

Abstract
Neonatal hypoxia-ischemia in the preterm human leads to selective injury to the subcortical developing white matter, which results in periventricular leukomalacia (PVL), a condition associated with abnormal neurodevelopment. Maturation-dependent vulnerability of late oligodendrocyte progenitors is thought to account for the cellular basis of this condition. A high frequency of cognitive and sensory deficits with decreasing gestational age suggests pervasive abnormalities of cortical development. In a neonatal rat model of hypoxic-ischemic injury that produces the characteristic pattern of subcortical injury associated with human PVL, selective subplate neuron death is seen. The premature subplate neuron death occurs after thalamic axons have reached their targets in cortex. Thus, as expected, thalamocortical connections form normally, including patterned connections to somatosensory cortex. However, deficits in motor function still occur, as in babies with PVL. Subplate neuron cell death in PVL provides another mechanism for abnormal neurodevelopment after neonatal hypoxia-ischemia.
Keywords: periventricular leukomalacia, visual, cortex, development, premature infant, thalamocortical
Introduction
Hypoxia-ischemia (H-I) results in selective damage to different brain structures depending on the developmental stage at which it occurs (Johnston, 1998). H-I in the preterm human [gestational week (GW) 23–32] causes damage to subcortical developing white matter, a condition known as periventricular leukomalacia (PVL) (Volpe, 2001b). Developmental immaturity of the cerebral vasculature is thought to account for this characteristic subcortical distribution (Volpe, 2001a). Other mechanisms include selective cellular vulnerability, which relates to intrinsic properties of the vulnerable cell type. Cells of the oligodendrocyte lineage manifest stage-specific vulnerability to H-I (Back et al., 2002) through mechanisms of oxidative stress (Oka et al., 1993; Back et al., 1998) and excitotoxicity (Matute et al., 1997; Fern and Moller, 2000; Follett et al., 2000). Subplate neurons, a transient cell type located beneath the cortical plate (Chun et al., 1987), are also vulnerable to stage-specific excitotoxicity (Chun and Shatz, 1988).
H-I in the preterm infant disrupts normal development and results in significant cerebral injury. Neurological disability is observed in 51% of premature infants (GW <25 weeks) examined at 30 months of age (Wood et al., 2000) and persists into adulthood (Hack et al., 2002). Deficits are found in motor, perceptual, and cognitive systems (Volpe, 2001b). These widespread abnormalities of cerebral development can be measured quantitatively with advanced magnetic resonance imaging (Miller et al., 2002). Cognitive impairment is associated significantly with decreasing gestational age (Piecuch et al., 1997). Cortical visual impairment (i.e., visual loss caused by impairment of posterior visual pathways) is particularly common, especially in infants with PVL, in whom estimates range from 66% (Lanzi et al., 1998) to 94% in infants with severe PVL (Cioni et al., 1997).
Subplate neurons are required for normal visual cortical development (for review, see Allendoerfer and Shatz, 1994). They are among the first generated cells of neocortex (Luskin and Shatz, 1985) and come to lie beneath the developing cortical plate, where they participate in the earliest neocortical circuitry (Friauf and Shatz, 1991). Subplate neurons undergo programmed cell death and are primarily absent from adult neocortex (Chun et al., 1987). In humans, the subplate zone peaks in size at GW 24, when the subplate is four times the width of the developing cortical plate (Kostovic and Rakic, 1990), and declines thereafter. The peak of subplate development coincides with the gestational age associated with the highest incidence of PVL. Given their critical role in normal visual cortical development, the death of subplate neurons after H-I would illuminate mechanisms that account for the high incidence of cortical visual impairment observed in PVL and provide a general model for abnormal cortical development after H-I. In the present study, we examine a neonatal rodent model of H-I brain injury (Sheldon et al., 1998) to determine whether subplate neurons die after neonatal H-I.
Materials and Methods
Animals. Timed-pregnant Sprague Dawley rats (Simonsen, Gilroy, CA) were allowed food and waterad libitum. All animal research was approved by the University of California San Francisco Committee on Animal Research and was performed in accordance with standards of humane care set forth in the Policy on Humane Care and Use of Laboratory Animals.
Hypoxia-ischemia. The manipulation was performed at postnatal day 1 (P1) or P2 (day of birth = P0) as described previously (Sheldon et al., 1996). The first three litters received H-I at P1, and mortality was 31% (10 of 32). These animals were used forin situ end labeling (ISEL) and bromodeoxyuridine (BrdU) staining (Figs. 1-3). Because mortality was lower (19%), and the pattern of injury by ISEL staining was unchanged, subsequent litters received H-I at P2. These animals were used for analysis of thalamocortical innervation and motor testing (Figs. 4, 5). Total mortality was 25% (16 of 64). To produce ischemia, pups were anesthetized with nitrous oxide, halothane, and oxygen. A midline incision was made in the neck; the right common carotid artery was dissected from the jugular vein and permanently coagulated with a bipolar coagulator [common carotid ligation (CCL)]. Sham-operated animals received the same operation, except that the carotid artery was not coagulated. Animals were returned to the dam for 1–2 hr. Subsequently, pups receiving hypoxia were placed in 5.6% oxygen in chambers floating in a 37°C water bath for 3 hr. One pup from each litter was monitored with a skin surface temperature probe to ensure consistency between litters and that the animals did not overheat. The body surface temperature was kept constant at 34°C (normal P7 core temperature, 35–37°C). Six litters of rodents, representing 64 animals, were used for the study. Rats were killed with pentobarbital, 100 mg/kg, given by intraperitoneal injection.
Fig. 1.
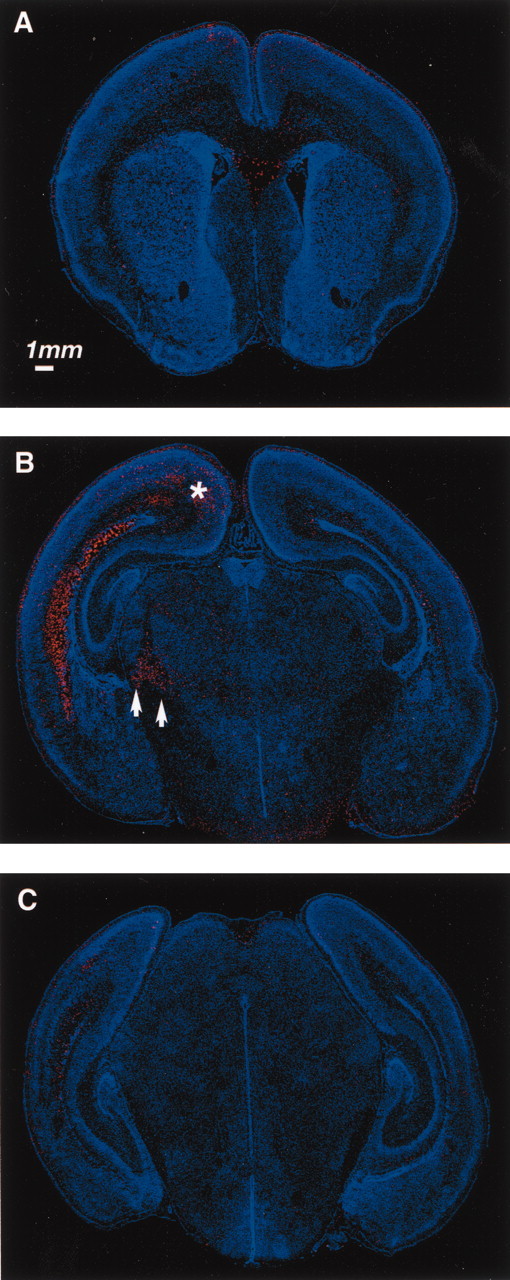
Subcortical topography of cell death 12 hr after P1 H-I. Dying cells with DNA strand breaks are detected with ISEL 12 hr after H-I at P1. A–C, ISEL signal (red) and bis-benzimide nuclear counterstain (blue) are shown alone in coronal sections from frontal (A), parietal (B), and occipital (C) regions. Arrowheads mark cell death in reticular thalamus. Asterisk marks cell death in retrosplenial cortex.
