

Abstract
Microglia motility plays a crucial role in response to lesion or exocytotoxic damage of the cerebral tissue. We used two in vitro assays, a wound-healing model and a chemotaxis assay, to show that the neuropeptide neurotensin elicited the migration of the human microglial cell line C13NJ by a mechanism dependent on both phosphatidylinositol 3-kinase (PI 3-kinase) and mitogen-activated protein (MAP) kinase pathways. The effect of neurotensin on cell migration was blocked by the neurotensin receptor-3 propeptide, a selective ligand of this receptor. We demonstrate, by using RT-PCR, photoaffinity labeling, and Western blot analysis, that the type I neurotensin receptor-3 was the only known neurotensin receptor expressed in these microglial cells and that its activation led to the phosphorylation of both extracellular signal-regulating kinases 1/2 and Akt. Furthermore, the effect of neurotensin on cell migration was preceded by a profound modification of the F-actin cytoskeleton, particularly by the rapid formation of numerous cell filopodia. Both the motility and the filopodia appearance induced by neurotensin were totally blocked by selective inhibitors of MAP kinases or PI 3-kinase pathways. This demonstrates that the neurotensin receptor-3 is functional and mediates the migratory actions of neurotensin.
Keywords: human microglia, neurotensin receptor-3, signaling, sortilin, migration, PI 3-kinase, MAP kinase
Introduction
Brain macrophage microglia are considered to function as monocytes or tissue macrophages of the somatic immune system (Gehrmann et al., 1995). These cells, when activated after a brain injury, proliferate in situ(Marty et al., 1991) and display directed migration to eliminate damaged neurons or invading microorganisms (Brockhaus et al., 1996). Several neuropeptides have been described to modulate the immune responses in the peripheral system (De la Fuente et al., 1998; Delgado et al., 1999). Thus, the neuropeptide neurotensin (NT) has been reported to regulate some cellular functions of the peripheral immune system (Koff and Dunegan, 1985; Garrido et al., 1992; De la Fuente et al., 1993), including the cutaneous inflammatory process (Ramez et al., 2001). However, the mechanism of NT action and the receptor type involved in the effects of the peptide on central macrophages remain unknown. NT responses are known to be mediated through at least three receptors identified to date (for review, see Vincent et al., 1999). The first two NT receptors (NTR1 and NTR2) belong to the family of G-protein-coupled receptors (GPCRs) (Tanaka et al., 1990; Chalon et al., 1996; Mazella et al., 1996). The third NT receptor, NTR3 (Mazella et al., 1998), also called sortilin (Petersen et al., 1997), is a type I receptor belonging to the new family of receptors sharing an N-terminal luminal domain related to the yeast sorting receptor Vps10p (Marcusson et al., 1994). The role of the two GPCRs NTR1 and NTR2 in the effects of NT has been partly elucidated by using the NT antagonist SR48692, which is selective for the NTR1 (Betancur et al., 1997; Vincent et al., 1999). The NTR1 is functionally coupled to phospholipase C (Hermans et al., 1992; Chabry et al., 1994), it induces MAP kinase phosphorylation (Poinot-Chazel et al., 1996), and it is also described as being responsible for the NT-induced modulation of dopaminergic transmission (Nemeroff, 1986). The NTR2 for which the cellular coupling remains to be clarified seems to be involved in the analgesic effect of the peptide (Dubuc et al., 1999). The role of the recently cloned NTR3 as an NT receptor remains speculative because this protein has been shown to bind several other ligands such as the receptor associated protein (Petersen et al., 1997) or lipoprotein lipase (Nielsen et al., 1999). However, this NT binding protein is the only one that is present in all of the cancer cell lines on which NT exerts a proliferative action (Dal Farra et al., 2001). When stably expressed into Chinese hamster ovary (CHO) cells, this receptor responds to NT in the thymidine incorporation assay (Dal Farra et al., 2001) and is maintained at the cell surface, although it efficiently internalizes the peptide (Navarro et al., 2001).
In this study we investigated the role of NT in the growth and migration of the human microglial cell line C13NJ. These immortalized cells possess the macrophagic characteristics of adherence and phagocytosis and express several macrophagic antigens (Janabi et al., 1995). We found that only NTR3 was expressed in these cells. Functional studies indicated that NT which is not synthesized in this cell line did not induce cell growth but efficiently stimulated the migration of cells at concentrations ranging between 1 and 10 nm. The chemoattractive effect of NT was totally blocked in the presence of the NTR3 propeptide. Finally, we showed that activation of C13NJ cells by NT led to a rapid phosphatidylinositol 3 (PI 3)-kinase-dependent formation of filopodia as shown by F-actin labeling.
Materials and Methods
Materials
Neurotensin was purchased from Peninsula Laboratories and NT (2–13) was synthesized by Neosystem.125I-Tyr3-NT and α-azidobenzoyl-125I-Tyr3-NT(2–13) (azido 125I-NT) were prepared and purified as described previously (Sadoul et al., 1984; Mazella et al., 1985). Levocabastine was generously provided by A. Schotte (Janssen Laboratories, Belgium). SR48692 was a gift from Sanofi-Synthelabo. DMEM was from Invitrogen (Gaithersburg, MD), and fetal calf serum was from BioWest. Gentamicin, 1–10-phenanthroline, bovine serum albumin (BSA), mowiol, paraformaldehyde, PD98059, mammalian protease, and phosphatase inhibitor mixtures were from Sigma. LY294002 and wortmannin were from Calbiochem (La Jolla, CA).Taq polymerase was from Appligen, and the Reverse Transcription system kit was from Promega (Madison, WI). The mouse monoclonal anti-syntaxin-6 antibody was purchased from Transduction Laboratories (Lexington, KY). Antibodies against the phosphorylated or total forms of extracellular signal-regulated kinase (ERK) 1/2 were from Santa Cruz Laboratory (Santa Cruz, CA). The rabbit polyclonal phospho-Akt antibody was from Cell Signaling. FITC-conjugated donkey anti-rabbit and Cy-5-conjugated donkey anti-mouse antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Texas Red-X phalloidin was from Molecular Probes (Eugene, OR). HRP-conjugated goat anti-rabbit or anti-mouse were from Amersham Biosciences (Arlington Heights, IL). Oligodeoxynucleotides and the NTR3 propeptide resulting from the furin cleavage of the precursor form of NTR3/sortilin (Munck Petersen et al., 1999) were synthesized by Eurogentec. The antiserum against the luminal domain of NTR3/sortilin was a generous gift from Dr. C. M. Petersen (University of Aarhus, Aarhus, Denmark). The human microglial cell line C13NJ was kindly provided by the Dr. M. Tardieu (Université Paris sud, le Kremlin-Bicêtre, France).
