

Abstract
After brief periods of heightened stimulation, calcium entry through L-type calcium channels leads to activation of the transcription factor cAMP response element-binding protein (CREB) and CRE-dependent transcription. Many of the details surrounding the mechanism by which L-type calcium channels are privileged in signaling to CREB, to the exclusion of other calcium entry pathways, has remained unclear. We hypothesized that the PDZ interaction sequence contained within the last four amino acids of the calcium channel α1C (CaV1.2) subunit [Val-Ser-Asn-Leu (VSNL)] is critical for L-type calcium channels (LTCs) to interact with the signaling machinery that triggers activity-dependent gene expression. To disrupt this interaction, hippocampal CA3–CA1 pyramidal neurons were transfected with DNA encoding for enhanced green fluorescent protein tethered to VSNL (EGFP-VSNL). EGFP-VSNL significantly attenuated L-type calcium channel-induced CREB phosphorylation and CRE-dependent transcription, although somatic calcium concentrations after stimulation remained unchanged. The effect of EGFP-VSNL was specific to the actions of L-type calcium channels, because CREB signaling after NMDA receptor stimulation remained intact. The importance of the PDZ interaction sequence was verified using dihydropyridine (DHP)-insensitive α1C subunits. Neurons transfected with α1C lacking the terminal five amino acids (DHP–LTCnoPDZ) exhibited attenuated CREB responses in comparison with cells expressing the full-length subunit (DHP–LTC). Collectively, these data suggest that localized calcium responses, regulated by interactions with PDZ domain proteins, are necessary for L-type calcium channels to effectively activate CREB and CRE-mediated gene expression.
Keywords: NIL-16, CREB, NFAT, L-type calcium channel, α1C, CaV1.2, CIPP, NMDA
Introduction
Activity-dependent gene expression is critical for the changes that occur during development, learning and memory, induction of chronic pain, and the use of abusive drugs (Morgan and Curran, 1991; Bonni and Greenberg, 1997; Nestler and Aghajanian, 1997; Lanahan and Worley, 1998; Melzack et al., 2001). The transcription factor cAMP response element-binding protein (CREB) plays an important role in each of these processes (Finkbeiner et al., 1997;Ji and Rupp, 1997; Carlezon et al., 1998; Milner et al., 1998; Silva et al., 1998; Anderson and Seybold, 2000). Within the hippocampus and other brain regions, CREB activation after brief stimulation (≤3 min) is primarily mediated by calcium entry via L-type calcium channels (with a smaller component attributable to NMDA receptors) (Sheng et al., 1991; Bading et al., 1993; Deisseroth et al., 1996; Mermelstein et al., 2000). Calcium influx through L-type channels activates calmodulin (CaM), leading to phosphorylation of CREB on Ser133 by CaM-dependent protein kinases (Enslen et al., 1995; Tokumitsu et al., 1995; Bito et al., 1996; Ahn et al., 1999; Ho et al., 2000; Ribar et al., 2000; Kang et al., 2001). Phosphorylation of CREB promotes its interaction with CREB binding protein (CBP) or a similar coactivator, leading to CRE-dependent transcription (Parker et al., 1996). Yet after various forms of stimulation, L-type channels contribute only a small percentage to the overall rise in cytoplasmic calcium (Deisseroth et al., 1998; Dolmetsch et al., 2001). Thus the mechanism by which calcium entry specifically through L-type channels mediates CREB activation is not completely understood.
Several lines of evidence suggest that the localization of L-type channels is an important determinant in their ability to signal to CREB. Using various calcium chelators, a recent study proposed that subcellular microdomains adjacent to the plasma membrane contain the calcium-sensitive signaling machinery necessary for CREB phosphorylation (Deisseroth et al., 1996). To play a privileged role in activity-dependent gene expression, L-type calcium channels would be localized to these regions, to the exclusion of other calcium entry routes. α1C (CaV1.2), the principal subunit of a large proportion of L-type calcium channels expressed in brain, ends with the PDZ interaction sequence Val-Ser-Asn-Leu (VSNL) (Koch et al., 1990; Snutch et al., 1991). VSNL promotes α1C interactions with at least two PDZ domain proteins, channel-interacting PDZ domain protein and neuronal interleukin-16 (Kurschner et al., 1998; Kurschner and Yuzaki, 1999). Because PDZ domain proteins are critical for linking channels and receptors to specific second messenger systems (Craven and Bredt, 1998; Sheng and Pak, 2000), we hypothesized that the interactions between α1C and PDZ domain proteins are required for L-type calcium channels to efficiently activate CREB.