Fig. 2.
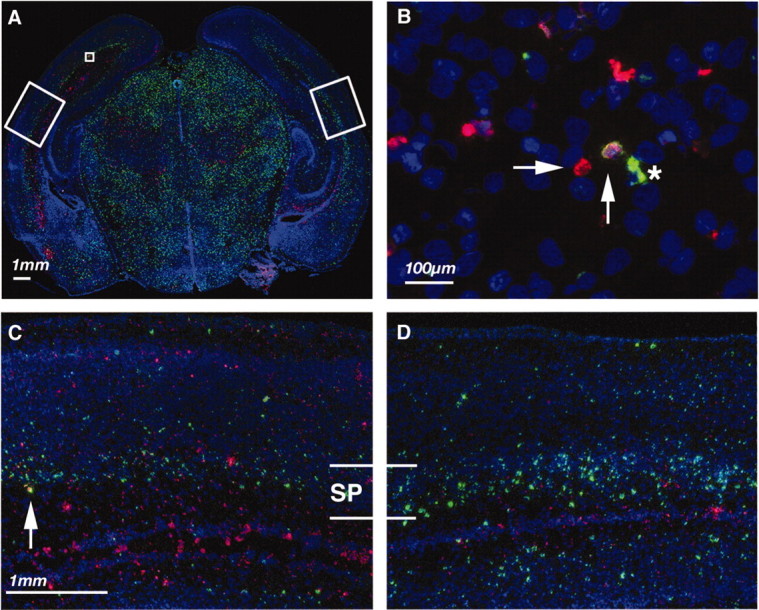
Cell death and BrdU staining for subplate neurons 24 hr after P1 H-I. ISEL (red), BrdU immunohistochemistry (green), and bis-benzimide nuclear counterstain (blue) in coronal sections taken from parietal cortex 24 hr after H-I at P1.A, Low-magnification views showing both hemispheres. Large white boxes in A delineate higher-magnification views inD of contralateral hemisphere (hypoxia) and in Cof ipsilateral hemisphere (H-I), and small white box indicates region shown in B that demonstrated single ISEL-positive (horizontal arrow), BrdU-positive (asterisk), and double-positive (vertical arrow) cells. SP, Subplate.
Fig. 3.

Quantification of BrdU-positive subplate neurons 4 d after H-I at P1. A, Plot of subplate neuron cell death (see Materials and Methods) comparing animals receiving H-I with controls (sham-operated and unmanipulated littermate;p = 0.006). Severely (B), moderately (C), and mildly (D) affected examples of subplate neuron BrdU immunohistochemistry and cresyl violet staining from sections used for quantification of subplate neuron death are shown. Plotted percent cell death for each example (B, C, D) is indicated by letter in A. SP, Subplate.
Fig. 4.
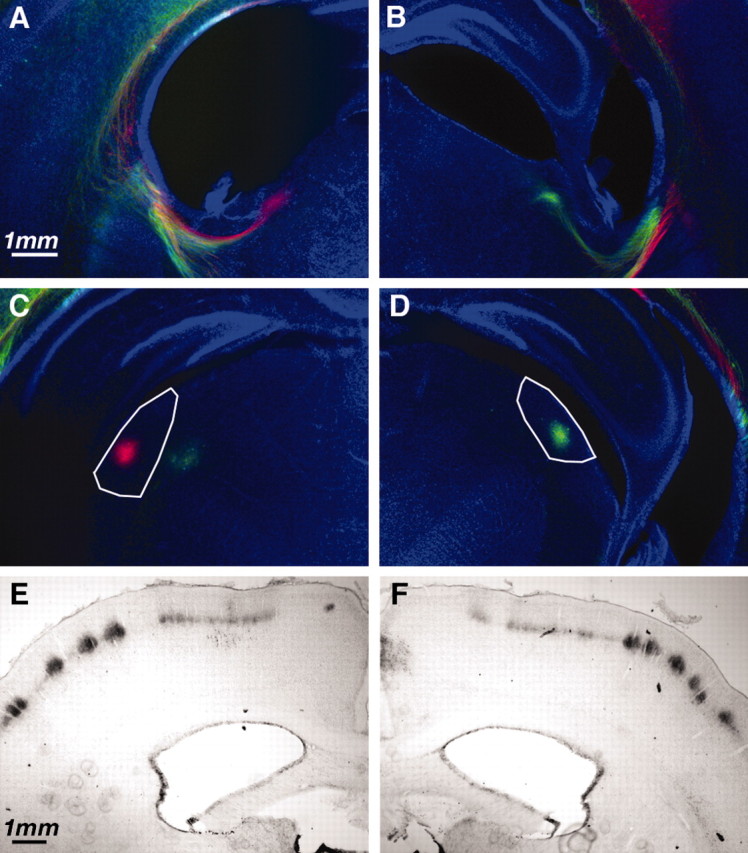
Thalamocortical projection to somatosensory and visual cortex after P2 H-I. A–D, Thalamocortical and corticothalamic axons labeled with DiI and DiD crystals placed in auditory or visual cortex. Coronal sections at the level of internal capsule (A, B) or lateral geniculate nucleus (C, D) demonstrate labeled fibers and cell bodies. To distinguish ipsilateral/H-I hemisphere from contralateral/hypoxic hemisphere, the dyes were placed in opposite configuration. For this example, the ipsilateral hemisphere (A, C) had DiI (red) placed in visual cortex and DiD (green) placed in auditory cortex. The contralateral hemisphere (B, D) had DiD (green) placed in visual cortex and DiI (red) placed in auditory cortex. Axons can be seen traversing the internal capsule (A, B), and neurons in lateral geniculate (visual thalamus) are back-labeled (C, D) in an identical manner in the two hemispheres. E, F, Cytochrome oxidase staining of sensory thalamocortical axons in patchy representation of whisker barrels in hemisphere receiving H-I (E) and hypoxia alone (F).
Fig. 5.