RT-PCR analysis
Total RNAs were extracted from human microglial C13NJ cells by the method of Chomczynski and Sacchi (1987) using the RNAble kit (Promega). Briefly, 2 μg of total RNA was reverse transcribed using 1 μg of oligo-dT15 primer and 30 U of avian myoblastosis virus reverse transcriptase as described previously (Dal Farra et al., 2001). PCR amplification was then performed with a first cycle at 94°C for 3 min, 52°C for 2 min, 72°C for 1 min, followed by 29 cycles, 94°C for 40 sec, 52°C for 1 min, 72°C for 1 min, and a final extension step at 72°C for 5 min. PCR products were analyzed on a 2% agarose gel. Three sets of oligonucleotides were used. A first pair composed of sense primer (5′-TCATCGCCTTTGTGGTCTGCT-3′) and antisense primer (5′-TGGTTGCTGGACACGCTGTCG-3′) was directed against the human NTR1; the second pair composed of sense primer (5′-GTCTCCTCAGCTTCATCGTAT-3′) and antisense primer (5′-TCCCCAAAGCCTGAAGCTGTA-3′) was directed against the human NTR2; and the third pair composed of sense primer (5′-AGAATGGTCGAGACTATGTTG-3′) and antisense primer (5′-AAGAGCTATTCCAAGAGGTCC-3′) was directed against the human NTR3. The predicted sizes of amplified fragments were 291, 429, and 552 bp for human NTR1, NTR2, and NTR3, respectively. Plasmid controls were generated by amplifying in parallel the cDNA fragment encoding the human NTR1, NTR2, or NTR3 subcloned into the expression vector.
Cell culture and binding experiments
The human microglial cell line C13NJ was maintained in DMEM supplemented with 10% fetal calf serum (FCS) and 50 μg/ml gentamicin at 37°C under 5% CO2.
Saturation experiments were performed on whole cells plated in 22-mm-well culture dishes as described previously (Botto et al., 1998). Briefly, cells were equilibrated at 37°C in an Earle's Tris-HEPES buffer containing 0.8 mm 1–10-phenanthroline and 0.2% bovine serum albumin (binding buffer) for 10 min. Cells were then incubated in the same buffer for 30 min with 0.5 nm125I-Tyr3-NT (2000 Ci/mmol) in the absence or presence of various concentrations of unlabeled neurotensin, levocabastine, SR48692, or NTR3 propeptide. At the end of incubations, cells were washed twice with binding buffer, harvested with 1 ml of 0.1N NaOH, and counted in a gamma counter (counting efficiency, 80%). Nonspecific binding was determined in the presence of 1 μm unlabeled NT and represented <10% of the total binding.
Photoaffinity labeling on whole cells and Western blot analysis
C13NJ cells were first equilibrated in binding buffer and then incubated with 0.5 nmα-azidobenzoyl-125I-Tyr3-NT(2–13) in binding buffer for 30 min at 37°C. Cells were then washed three times with Earle's buffer and directly irradiated on ice for 2 min using an XL-1500 UV Crosslinker (Spectronics Corporation). Cells were lysed with denaturing Laemmli buffer, sonicated, and analyzed by SDS-PAGE. Radiolabeled bands were detected on the dried gel using a phosphorimager (Fuji 2500 BAS). Nonspecific labeling was determined in the presence of 1 μm NT. Alternatively, cell homogenates were transferred onto nitrocellulose after SDS-PAGE, and the NTR3 was detected using an antibody directed against the luminal domain of the protein.
Confocal microscopy experiments
Dual detection of the NTR3 and trans-Golgi network marker syntaxin-6 in human C13NJ cells. Human microglial cells were plated on glass coverslips 24 hr before experiments. Cells were first washed for 5 min in PBS and then fixed with 3% paraformaldehyde in PBS for 20 min at room temperature. Coverslips were rinsed twice with PBS and incubated with 50 mmNH4Cl in PBS for 10 min to quench an excess of free aldehyde groups. After 20 min in PBS containing 10% horse serum (HS), cells were double labeled with both a rabbit polyclonal anti-NTR3 antibody (1:300) and a mouse monoclonal anti-syntaxin-6 antibody (3 μg/ml), a marker of the trans-Golgi network (TGN), for 1 hr at room temperature in PBS containing 5% HS. Cells were rinsed three times in PBS and then incubated at room temperature with both an FITC-conjugated donkey anti-rabbit antibody (1:600) and a Cy-5-conjugated donkey anti-mouse antibody (1:600) in PBS containing 5% HS for 30 min. After two washes with PBS and one with water, coverslips were mounted on glass slides with mowiol for confocal microscopy examination.
Texas Red–phalloidin staining in human C13NJ cells. Human resting C13NJ cells grown on 12 mm coverslips were preincubated or not with 1 μm wortmannin for 5 hr or with 24 μm PD98059 for 2 hr and then incubated at 37°C in Earle's Tris-HEPES buffer in the presence or absence of 10 nm NT for 10 min. After a rapid washing step, cells were fixed, permeabilized, stained with Texas Red-X phalloidin for 20 min at room temperature, and then mounted as described above.
Confocal microscopy. Labeled cells were visualized under a Leica laser scanning confocal microscope equipped with an inverted microscope and an argon-krypton laser. Samples were scanned at 488 nm (for FITC-conjugated donkey anti-rabbit antibody), 568 nm (for Texas Red-X phalloidin), and 647 nm (for Cy-5-conjugated donkey anti-mouse antibody) wavelength excitations as necessary. Images were acquired as a single transcellular optical section and averaged over 32 scans per frame.
Detection of phosphorylated Akt and MAP Kinase ERK1/2
Cells at ∼70% confluency incubated overnight in serum-free medium were preincubated or not for 2 hr with 24 μm of the specific ERK1/2 inhibitor PD98059 or for 5 hr with 1 μm of the PI 3-kinase inhibitor wortmannin or 50 μm LY294002. Cells were then stimulated with various concentrations of NT for the indicated periods of time at 37°C and then lysed with a buffer containing 50 mm Tris, pH 7.5, 100 mm NaCl, 5 mm EDTA, 0.5% Na-deoxycholate, 1% NP-40, and 0.1% SDS in the presence of phosphatase and protease inhibitor mixtures (1:100). Identical amounts of solubilized protein (∼40 μg) were analyzed on SDS-PAGE, electrotransferred onto nitrocellulose, and subjected to immunoblotting using an antibody directed against the active phosphorylated forms of ERK1/2 or Akt. Results were standardized within the same blot using the anti-NTR3 antibody and the total anti-ERK1/2 antibodies. The amount of phospho-ERK, phospho-Akt, total ERK, and NTR3 was quantified by scanning the optical density of each band revealed by the Lumilight enhanced chemiluminescence method (Roche Diagnostics) using NIH Image 1.62 software. The stimulation was calculated from the ratio between the density of each band and the density of the band obtained in the control condition.
Cell migration assays
In vitro wound-healing assay. Microglial C13NJ cells cultured in six-well plates were washed three times in serum-free medium and wounded with a sterile plastic tip producing scars of ∼250–300 μm. Three parallel scars were made per well. Wounded cells were washed twice with serum-free medium and incubated for 48 hr with DMEM-free medium (basal migration control), with 10 nmNT, 10% FCS (maximal migration control), or 10 nm NT plus 1 μm propeptide. For each condition, four representative fields (250 × 500 μm), within the scar and avoiding cell borders, were photographed through an inverted phase-contrast Leica DM IL microscope, and the number of migrated cells by normalized area (0.125 mm2) was counted and expressed as percentage of FCS migrated cells. We tested the ability of MAP kinase or PI 3-kinase inhibitors to block the effect of NT. Unfortunately, both inhibitors were highly toxic at the concentrations used in this study when incubated for 48 hr with C13NJ cells. Two wells were analyzed for each experimental condition, and experiments were repeated at least three times. Statistical analysis between each experimental condition was made from the number of wells (n = 6) by using the Student's t test. Differences were considered significant for p < 0.05.
