPSCs were recorded for 2 hr after tetanization or until voltage-clamp fidelity was lost by electrode clogging, membrane resealing, or loss of tight seals. The initial holding current (Ihold) varied from cell to cell (−150 to −300 pA; the holding potential was −90 mV in these experiments) and was monitored continuously during recording to detect electrode blockage, membrane resealing, or loss of tight seals. Series and input resistances were assessed every 5 min by measuring the steady-state currents in response to −5 mV, 50 msec hyperpolarizing steps. An initial series resistance of 11–15 MΩ was deemed acceptable to begin baseline data acquisition; no electronic series resistance compensation was used. For this reason, we terminated experiments that showed a rise or fall in series resistance >15% of the value observed during baseline acquisition.
For cells in which the PKA inhibitors Rp-cAMPS and PKI6–22 were included in the patch pipette, data were rejected by the same criterion. However, the rate of LTP failures was not markedly different from controls (4 of 29 cells), so an increase in failures cannot account for the data presented. For these figures, only cells held for at least 1 hr were included, and all showed a slow return of EPSC amplitude to the pretetanic baseline. In experiments using the catalytic subunit Cα, a slow rise in holding current was always observed that ceased within 10–20 min.
Drugs. The non-NMDA glutamate receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX; Sigma, St. Louis, MO) was added to a final concentration of 20 μmfrom 200 mm stocks in dimethylsulfoxide (DMSO; Sigma). The NMDA receptor antagonist 2-amino-5-phosphonopentanoic acid (APV; Sigma) and the GABAA antagonist bicuculline methiodide (Sigma) were added to superfusate from 50 mm aqueous stocks to final concentrations of 50 and 10 μm, respectively. The AC activator FSK (Sigma) was added to ACSF from a 50 or 100 mm stock in DMSO together with the cyclic nucleotide phosphodiesterase inhibitor 3-isobutyl-1-methyl-xanthine (IBMX; Sigma) from a freshly prepared 10 mm stock in water. PKA inhibitors Rp-cAMPS (Sigma) and PKI6–22 (6–22 amide; Biomol, Plymouth Meeting, PA) (Glass et al., 1989;Dostmann, 1995) were added from concentrated aqueous stock solutions to the pipette solution for final concentrations of 50 and 20 μm, respectively. PKI6–22 was kept frozen in small aliquots at −20°C before use, and the pipette solution with PKI6–22 was back-filled into patch pipettes. To inactivate the inhibitory peptide PKI6–22, we pretreated 20 mm stocks with 300 μm trypsin for 12 hr and then heated the mixture to 95°C in sealed tubes for 2 hr. The PKA catalytic subunit Cα (Sigma) was first dissolved at 500 U/ml in ice-cold distilled water with 3 mg/ml dithiothreitol and stored at 4°C in accordance with the manufacturer's instructions. No loss of activity, as indicated by a rise in EPSC amplitude after patch rupture, was noted over 2–3 weeks. The Cα stock solution was then diluted in an ice-cold pipette solution to a final concentration of 5 U/ml before back-filling the patch pipettes. Inactivated catalytic subunit was prepared by heating aliquots to 90°C for 90 min (Castellucci et al., 1980). In some paired-pulse facilitation (PPF) recordings, cyclothiazide (CTZ; Sigma), an inhibitor of AMPA receptor inactivation (Colquhoun et al., 1992; Trussell et al., 1993), was added to ACSF from a 100 mm stock in methanol (50 μm final concentration). The matching control experiments were performed in 0.02% methanol. PPF was elicited by the delivery of twin stimulus pulses spaced 100 msec apart.
Statistical analysis. Statistical analysis for paired data sets (control LTP vs LTP in the presence of postsynaptic PKA modulators) used Student's t test. To compare multiple data sets, we used two-way ANOVA and a subsequent post hocTukey–Kramer test.
Results
Evaluation of glutamatergic and GABAergic contributions to postsynaptic currents evoked by Schaffer collateral stimulation
Stimulation of the CA1 Schaffer collateral pathway with multiple trains of 100 Hz (spaced 5–10 min apart) induces long-lasting LTP that requires activation of the cAMP–PKA pathway for its full expression and persistence (Huang and Kandel, 1994; Blitzer et al., 1995; Impey et al., 1996; Abel et al., 1997; Wong et al., 1999; Nguyen et al., 2000;Otmakhova et al., 2000; Woo et al., 2000, 2002, 2003). There is genetic and pharmacological evidence that this spaced stimulation protocol preferentially engages PKA-dependent, long-lasting LTP in area CA1 (Woo et al., 2003), and that hippocampal long-term memory in mice is strongly correlated with this type of LTP in area CA1 (Abel et al., 1997). The PKA dependence of LTP has been studied almost exclusively by measuring extracellular field potentials under conditions that preserve the electrophysiological properties of CA3 and CA1 neurons. In this study, we used whole-cell patch clamping and intracellular perfusion of various PKA modulators to examine the role of postsynaptic PKA in long-lasting LTP of glutamatergic transmission. Because the induction of PKA depends on the temporal pattern of presynaptic activity and postsynaptic depolarization, we sought to preserve physiological conditions by using multiple bursts of 100 Hz stimulation in the absence of GABA receptor antagonists, with K+ as the dominant intracellular cation. Indeed, we found that the blockade of GABAA-mediated inhibition with the antagonist bicuculline (10 μm) greatly potentiated the peak magnitude of the postsynaptic depolarization evoked by 100 Hz stimulation (46 ± 5 mV, n = 4 cells in bicuculline; 22 ± 3 mV, n = 5 cells under control conditions) (Fig. 1A). This result underscores the importance of intact inhibition in reducing the postsynaptic response to LTP-inducing presynaptic stimulation.
Fig. 1.