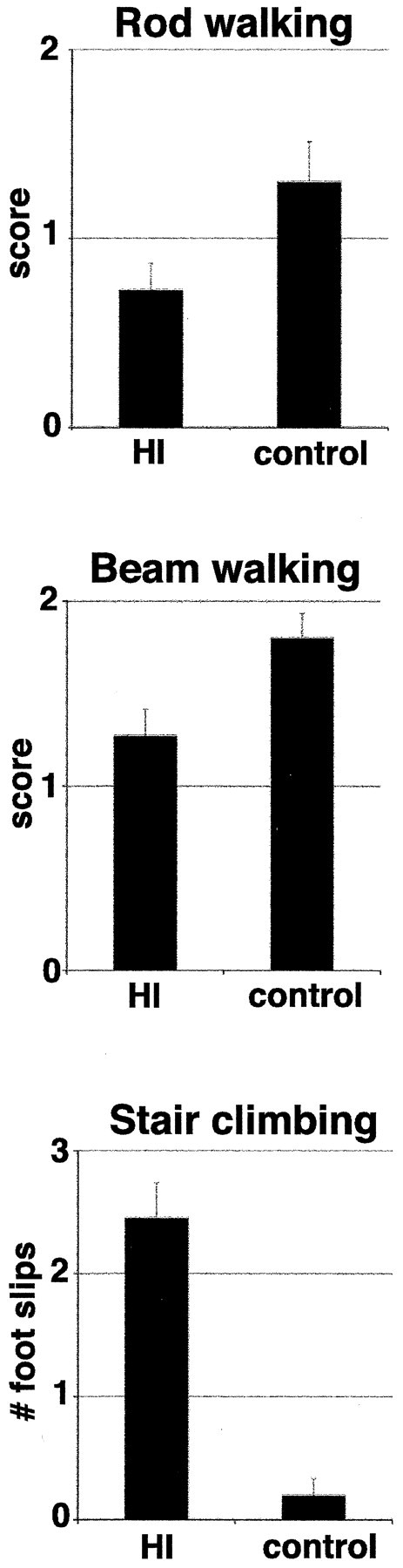
Motor deficits and abnormal myelination in mature animals after H-I at P2. Animals receiving H-I (n = 11) performed significantly worse on motor testing (rod walking, tied p = 0.04; beam walking, tied p = 0.02; and stair climbing,p < 0.0001) than controls [unmanipulated littermate (n = 8) and sham-operated (n = 2)]. For rod and beam walking, score 2, normal; 1, impaired; and 0, unable to perform task. For stair climbing, animals were scored for the number of foot slips while climbing. Values are mean of the median score ± SEM. HI, Hypoxia-ischemia.
Subplate neuron BrdU birth dating. Timed-pregnant Sprague Dawley rats received an intraperitoneal injection of BrdU, 30 mg/kg (Sigma, St. Louis, MO), at embryonic day 12.5 (E12.5; plug date, E0.5) to label subplate neurons in neocortex (Bayer and Altman, 1990).
Fluorescent BrdU staining. BrdU-labeled subplate neurons were visualized as described previously (McQuillen et al., 2002). Brains were removed rapidly from the cranium and flash-frozen in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA) in a dry ice–95% ethanol bath. Coronal cryostat sections (10 μm thick) were fixed in 0.1 m sodium phosphate-buffered 4% paraformaldehyde, extracted with 0.6% Triton X-100, acetylated, quenched in 3% hydrogen peroxide, and dehydrated through graded alcohols. To expose incorporated BrdU, the sections were microwaved for 10 min in 0.1 m sodium citrate, pH 5.0. Anti-BrdU antibody (IU4; Caltag, Burlingame, CA) was applied at 1:20,000 with exonuclease III (ExoIII) 100 U/μl (Roche Molecular Biochemicals, Indianapolis, IN) in ExoIII buffer supplemented to 100 mm NaCl with 1% bovine serum albumin at 37°C for 1 hr. After washing, horseradish peroxidase-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA) was applied at 1:200 in blocking solution (supplied by the manufacturer) for 30 min, followed by tyramide signal amplification (TSA) (Direct-green; PerkinElmer Life Sciences, Boston, MA). The sections were counterstained with 0.001% bis-benzimide.
ISEL staining. To visualize dying cells, we used a modified version of the ISEL+ method (Blaschke et al., 1996), performed as described previously (McQuillen et al., 2002). A reaction mixture containing 1 μm biotin-deoxyuridine 5′-triphosphate (Roche Molecular Biochemicals), 0.15 U/ml terminal transferase (Invitrogen, Rockville, MD), 1× terminal transferase buffer, and 1% bovine serum albumin was applied, and the sections were incubated for 1 hr at 37°C. Sections were washed and then incubated with NeutraLite avidin–horseradish peroxidase (Molecular Probes, Eugene, OR) at 1:1000 in blocking solution (supplied with the TSA Direct kit) for 30 min. The sections were washed and developed with TSA Direct-Cy3 (PerkinElmer Life Sciences). Double labeling was performed with ISEL reacted before microwave treatment and BrdU primary antibody incubation. Visualization with direct TSA was performed sequentially, with inactivation of peroxidase between development of ISEL and BrdU immunohistochemistry. Sections of neonatal rat thymus (positive) and liver (negative) were analyzed as controls for the sensitivity of ISEL staining.
DAB immunohistochemistry. Animals were perfused transcardially with 0.1 m phosphate buffer followed by cold 4% paraformaldehyde/0.1 mphosphate buffer. Perfused brains were cryoprotected in 25% sucrose in 0.1 m phosphate buffer before being sectioned on a sliding microtome. Fifty micrometer sections were quenched with 3% hydrogen peroxide, washed, and blocked (5% horse serum, 5% fish skin gelatin, and 0.1% Triton X-100 in 0.1 mTris-buffered saline). For BrdU staining, DNA was denatured by incubating sections in 2N HCl at 37°C for 30 min and then washed in 0.1 m sodium borate, pH 8.5, for 10 min as described previously (Parent et al., 1999). Primary antibody dilutions used were 1:200 for anti-GFAP (G-A-5; Sigma) and 1:1000 for BrdU (mouse monoclonal; Roche Molecular Biochemicals). Primary antibody was applied overnight at room temperature. Secondary antibody (biotinylated donkey anti-mouse; Jackson ImmunoResearch) was applied for 1 hr at room temperature. The sections were washed and developed with a mouse Elite ABC kit (Vector Laboratories, Burlingame, CA).
BrdU counting. The subplate zone was identified as described previously (McQuillen et al., 2002) by accepted criteria (Boulder Committee, 1970) and cytoarchitectonic features of neocortex (Bayer and Altman, 1990). Specifically, in the radial domain, the subplate (layer VIB) was localized at the base of the cortical plate, immediately below layer VI neurons, and contained characteristic pyramidal neurons. The borders in the coronal plane were determined by the characteristic six-layered neocortex and extended to cingulate cortex in the medial direction and entorhinal cortex laterally. Only heavily BrdU-labeled cells that fell into this region were counted. As in previous studies (Price et al., 1997), heavily labeled cells were defined as cells in which more than half the nucleus was labeled. Littermates were analyzed for quantification of subplate neuron death. Brains from animals receiving H-I at P1 (n = 6), sham-operated animals (n = 3), or unmanipulated animals (n = 2) were analyzed at P5. Despite the use of timed-pregnant rats, significant variability occurs between litters in BrdU uptake and labeling of subplate neurons. Therefore, no attempt was made to compare absolute numbers of BrdU-positive neurons among experimental groups. Cell counts were expressed as the interhemispheric ratio of BrdU-labeled subplate neurons (BrdU-positive cells ipsilateral to CCL/BrdU-positive cells contralateral to CCL). Cell death is expressed as a percentage derived from this ratio (100 − ipsilateral/contralateral × 100). The brains were sectioned entirely, and a random series representing every 10th section was selected and analyzed. There were no differences between groups in the number of sections per animal (85 ± 8.4 vs 82 ± 8.4 sections per brain, mean ± SD; p = 0.57); therefore, average cell death per section is reported. The subplate was identified as described above in digital images, and heavily labeled BrdU-positive cells were counted in each hemisphere in each section with the cell-counting macro (ftp://rsbweb.nih.gov/pub/nih-image/user-macros/).