Two separate lines of experiments were pursued to test this hypothesis. First, VSNL was expressed in hippocampal pyramidal neurons. In theory, VSNL would compete with endogenous L-type calcium channels for the PDZ domain proteins with which α1C typically interacts, resulting in the displacement of these channels from the calcium-sensing microdomains normally involved in CREB activation. To visualize VSNL, the peptide was added to the C terminus of enhanced green fluorescent protein (EGFP-VSNL), generating a fluorescent protein that in many respects would mimic α1Clocalization. Thus, the effect of EGFP-VSNL on L-type calcium channel-dependent CREB signaling was first determined. In the second set of experiments, neurons were transfected with DNA encoding for either full-length α1C subunits that generate dihydropyridine (DHP)-insensitive L-type calcium channels (DHP-LTC) (Dolmetsch et al., 2001) or a dihydropyridine-insensitive α1C subunit containing a premature stop codon, resulting in the deletion of the PDZ interaction sequence (DHP-LTCnoPDZ). The results from both studies suggest that the VSNL sequence on α1C plays a critical role in L-type calcium channel-mediated CREB phosphorylation and CRE-dependent transcription.
Materials and Methods
Cell culture and transfection. Hippocampal pyramidal neurons from 1- to 2-d-old rats were cultured as described previously (Mermelstein et al., 2000). Briefly, after decapitation and brain removal, the CA3–CA1 region of the hippocampus was isolated in an ice-cold modified HBSS solution containing 20% fetal bovine serum (FBS) (Hyclone, Logan, UT) and (in mm): 4.2 NaHCO3, 1 HEPES, pH 7.35, 300 mOsm. All chemicals were obtained from Sigma (St. Louis, MO) unless stated otherwise. After dissection, the hippocampi were washed and digested for 5 min with 10 mg/ml Trypsin (type XI) in a solution that contained (in mm): 137 NaCl, 5 KCl, 7 Na2HPO4, 25 HEPES, and DNase (1500 U), pH 7.2, 300 mOsm. The tissue was washed and then dissociated using a series of Pasteur pipettes of decreasing diameter. The cell suspension was pelleted twice to remove contaminants, plated on 10 mm coverslips, and allowed to adhere for 15 min before addition of 2 ml MEM (Invitrogen, Grand Island, NY) containing (in mm): 28 glucose, 2.4 NaHCO3, 0.0013 transferrin (Calbiochem, La Jolla, CA), 2 glutamine, 0.0042 insulin, and 10% FBS, pH 7.35, 300 mOsm. Twenty-four hours after plating, 1 ml of media was replaced with a similar solution containing 4 μm cytosine 1-β-d-arabinofuranoside and 5% FBS. Three days later, 1 ml of media was replaced with modified MEM containing 5% FBS. Media solutions contained 2 μg/ml gentamicin (Invitrogen, Carlsbad, CA) to prevent bacterial growth.
Cultured neurons were transfected 7–9 d in vitro (DIV) using a calcium phosphate-based technique (Graef et al., 1999). African Green Monkey kidney (COS-7) cells and human embryonic kidney (HEK)-293 cells were maintained in a DMEM-F12 solution (Life Technologies) containing 2 μg/ml gentamicin. Transfections were performed at 70–80% confluency using Lipofectamine 2000 following the manufacturer's instructions (Invitrogen).
DNA subcloning and immunocytochemistry. pEGFP-C1 was obtained from Clontech (Palo Alto, CA). The sequences encoding for VSNL and Ile-Thr-Thr-Leu (ITTL) were inserted into the multiple cloning site using standard procedures and verified by direct sequencing. NIL-16 was a gift from C. Kurschner (Myriad Proteomics); the DHP–LTC construct was a gift from M. Greenberg (Harvard University) and R. Dolmetsch (Stanford University). Generation of the DHP–LTCnoPDZ construct was the result of a single round of QuickChange mutagenesis following the manufacturer's instructions (Stratagene, La Jolla, CA), replacing the DNA sequence that encodes for the final tyrosine immediately before VSNL in α1C with a premature stop codon. The mutagenesis was verified by direct sequencing.
For experiments measuring CREB phosphorylation, ∼20 hr after transfection, cells were preincubated for 3 hr in a standard Tyrode solution containing 1 μm TTX. When applicable, 5 μm nifedipine and 25 μmd-AP5 (Tocris, Ellisville, MO) were used to block L-type calcium channels and NMDA receptors. KN-93 (2 μm;Calbiochem) was used to block CaM-dependent protein kinases, and the inactive analog KN-92 (10 μm) was used because a control. Diltiazem (100 μm) was used to block the dihydropyridine-insensitive calcium channels. AP5, nifedipine, KN-93, KN-92, and diltiazem were present 30 min before and throughout the stimulation. Three minute application of 50 μm NMDA was used to initiate NMDA receptor-mediated CREB phosphorylation.