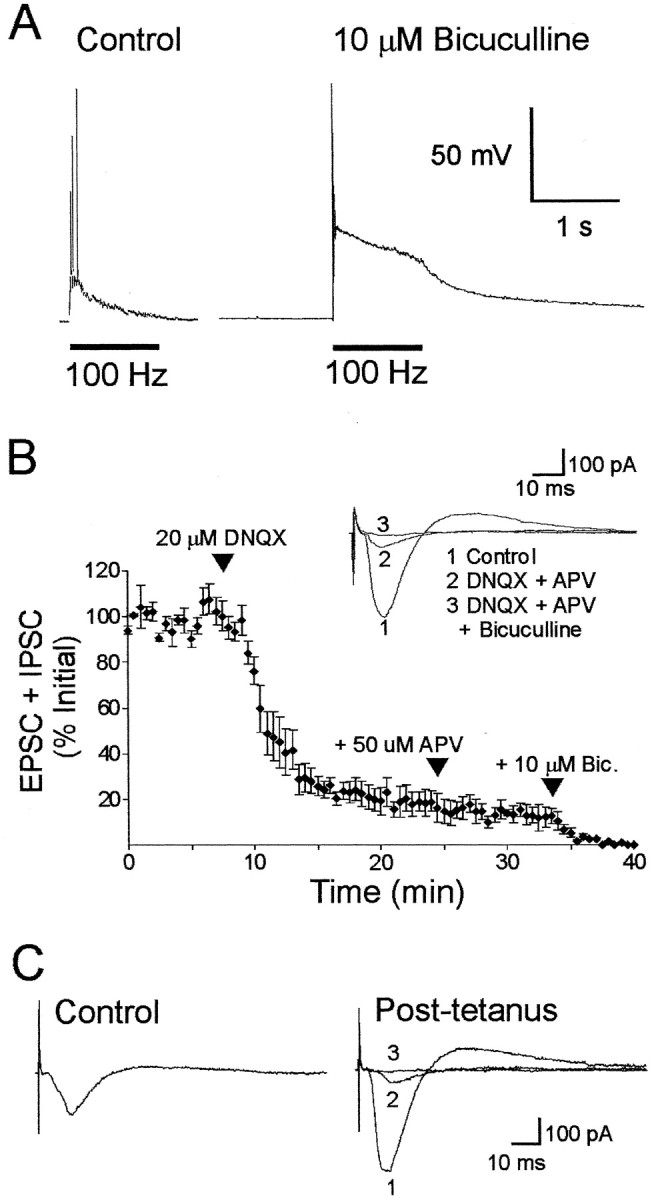
GABAA currents reduce the depolarization in CA1 neurons resulting from high-frequency (100 Hz) stimulation of the Schaffer collaterals, but whole-cell currents in response to low-frequency test stimulation are mediated primarily by glutamate receptors. A, Extracellular bicuculline greatly amplified the postsynaptic depolarization measured under current clamp in response to 100 Hz synaptic stimulation, indicating that GABAA activity reduces the depolarization that induces LTP. B, Postsynaptic currents evoked by low-frequency test stimulation were reduced by 81% after the application of the glutamate receptor antagonist DNQX. The NMDA antagonist APV and the GABAA antagonist bicuculline (Bic.) blocked the remaining current. The GABAA-mediated IPSC comprises only a small fraction (15%) of the total current. C, Postsynaptic currents evoked by test stimulation after LTP induction (4 trains of 1 sec and 100 Hz separated by 5 min) were almost completely blocked by the glutamatergic antagonist DNQX and APV (average, 89%), whereas the remaining current was blocked by bicuculline.
To determine whether GABAergic IPSCs contaminate EPSCs, we measured evoked currents in the presence of glutamate and GABA receptor antagonists (Fig. 1B,C). Application of the non-NMDA glutamate receptor antagonist DNQX (20 μm) reduced the amplitude of the inward current by >80%, and coapplication of DNQX and the NMDA receptor antagonist APV (50 μm) reduced currents by 85%. The remaining current was blocked by the GABAA receptor antagonist bicuculline (average of five cells) (Fig.1B). Ten minutes after the last of four trains used to induce LTP (1 sec, 100 Hz, 5 min apart), this GABAA component was only 11 ± 4% of the peak (average from four cells; sample recording shown in Fig.1C). Thus, the underlying IPSCs made only a small contribution to the peak current and did not confound interpretation of the results presented in the following sections. In subsequent figures, evoked currents are referred to as EPSCs.
Repeated stimulation elicits LTP of EPSCs that is attenuated by a membrane-permeant inhibitor of PKA
The cellular locus of PKA activation critical for the expression of long-lasting LTP remains unidentified. As an initial step toward this end, we first tested the effects of postsynaptic application of a widely used cell-permeant inhibitor of PKA, Rp-cAMPS (Dostmann, 1995). Previous studies have shown that Rp-cAMPS inhibits the expression of long-lasting LTP in area CA1 when it is either bath-applied (Frey et al., 1993; Blitzer et al., 1995; Woo et al., 2000) or injected into postsynaptic CA1 pyramidal neurons (Blitzer et al., 1995; Otmakhov and Lisman, 2002). We found that four 100 Hz trains, separated by 5 min (4 × 100 Hz in all figures), evoked a large, nondecaying potentiation of EPSC amplitudes (Fig. 2,circles). When Rp-cAMPS (50 μm) was included in the patch pipette, LTP decayed to near the pretetanization baseline in every cell held for at least 1.5 hr (Fig. 2,triangles). At 1.5 hr after the last 100 Hz train, mean EPSC sizes were as follows: controls, 200 ± 17%, n = 10 cells; Rp-cAMPS, 119 ± 16%, n = 7 cells (p < 0.01). Postsynaptic infusion of Rp-cAMPS did not reduce baseline EPSC amplitudes (Fig. 2, squares) (n = 5) or the initial magnitude of the potentiation (Fig. 2, triangle at 35 min). Mean EPSC sizes measured immediately after the last 100 Hz train were as follows: controls, 212 ± 18%; Rp-cAMPS, 218 ± 32% (p> 0.1). Thus, Rp-cAMPS inhibits the expression of long-lasting LTP without significantly altering the magnitude of initial potentiation in CA1 pyramidal neurons. For cells treated with Rp-cAMPS, theIhold during the first 110 min after the fourth 100 Hz train was 110 ± 3% (147 measurements inn = 7 cells) of pretetanization baseline values (p < 0.05). This small but statistically significant increase in Iholdby Rp-cAMPS is consistent with similar effects reported by Otmakhov and Lisman (2002). In contrast, Rp-cAMPS did not significantly alter input resistance: treated cells had a mean input resistance during the first 110 min after tetanization that was 108 ± 7% (147 measurements in n = 7 cells) of pretetanization baseline values (p > 0.2). The series resistance also did not change significantly in drug-treated cells during the post-tetanization period: the mean value in treated cells was 102 ± 1% (147 measurements in n = 7 cells) of pretetanization baseline values (p > 0.2).
Fig. 2.