Perl's iron stain. Free-floating sections were stained in a 1:1 mixture of 2% v/v HCl and 2% potassium ferrocyanide for 30 min at room temperature (Connor et al., 1995). The sections were rinsed in distilled water twice for 10 min, and then precipitated iron was visualized with DAB, 0.5 mg/ml, in 0.1 mphosphate buffer with 0.07% hydrogen peroxide for 5–10 min. The sections were rinsed in 0.1 m PBS and mounted on slides, dehydrated, and put under cover glass.
Cytochrome oxidase staining. Free-floating sections were stained for cytochrome oxidase activity (Anderson et al., 1975) in a reaction mixture that contained 5 mg of DAB, 10 mg of cytochrome C (Sigma), 750 mg of sucrose, 20 mg of catalase (Sigma), and 9 ml of 0.05 m sodium phosphate buffer, pH 7.4. Sections were incubated overnight at 37°C, rinsed in 0.1 m PBS, mounted on slides, dehydrated, and put under cover glass.
Carbocyanine dye labeling. Rats were fixed by transcardial perfusion with 0.1 m sodium phosphate-buffered 4% paraformaldehyde or immersion-fixed in the same fixative (embryonic ages). Brains were removed and stored in fixative with 0.02% sodium azide. Small (∼100 μm), similar-sized crystals of DiI (D-282; Molecular Probes) or 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD; D-307; Molecular Probes) were placed into visual cortex. Alternate configurations of dyes were placed in each hemisphere to distinguish ipsilateral from contralateral hemispheres. The dye was allowed to transport at 37°C for 3 weeks. Coronal sections were cut at 50–100 μm on a vibratome. Sections were counterstained with 0.001% bis-benzimide.
Imaging. Digital images were acquired with a Nikon (Mellville, NY) Eclipse 800 microscope and a cooled CCD camera (Spot2; Diagnostic Instruments, Sterling Heights, MI). Digital images were analyzed on an Apple (Cupertino, CA) G4 computer with the public domain NIH Image program (developed at the National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
Confocal montage imaging. Fluorescent cryosections (BrdU/ISEL) were imaged on a Leica (Cambridge, UK) confocal microscope. Low-magnification montages were reconstructed from 5 or 10× fields with Leica Qwin montaging software and a motorized stage. Complete sections represent a montage of 8–10 separate fields.
Motor testing. Animals were raised to 3 months of age and then assessed for motor deficits (Crawley, 2000). Each animal was observed for gross deficits with open-field locomotion. Then animals were scored on a three-point scale (2, normal; 1, impaired; and 0, incapable of performing task) for rod and beam walking (1-inch-diameter rod, 1-inch-wide beam) and for the number of foot faults on stair climbing. Each animal was tested three times on each test, and the median value was analyzed for significance. Finally, gait analysis was performed with footprint pattern. Animals receiving H-I were compared with controls consisting of both unmanipulated littermates and sham-operated animals.
Statistics. Normally distributed data are reported as mean ± SD. Hypothesis testing for differences between groups was performed with an unpaired, two-tailed t test. Nonparametric data and percents are reported as mean ± interquartile range (subplate cell death) or mean of median scores (motor testing) ± SEM. Differences between groups were determined with Mann–WhitneyU test and corrected for ties.
Results
P1 rodent H-I results in subcortical, periventricular cell death
To determine the pattern of cell death after CCL and hypoxia at P1 (H-I), we used fluorescent ISEL for detection of DNA strand breaks associated with dying cells, combined with a nuclear counterstain to visualize anatomy. After H-I, cells may die with morphologic features of necrosis or apoptosis (Northington et al., 2001). We use ISEL staining solely as a sensitive indicator of cell death, with the knowledge that cells dying with either phenotype may display an ISEL signal (Charriaut-Marlangue and Ben-Ari, 1995). DNA fragmentation can be detected normally in the brains of postnatal rats (Spreafico et al., 1995) and represents naturally occurring cell death during development. This normal pattern of cell death can be appreciated in sham-operated and unmanipulated littermates (data not shown) in the same pattern observed in the hemisphere contralateral to the carotid ligation in this model (Fig. 1A–C, right hemispheres). As has been noted previously (Vannucci, 1990), hypoxia or ischemia alone does not result in any detectable increase in cell death. Superimposed on this naturally occurring cell death in the hemisphere ipsilateral to CCL is cell death that resulted from H-I (Fig. 1A–C, left hemispheres). At 12 hr after manipulation, cell death caused by H-I peaked, and a broad band of cell death could be appreciated in the subcortical regions that contained subplate, intermediate, and subventricular zones (Fig.1B,C). The ventricular zone was affected but to a lesser degree. A smaller amount of cell death was also apparent scattered throughout the neocortex, and retrosplenial cortex was especially affected (Fig. 1B, asterisk). Within the thalamus, the reticular nucleus and internal capsule showed increased cell death (Fig. 1B, arrowheads). In the hippocampus, dying cells were not observed in the pyramidal layer in any hippocampal subdivision. Scattered dying cells were noted in the hippocampal subgranular layer in both hemispheres. This pattern of cell death was present throughout the rostral-caudal extent of cerebral cortex, beginning at the anterior commissure and extending into occipital cortex (Fig. 1A–C). Cell death in the intermediate zone was most intense in parietal cortex at the level of thalamus but extended to more caudal brain regions, including visual and visual association areas, following the dorsolateral aspect of the lateral ventricles (Fig. 1A–C). Temporally, ISEL attributable to H-I could be detected as early as the end of the hypoxic period in animals that died during the procedure (data not shown). By 12 hr after hypoxia, the ISEL signal peaked (Fig. 1); it diminished by 24 hr (Fig. 2), and by 4 d after H-I, increased cell death was not detectable (data not shown). Cell death was not assayed at time points beyond 96 hr.
To confirm this pattern of subcortical injury with other methods, we performed GFAP immunohistochemistry and Perl's iron staining 4 d after H-I at P1 (data not shown). Both GFAP immunohistochemistry and iron staining were increased in the subplate and intermediate zones in exactly the distribution of the heaviest ISEL staining. Cresyl violet staining of neocortex was remarkably normal despite the presence of scattered ISEL, and the histologic appearance of cortex did not reveal prominent cell loss (Fig.3B–D). Animals with severe injury did show some loss of lower-layer neurons in neocortex (Fig.3B).
Subplate neurons die after neonatal H-I
To determine whether subplate neurons specifically were among dying cells, we permanently labeled subplate neurons as they were generating from dividing neuroblasts with a labeled nucleotide, BrdU (BrdU birth dating). Subplate neurons are generated from dividing neuroblasts from E10.5 to E12.5 (Bayer and Altman, 1990). A pulse of BrdU is taken up by dividing neuroblasts in S-phase. Only neurons generated from the next round of cell division (i.e., subplate and marginal zone neurons) receive heavy BrdU label. The BrdU is diluted with each successive round of cell division, so that later-generated neurons (e.g., layer VI-II) receive progressively lighter label. Immunohistochemistry can then detect BrdU in heavily labeled subplate neurons at any age. BrdU birth dating is currently the optimal method for identifying subplate neurons (Allendoerfer and Shatz, 1994).