After stimulation, cells were fixed for 20 min with ice-cold 4% paraformaldehyde (Electron Microscopy Sciences, Ft. Washington, PA) in PBS containing 4 mm EGTA. Cells were washed three times with PBS and then permeabilized for 5 min with 0.1% Triton X-100 (VWR, West Chester, PA). After PBS wash, cells were incubated for 0.5 hr at 37°C in a PBS-based block solution containing 1% BSA and 2% goat serum (Jackson ImmunoResearch, West Grove, PA). Afterward, coverslips were exposed to the primary antibody [1:1000 dilution of the polyclonal anti-pCREB antibody (Upstate Biotechnology, Lake Placid, NY)] for 1 hr at 37°C. For detecting α1C, neurons were fixed for ∼12 hr after transfection with EGFP, EGFP-VSNL, or EGFP-ITTL. In these experiments, the preincubation and stimulation steps were omitted, and the primary antibody (1:100; Alomone Labs, Jerusalem, Israel) incubation was performed overnight at 37°C. After exposure to the primary antibody, cells were washed with PBS and incubated for 1 hr with a Cy3-conjugated secondary antibody (Jackson ImmunoResearch). Cells were washed again and mounted using the anti-quenching reagent Citifluor (Ted Pella, Redding, CA). Acquisition and quantification of fluorescent intensities (n = ∼15 per group) were determined using a Bio-Rad confocal workstation (MRC 1024) and Metamorphsoftware (version 4.6). Transfected neurons were compared with untransfected cells on the same coverslip, often in the same image. To verify consistency across cover slides, multiple coverslips (two to three) were prepared following the same experimental conditions. Experiments were also replicated multiple times to verify results. Immunocytochemistry on COS-7 cells followed similar protocols. NIL-16 was detected with an antibody (1:100) from PharMingen (San Diego, CA) (Kurschner and Yuzaki, 1999). Expression of DHP–LTC and DHP–LTCnoPDZ was determined with an Xpress antibody (1:1000) from Invitrogen.
For quantification of α1C and EGFP-VSNL staining, confocal images were taken under equivalent magnification and laser intensity. In addition, the confocal section that included the primary dendrite was used for analysis. For the analysis of α1C clustering, a single fluorescence threshold was set and applied equally across groups to minimize the inclusion of background staining. On the basis of a Gaussian distribution, individual puncta were defined as fluorescent regions of two to six adjacent pixels (∼0.5 μm), all above the threshold value. Fluorescent regions larger than six pixels were considered to be multiples. The total number of fluorescent aggregations within a 7 μm radius from the center of the nucleus was included in the analysis. Because of the inherent differences in transfection level and EGFP expression, no single background threshold could be applied for the quantification of EGFP-VSNL puncta. Thus, individual thresholds were set for each cell, and puncta were counted according to the methods described above. Multiple observers (n = 4) performed analyses of each cell without knowledge of experimental hypotheses, and the average counts were used for the final analysis.
Coimmunoprecipitation. HEK-293 or COS-7 cells transfected with NIL-16 and either EGFP or EGFP-VSNL were washed once with 4°C PBS and then lysed in ice-cold modified radioimmunoprecipitation (RIPA) buffer without SDS (Harlow and Lane, 1988) that contained (in mm): 1 EDTA, 200 PMSF, 200 Na3VO4, and 200 NaF plus 1 μg/ml of aprotinin, leupeptin, and pepstatin. Cells were collected in 1.5 ml Eppendorf tubes and spun in a microcentrifuge at 14,000 rpm for 10 min. The supernatant was transferred to a separate 1.5 ml tube containing 30 μl of protein-G beads and agitated for 1 hr at 4°C. After centrifugation at 2000 rpm for 2 min, the supernatant was removed and added to a fresh 1.5 ml tube containing 30 μl of protein-G beads and 5 μg of the anti-NIL-16 antibody. After agitation for 1 hr at 4°C, the solution was centrifuged at 2000 rpm for 2 min. The pellet was washed three times with RIPA buffer and prepared for Western blotting using commercially available reagents (Invitrogen). After blocking in a Tris-buffered saline solution containing 10% milk and 1% BSA, the blot was incubated with a chicken polyclonal anti-EGFP antibody (1:1000; Upstate Biotechnology) overnight at 4°C. Both the primary and secondary antibodies were diluted in Tris-buffered saline containing 1% milk, 1% BSA, and 0.1% Tween 20. The membrane was washed in 0.1% Tween–Tris-buffered saline and probed with an anti-chicken-HRP antibody (1:25,000) (Pierce, Rockford, IL) for 1 hr at 25°C and then exposed to X-Omat XB-1 film (Kodak, Rochester, NY). Direct loading of the cell lysis solution was used as a positive control for transfection efficiency.