Chemotaxis assay. C13NJ chemotaxis assays were performed in 24-well modified Boyden transwell microchambers containing 8 μm pore size polyethylene terephthalate (Falcon). The lower surface of each filter was coated with PBS containing 6.5 μg/ml rat fibronectin (30 min at 37°C) and then blocked with 1% BSA in serum-free DMEM for 30 min at 37°C. For each assay, 104cells pretreated or not with MAP kinase or PI 3-kinase inhibitors as described above were resuspended in 200 μl of DMEM supplemented with 0.5% FCS and plated on the upper chamber, and lower wells were filled with either DMEM containing 0.5% FCS (background migration), with 1% BSA (nonspecific migration), with 10 nm NT in 0.5% FCS in DMEM, with 10 nm NT in 0.5% FCS in the presence of increasing concentration of NTR3 propeptide, or with DMEM supplemented with 10% FCS (maximal migration control) and incubated at 37°C for 6 hr. Cells remaining in the upper surface of the filter were removed with a cotton swab, and cells that had migrated (lower surface) were fixed in 3% paraformaldehyde and stained for 10 min at room temperature with 0.5% crystal violet (in PBS–methanol 10% v/v). Filters were mounted on glass slides, and stained migrated cells were counted (10 fields per filter) under an inverted Leica DM IL microscope. The number of cells that migrated through the filter was expressed as the percentage of migrated cells measured in the presence of 10% FCS. Statistical analysis between each experimental condition was made from the number of filters (n = 6) by using the Student's t test. Differences were considered significant for p < 0.05.
Results
Human microglial cells express the neurotensin receptor-3
To determine which mRNAs of NT receptors types are expressed by the microglial cell line C13NJ, we isolated RNA from microglia cultures and performed RT-PCR using specific oligodeoxynucleotides for each receptor. All of the three known NTRs are expressed in the brain (Vincent et al., 1999), but only NTR3 expression was detected in microglia that contained neither NTR1 nor NTR2 (Fig.1A). To confirm the specificity of NTR3 RT-PCR generated products, we cloned the 552 bp fragment into the p-Target vector. Sequencing of the recombinant plasmid definitively identified the PCR product as the human NTR3.
Fig. 1.

Expression of neurotensin receptor-3 by microglial cells. A, RNA from cultured C13NJ cells was reverse transcribed, and cDNA was amplified by PCR using sense and antisense NTR-specific primers (lane 1). Lane 2, H2O with both sense and antisense primers; lane 3, RNA with both primers; lane 4, RT product with sense primers alone; lane 5, RT product with antisense primers alone; lane 6, PCR with plasmids controls. B, Whole cells were photolabeled using α-azidobenzoyl-125I-Tyr3-NT(2–13) (0.5 nm) as described in Materials and Methods in the absence (lane 1) or presence (lane 2) of 1 μm unlabeled NT. Proteins from cell homogenates were then separated by SDS-PAGE (8% acrylamide); the gel was fixed and air dried before phosphorimager analysis. Lane 3, Western blot analysis of cell homogenate using the NTR3 antibody. C, D, andC/D show that the fluorescence immunolabeling of NTR3 (C) and syntaxin-6 (D) in C13NJ cells is colocalized primarily (C/D) in the trans-Golgi compartment and processes. Scale bar, 10 μm.
The ability of the NTR3 expressed in human microglia to specifically bind NT was assessed by photoaffinity cross-linking on whole cells using azido125I-NT. As shown in Figure1B, a single protein band with a molecular weight of 110 kDa was specifically photolabeled because its labeling was totally abolished in the presence of 1 μm unlabeled NT. Western blot analysis of microglia protein extracts using the anti-NTR3 antibody also revealed a single band with a Mr of 110 kDa (Fig. 1B). Saturation binding experiments performed with increasing concentrations of 125I-NT on whole microglial cells indicated a Kd value for NT of 2.5 ± 0.48 nm and a maximal binding capacity (Bmax) of 30 ± 6 fmol/mg. We also measured by competition experiments the potencies of ligands selective for the NTR1 (SR48692), the NTR2 (levocabastine), or the NTR3 (propeptide) to displace the binding of125I-NT on C13NJ cells and on CHO cells transfected with the human NTR3 (Table1). Neither levocabastine nor SR48692 efficiently competed with NT (IC50 of 0.2 and 10 μm) both on C13NJ and CHO cells, thus confirming the absence of NTR1 and NTR2 in microglia. The propeptide resulting from the maturation of the precursor form of sortilin/NTR3 (Munck Petersen et al., 1999) bound almost as efficiently as NT on C13NJ (IC50 = 8.8 nm). By contrast, the IC50 values of both NT and the propeptide were 50–100 times higher on CHO cells expressing the human NTR3 (Table 1). Note that unrelated peptides such as vasoactive intestinal peptide or somatostatin did not displace the binding of125I-NT to microglial cells (data not shown).
Table 1.
Apparent affinities of NT and specific NT receptor ligands for binding to NTR3-expressing cells
| IC50 (nm) | ||
|---|---|---|
| Microglial cell line C13NJ | CHO-hNTR3 transfected cells | |
| NT | 2.51 ± 0.48 | 270 ± 68 |
| Levocabastine | >10,000 | >10,000 |
| SR48692 | 238 ± 46 | >10,000 |
| NTR3-propeptide | 8.81 ± 0.78 | 461 ± 87 |
IC50 values were determined in competition experiments involving constant amounts of cells and125I-Tyr3-NT and increasing concentrations of the indicated unlabeled competitors. The IC50 is the concentration of unlabeled competitor that induces half-displacement of125I-Tyr3-NT bound to cells. Levocabastine, SR48692, and the NTR3 propeptide are selective ligands of the NTR2, the NTR1, and the NTR3, respectively. Values are the mean ± SEM of two to four experiments performed in duplicate.
To visualize the subcellular location of the NTR3, we performed fluorescent immunocytochemistry and confocal imaging on human microglial cells. As shown in Figure 1C, the bulk of NTR3 labeling was intracellular, essentially concentrated in a syntaxin-6-positive TGN compartment (Fig. 1D). This labeling was expected for the NTR3/sortilin protein and confirms that the bulk of NTR3 was intracellular, as already observed by Petersen and colleagues (1997). A more diffuse labeling was also observed on cellular neurite-like processes, suggesting the presence of a low amount of receptor at the cell surface.
Functional signaling of the neurotensin receptor-3 in microglia
NT (10 nm) rapidly and transiently stimulated the phosphorylation of MAP kinases ERK1/2 in microglia cells with a maximal effect after 2.5 min that was maintained up to 5 min to finally recover the basal level after 10 min (Fig.2A). Standardization of the same blots using the anti-NTR3 antibodies indicated a maximal 3.5 to 4-fold stimulation of phospho-ERK at 2.5 min (Fig. 2C). The concentration–response of ERK phosphorylation showed a maximal effect at 10 nm NT and a peptide concentration inducing 50% of the maximal effect (EC50) of ∼5 nm (Fig.2B,D). The specificity of ERK1/2 activation was demonstrated by the total inhibition of the NT effect in the presence of the MAP kinase inhibitor PD98059 (Fig.2B).
Fig. 2.