Postsynaptic application of a cell-permeant PKA inhibitor blocks the expression of long-lasting LTP. Four 100 Hz trains 5 min apart evoked potentiation of EPSCs in CA1 pyramidal neurons (circles). Inclusion of a PKA inhibitor, Rp-cAMPS, in the patch pipette caused EPSCs to decay to pretetanization amplitudes within 2 hr after tetanization (triangles). The postsynaptic application of Rp-cAMPS had no significant effect on EPSC amplitudes evoked at test stimulus strength (0.033 Hz;squares). Sample paired EPSC traces from two experiments (top) were measured 20 min before and 1.5 hr after the final 100 Hz train. Rp-cAMPS was applied for the duration of these experiments.
These results show that stable LTP, lasting for >100 min and elicited by repeated high-frequency tetanization of the Schaffer collateral pathway, can be recorded in the whole-cell configuration in mouse hippocampal slices, and that a cell-permeant PKA inhibitor blocks expression of persistent LTP when it is perfused into postsynaptic CA1 pyramidal neurons (Blitzer et al., 1995).
Activation of postsynaptic PKA is necessary for long-lasting LTP in area CA1
Because Rp-cAMPS is cell-permeant, leakage of this drug out of the postsynaptic cell can affect presynaptic and glial PKA activity. Also, there is evidence that the inhibitory effect of Rp-cAMPS on LTP expression in hippocampal slices may occur through its actions on molecular targets that are independent of PKA (Otmakhov and Lisman, 2002). Thus, definitive identification of the cellular locus of PKA activation that is critical for the expression of long-lasting LTP requires the use of a cell-impermeant, highly specific inhibitor of PKA.
To determine whether the activation of postsynaptic PKA is necessary for the expression of long-lasting LTP, we infused PKI6–22, a cell-impermeant and highly specific inhibitory peptide that binds to catalytic subunits of PKA (Glass et al., 1989), into CA1 pyramidal cells (Fig.3A,B). We found that in all experiments that included 20 μmPKI6–22 in the pipette and that were maintained for at least 1.5 hr after tetanic stimulation, the LTP evoked by four 100 Hz trains decayed to near pretetanization baseline levels (mean EPSC sizes at 1.5 hr: controls, 208 ± 18%,n = 7; PKI6–22, 105 ± 15%, n = 6; p < 0.01). PKI6–22 did not affect LTP when it was pretreated with trypsin, a protease that cleaves the peptide (Fig.3C). Mean EPSC sizes measured 40 min after tetanization were as follows: controls, 198 ± 16%; intact PKI6–22, 154 ± 13%; digested PKI6–22, 231 ± 21% (n = 7; p > 0.05 for comparison between digested PKI and controls). Intact PKI6–22 did not significantly alter EPSCs in cells that were not tetanized (Fig. 3B,squares) (n = 6; p > 0.1). It also did not significantly alterIhold, input resistance, or series resistance during the first 100 min after tetanization (means:Ihold, 104 ± 26%; input resistance, 107 ± 8%; series resistance, 104 ± 5% of pretetanization values; p > 0.1 for all parameters; 120 measurements for each parameter from n = 6 cells treated with intact PKI6–22). Finally, the incidence of LTP failures (LTP not lasting 15 min after tetanus; see Materials and Methods) that were excluded from the data pool was not markedly different for either Rp-cAMPS or PKI6–22 (4 of 29 recordings, or 14%, with inhibitors; 18% for controls). Therefore, the reduced stability of long-lasting LTP in the presence of PKA inhibitors represents a disruption of LTP maintenance rather than reduced induction probability. These data provide definitive evidence for a critical requirement for activation of postsynaptic PKA in the expression of stable LTP of excitatory synaptic transmission lasting for up to 100 min in CA1 pyramidal neurons.
Fig. 3.
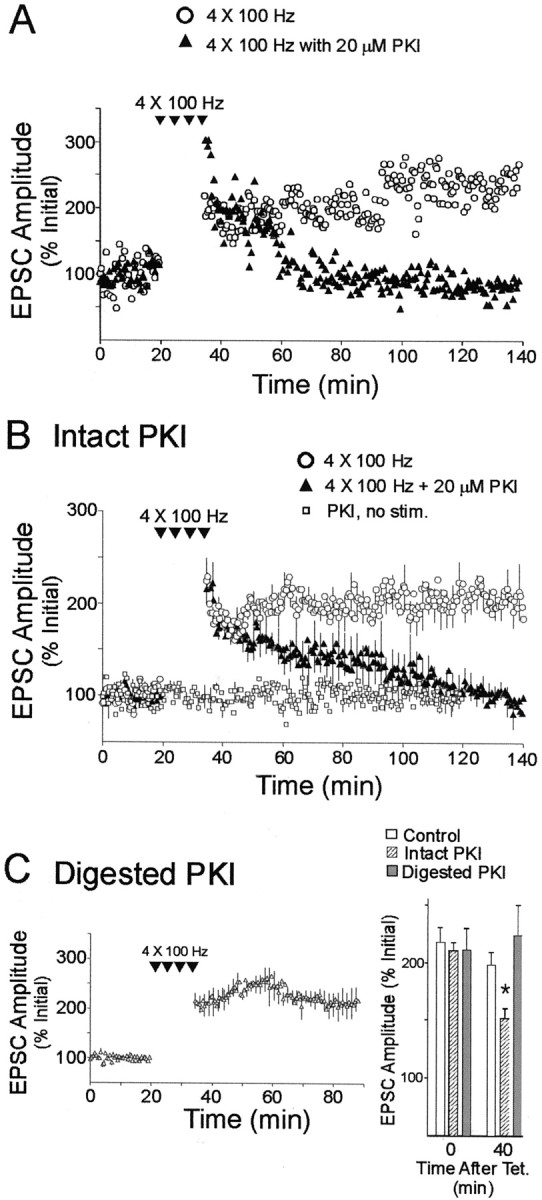
Expression of long-lasting LTP is impaired by the postsynaptic application of a cell-impermeant inhibitory peptide of PKA, PKI6–22 (20 μm). A, Plot of LTP data from two cells, recorded in the absence (circles) and presence (triangles) of postsynaptically applied PKI6–22. B, Averaged data from all experiments. In the presence of postsynaptic PKI6–22, EPSC amplitudes after tetanization decayed to pretetanization values within 100 min after tetanization (triangles). Postsynaptic PKI6–22 did not significantly affect EPSCs evoked at 0.033 Hz, elicited at test stimulus strength (squares). C, Digestion of PKI6–22 with trypsin blocked its inhibition of long-lasting LTP. The average EPSC amplitude measured 40 min after tetanization in the presence of intact PKI6–22 was significantly smaller than that measured with digested PKI6–22 (*p < 0.05; post hoc Tukey–Kramer test).