Electrophysiology. Whole-cell patch-clamp recordings of cultured hippocampal neurons (∼10 DIV, 24 hr after transfection) were performed at room temperature using standard techniques. Warner GC120T-10 borosilicate glass electrodes (Warner Instrument Corp., Hamden, CT) were pulled on a Flaming/Brown p-97 puller (Sutter Instrument Co., Novato, CA) and fire polished with an MF-830 microforge (Narishige, Hempstead, NY). The intracellular recording solution contained (in mm): 190N-methyl-d-glucamine, 40 HEPES, 5 BAPTA, 4 MgCl2, 12 phosphocreatine, 3 Na2ATP, and 0.2 Na3GTP, pH 7.2, 275 mOsm. The external recording solution contained (in mm): 135 NaCl, 20 CsCl, 1 MgCl2, 10 HEPES, 0.0001 TTX, and 5 mm BaCl2. Bath solution contained (in mm): 140 NaCl, 2 KCl, 23 glucose, 15 HEPES, 1 CaCl2, 2 MgCl2, and 0.01 glycine. All reagents were obtained from Sigma except ATP and GTP (Boehringer Mannheim, Indianapolis, IN) and BAPTA (Calbiochem). The junction potential (<2 mV) was not compensated. Recordings were obtained using an Axopatch 200B amplifier (Axon Instruments, Union City, CA) controlled by a PC running pCLAMP software (version 8.0) using a 125 kHz interface. Electrode resistances were ∼3 MΩ. The series resistance was compensated >70%.
Gene expression assay. Cultured neurons (∼7–9 DIV) were transfected with a luciferase-based reporter for CRE- or nuclear factor of activated T-cells (NFAT)-dependent transcription and EGFP or EGFP-VSNL. After transfection, 2 μm TTX was added to the media. The next day, cells were washed with DMEM, and half were stimulated for 3 min in the high K+solutions indicated. For the CRE-luciferase assays, all cells were pretreated for 30 min with 25 μm AP5, which was also present during stimulation. Afterward, the original media was supplemented with 2 μm TTX and reapplied to the coverslips. Sixteen hours after stimulation, cells were lysed and cellular protein was isolated. Luciferase expression was measured using standard procedures. When measuring the L-type calcium channel component of synaptically mediated NFAT-dependent transcription, half of the neurons were treated with 5 μmnifedipine after transfection. As a control for cellular viability, a constitutively active reporter (PBJ5-luciferase) was transfected with either EGFP or EGFP-VSNL. In separate experiments, the CRE-luciferase reporter was transfected with either DHP–LTC or DHP–LTCnoPDZ. The following day, 25 μm AP5 and 5 μm nifedipine were added to the cell media. Half of the coverslips were also exposed to 100 μm diltiazem. Thirty minutes later, coverslips were stimulated with a modified cell media solution containing 20 mm K+ and either AP5 and nifedipine or AP5, nifedipine, and diltiazem. Three hours later, cells were lysed and luciferase expression was measured. Luciferase expression was not significantly different across groups in unstimulated neurons. All experiments were replicated to verify results.
Calcium photometry. Internal calcium concentrations were determined using the calcium indicator Indo-1 (Grynkiewicz et al., 1985), using methods published previously (Werth et al., 1996). Briefly, ∼24 hr after transfection, cells were loaded with 2 μm Indo-1 AM ester at room temperature for 40 min. During recording, EGFP- and EGFP-VSNL-transfected cells were superfused with a Tyrode solution containing 1 μm TTX and 25 μm AP5 at a rate of 1–2 ml/min. In separate experiments, 5 μm nifedipine was also added to the Tyrode solution. After acquisition of a stable baseline, the perfusate was switched for 3 min to a 20 mmK+ solution containing the appropriate channel blockers. Complete solution exchanges occurred within 10 sec.