Effect of NT on the phosphorylation of MAP kinases ERK1/2 in C13NJ cells. Microglial cells were stimulated either with 10 nm NT for various times (A) or with increasing concentrations of NT for 2.5 min at 37°C (B). The phosphorylation of MAP kinases (p-Erk) was determined by immunoblotting using an antibody directed against the phosphorylated active form of ERK. Immunoblots shown in A and B are representative of typical experiments. C,D, Data were standardized from three different experiments using the labeling obtained on the same blot with the anti-NTR3 antibody and the total ERK antibody and expressed as means ± SEM. PD98059 is the specific MAP kinase inhibitor.
We next investigated the influence of NT on the PI 3-kinase pathway by measuring the phosphorylation level of Akt as a function of both time and peptide concentration. The effect of NT was much more pronounced (10-fold stimulation) and more long-lasting with a maximal phosphorylation obtained at 30 min (Fig.3A). When cells were treated for 30 min with increasing concentrations of NT, the maximal stimulation (10-fold increase) was observed between 1 and 10 nm, whereas higher concentrations of NT were without effect on the phosphorylation of Akt (Fig. 3B). As expected from the PI 3-kinase pathway, when cells were pretreated with the PI 3-kinase inhibitors wortmannin or LY294002, the phosphorylation of Akt induced by NT was totally abolished (Fig. 3B).
Fig. 3.

Effect of NT on the phosphorylation of Akt in microglial cells. Cells were stimulated either with 10 nmNT for various times (A) or with increasing concentrations of the peptide for 30 min at 37°C (B). The phosphorylation of Akt was determined by immunoblotting using an antibody directed against the phosphorylated active form of Akt. A, B, Immunoblots representative of typical experiments. C,D, Data were standardized using the labeling obtained on the same blot with the anti-NTR3 antibody and expressed as means ± SEM from three independent experiments. Wortmannin and LY294002 are PI 3-kinase inhibitors.
NT induces the migration of human microglia
To study the role of NTR3 in microglia, we first tested the effect of NT on cell proliferation by measuring3H-thymidine incorporation. The incubation of cells with 1, 10, or 100 nm NT did not increase3H-thymidine incorporation after 24 hr, whereas serum markedly stimulated this incorporation on microglial cells (data not shown).
To assess the role of NT on cell migration, we used an in vitro wound-healing assay. A cell-free zone was created within a semiconfluent monolayer of microglial culture by scratching cells off with a pipette tip. The cell-free zone was commonly ∼250–300 μm wide and several millimeters long. Subsequently, we analyzed by time-lapse microscopy how the cells repopulated the cell-free zone. In control conditions, when cells were incubated in the absence of FCS, a negligible number of cells were observed in the wound area after 48 hr (Fig.4Aa,a′,B). When cells were incubated in the presence of 10% FCS, we observed first an invasion of the cell-free zone by extension of cells located in the immediate vicinity of the wound margin followed by a translocation of the cell bodies (Fig.4Ab,b′). Note that cells migrated and proliferated in the presence of serum. When cells were incubated in the absence of serum but in the presence of 10 nm NT, a significant amount of cells were translocated in the wounded zone, although the total number of cells did not increase (Fig.4Ac,c′,B). We counted the number of migrating cells as a function of the surface from the original cell-free area (250 × 500 μm = 0.125 mm2). In the presence of 10 nm NT, the number of cells that migrated corresponded to 33.7 ± 1.8% of the number of migrating cells in the presence of 10% FCS (Fig. 4B), whereas without stimulation only 3.4 ± 1% of cells migrated (Fig.4A,B). Interestingly, the translocation of cells induced by 10 nm NT was totally abolished in the presence of 1 μm NTR3 propeptide and remained to the basal level (6.5 ± 1.1%) (Fig.4Ad,d′,B).
Fig. 4.
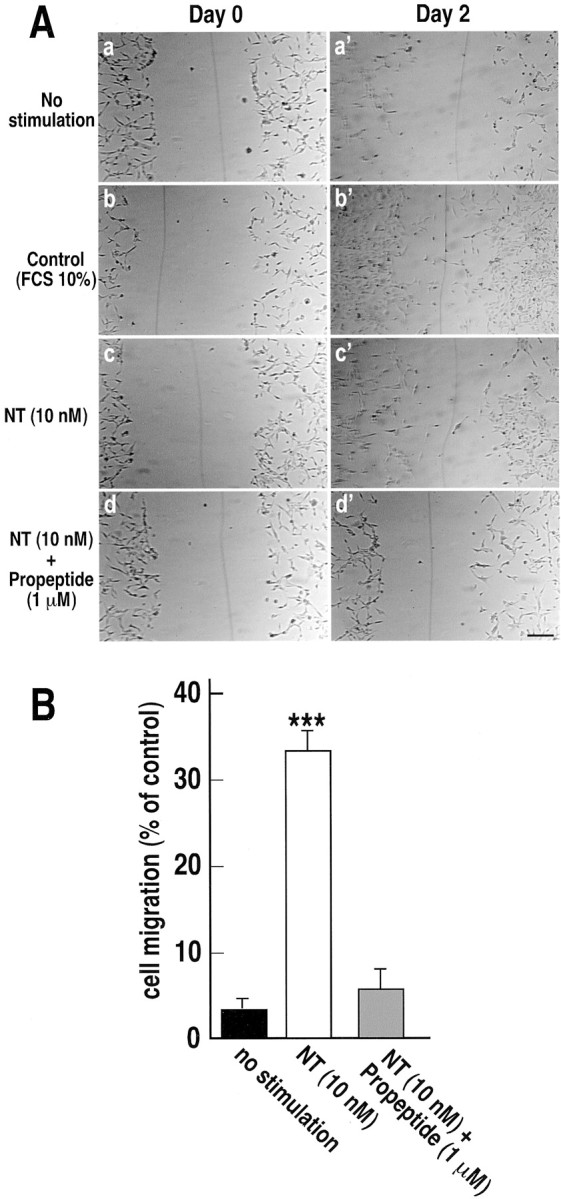
Microglia migration into a cell-free area.A, The monolayer of microglia was scratched. Images were taken at 0 (Day 0) and 48 hr (Day 2) after wounding. Cells were incubated in the absence (No stimulation, a, a′) or presence (Control, b, b′) of 10% serum and compared with cells grown in the absence of serum and 10 nm NT (c, c′) or 10 nm NT and 1 μm propeptide (d,d′). B, Four representative fields were counted from normalized areas corresponding to each condition and expressed as the percentage ± SEM of cells that migrated in the presence of 10% serum. ***p < 0.001 forn = 6 (number of wells) when compared with nonstimulated cells. Scale bar, 50 μm.
To confirm the effect of NT as a chemoattractant on microglial cells, we also used the chemotaxis assay in modified Boyden chambers. The amount of cells that passed through the filters after 6 hr with 10 nm NT reached ∼35% of the number of cells stimulated with serum (Fig. 5A). In the absence of NT or in the presence of 1% bovine serum albumin, only 10% of cells migrated; this value represents the background of the experiment. Because NT stimulated both ERK1/2 and PI 3-kinase pathways, we tested the effect of the specific MAP kinase inhibitor PD98059 and of the PI 3-kinase inhibitors LY294002 and wortmannin on NT-induced migration. As expected, both inhibitors blocked the migration elicited by the peptide (Fig. 5A). When cells were treated with 10 nm NT in the presence of increasing concentrations of NTR3 propeptide, the amount of migrated cells decreased progressively to reach the basal level at 1 μm propeptide (Fig. 5B).