The requirement for activation of postsynaptic PKA is activity dependent
Studies with cell-permeant inhibitors of PKA have suggested that PKA activation is necessary for the expression of long-lasting forms of LTP induced by multitrain stimulus protocols (Frey et al., 1993; Huang and Kandel, 1994; Woo et al., 2000), whereas a more rapidly decaying form of LTP, elicited by a single 100 Hz train (“single-train LTP”), is believed to be less dependent on PKA (Huang and Kandel, 1994; Blitzer et al., 1995; Otmakhova et al., 2000; Duffy et al., 2001). It is unclear whether the dependence of LTP on activation of postsynaptic PKA is activity dependent. Indeed, Blitzer et al. (1995)have shown that postsynaptic injection of Rp-cAMPS did not affect single-train LTP. However, as stated above, Rp-cAMPS is cell-permeant, and it may affect other cellular loci and processes independent of postsynaptic PKA (Otmakhov and Lisman, 2002). Therefore, like our experiments on multitrain LTP, we used the highly specific and cell-impermeant PKA inhibitor PKI6–22 to test the hypothesis that the activation of postsynaptic PKA is not needed for the expression of single-train LTP.
In support of this idea, we found that infusion of PKI6–22 did not reduce LTP induced by a single 100 Hz train. There was no significant difference between mean EPSC sizes measured in control and PKI-treated cells: 1 hr after tetanization, the mean EPSC amplitude in control cells was 114 ± 12% (n = 5), whereas the mean EPSC size in cells injected with PKI was 121 ± 16% (n = 6;p > 0.1). Moreover, no significant effects on potentiation immediately after tetanization were seen (data not shown). Thus, the dependence of LTP on postsynaptic PKA is activity dependent. Our data are consistent with previous studies showing a relative insensitivity of single-train LTP to inhibitors of PKA (Huang and Kandel, 1994; Blitzer et al., 1995; Otmakhova et al., 2000; Duffy et al., 2001); they provide definitive evidence that activation of postsynaptic PKA differentially regulates the expression of two key forms of LTP in an activity-dependent manner.
PPF is reduced only during the first 5 min of long-lasting LTP
It is unclear whether enhanced presynaptic transmitter release contributes to long-lasting LTP induced by multiple trains of 100 Hz stimulation. It is possible that enhanced presynaptic transmitter release may be evident for only a limited time after repeated tetanization. There is evidence that the induction of a long-lasting LTP by chemical activation of the cAMP–PKA pathway is associated with an increase in the probability of transmitter release and an enhancement of the number of active presynaptic terminals (Bolshakov et al., 1997; Ma et al., 1999; Sokolov et al., 2002). However, these studies did not address the question of whether alterations in the probability of presynaptic transmitter release contribute to PKA-dependent, tetanus-induced LTP.
To address this issue, we examined PPF after the induction of multitrain LTP. PPF is an increase in the amplitude of the second of two EPSCs evoked by closely spaced stimuli; it is believed to result from an increase in presynaptic transmitter release (Zucker, 1999) [for PPF data during LTP, see Zalutsky and Nicoll (1990), Christie and Abraham (1994), Kuhnt and Voronin (1994), Schulz et al. (1994), andWeisskopf et al. (1994)]. PPF should decrease if the probability of presynaptic transmitter release increases. We observed that PPF was significantly reduced during the first 5 min after repeated 100 Hz stimulation (Fig.4A,B), suggesting that presynaptic transmitter release increased during this period. However, PPF returned to pretetanization ratios thereafter (Fig.4A,B). The mean ratios of paired EPSC sizes measured at 0–5, 5–10, and 10–15 min after tetanization were 49 ± 10% (n = 11 cells; 10 paired-pulse measurements per cell), 83 ± 10% (n = 10 cells), and 89 ± 8% (n = 10 cells), respectively, of mean pretetanization ratios (Fig. 4A) (p < 0.05 for the 0–5 min time bin only). At time points after 5 min post-tetanization, no significant differences were observed between mean PPF ratios measured before and after tetanization (Fig.4A) (n = 7 cells per point;p > 0.1).
Fig. 4.

PPF is transiently depressed immediately after tetanization. A, Measurements of PPF before and after tetanization (4 × 100 Hz, 5 min between trains). PPF is transiently depressed during the first 5–10 min after tetanization but recovered to pretetanization amplitudes within 15 min after tetanization. The dotted line indicates the level of PPF before tetanization. B, Sample EPSC traces from one cell showing PPF before (a) and 30 min after (b) induction of multitrain LTP.C, Comparison of post-tetanization PPF in the presence of bath-applied CTZ or vehicle (0.05% methanol). CTZ had no significant effect on PPF after repeated tetanization, compared with controls treated with vehicle (p > 0.05).
These findings suggest that the enhanced probability of presynaptic transmitter release contributes to the first 5 min of long-lasting LTP induced by multitrain stimulation, and that the maintained potentiation of EPSCs seen thereafter may result from postsynaptic mechanisms. The time course of decay of LTP seen after the postsynaptic infusion of PKI6–22 (Fig. 3) supports the latter hypothesis: The potentiation started to decay ∼3–8 min after tetanization in the presence of postsynaptically infused PKI6–22.
It is possible that PPF may be reduced during LTP because of an increase in postsynaptic AMPA receptor desensitization, with little or no increase in the probability of presynaptic transmitter release (Colquhoun et al., 1992; Arai and Lynch, 1996). To address this hypothesis, we examined PPF in the presence of 50 μm CTZ, an inhibitor of AMPA receptor desensitization (Trussell et al., 1993). In the presence of CTZ, the PPF seen within the first 5 min after tetanization (n = 6) was not significantly different from PPF in drug-free vehicle (n = 8) (Fig.4C, striped and graybars at 0–5 min) (p > 0.1), suggesting that the post-tetanic reduction in PPF did not result from the desensitization of postsynaptic AMPA receptors. In the presence of CTZ, PPF at 0–5 min after tetanization was still significantly less than pretetanic PPF in CTZ (61 ± 4%; n = 6; p < 0.05) (Fig. 4C, stripedbar at 0–5 min). In contrast, PPF measured in CTZ 15–20 min after tetanization (112 ± 15%; n = 4) was not significantly different from either pretetanic PPF in CTZ (Fig. 4C, 15–20 min, stripedbar) (p > 0.1) or PPF measured at 15–20 min after tetanization in drug-free vehicle (92 ± 10%; n = 5; p > 0.1) (Fig. 4C, graybar). Thus, AMPA receptor desensitization does not significantly contribute to the depression of PPF seen during the first 5 min of long-lasting LTP induced by multitrain stimulation.