At 24 hr after H-I, a comparison of BrdU staining in the hemisphere ipsilateral to CCL with the contralateral hemisphere showed a significant decrease in BrdU-labeled subplate neurons (compare Fig. 2, compare C, D). There was a corresponding increase in ISEL-labeled dying cells in the subplate and intermediate zone ipsilateral to CCL (Fig. 2A,C), although the amount of cell death was less than at 12 hr after manipulation (Fig. 1). Using BrdU immunohistochemistry in combination with ISEL labeling, we identified double-labeled cells that indicated dying subplate neurons (Fig. 2B,C, vertical arrows). These cells were noted frequently only in the hemispheres receiving H-I. In animals receiving H-I in which double labeling was performed (n = 4), we found significantly more double-labeled dying subplate neurons ipsilateral to CCL than contralateral (4.5 ± 1.9 vs 0.4 ± 0.5 cells per section; p = 0.0001).
Quantification of subplate neuron cell death after H-I
There is significant interanimal variability in the amount of damage after neonatal H-I (Rice et al., 1981; Sheldon et al., 1996,1998; Towfighi et al., 1997). To quantify the extent and variability of subplate neuron cell death after H-I, we examined BrdU-positive cell density 4 d after H-I at P1 using systematic random sampling and digital imaging to quantify subplate neuron cell death per section (see Materials and Methods). Subplate neuron cell death was increased significantly by H-I (45 ± 48 vs 2 ± 9%, mean ± interquartile range; p = 0.006, Mann–WhitneyU test; Fig. 3A). The severity of injury in any given animal was readily apparent from inspection of either the BrdU labeling or cresyl violet staining and ranged from mildly increased death (Fig. 3D) to nearly complete death of heavily BrdU-labeled subplate neurons (Fig. 3B). The variable neuronal injury that resulted from this manipulation, with injury to subplate neurons even in the mildest cases, confirms the selective vulnerability of subplate neurons relative to other neuronal populations at this age.
Thalamocortical connections form normally after neonatal H-I
BrdU labeling suggests that significant numbers of subplate neurons die prematurely after neonatal H-I. To determine the effects of this premature subplate neuron cell death on thalamocortical development, it was first necessary to determine whether thalamocortical connections formed normally. Given the significant cell death in the intermediate and subventricular zones, it is possible that there was significant injury to the developing thalamocortical and corticofugal axons. Excitotoxic ablation of subplate neurons in cat, before ingrowth of thalamic axons into visual cortex, prevents normal innervation of their targets in layer IV (Ghosh et al., 1990). However, ablation immediately after innervation does not result in the absence of thalamic innervation of layer IV (Ghosh and Shatz, 1992). In rat, thalamic axons have reached neocortex by P0 and innervate appropriate targets over the subsequent days (Catalano et al., 1996). On the basis of these observations, we predicted that H-I at P2 would not disrupt visual thalamocortical innervation. Indeed, this is what we observed using the lipophilic carbocyanine dyes DiI and DiD to trace thalamocortical connections at P7 after H-I at P2. At these ages, label in thalamus resulting from dye placement in cortex is a combination of retrograde labeling of thalamic neurons and anterograde labeling of the descending corticothalamic projections. After dye crystal placement into visual cortex (n = 5), there was robust label in the lateral geniculate nucleus (visual thalamus) in both hemispheres (Fig. 4C,D). Furthermore, despite extensive cell death observed in the internal capsule (Fig.1B), labeled fibers could be observed coursing normally through the intermediate zone and turning medially into the internal capsule before entering thalamus (Fig.4A,B).
Ablation of subplate neurons immediately after innervation of layer IV prevents the activity-dependent refinement of thalamocortical connections into ocular dominance columns (Ghosh and Shatz, 1992). Rodent visual cortex contains monocular and binocular zones but no finer organization of ocular dominance (Antonini et al., 1999). To assess the development of patterned thalamocortical connections after H-I at P2, we visualized the topographical whisker barrel representation in somatosensory cortex at P10. The whisker barrel representation is consolidated over the first postnatal week (O'Leary et al., 1994), and genetic manipulations of glutamatergic (Cases et al., 1996) and serotonergic (Vitalis et al., 1998) neurotransmission disrupt barrel formation. In every case we examined at P10 (n = 4), after H-I at P2, well formed cytochrome oxidase patches appeared in layer IV of somatosensory neocortex ipsilateral to CCL (Fig. 4E) that were identical to those observed in the contralateral hemisphere (Fig.4F).
We conclude from these analyses that P2 H-I does not disrupt initial thalamocortical pathfinding and innervation of somatosensory and visual cortex. Moreover, the initial development of patterned connections in somatosensory cortex proceeds normally. Our analysis does not address subsequent refinement and plasticity of sensory thalamocortical connections.
P2 H-I results in motor deficits
Human PVL is characterized by dysmyelination and spastic diplegia, a static motor deficit with more pronounced involvement of lower extremities than of upper extremities (Volpe, 2001b). To determine whether H-I at P2 leads to motor deficits, we allowed animals from two litters to develop to maturity after H-I at P2. Animals were then observed during open-field locomotion and gait analysis, but they did not display any observable deficits (data not shown). However, when tested on rod and beam walking, animals receiving H-I performed significantly worse than controls (Fig.5; rod walking score, 1.3 ± 0.2 vs 0.7 ± 0.1, control vs H-I, mean of median scores ± SEM; tied p = 0.04, Mann–Whitney U test; beam walking score, 1.8 ± 0.1 vs 1.3 ± 0.1; tiedp = 0.02). To confirm this observation, rats were tested for foot faults while they climbed an inclined stair. H-I-treated animals had more faults than controls (Fig. 5; 2.4 ± 0.3 vs 0.2 ± 0.1; p < 0.0001, Mann–WhitneyU test). Although faults were not recorded by extremity, qualitatively we observed faults of the left rear paw most frequently in animals receiving right CCL and hypoxia. These observations indicate that despite normal-appearing open-field locomotion, P2 H-I leads to significant motor deficits. The relationship of these motor deficits to subplate cell death remains to be elucidated.
Discussion
These results demonstrate that a rat model of early neonatal H-I leads to significant, premature subplate neuron cell death. Despite intense subcortical injury to the developing subplate and intermediate zones, thalamocortical connections to somatosensory and visual cortex form normally. This manipulation results in measurable motor deficits in mature animals. These data, combined with observations of injury to oligodendrocyte progenitors (Back et al., 2002), support H-I at P2 in rat as a model of human PVL. This model is useful for testing the hypothesis that neonatal H-I disrupts neurodevelopment through effects on cortical plasticity and the refinement of cortical connections as a result of subplate neuron cell death.