Fig. 5.
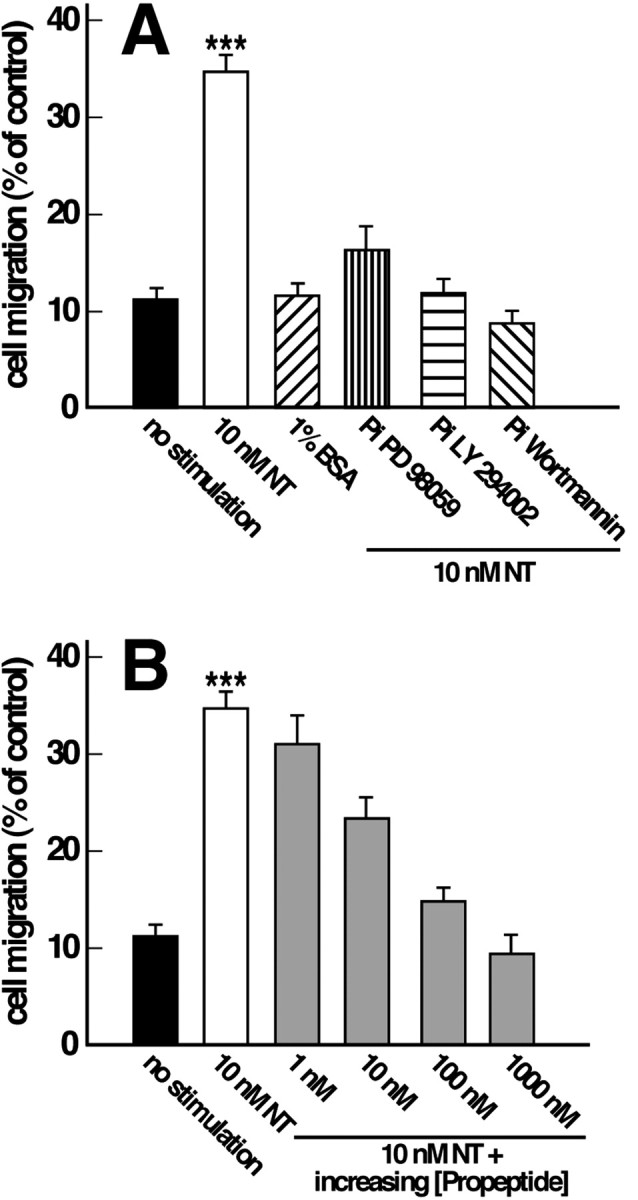
Microglia migration in modified Boyden chamber. Microglia migration in response to various experimental conditions was tested in the modified Boyden chamber. The average number of migrating cells was expressed as the percentage of migrating cells counted in the presence of 10% serum. A, The number of cells migrating across the membrane was determined after incubation with either 10 nm NT or 1% bovine serum albumin (1% BSA) or 10 nm NT after preincubation with 24 μmPD98059 or 50 μm LY294002 or 1 μmwortmannin. B, The number of cells migrating across the membrane was determined after incubation with either 10 nmNT alone or with 10 nm NT in the presence of indicated concentrations of propeptide. ***p < 0.001 forn = 6 (number of wells) when compared with nonstimulated cells.
Effect of NT on actin cytoskeleton
The effect of NT on microglia migration prompted us to investigate a possible action of the peptide on F-actin. Confocal imaging of C13NJ cells stained with Texas Red-X phalloidin is shown in Figure6. In untreated cells, F-actin staining allowed us to visualize the cytoskeleton of cells (Fig.6a,a′). The cell shape was representative of microglial cells with a sustained concentration of actin at the plasma membrane. In cells treated for 10 min with 10 nm NT, we first observed an increase in F-actin staining with a greater number of visible stress fibers (Fig.6b). Interestingly, NT also promoted the formation of numerous filopodia rich in actin and visible in Figure6B′.
Fig. 6.
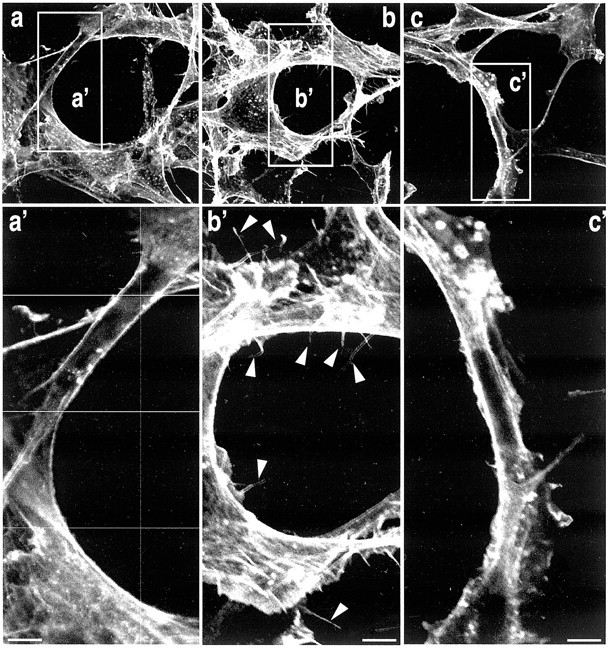
NT induces rapid formation of membrane filopodia. Human microglial cells were grown on glass coverslips, pretreated (c, c′) or not (a,b,a′,b′) with 1 μm wortmannin for 5 hr, and incubated in the absence (a,a′) or presence (b,b′) of 10 nm NT for 10 min at 37°C. Cells were labeled with Texas-Red phalloidin to detect F-actin. Control cells contain actin stress fibers throughout the cells body and processes. In the presence of NT, a significant increase of filopodia formation was observed (arrowheads); this formation was blocked in the presence of wortmannin (c,c′). a′, b′, andc′ are enlargements of a,b, and c images. Scale bar, 2 μm.
As expected, preincubation of cells with wortmannin before and during incubation with NT totally abolished filopodia formation induced by the peptide (Fig. 6c). The same result was obtained when the MAP kinase inhibitor PD98059 was used (data not shown), indicating a crucial role of both PI 3-kinase and ERK1/2 pathways in this process.
Discussion
Microglia express the neurotensin receptor-3
In the present study, we provide evidence that human microglia express only one known member of the NT receptor family, namely NTR3. Indeed, both RT-PCR experiments using specific primers distinguishing between the three known NTR subtypes and specific photoaffinity labeling of a single 110 kDa protein demonstrated that the NTR3 is expressed and binds NT in human C13NJ microglial cells. Neither the NTR1 nor the NTR2 has been detected using both experiments. The binding properties of NT have been characterized by using iodinated NT on whole cells, whereas the expression of the NTR3 protein has been revealed by Western blotting of cell homogenates. The binding properties of NT and the NTR3 propeptide on C13NJ cells do not match those measured on CHO cells transfected with the NTR3 (Table 1). This discrepancy could be caused by the consequence of membrane environment of the NTR3, which is certainly different in microglial and CHO cells. Thus, NT-induced activation of MAP kinases (Martin et al., 2002) and Akt was observed in C13NJ cells but not in CHO cells when compared with the NT effects. Another possible explanation would be that the endogenous receptor is correctly matured in C13NJ cells, leading to a membrane protein with a high affinity for its ligands, whereas the exogenous receptor expressed in a heterologous system is not matured or is only partly matured and thus devoid of high affinity for its ligands (Mazella et al., 1998;Munck Petersen et al., 1999).