Maintenance of synaptic facilitation evoked by FSK is attenuated by PKI6–22
The results presented above indicate that postsynaptic PKA activation is necessary for the maintenance of long-lasting LTP. Coapplication of the adenylyl cyclase activator FSK and the cyclic nucleotide phosphodiesterase inhibitor IBMX can occlude long-lasting LTP evoked by subsequent tetanic stimulation (Frey et al., 1993; Huang and Kandel, 1994), suggesting that PKA activation is both necessary and sufficient to induce long-lasting LTP. However, the extracellular application of FSK and IBMX could activate PKA both presynaptically and postsynaptically, and FSK has been shown to enhance non-NMDA receptor-mediated currents in cultured hippocampal neurons (Greengard et al., 1991; Wang et al., 1991). To determine the locus of the FSK response, we compared the response of evoked EPSCs to FSK and IBMX coapplication in the presence or absence of intracellular postsynaptic PKI6–22.
In control neurons, the application of FSK and IBMX evoked a large increase in the EPSC that was maintained after washout of the drugs (Fig. 5A, circles) (n = 5 cells). In CA1 neurons in which PKI6–22 was infused for 30–35 min before the application of FSK/IBMX, the initial rise in EPSC amplitude was unaffected, whereas its persistence was significantly attenuated (n = 4 cells). In agreement with previous studies (Bolshakov et al., 1997; Carroll et al., 1998), this result suggests that FSK increases the EPSC amplitude by activating predominantly, but not exclusively, postsynaptic PKA [see Woo et al. (2002) for Rp-cAMPS block of FSK-induced facilitation].
Fig. 5.

Activation of PKA in postsynaptic CA1 pyramidal neurons is sufficient to elicit persistent synaptic facilitation.A, Infusion of PKI6–22 into the postsynaptic CA1 pyramidal neuron attenuates persistence of the increase in evoked EPSCs induced by bath application of FSK and IBMX. Under control conditions, FSK and IBMX caused a rapid increase in EPSC amplitude that was maintained after drug washout. The infusion of PKI6–22 had no effect on the early increase, but it attenuated persistence of the facilitation of EPSCs. B, Postsynaptic application of constitutively active PKA catalytic subunits (Cα) caused a rapid increase in the amplitude of EPSCs (circles) that was not seen in the presence of 20 μm DNQX and 50 μm APV (squares), indicating that Cα augmented the EPSC and not the smaller underlying IPSC. Heat-denatured Cα had no significant effect on EPSC amplitudes (diamonds). The facilitation was independent of the strength of the test stimulation used to elicit EPSCs that were 20 or 40% of maximum evoked amplitudes (circles, triangles). The first point on the graph was measured 1 min after break-in, and the first four EPSCs measured thereafter were used as baseline control amplitudes. This was necessitated by the rapidity of onset of EPSC facilitation seen after break-in with pipettes containing active catalytic subunits. Each pair of sample EPSC traces was measured at 2 and 24 min. The last two pairs of traces share the same calibration bars: 200 pA, 20 msec.
Postsynaptic application of the catalytic subunit of PKA induces facilitation of EPSCs
We have shown that postsynaptic infusion of a cell-impermeant PKA inhibitor blocks the expression of long-lasting LTP. It is unclear whether postsynaptic protein phosphorylation mediated by PKA alone is sufficient for eliciting long-lasting synaptic facilitation. One way to mimic PKA phosphorylation is to infuse catalytic subunits of PKA into neurons (Castellucci et al., 1980). We observed that infusion of constitutively active PKA catalytic subunits (type Cα) into CA1 pyramidal neurons evoked a large and sustained increase in EPSC sizes (Fig. 5B, circles) (n = 9) but had no effect on the IPSC seen in the presence of DNQX and APV (Fig.5B, squares) (mean IPSC size, 100 ± 17%;n = 7). Thus, postsynaptic perfusion of Cα specifically augments the glutamatergic EPSC. In contrast, significantly less enhancement of EPSCs was observed with heat-denatured catalytic subunits (135 ± 24%; n= 9; p < 0.01), indicating that catalytic activity is required for augmentation of the EPSCs (Fig. 5B,diamonds). Moreover, the facilitation was not affected by the strength of the test stimulation. With intact subunits, the mean EPSC size measured 24 min after whole-cell break-in was 344 ± 52% (n = 9) using a stimulus strength that evoked EPSCs that were 40% of maximum amplitudes. With evoked EPSCs that were 20% of maximum sizes, the mean size measured 24 min after break-in with active catalytic subunits was 353 ± 17% (n= 8; p > 0.1). Moreover, the facilitations induced by active catalytic subunits using both stimulus strengths were persistent (for 40% maximum amplitudes: 326 ± 55%, n = 9; for 20% maximum amplitudes: 299 ± 22%, n = 5; both measured at 45 min; data not shown). For both intact and denatured catalytic subunits, no significant changes inIhold, input resistance, and series resistance were seen during the first 20 min after whole-cell break-in (mean values, 131 ± 29, 129 ± 32, and 105 ± 12%, respectively, of mean preinfusion values; n = 9 cells; 20 measurements for each parameter).
Our collective data show that the activation of postsynaptic PKA in CA1 pyramidal neurons is necessary for maintaining long-lasting LTP, and that it is sufficient for initiating persistent synaptic facilitation.
Discussion
Identifying the cellular locus of action of particular protein kinases is an important step toward elucidating the molecular mechanisms of synaptic plasticity. Acute intracellular application of cell-impermeant, highly specific modifiers of PKA has been used successfully in other preparations to directly implicate protein kinases in modulating synaptic plasticity. In Aplysia, injection of PKI into sensory neurons innervating the gill blocked presynaptic facilitation induced by serotonin (Castellucci et al., 1982), whereas the injection of PKA catalytic subunits into the same neurons simulated presynaptic facilitation (Castellucci et al., 1980). In crayfish, injection of PKI into motoneurons blocked the late phase of long-term facilitation of neuromuscular transmission induced by tetanic stimulation (Dixon and Atwood, 1989). In rodent hippocampal slices, injection of peptide inhibitors of PKC and calcium/calmodulin-dependent protein kinase II (CaMKII) into postsynaptic CA1 pyramidal neurons blocked the induction of LTP (Malinow et al., 1989). Postsynaptic injection of PKI also depressed synaptic transmission and occluded the induction of long-term depression (LTD) in CA1 pyramidal cells (Kameyama et al., 1998).