Maturation-dependent topography of H-I cerebral injury
H-I brain injury produces age-dependent and region-specific injury. Many factors contribute to the evolution of this pattern, including maturation of cerebral oxygen and substrate delivery through development of the cerebral vasculature and autoregulation (Volpe, 2001a), selective cellular vulnerability related to oxidative metabolism (Ferriero, 2001), excitatory amino acid signaling (Johnston et al., 2001; Jensen, 2002), and programmed cell death (Han et al., 2000; Northington et al., 2001). The Levine model [i.e., right CCL followed by hypoxia (Levine, 1960)] performed on immature rats (Rice et al., 1981) at selected ages (Sheldon et al., 1996; Towfighi et al., 1997) also produces age-dependent and region-specific brain injury. Analyzing H-I at P1 with ISEL, we clearly show a subcortical pattern of cell death similar to human PVL. In agreement with others (Towfighi et al., 1997), we did not see significant hippocampal injury at this age, a distinct difference from H-I at P7 and later, when hippocampal region CA3 becomes vulnerable to injury before the adult pattern of CA1 injury occurs beginning at P21. With this sensitive assay for cell death, we did note low levels of diffuse cell death in neocortex and retrosplenial cortex. In the most severely affected animals, lower cortical layers begin to show laminar cell loss. In even the mildest cases, subplate neuron cell death occurred, which confirms the selective vulnerability of these neurons relative to other neuronal populations. We did not note columnar patterns of cell death that are frequently observed at P7 or laminar injury to cortical layers III and V as occurs at P13 (Towfighi et al., 1997). Most descriptions of the pathology of PVL indicate that neocortex is spared (Banker and Larroche, 1962). However, using sensitive quantitative volumetric techniques with magnetic resonance imaging of human infants with PVL, several studies (Inder et al., 1999; Peterson et al., 2000) noted marked reduction in cerebral cortical gray matter volume throughout the brain, which raises the possibility that neuronal cell death in neocortex may be more widespread than has been appreciated in human PVL. Alternatively, this could represent altered cortical development after injury to subcortical areas.
Selective cellular vulnerability: subplate neurons
Many authors have speculated that changing patterns of brain injury during development relate to intrinsic properties of the affected cell type, i.e., selective cellular vulnerability (Mattson et al., 1989; Johnston, 1998; Volpe, 2001a). Late oligodendrocyte progenitors are the prototypical examples of such a cell type, because they manifest stage-specific vulnerability in vitro to excitotoxicity (Matute et al., 1997; Fern and Moller, 2000), oxidative stress (Oka et al., 1993; Back et al., 1998), and oxygen–glucose deprivation (Fern and Moller, 2000) and in vivo to H-I (Back et al., 2002). In humans, preoligodendrocytes are the predominant cell type in the oligodendrocyte lineage during the developmental period of peak incidence of PVL (Back et al., 2001).
Subplate neurons share many of these same properties. Subplate neurons are a transient cell population (Chun et al., 1987; Al-Ghoul and Miller, 1989; Price et al., 1997), and they undergo programmed cell death in the first postnatal week in mice (McQuillen et al., 2002). Subplate neurons are located beneath the developing neocortex (Luskin and Shatz, 1985; Kostovic et al., 2002) near areas of diffuse white matter signal abnormality seen on magnetic resonance imaging in preterm human infants (Maalouf et al., 2001) at risk for the diffuse type of PVL (Volpe, 2001b). In humans, the subplate zone peaks at the onset of the developmental window of vulnerability to PVL (GW 24) and undergoes dissolution during the third trimester, and subplate neurons are largely absent after 6 months of postnatal age (Kostovic and Rakic, 1990). Subplate neurons undergo programmed cell death to a much greater extent than other cortical neurons (Price et al., 1997). Subplate neurons are vulnerable to excitotoxic cell death (Chun and Shatz, 1988), which allows for the selective ablation of subplate neurons after injection of the glutamate agonist kainate into embryonic and postnatal kittens (Ghosh et al., 1990; Ghosh and Shatz, 1992). At later time points, cortical neurons become sensitive to kainate, and the injections no longer result in a selective ablation of subplate neurons (Ghosh and Shatz, 1994). Subplate neurons become incorporated into mature synaptic networks, receiving excitatory input from thalamus and making excitatory connections with neurons in layer IV of neocortex, as well as sending recurrent collateral projections back to thalamus (Friauf and Shatz, 1991). In the present study, we have demonstrated that P2 H-I leads to moderate to near-complete subplate neuron cell death, whereas most cortical neurons are left intact. The mechanism of this selective vulnerability of subplate neurons to H-I is unknown but may relate to early cellular maturation (Chun and Shatz, 1989), with a developmentally related increase in glutamate receptor expression, including NMDA receptor 1 (Catalano et al., 1997) and AMPA and kainate receptors (Furuta and Martin, 1999). Notably, most of these observations have been made in vivo. We have described recently a method for purifying subplate neurons that will be useful in elucidating cellular mechanisms of subplate neuron cell death in vitro (DeFreitas et al., 2001).
Abnormal cortical development after neonatal H-I: role of subplate neuron cell death
The involvement of subplate neurons in neonatal H-I brain injury is significant in light of the role that subplate neurons play in normal cortical development (for review, see Allendoerfer and Shatz, 1994). Human thalamocortical development begins at the time of the development of layers in the lateral geniculate nucleus at GW 22–25 (Hitchcock and Hickey, 1980). Synaptogenesis in human visual cortex occurs between GW 28 and birth (Huttenlocher et al., 1982). In rodent, somatosensory thalamic afferents have reached cortex at P0 (Catalano et al., 1996) and innervate their targets in layer IV soon thereafter. Thus, our model and human PVL occur at a stage of visual cortical development most analogous to the early postnatal cat when thalamic afferents have entered visual cortex but have not segregated into ocular dominance columns. Ablation at this point in development disrupts the activity-dependent refinement of thalamocortical connections into mature ocular dominance columns (Ghosh and Shatz, 1992). Ocular dominance columns form through an activity-dependent competition (for review, see Goodman and Shatz, 1993), possibly for neurotrophins. Although the cellular and molecular basis of the disruption of column formation after subplate neuron ablation is not known, one possibility was suggested by the effects of subplate neuron ablation on BDNF expression (Lein et al., 1999). Kainate injection leads to a long-lasting increase in BNDF expression localized to the region of subplate ablation. Increased BDNF is associated with alterations in the phenotype of cortical inhibitory neurons, leading to the suggestion that subplate neurons modulate activity-dependent competition by regulating levels of neurotrophins and excitability in the developing cortex.
Abnormal or delayed myelination is the hallmark of PVL. H-I at P7 depletes the subventricular zone of oligodendrocyte progenitors (Levison et al., 2001). However, H-I at P2 and P7 results in the proliferation of reactive late oligodendrocyte progenitors (Back et al., 2002), and decreased myelin basic protein expression is only transient after H-I at P7 (Liu et al., 2002). Although cortical visual impairment is associated frequently with abnormalities on magnetic resonance imaging, 71% of premature infants with moderate PVL during the neonatal period were found to have at least one abnormality of visual testing at 1 year of age, and yet 66% of these children had normal optic radiations, and all had normal-appearing visual cortex (Cioni et al., 1997). These findings underscore the need to examine fully the neurobiology of neonatal H-I and its impact on cortical development. Subplate neuron injury, alone or coincident with oligodendrocyte injury, could explain these observations.
Footnotes
This research was supported by National Eye Institute Grants EY02858 (C.J.S.) and NS35902 (D.M.F.) and National Institutes of Health Grant K08 HD01396-03 (P.S.M.). We thank Cynthia Cowdrey for preparing cryostat sections and Michael DeFreitas and Gabriel Zada for assistance with bromodeoxyuridine, in situ end labeling, and double staining.
Correspondence should be addressed to Dr. P. S. McQuillen, Department of Pediatrics, Box 0106, University of California San Francisco Medical Center, San Francisco, CA 94143-0106. E-mail:psmcq@itsa.ucsf.edu.