The expression of the NTR3 alone in microglia is of special interest because to date the NTR3 was almost always detected in cells or tissues bearing at least one other NT receptor. Indeed, the NTR3 was shown to be present with the NTR1 in neuronal cells (Chabry et al., 1993) and in almost all human cancer cells from prostate, pancreas, and colon (Dal Farra et al., 2001; Martin et al., 2002) and also with the NTR2 in rat astrocytes (Nouel et al., 1999). The presence of the NTR3 with another NT receptor makes it difficult to assess which receptor is responsible for the biological or cellular effect of NT. By contrast, the fact that the NTR3 is the only known NT receptor endogenously expressed in human microglia allowed us to demonstrate for the first time its functional role. The NTR3 is specifically involved in the microglial cell migration induced by NT, and this effect is mediated both by ERK1/2 and PI 3-kinase pathways.
Neurotensin activates both ERK1/2 and PI 3-kinase pathways
Because NT specifically binds the NTR3 in the human C13NJ cells, we attempted to define the effect of the peptide on both the MAP kinase and the PI 3-kinase pathways. Interestingly, NT stimulates the phosphorylation of ERK1/2 and Akt in a time and a concentration-dependent manner. However, the phosphorylation of ERK1/2 is transient with a maximal stimulation at 2.5 min and rapidly returns to the basal level between 5 and 10 min, whereas the phosphorylation of Akt is more persistent in time with a maximal effect at 30 min. More surprising is the bell-shaped dose–response curve obtained with phospho-Akt. Indeed, the maximal response was measured between 1 and 10 nm NT, and increasing concentrations of peptide resulted in an absence of response. This may reflect the occurrence of a desensitization process in the transduction system and indicate that a fractional occupancy is sufficient to downstream signaling leading to the final effect. The involvement of both the ERK1/2 and the PI 3-kinase pathways was demonstrated by the loss of NT effect observed in the presence of specific inhibitors such as PD98059 and wortmannin or LY294002.
Neurotensin stimulates microglia mobility and migration
During activation in response to injury and inflammation, microglial cells can actively migrate into the damaged region of the brain. To fulfill this function, microglia bear receptors for motility factors such as IL-10, epidermal growth factor, complement 5A, and hyaluronan (Turley et al., 1994; Nolte et al., 1996, 1997; Huettner et al., 1997). C13NJ microglial cells represent an interesting model to study cell migration because wound healing is rapidly achieved when chemoattractant factors that are present in 10% serum are added. However, to avoid the possible contribution of cell proliferation to the migration, we performed the assay in the absence of serum. Under these conditions, NT (10 nm) induces a marked activation of cell migration with an effect representing ∼35% of that measured in the presence of serum. An identical result was obtained by using the modified Boyden chamber. Although the NT effect on cell migration defined using the chemotaxis assay was totally blocked by both PI 3-kinase and MAP kinase inhibitors, we could not confirm the blocking action of these inhibitors, which were toxic with time in the wound-healing assay on C13NJ cells. More interesting is the observation that the NTR3 propeptide resulting from the furin maturation of the precursor form of NTR3/sortilin (Munck Petersen et al., 1999) acts as an antagonist of NT on the NTR3. Indeed, the propeptide efficiently abolished the migration effect of NT on both assays. If the antagonist action of the propeptide is further confirmed on other cell systems or in vivo, it will become a new useful tool in the attribution of specific NT effects to the NTR3. PI 3-kinase activation has been described as participating in the ligand-induced migration of several cell types, including renal epithelial cells (Royal et al., 1997) and vascular smooth muscle cells (Duan et al., 2000). MAP kinase activation has been shown to trigger migration of gastric epithelial cells (Hollande et al., 2001) and endothelial cells (Pintucci et al., 2002). However, the specific involvement of these pathways either in the cell dissociation or in the motility has not been clearly defined. Recent results obtained on HepG2 human hepatoma cells showed that PI 3-kinase was involved in cell dissociation, whereas inhibition of MAP kinases blocked the motility response to growth factors (Sipeki et al., 1999). More recently, it was demonstrated that glycine-extended gastrin-induced migration of gastric epithelial cells was correlated with a long-term activation of MAP kinases (Hollande et al., 2001). However, in our system, the migration induced by NT seems to be dependent on a rapid and reversible activation of ERK1/2 because the overall migration process is totally blocked in the presence of MAP kinase inhibitor (Fig. 5).
The migration of microglial cells elicited by NT, observed after several hours, is preceded by a profound modification of the actin cytoskeleton, the dynamic properties of which provide the driving force for cells to move and divide. The effect of NT on the rapid formation of filopodia requires PI 3-kinase as demonstrated by the absence of F-actin modification in the presence of wortmannin (Fig.6c). As initially mentioned for cell migration, many previously reported experiments showed the involvement of the PI 3-kinase pathway in F-actin cytoskeleton modifications using either wortmannin, blocking antibodies, or dominant negative forms of PI 3-kinase (Martin et al., 1996; Siddhanta et al., 1998; Wang et al., 1999; Khayat et al., 2000). However, in the case of insulin signaling, which causes cortical F-actin polymerization in 3T3-L1 adipocytes, the effects were unaffected by the PI 3-kinase inhibitor wortmannin, although the kinase is activated (Jiang et al., 2002). In the present work, the possible involvement of several intracellular regulators of the actin cytoskeleton-like members of the Rho family and various integrins remains to be determined.
The presence of functional NTR3 in microglial cells and the effect of NT on migration could be correlated to the role initially ascribed for the peptide in the inflammation process. Indeed, NT has been shown to stimulate the expression of IL-8 through an ERK-dependent pathway in human colonocytes (Zhao et al., 2001). NT also modulates both adherence and chemotaxis capacities of murine lymphocytes (Garrido et al., 1992) and stimulates the phagocytic function of mouse macrophages (De la Fuente et al., 1993). However, this work further suggests that NT plays an important role in response to inflammation or lesion in the CNS, in agreement with previous results demonstrating that the NTR2 expression is upregulated in astrocytes in response to a brain stab wound (Nouel et al., 1999). It has also been shown that the cerebral NT content was increased after ischemia or exocytotoxic lesions (Cheung and Cechetto, 1995) and therefore could stimulate the cerebral healing process by activation of neuropeptide receptors.
In conclusion, we demonstrate that human microglial cells express a functional NTR3. The activation of this receptor by NT triggers migration of cells that are considered to be brain macrophages. These findings may have important implications in the understanding of new pathways of inflammation mechanisms and in the development of novel therapeutic strategies for the treatment of human brain damage.
Footnotes
S.M. is fellowship recipient of the Association pour la Recherche contre le Cancer. We thank Dr. Claus Petersen and Dr. Morten S. Nielsen (Aarhus, Denmark) for the generous gift of NTR3 antibodies.
Correspondence should be addressed to Jean Mazella, Institut de Pharmacologie Moléculaire et Cellulaire, Unité Mixte de Recherche 6097 du Centre National de la Recherche Scientifique, 660 Route des Lucioles, Sophia Antipolis, 06560 Valbonne, France. E-mail:mazella@ipmc.cnrs.fr.