Ours is the first study to show that postsynaptic PKA is necessary for the expression of a form of long-lasting LTP that appears to resemble a late phase of LTP characterized previously by Kandel and colleagues (for review, see Huang et al., 1996; Kandel, 2001). Our low success rate in obtaining temporally extended whole-cell recordings in mouse hippocampal slices (on the time scale of 90–150 min) is explained by a number of serendipitous factors, including the obstruction of recording pipettes and presumed electrode drift. Initially, we tried perforated patch recording using nystatin, but we did not have significant success with this technique, because electrode occlusion by nystatin often occurred well before the planned end points of our recording sessions.Wang et al. (1991) have shown that ATP-regenerating pipette solutions are critical for maintaining robust whole-cell kainate–AMPA currents in hippocampal neurons. Our electrode solution contained, in addition to ATP and GTP, phosphocreatine and creatine phosphokinase, agents that were probably critical for recording over periods of 90–150 min. Indeed, we did not observe decreased baseline EPSC amplitudes when PKI was infused postsynaptically, unlike in a previous study by Kameyama et al. (1998). In this latter study, the rundown of EPSPs was proposed to result from the dephosphorylation of AMPA receptors (Rosenmund et al., 1994). The sharp electrodes used by Kameyama et al. (1998) did not contain ATP-regenerating agents. In our present study, we could frequently maintain recordings for 60–80 min and, much less frequently, for 90–150 min. Thus, despite our very low success rate (12%) in whole-cell recording of EPSCs for 90–150 min, such recordings are possible under certain conditions.
The blockade of PKA using PKI6–22 has been shown to attenuate, but not block, LTP evoked by a stimulation protocol consisting of only 40 pulses (Otmakhova et al., 2000). The PKI6–22 concentration used in that study was significantly higher than that used in our present experiments (2 mm vs 20 μm). The high potency of PKA inhibition by PKI6–22(Ki = 1.7 nm) (Glass et al., 1989) suggests that, in both studies, there should be substantial inhibition of PKA activity in injected cells. Measuring PKA activity in single cells is difficult, but PKI has been shown to inhibit basal and cAMP-evoked PKA activity in mouse hippocampal slice extracts by >90% (Abel et al., 1997). Our data reveal that a relatively low concentration of PKI6–22 in the pipette solution (20 μm) is sufficient for assessing the necessity for postsynaptic PKA activation in long-lasting LTP. It is noteworthy that Kameyama et al. (1998) also used 20 μm PKI6–22 to occlude LTD induction in postsynaptic CA1 neurons in slices. Diffusion of PKI6–22 to the apical dendrites of pyramidal cells has been estimated to take ∼40 min or longer (Otmakhova et al., 2000). This factor was considered in our experiments: our recording period before the end of tetanization was 35 min in duration for the PKI6–22 experiments. Although the effective concentration of applied PKI6–22 at synaptic sites is unknown, it is clear that it is not necessary to use millimolar pipette concentrations of PKI6–22 to achieve inhibition of some types of synaptic modifications in hippocampal neurons.
Single-train LTP was not blocked by postsynaptic application of PKI6–22, whereas long-lasting LTP was impaired by the postsynaptic application of PKI6–22. This suggests that activation of postsynaptic PKA is sensitive to the amount of imposed stimulation. These findings are consistent with previous studies that have shown selective impairment of multitrain LTP (but not of single-train LTP) by pharmacological and genetic inhibition of PKA (Frey et al., 1993; Matthies and Reymann, 1993; Huang and Kandel, 1994;Blitzer et al., 1995; Impey et al., 1996; Abel et al., 1997). In these studies, the blockade of PKA by cell-permeant inhibitors, or genetic inhibition of PKA in hippocampal neurons, did not permit definitive identification of the critical cellular locus of PKA activation. Our present findings with PKI6–22 suggest that the critical site of PKA activation in these previous studies may likely be the pyramidal cells of CA1. Also, Woo et al. (2003) have shown, by using field recordings in combination with genetic and pharmacological manipulations of PKA, that multitrain spaced stimulation (identical to that used here) preferentially elicits PKA-dependent forms of long-lasting LTP in mouse hippocampal slices. Thus, from a broader perspective, the present data consolidate the idea that the engagement of protein kinase-dependent forms of synaptic plasticity is sensitive to specific amounts of synaptic stimulation.
Our data also underscore possible mechanistic differences between chemically induced and tetanus-induced forms of hippocampal synaptic potentiation. The pharmacological activation of PKA by FSK or by cAMP analogs increases presynaptic transmitter release (Chavez-Noriega and Stevens, 1992; Carroll et al., 1998). cAMP analogs that activate PKA, such as Sp-cAMPS, can induce long-lasting LTP that correlates with the recruitment of previously silent release sites in presynaptic terminals of cultured hippocampal neurons (Bolshakov et al., 1997; Ma et al., 1999; Bozdagi et al., 2000; Zakharenko et al., 2001). Also, chemical activation of the cAMP–PKA pathway can directly modulate transmitter secretion in a calcium-independent manner in hippocampal presynaptic terminals (Trudeau et al., 1996). In the present study, we have examined only tetanus-induced LTP and found that PPF was significantly depressed only during the first 5 min after tetanization. This suggests that increased probability of transmitter release only briefly contributes to the induction of long-lasting LTP induced by repeated tetanization. This is consistent with published data supporting postsynaptic mechanisms for LTP induction in CA1 (Malinow et al., 1989;Wu and Saggau, 1994; for review, see Malenka and Nicoll, 1999). Our data do not rule out presynaptic contributions to the expression of tetanus-induced LTP at later time points after tetanization. For example, the development of new boutons (Ma et al., 1999) could increase total glutamate release without affecting PPF, and quantal analysis of EPSPs has suggested some presynaptic contribution for late LTP (Sokolov et al., 2002). In other hippocampal pathways, such as the mossy fiber–CA3 pathway, tetanus-induced LTP is cAMP dependent and appears to require presynaptic mechanisms (Weisskopf et al., 1994). However, there is evidence to support a postsynaptic locus of expression for PKA-dependent LTP induced by tetanization. Long-lasting LTP, induced by a stimulation regimen similar to ours, was blocked when apical dendrites were severed from the cell bodies of CA1 pyramidal neurons in slices (Frey et al., 1989). Because LTP induced by this multitrain stimulation regimen is PKA dependent (Frey et al., 1993;Matthies and Reymann, 1993) and also requires the PKA-dependent synthesis of postsynaptic AMPA receptors for its expression (Nayak et al., 1998), these considerations support our hypothesis that postsynaptic mechanisms, including the activation of postsynaptic PKA, are critical for the expression of long-lasting, tetanus-induced LTP.