References
- 1.Al-Ghoul WM, Miller MW. Transient expression of Alz-50 immunoreactivity in developing rat neocortex: a marker for naturally occurring neuronal death? Brain Res. 1989;481:361–367. doi: 10.1016/0006-8993(89)90815-9. [DOI] [PubMed] [Google Scholar]
- 2.Allendoerfer KL, Shatz CJ. The subplate, a transient neocortical structure: its role in the development of connections between thalamus and cortex. Annu Rev Neurosci. 1994;17:185–218. doi: 10.1146/annurev.ne.17.030194.001153. [DOI] [PubMed] [Google Scholar]
- 3.Anderson WA, Bara G, Seligman AM. The ultrastructural localization of cytochrome oxidase via cytochrome. J Histochem Cytochem. 1975;23:13–20. doi: 10.1177/23.1.163854. [DOI] [PubMed] [Google Scholar]
- 4.Antonini A, Fagiolini M, Stryker MP. Anatomical correlates of functional plasticity in mouse visual cortex. J Neurosci. 1999;19:4388–4406. doi: 10.1523/JNEUROSCI.19-11-04388.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banker BQ, Larroche JC. Periventricular leukomalacia of infancy. Arch Neurol. 1962;7:386–410. doi: 10.1001/archneur.1962.04210050022004. [DOI] [PubMed] [Google Scholar]
- 9.Bayer SA, Altman J. Development of layer I and the subplate in the rat neocortex. Exp Neurol. 1990;107:48–62. doi: 10.1016/0014-4886(90)90062-w. [DOI] [PubMed] [Google Scholar]
- 10.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 11.Boulder Committee. Embryonic vertebrate central nervous system: revised terminology. Anat Rec. 1970;166:257–261. doi: 10.1002/ar.1091660214. [DOI] [PubMed] [Google Scholar]
- 12.Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron. 1996;16:297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 13.Catalano SM, Robertson RT, Killackey HP. Individual axon morphology and thalamocortical topography in developing rat somatosensory cortex. J Comp Neurol. 1996;367:36–53. doi: 10.1002/(SICI)1096-9861(19960325)367:1<36::AID-CNE4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 14.Catalano SM, Chang CK, Shatz CJ. Activity-dependent regulation of NMDAR1 immunoreactivity in the developing visual cortex. J Neurosci. 1997;17:8376–8390. doi: 10.1523/JNEUROSCI.17-21-08376.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charriaut-Marlangue C, Ben-Ari Y. A cautionary note on the use of the TUNEL stain to determine apoptosis. NeuroReport. 1995;7:61–64. [PubMed] [Google Scholar]
- 16.Chun JJ, Shatz CJ. A fibronectin-like molecule is present in the developing cat cerebral cortex and is correlated with subplate neurons. J Cell Biol. 1988;106:857–872. doi: 10.1083/jcb.106.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chun JJ, Shatz CJ. The earliest-generated neurons of the cat cerebral cortex: characterization by MAP2 and neurotransmitter immunohistochemistry during fetal life. J Neurosci. 1989;9:1648–1667. doi: 10.1523/JNEUROSCI.09-05-01648.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun JJ, Nakamura MJ, Shatz CJ. Transient cells of the developing mammalian telencephalon are peptide-immunoreactive neurons. Nature. 1987;325:617–620. doi: 10.1038/325617a0. [DOI] [PubMed] [Google Scholar]
- 19.Cioni G, Fazzi B, Coluccini M, Bartalena L, Boldrini A, van Hof-van Duin J. Cerebral visual impairment in preterm infants with periventricular leukomalacia. Pediatr Neurol. 1997;17:331–338. doi: 10.1016/s0887-8994(97)00152-5. [DOI] [PubMed] [Google Scholar]
- 20.Connor JR, Pavlick G, Karli D, Menzies SL, Palmer C. A histochemical study of iron-positive cells in the developing rat brain. J Comp Neurol. 1995;355:111–123. doi: 10.1002/cne.903550112. [DOI] [PubMed] [Google Scholar]
- 21.Crawley JN. What's wrong with my mouse? Behavioral phenotyping of transgenic and knockout mice. Wiley-Liss; New York: 2000. [Google Scholar]
- 22.DeFreitas MF, McQuillen PS, Shatz CJ. A novel p75NTR signaling pathway promotes survival, not death, of immunopurified neocortical subplate neurons. J Neurosci. 2001;21:5121–5129. doi: 10.1523/JNEUROSCI.21-14-05121.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferriero DM. Oxidant mechanisms in neonatal hypoxia-ischemia. Dev Neurosci. 2001;23:198–202. doi: 10.1159/000046143. [DOI] [PubMed] [Google Scholar]
- 25.Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friauf E, Shatz CJ. Changing patterns of synaptic input to subplate and cortical plate during development of visual cortex. J Neurophysiol. 1991;66:2059–2071. doi: 10.1152/jn.1991.66.6.2059. [DOI] [PubMed] [Google Scholar]
- 27.Furuta A, Martin LJ. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J Neurobiol. 1999;39:67–80. [PubMed] [Google Scholar]
- 28.Ghosh A, Shatz CJ. Involvement of subplate neurons in the formation of ocular dominance columns. Science. 1992;255:1441–1443. doi: 10.1126/science.1542795. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh A, Shatz CJ. Segregation of geniculocortical afferents during the critical period: a role for subplate neurons. J Neurosci. 1994;14:3862–3880. doi: 10.1523/JNEUROSCI.14-06-03862.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh A, Antonini A, McConnell SK, Shatz CJ. Requirement for subplate neurons in the formation of thalamocortical connections. Nature. 1990;347:179–181. doi: 10.1038/347179a0. [DOI] [PubMed] [Google Scholar]
- 31.Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell [Suppl] 1993;72:77–98. doi: 10.1016/s0092-8674(05)80030-3. [DOI] [PubMed] [Google Scholar]
- 32.Hack M, Flannery DJ, Schluchter M, Cartar L, Borawski E, Klein N. Outcomes in young adulthood for very-low-birth-weight infants. N Engl J Med. 2002;346:149–157. doi: 10.1056/NEJMoa010856. [DOI] [PubMed] [Google Scholar]
- 33.Han BH, D'Costa A, Back SA, Parsadanian M, Patel S, Shah AR, Gidday JM, Srinivasan A, Deshmukh M, Holtzman DM. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000;7:38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- 34.Hitchcock PF, Hickey TL. Prenatal development of the human lateral geniculate nucleus. J Comp Neurol. 1980;194:395–411. doi: 10.1002/cne.901940207. [DOI] [PubMed] [Google Scholar]
- 35.Huttenlocher PR, de Courten C, Garey LJ, Van der Loos H. Synaptogenesis in human visual cortex: evidence for synapse elimination during normal development. Neurosci Lett. 1982;33:247–252. doi: 10.1016/0304-3940(82)90379-2. [DOI] [PubMed] [Google Scholar]
- 36.Inder TE, Huppi PS, Warfield S, Kikinis R, Zientara GP, Barnes PD, Jolesz F, Volpe JJ. Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Ann Neurol. 1999;46:755–760. doi: 10.1002/1531-8249(199911)46:5<755::aid-ana11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 37.Jensen F. The role of glutamate receptor maturation in perinatal seizures and brain injury. Int J Dev Neurosci. 2002;20:339–347. doi: 10.1016/s0736-5748(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 38.Johnston MV. Selective vulnerability in the neonatal brain. Ann Neurol. 1998;44:155–156. doi: 10.1002/ana.410440202. [DOI] [PubMed] [Google Scholar]