References
- 1.Betancur C, Azzi M, Rostène W. Nonpeptide antagonists of neuropeptide receptors: tools for research and therapy. Trends Pharmacol Sci. 1997;18:372–383. doi: 10.1016/s0165-6147(97)01109-7. [DOI] [PubMed] [Google Scholar]
- 2.Botto JM, Chabry J, Sarret P, Vincent JP, Mazella J. Stable expression of the mouse levocabastine-sensitive neurotensin receptor in HEK 293 cell line. Binding properties, photoaffinity labeling and internalization mechanism. Biochem Biophys Res Commun. 1998;243:585–590. doi: 10.1006/bbrc.1997.8071. [DOI] [PubMed] [Google Scholar]
- 3.Brockhaus J, Moller T, Kettenmann H. Phagocytozing ameboid microglial cells studied in a mouse corpus callosum slice preparation. Glia. 1996;16:81–90. doi: 10.1002/(SICI)1098-1136(199601)16:1<81::AID-GLIA9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 4.Chabry J, Gaudriault G, Vincent JP, Mazella J. Implication of various forms of neurotensin receptors in the mechanism of internalization of neurotensin in cerebral neurons. J Biol Chem. 1993;268:17138–17144. [PubMed] [Google Scholar]
- 5.Chabry J, Labbé-Jullié C, Gully D, Kitabgi P, Vincent JP, Mazella J. Stable expression of the cloned rat brain neurotensin receptor into fibroblasts: binding properties, photoaffinity labeling, transduction mechanisms, and internalization. J Neurochem. 1994;63:19–27. doi: 10.1046/j.1471-4159.1994.63010019.x. [DOI] [PubMed] [Google Scholar]
- 6.Chalon P, Vita N, Kaghad M, Guillemot M, Bonnin J, Delpech B, Le Fur G, Ferrara P, Caput D. Molecular cloning of a levocabastine-sensitive neurotensin binding site. FEBS Lett. 1996;386:91–94. doi: 10.1016/0014-5793(96)00397-3. [DOI] [PubMed] [Google Scholar]
- 7.Chomczynski P, Sacchi N. Single step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 8.Cheung RT, Cechetto DF. Neuropeptide changes following excitotoxic lesion of the insular cortex in rats. J Comp Neurol. 1995;362:535–550. doi: 10.1002/cne.903620408. [DOI] [PubMed] [Google Scholar]
- 9.Dal Farra C, Sarret P, Navarro V, Botto JM, Mazella J, Vincent JP. Involvement of the neurotensin receptor subtype NTR3 in the growth effect of neurotensin on cancer cell lines. Int J Cancer. 2001;92:503–509. doi: 10.1002/ijc.1225. [DOI] [PubMed] [Google Scholar]
- 10.De la Fuente M, Garrido JJ, Arahuetes RM, Hernanz A. Stimulation of phagocytic function in mouse macrophages by neurotensin and neuromedin N. J Neuroimmunol. 1993;42:97–104. doi: 10.1016/0165-5728(93)90216-l. [DOI] [PubMed] [Google Scholar]
- 11.De la Fuente M, Carrasco M, Del Rio M, Hernanz A. Modulation of murine lymphocyte functions by sulfated cholecystokinin octapeptide. Neuropeptides. 1998;32:225–233. doi: 10.1016/s0143-4179(98)90041-5. [DOI] [PubMed] [Google Scholar]
- 12.Delgado M, Munoz-Elias EJ, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide enhance IL-10 production by murine macrophages: in vitro and in vivo studies. J Immunol. 1999;162:1707–1716. [PubMed] [Google Scholar]
- 13.Duan C, Bauchat JR, Hsieh T. Phosphatidylinositol 3-kinase is required for insulin-like growth factor-I-induced vascular smooth muscle cell proliferation and migration. Circ Res. 2000;86:15–23. doi: 10.1161/01.res.86.1.15. [DOI] [PubMed] [Google Scholar]
- 14.Dubuc I, Sarret P, Labbé-Jullié C, Botto JM, Honoré E, Bourdel E, Martinez J, Costentin J, Vincent JP, Kitabgi P, Mazella J. Identification of the receptor subtype involved in the analgesic effect of neurotensin. J Neurosci. 1999;19:503–510. doi: 10.1523/JNEUROSCI.19-01-00503.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrido JJ, Arahuetes RM, Hrenanz A, De la Fuente M. Modulation by neurotensin and neuromedin N of adherence and chemotaxis capacity of murine lymphocytes. Regul Pept. 1992;41:27–37. doi: 10.1016/0167-0115(92)90511-r. [DOI] [PubMed] [Google Scholar]
- 16.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immunoeffector cell of the brain. Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 17.Hermans E, Maloteaux JM, Octave JN. Phospholipase C activation by neurotensin and neuromedin N in Chinese hamster ovary cells expressing the rat neurotensin receptor. Brain Res Mol Brain Res. 1992;15:332–338. doi: 10.1016/0169-328x(92)90126-v. [DOI] [PubMed] [Google Scholar]
- 18.Hollande F, Choquet A, Blanc EM, Lee DJ, Bali JP, Baldwin GS. Involvement of phosphatidylinositol 3-kinase and mitogen-activated protein kinases in glycine-extended gastrin-induced dissociation and migration of gastric epithelial cells. J Biol Chem. 2001;276:40402–40410. doi: 10.1074/jbc.M105090200. [DOI] [PubMed] [Google Scholar]
- 19.Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases cell proliferation and motility in vitro. Anticancer Res. 1997;17:3217–3224. [PubMed] [Google Scholar]
- 20.Janabi N, Peudenier S, Héron B, Heng Ng K, Tardieu M. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cell with SV40 large T antigen. Neurosci Lett. 1995;195:105–108. doi: 10.1016/0304-3940(94)11792-h. [DOI] [PubMed] [Google Scholar]
- 21.Jiang ZY, Chawla A, Bose A, Way M, Czech MP. A phosphatidylinositol 3-kinase-independent insulin signaling pathway to N-WASP/Arp2/3/F-actin required for GLUT4 glucose transporter recycling. J Biol Chem. 2002;277:509–515. doi: 10.1074/jbc.M108280200. [DOI] [PubMed] [Google Scholar]
- 22.Khayat ZA, Tong P, Yaworsky K, Bloch RJ, Klip A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J Cell Sci. 2000;113:279–290. doi: 10.1242/jcs.113.2.279. [DOI] [PubMed] [Google Scholar]
- 23.Koff WC, Dunegan MA. Modulation of macrophage-mediated tumoricidal activity by neuropeptides and neurohormones. J Immunol. 1985;135:350–354. [PubMed] [Google Scholar]
- 24.Marcusson EG, Horazdovsky BF, Cereghino JL, Gharakhanian E, Emr SD. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- 25.Martin S, Navarro V, Vincent JP, Mazella J. Neurotensin receptor-1 and –3 complex modulates the cellular signaling of neurotensin in the HT29 cell line. Gastroenterology. 2002;123:1135–1143. doi: 10.1053/gast.2002.36000. [DOI] [PubMed] [Google Scholar]
- 26.Martin SS, Haruta T, Morris AJ, Klippel A, Williams LT, Olefsky JM. Activated phosphatidylinositol 3-kinase is sufficient to mediate actin rearrangement and GLUT4 translocation in 3T3–L1 adipocytes. J Biol Chem. 1996;271:17605–17608. doi: 10.1074/jbc.271.30.17605. [DOI] [PubMed] [Google Scholar]