Which postsynaptic targets are phosphorylated by PKA and required for long-lasting LTP? The list of potential candidates is long, but PKA-mediated phosphorylation of glutamatergic receptor subunit 1 (GluR1) at serine-845 is important for the bidirectional regulation of hippocampal synaptic plasticity (Banke et al., 2000; Lee et al., 2000) [for GluR4 data, see Carvalho et al. (1999)], and PKA activators can increase EPSC amplitudes by promoting PKA-mediated phosphorylation of AMPA receptors (Wang et al., 1991). NMDA receptor-mediated currents are also increased by PKA-mediated phosphorylation (Raman et al., 1996;Leonard and Hell, 1997). PKA-mediated phosphorylation of a protein phosphatase inhibitor, I-1, could allow for the suppression of protein phosphatase-1, and the subsequent disinhibition of CaMKII may gate the expression of long-lasting LTP at postsynaptic sites (Blitzer et al., 1995, 1998; Brown et al., 2000). Finally, fusion proteins, consisting of PKI and nuclear localization signals, are capable of blocking long-lasting LTP in hippocampal slices when they are delivered into the nuclear compartment of neurons (Matsushita et al., 2001). This block of LTP was caused by the inhibition of cAMP response element-binding protein (CREB) phosphorylation (Matsushita et al., 2001). Thus, CREB may be another postsynaptic target for PKA that is critical for the expression of long-lasting LTP in CA1 pyramidal neurons (Impey et al., 1996).
Footnotes
This work was supported by equipment and operating grants from the Alberta Heritage Foundation for Medical Research (P.V.N.), the Canadian Institutes of Health Research (formerly the Medical Research Council of Canada) (P.V.N.), the Alberta Paraplegic Foundation (P.V.N.), and the Natural Sciences and Engineering Research Council of Canada (P.V.N.). S.D. held a postdoctoral fellowship from the Alberta Heritage Foundation for Medical Research (AHFMR). P.V.N. is an AHFMR Scholar and a Medical Research Council Scholar.
Correspondence should be addressed to Dr. S. Duffy, Department of Physiology, University of Alberta, Medical Sciences Building, Edmonton, Alberta, T6G 2H7, Canada. E-mail: sduffy@ualberta.ca.
References
- 1.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Abraham WC, Logan B, Greenwood JM, Dragunow M. Induction and experience-dependent consolidation of stable long-term potentiation lasting months in the hippocampus. J Neurosci. 2002;22:9626–9634. doi: 10.1523/JNEUROSCI.22-21-09626.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arai A, Lynch G. Response to repetitive stimulation of AMPA receptors in patches excised from fields CA1 and CA3. Brain Res. 1996;716:202–206. doi: 10.1016/0006-8993(96)00030-3. [DOI] [PubMed] [Google Scholar]
- 4.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernabeu R, Bevilaqua L, Ardenghi P, Bromberg E, Schmitz P, Bianchin M, Izquierdo I, Medina JH. Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning of rats. Proc Natl Acad Sci USA. 1997;94:7041–7046. doi: 10.1073/pnas.94.13.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 7.Bliss TVP, Lømo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 9.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1943. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 10.Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during cAMP-dependent late phase LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 11.Bozdagi O, Shan W, Tanaka H, Benson DL. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- 12.Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM. Long-term potentiation induced by theta-frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci. 2000;20:7880–7887. doi: 10.1523/JNEUROSCI.20-21-07880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll RC, Nicoll RA, Malenka RC. Effects of PKA and PKC on miniature excitatory postsynaptic currents in CA1 pyramidal cells. J Neurophysiol. 1998;80:2797–2800. doi: 10.1152/jn.1998.80.5.2797. [DOI] [PubMed] [Google Scholar]
- 14.Carvalho AL, Kameyama K, Huganir RL. Characterization of phosphorylation sites on the glutamate receptor 4 subunit of the AMPA receptors. J Neurosci. 1999;19:4748–4754. doi: 10.1523/JNEUROSCI.19-12-04748.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castellucci VF, Kandel ER, Schwartz JH, Wilson FD, Nairn AC, Greengard P. Intracellular injection of the catalytic subunit of cAMP-dependent protein kinase simulates facilitation of transmitter release underlying behavioural sensitization in Aplysia. Proc Natl Acad Sci USA. 1980;77:7492–7496. doi: 10.1073/pnas.77.12.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castellucci VF, Nairn AC, Greengard P, Schwartz JH, Kandel ER. Inhibitor of adenosine 3′:5′-monophosphate-dependent protein kinase blocks presynaptic facilitation in Aplysia. J Neurosci. 1982;2:1673–1681. doi: 10.1523/JNEUROSCI.02-12-01673.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chavez-Noriega LE, Stevens CF. Modulation of synaptic efficacy in field CA1 of the rat hippocampus by forskolin. Brain Res. 1992;574:85–92. doi: 10.1016/0006-8993(92)90803-h. [DOI] [PubMed] [Google Scholar]
- 18.Christie BR, Abraham W. Differential regulation of paired-pulse plasticity following LTP in the dentate gyrus. NeuroReport. 1994;5:385–388. doi: 10.1097/00001756-199401120-00003. [DOI] [PubMed] [Google Scholar]
- 19.Colquhoun D, Jonas P, Sakmann B. Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurons of rat hippocampal slices. J Physiol (Lond) 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon D, Atwood HL. Adenylate cyclase system is essential for long-term facilitation at the crayfish neuromuscular junction. J Neurosci. 1989;9:4246–4252. doi: 10.1523/JNEUROSCI.09-12-04246.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dostmann WR. (Rp)-cAMPS inhibits the cAMP-dependent protein kinase by blocking the cAMP-induced conformational transition. FEBS Lett. 1995;375:231–234. doi: 10.1016/0014-5793(95)01201-o. [DOI] [PubMed] [Google Scholar]
- 22.Duffy SN, Craddock KJ, Abel T, Nguyen PV. Environmental enrichment modifies the PKA-dependence of hippocampal LTP and improves hippocampus-dependent memory. Learn Mem. 2001;8:26–34. doi: 10.1101/lm.36301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frey U, Krug M, Brodemann R, Reymann K, Matthies H. Long-term potentiation induced in dendrites separated from rat's CA1 pyramidal somata does not establish a late phase. Neurosci Lett. 1989;97:135–139. doi: 10.1016/0304-3940(89)90152-3. [DOI] [PubMed] [Google Scholar]