- 39.Johnston MV, Trescher WH, Ishida A, Nakajima W. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr Res. 2001;49:735–741. doi: 10.1203/00006450-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Kostovic I, Rakic P. Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J Comp Neurol. 1990;297:441–470. doi: 10.1002/cne.902970309. [DOI] [PubMed] [Google Scholar]
- 41.Kostovic I, Judas M, Rados M, Hrabac P. Laminar organization of the human fetal cerebrum revealed by histochemical markers and magnetic resonance imaging. Cereb Cortex. 2002;12:536–544. doi: 10.1093/cercor/12.5.536. [DOI] [PubMed] [Google Scholar]
- 42.Lanzi G, Fazzi E, Uggetti C, Cavallini A, Danova S, Egitto MG, Ginevra OF, Salati R, Bianchi PE. Cerebral visual impairment in periventricular leukomalacia. Neuropediatrics. 1998;29:145–150. doi: 10.1055/s-2007-973551. [DOI] [PubMed] [Google Scholar]
- 43.Lein ES, Finney EM, McQuillen PS, Shatz CJ. Subplate neuron ablation alters neurotrophin expression and ocular dominance column formation. Proc Natl Acad Sci USA. 1999;96:13491–13495. doi: 10.1073/pnas.96.23.13491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:139–147. [PMC free article] [PubMed] [Google Scholar]
- 45.Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ. Hypoxia/ischemia depletes the rat perinatal subventricular zone of oligodendrocyte progenitors and neural stem cells. Dev Neurosci. 2001;23:234–247. doi: 10.1159/000046149. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Silverstein FS, Skoff R, Barks JD. Hypoxic-ischemic oligodendroglial injury in neonatal rat brain. Pediatr Res. 2002;51:25–33. doi: 10.1203/00006450-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Luskin MB, Shatz CJ. Studies of the earliest generated cells of the cat's visual cortex: cogeneration of subplate and marginal zones. J Neurosci. 1985;5:1062–1075. doi: 10.1523/JNEUROSCI.05-04-01062.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maalouf EF, Duggan PJ, Counsell SJ, Rutherford MA, Cowan F, Azzopardi D, Edwards AD. Comparison of findings on cranial ultrasound and magnetic resonance imaging in preterm infants. Pediatrics. 2001;107:719–727. doi: 10.1542/peds.107.4.719. [DOI] [PubMed] [Google Scholar]
- 49.Mattson MP, Guthrie PB, Kater SB. Intrinsic factors in the selective vulnerability of hippocampal pyramidal neurons. Prog Clin Biol Res. 1989;317:333–351. [PubMed] [Google Scholar]
- 50.Matute C, Sanchez-Gomez MV, Martinez-Millan L, Miledi R. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci USA. 1997;94:8830–8835. doi: 10.1073/pnas.94.16.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McQuillen PS, DeFreitas MF, Zada G, Shatz CJ. A novel role for p75NTR in subplate growth cone complexity and visual thalamocortical innervation. J Neurosci. 2002;22:3580–3593. doi: 10.1523/JNEUROSCI.22-09-03580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller SP, Vigneron DB, Henry RG, Bohland MA, Ceppi-Cozzio C, Hoffman C, Newton N, Partridge JC, Ferriero DM, Barkovich AJ. Serial quantitative diffusion tensor MRI of the premature brain: development in newborns with and without injury. J Magn Reson Imaging. 2002;16:621–632. doi: 10.1002/jmri.10205. [DOI] [PubMed] [Google Scholar]
- 53.Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis. 2001;8:207–219. doi: 10.1006/nbdi.2000.0371. [DOI] [PubMed] [Google Scholar]
- 54.O'Leary DD, Ruff NL, Dyck RH. Development, critical period plasticity, and adult reorganizations of mammalian somatosensory systems. Curr Opin Neurobiol. 1994;4:535–544. doi: 10.1016/0959-4388(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 55.Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parent JM, Tada E, Fike JR, Lowenstein DH. Inhibition of dentate granule cell neurogenesis with brain irradiation does not prevent seizure-induced mossy fiber synaptic reorganization in the rat. J Neurosci. 1999;19:4508–4519. doi: 10.1523/JNEUROSCI.19-11-04508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peterson BS, Vohr B, Staib LH, Cannistraci CJ, Dolberg A, Schneider KC, Katz KH, Westerveld M, Sparrow S, Anderson AW, Duncan CC, Makuch RW, Gore JC, Ment LR. Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA. 2000;284:1939–1947. doi: 10.1001/jama.284.15.1939. [DOI] [PubMed] [Google Scholar]
- 58.Piecuch RE, Leonard CH, Cooper BA, Kilpatrick SJ, Schlueter MA, Sola A. Outcome of infants born at 24–26 weeks' gestation, II: neurodevelopmental outcome. Obstet Gynecol. 1997;90:809–814. doi: 10.1016/S0029-7844(97)00429-8. [DOI] [PubMed] [Google Scholar]
- 59.Price DJ, Aslam S, Tasker L, Gillies K. Fates of the earliest generated cells in the developing murine neocortex. J Comp Neurol. 1997;377:414–422. [PubMed] [Google Scholar]
- 60.Rice JED, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 61.Sheldon RA, Chuai J, Ferriero DM. A rat model for hypoxic-ischemic brain damage in very premature infants. Biol Neonate. 1996;69:327–341. doi: 10.1159/000244327. [DOI] [PubMed] [Google Scholar]
- 62.Sheldon RA, Sedik C, Ferriero DM. Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res. 1998;810:114–122. doi: 10.1016/s0006-8993(98)00892-0. [DOI] [PubMed] [Google Scholar]
- 63.Spreafico R, Frassoni C, Arcelli P, Selvaggio M, De Biasi S. In situ labeling of apoptotic cell death in the cerebral cortex and thalamus of rats during development. J Comp Neurol. 1995;363:281–295. doi: 10.1002/cne.903630209. [DOI] [PubMed] [Google Scholar]
- 64.Towfighi J, Mauger D, Vannucci RC, Vannucci SJ. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain Res Dev Brain Res. 1997;100:149–160. doi: 10.1016/s0165-3806(97)00036-9. [DOI] [PubMed] [Google Scholar]
- 65.Vannucci RC. Experimental biology of cerebral hypoxia-ischemia: relation to perinatal brain damage. Pediatr Res. 1990;27:317–326. doi: 10.1203/00006450-199004000-00001. [DOI] [PubMed] [Google Scholar]
- 66.Vitalis T, Cases O, Callebert J, Launay JM, Price DJ, Seif I, Gaspar P. Effects of monoamine oxidase A inhibition on barrel formation in the mouse somatosensory cortex: determination of a sensitive developmental period. J Comp Neurol. 1998;393:169–184. doi: 10.1002/(sici)1096-9861(19980406)393:2<169::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 67.Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001a;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 68.Volpe JJ. Neurology of the newborn, Ed 4. Saunders; Philadelphia: 2001b. [Google Scholar]
- 69.Wood NS, Marlow N, Costeloe K, Gibson AT, Wilkinson AR. Neurologic and developmental disability after extremely preterm birth. EPICure Study Group. N Engl J Med. 2000;343:378–384. doi: 10.1056/NEJM200008103430601. [DOI] [PubMed] [Google Scholar]