- 27.Marty S, Dusart I, Peschanski M. Glial changes following and excitotoxic lesion in the CNS-I: microglia/macrophages. Neuroscience. 1991;45:529–539. doi: 10.1016/0306-4522(91)90268-s. [DOI] [PubMed] [Google Scholar]
- 28.Mazella J, Kitabgi P, Vincent JP. Molecular properties of neurotensin receptors in rat brain. Identification of subunits by covalent labeling. J Biol Chem. 1985;260:508–514. [PubMed] [Google Scholar]
- 29.Mazella J, Botto JM, Guillemare E, Coppola T, Sarret P, Vincent JP. Structure, functional expression, and cerebral localization of the levocabastine-sensitive neurotensin/neuromedin N receptor from mouse brain. J Neurosci. 1996;16:5613–5620. doi: 10.1523/JNEUROSCI.16-18-05613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazella J, Zsürger N, Navarro V, Chabry J, Kaghad M, Caput D, Ferrara P, Vita N, Gully D, Maffrand JP, Vincent JP. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. J Biol Chem. 1998;273:26273–26276. doi: 10.1074/jbc.273.41.26273. [DOI] [PubMed] [Google Scholar]
- 31.Munck Petersen C, Nielsen MS, Jacobsen C, Tauris J, Jacobsen L, Gliemann J, Moestrup SK, Madsen P. Propeptide cleavage conditions sortilin/neurotensin receptor-3 for ligand binding. EMBO J. 1999;18:595–604. doi: 10.1093/emboj/18.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navarro V, Martin S, Sarret P, Nielsen MS, Petersen CM, Vincent JP, Mazella J. Pharmacological properties of the mouse neurotensin receptor 3. Maintenance of cell surface receptor during internalization of neurotensin. FEBS Lett. 2001;495:100–105. doi: 10.1016/s0014-5793(01)02367-5. [DOI] [PubMed] [Google Scholar]
- 33.Nemeroff CB. The interaction of neurotensin with dopaminergic pathways in the central nervous system: basic neurobiology and implications for the pathogenesis and treatment of schizophrenia. Psychoneuroendocrinology. 1986;11:15–37. doi: 10.1016/0306-4530(86)90029-6. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen MS, Jacobsen C, Olivecrona G, Gliemann J, Petersen CM. Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J Biol Chem. 1999;274:8832–8836. doi: 10.1074/jbc.274.13.8832. [DOI] [PubMed] [Google Scholar]
- 35.Nolte C, Moller T, Walter T, Kettenmann H. Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience. 1996;73:1091–1107. doi: 10.1016/0306-4522(96)00106-6. [DOI] [PubMed] [Google Scholar]
- 36.Nolte C, Kirchhoff F, Kettenmann H. Epidermal growth factor is a motility factor for microglial cells in vitro: evidence for EGF receptor expression. Eur J Neurosci. 1997;9:1690–1698. doi: 10.1111/j.1460-9568.1997.tb01526.x. [DOI] [PubMed] [Google Scholar]
- 37.Nouel D, Sarret P, Vincent JP, Mazella J, Beaudet A. Pharmacological and molecular characterization of glial low affinity neurotensin receptors: up regulation in response to lesion. Neuroscience. 1999;94:1189–1197. doi: 10.1016/s0306-4522(99)00354-1. [DOI] [PubMed] [Google Scholar]
- 38.Petersen CM, Nielsen MS, Nykjaer A, Jacobsen L, Tommerup N, Rasmussen HH, Roigaard H, Gliemann J, Madsen P, Moestrup SK. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J Biol Chem. 1997;272:3599–3605. doi: 10.1074/jbc.272.6.3599. [DOI] [PubMed] [Google Scholar]
- 39.Pintucci G, Moscatelli D, Saponara F, Biernacki PR, Baumann FG, Bizekis C, Galloway AC, Basilico C, Mignatti P. Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. FASEB J. 2002;16:598–600. doi: 10.1096/fj.01-0815fje. [DOI] [PubMed] [Google Scholar]
- 40.Poinot-Chazel C, Portier M, Bouaboula M, Vita N, Pecceu F, Gully D, Monroe JG, Maffrand JP, Le Fur G, Casellas P. Activation of mitogen-activated protein kinase couples neurotensin receptor stimulation to induction of the primary response gene Krox-24. Biochem J. 1996;320:145–151. doi: 10.1042/bj3200145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramez M, Bagot M, Nikolova M, Boumsell L, Vita N, Chalon P, Caput D, Ferrara P, Bensussan A. Functional characterization of neurotensin receptors in human cutaneous T cell lymphoma malignant lymphocytes. J Invest Dermatol. 2001;117:687–693. doi: 10.1046/j.0022-202x.2001.01439.x. [DOI] [PubMed] [Google Scholar]
- 42.Royal I, Fournier TM, Park M. Differential requirement of Grb2 and PI3-kinase in HGF/SF-induced cell motility tubulogenesis. J Cell Physiol. 1997;173:196–201. doi: 10.1002/(SICI)1097-4652(199711)173:2<196::AID-JCP20>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 43.Sadoul JL, Mazella J, Amar S, Kitabgi P, Vincent JP. Preparation of neurotensin selectively iodinated on the tyrosine 3 residue. Biological activity and binding properties on mammalian neurotensin receptors. Biochem Biophys Res Commun. 1984;120:812–819. doi: 10.1016/s0006-291x(84)80179-5. [DOI] [PubMed] [Google Scholar]
- 44.Siddhanta U, McIlroy J, Shah A, Zhang Y, Backer JM. Distinct roles for the p110alpha and hVPS34 phosphatidylinositol 3′-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol. 1998;143:1647–1659. doi: 10.1083/jcb.143.6.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sipeki S, Bander E, Buday L, Farkas G, Bacsy E, Ways DK, Farago A. Phosphatidylinositol 3-kinase contributes to Erk1/Erk2 MAP kinase activation associated with hepatocyte growth factor-induced cell scattering. Cell Signal. 1999;11:885–890. doi: 10.1016/s0898-6568(99)00060-1. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka K, Masu M, Nakanishi S. Structure and functional expression of the cloned rat neurotensin receptor. Neuron. 1990;4:847–854. doi: 10.1016/0896-6273(90)90137-5. [DOI] [PubMed] [Google Scholar]
- 47.Turley EA, Hossain MZ, Sorokan T, Jordan LM, Nagy JI. Astrocyte and microglial motility in vitro is functionally dependent on the hyaluronan receptor RHAMM. Glia. 1994;12:68–80. doi: 10.1002/glia.440120109. [DOI] [PubMed] [Google Scholar]
- 48.Vincent JP, Mazella J, Kitabgi P. Neurotensin and neurotensin receptors. Trends Pharmacol Sci. 1999;20:302–309. doi: 10.1016/s0165-6147(99)01357-7. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR, Klip A. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol. 1999;19:4008–4018. doi: 10.1128/mcb.19.6.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao D, Keates AC, Kuhnt-Moore S, Moyer MP, Kelly CP, Pothoulakis C. Signal transduction pathways mediating neurotensin-stimulated interleukin-8 expression in human colonocytes. J Biol Chem. 2001;276:44464–44471. doi: 10.1074/jbc.M104942200. [DOI] [PubMed] [Google Scholar]