- 24.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 25.Glass DB, Lundquist LJ, Katz BM, Walsh DA. Protein kinase inhibitor-(6–22)-amide peptide analogs with standard and nonstandard amino acid substitutions for phenylalanine 10: inhibition of cAMP-dependent kinase. J Biol Chem. 1989;264:14579–14584. [PubMed] [Google Scholar]
- 26.Greengard P, Jen J, Nairn AC, Stevens CF. Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science. 1991;253:1135–1138. doi: 10.1126/science.1716001. [DOI] [PubMed] [Google Scholar]
- 27.Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- 28.Huang YY, Nguyen PV, Abel T, Kandel ER. Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn Mem. 1996;3:74–85. doi: 10.1101/lm.3.2-3.74. [DOI] [PubMed] [Google Scholar]
- 29.Impey S, Mark M, Villacres EC, Poser S, Chavki C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 30.Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic PKA substrate in expression of homosynaptic LTD. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- 31.Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 32.Kuhnt U, Voronin L. Interaction between paired-pulse facilitation and LTP in area CA1 at guinea pig hippocampal slices: application of quantal analysis. Neuroscience. 1994;62:391–394. doi: 10.1016/0306-4522(94)90374-3. [DOI] [PubMed] [Google Scholar]
- 33.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 34.Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- 35.Ma L, Zablow L, Kandel ER, Siegelbaum SA. Cyclic AMP induces functional presynaptic boutons in hippocampal CA3-CA1 neuronal cultures. Nat Neurosci. 1999;2:24–30. doi: 10.1038/4525. [DOI] [PubMed] [Google Scholar]
- 36.Malenka RC, Nicoll RA. Long-term potentiation: a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 37.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 38.Martin S, Grimwood P, Morris RGM. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 39.Matsushita M, Tomizawa K, Moriwaki A, Li ST, Terada H, Matsui H. A high-efficiency protein transduction system demonstrating the role of PKA in long-lasting LTP. J Neurosci. 2001;21:6000–6007. doi: 10.1523/JNEUROSCI.21-16-06000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthies H, Reymann KG. Protein kinase A inhibitors prevent the maintenance of hippocampal LTP. NeuroReport. 1993;4:712–714. doi: 10.1097/00001756-199306000-00028. [DOI] [PubMed] [Google Scholar]
- 41.Meffert MK, Parfitt KD, Doze VA, Cohen GA, Madison DV. Protein kinases and long-term potentiation. Ann NY Acad Sci. 1991;627:2–9. doi: 10.1111/j.1749-6632.1991.tb25909.x. [DOI] [PubMed] [Google Scholar]
- 42.Nayak A, Zastrow DJ, Lickteig R, Zahniser NR, Browning MD. Maintenance of late-phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature. 1998;394:680–683. doi: 10.1038/29305. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen PV, Kandel ER. A macromolecular synthesis-dependent late phase of long-term potentiation requiring cAMP in the medial perforant pathway of rat hippocampal slices. J Neurosci. 1996;16:3189–3198. doi: 10.1523/JNEUROSCI.16-10-03189.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen PV, Kandel ER. Brief theta-burst stimulation induces a transcription-dependent late phase of LTP requiring cAMP in area CA1 of the mouse hippocampus. Learn Mem. 1997;4:230–243. doi: 10.1101/lm.4.2.230. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen PV, Duffy SN, Young JZ. Differential maintenance and frequency-dependent tuning of LTP at hippocampal synapses of specific strains of inbred mice. J Neurophysiol. 2000;84:2484–2493. doi: 10.1152/jn.2000.84.5.2484. [DOI] [PubMed] [Google Scholar]
- 46.Otmakhov N, Lisman JE. Postsynaptic application of a cAMP analogue reverses LTP in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2002;87:3018–3032. doi: 10.1152/jn.2002.87.6.3018. [DOI] [PubMed] [Google Scholar]
- 47.Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE. Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2000;20:4446–4451. doi: 10.1523/JNEUROSCI.20-12-04446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raman IM, Tong G, Jahr CE. β-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 49.Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- 50.Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;378:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- 51.Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in LTP. J Neurosci. 1994;124:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- 53.Sokolov MV, Rossokhin AV, Astrelin AV, Frey JU, Voronin LL. Quantal analysis suggests strong involvement of presynaptic mechanisms during the initial 3 h maintenance of LTP in rat hippocampal CA1 area in vitro. Brain Res. 2002;957:61–75. doi: 10.1016/s0006-8993(02)03600-4. [DOI] [PubMed] [Google Scholar]
- 54.Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- 55.Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- 56.Vargas G, Yeh T-Y, Blumenthal DK, Lucero MT. Common components of patch-clamp internal recording solutions can significantly affect protein kinase A activity. Brain Res. 1999;828:169–173. doi: 10.1016/s0006-8993(99)01306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang LY, Salter MW, MacDonald JF. Regulation of kainate receptors by cAMP-dependent protein kinase and phosphatases. Science. 1991;253:1132–1135. doi: 10.1126/science.1653455. [DOI] [PubMed] [Google Scholar]
- 58.Weisskopf MG, Castillo PE, Zalutski RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 59.Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- 60.Woo NH, Duffy SN, Abel T, Nguyen PV. Genetic and pharmacological demonstration of differential recruitment of cAMP-dependent protein kinases by synaptic activity. J Neurophysiol. 2000;84:2739–2745. doi: 10.1152/jn.2000.84.6.2739. [DOI] [PubMed] [Google Scholar]
- 61.Woo NH, Abel T, Nguyen PV. Genetic and pharmacological demonstration of a role for cyclic AMP-dependent protein kinase-mediated suppression of protein phosphatases in gating the expression of late LTP. Eur J Neurosci. 2002;16:1871–1876. doi: 10.1046/j.1460-9568.2002.02260.x. [DOI] [PubMed] [Google Scholar]
- 62.Woo NH, Duffy SN, Abel T, Nguyen PV (2003) Temporal spacing of synaptic stimulation critically modulates the dependence of LTP on cyclic AMP-dependent protein kinase. Hippocampus, in press. [DOI] [PubMed]
- 63.Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- 65.Zalutsky RA, Nicoll RA. Comparison of two forms of LTP in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- 66.Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]