To determine which cells were transfected with the DHP–LTC or DHP–LTCnoPDZ, neurons were cotransfected with EGFP (5:1 ratio of channel to EGFP). Similar to the findings reported by Dolmetsch et al. (2001), under these conditions, >90% of the neurons positive for EGFP also exhibited Xpress-tagged calcium channels (n > 50). For neurons transfected with dihydropyridine-insensitive L-type calcium channels, recording solutions contained TTX, AP5 and nifedipine. To determine the contribution of DHP–LTCs and DHP–LTCnoPDZs to the overall increase in [Ca2+]i after 20 mm K+depolarization, diltiazem (100 μm) was added to the recording solutions in a subset of experiments (n = 8). Differences in the residual increase in [Ca2+]i after diltiazem exposure was not significantly different between groups (0.4 nm;p > 0.05). After completion of each experiment, background light levels were determined. Records were later corrected for background and converted to [Ca2+]i by the equation [Ca2+]i =Kd β(R −Rmin)/(Rmax− R), in which R is the 405/490 nm fluorescence ratio. The dissociation constant (Kd) used for Indo-1 was 250 nm.Rmin,Rmax, and β were determined in ionomycin-permeabilized cells in calcium-free (1 mm EGTA) and 5 mmCa2+ buffers. Values ofRmin,Rmax, and β were 0.19, 2.3, and 3.5, respectively.
Results
Interactions between EGFP-VSNL and PDZ domain proteins
Previous reports have shown that α1C is expressed in a punctate pattern, heavily concentrated in the cell soma and proximal dendrites of neurons (Westenbroek et al., 1990; Hell et al., 1993). Using immunocytochemistry and confocal microscopy techniques, we found that expression of EGFP alone did not affect the normal distribution of α1C (Fig.1A). Three-dimensional reconstructions of immunolabeled neurons indicated that the vast majority of α1C expression was localized to the membrane surface. We hypothesized that the clustering of L-type calcium channels was based on α1C binding to PDZ domain proteins. Because the last four amino acids of α1C are believed to promote its interaction with PDZ domain proteins, we overexpressed EGFP-VSNL in an attempt to out-compete endogenous α1C protein for PDZ binding sites. In neurons transfected with EGFP-VSNL, the distribution of endogenous α1C was markedly altered (Fig.1B). The number of intense clusters of fluorescence generated by immunolabeled α1C was significantly decreased (Fig. 1D) (n= ∼15 per group; p < 0.001). Furthermore, the α1C immunofluorescence in EGFP-VSNL-expressing neurons was reminiscent of the staining observed in glial cells (Fig.1B, arrowheads) (Agrawal et al., 2000; Chung et al., 2001; Latour et al., 2001).
Fig. 1.
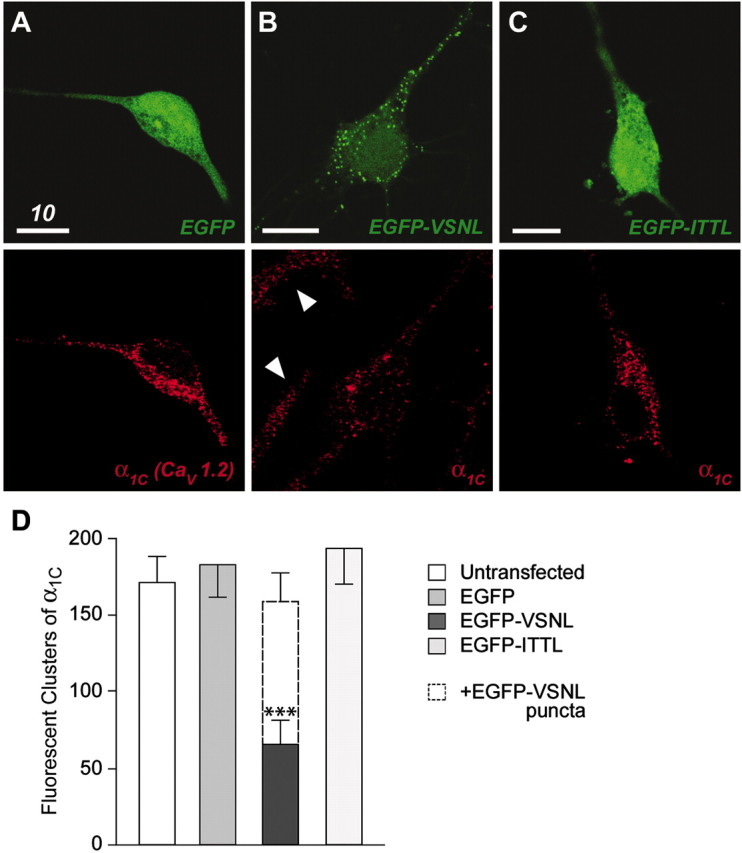
Disruption of endogenous α1Cclustering in neurons transfected with EGFP-VSNL. A, Paired confocal images of a neuron transfected with EGFP (green) and stained using an antibody for the calcium channel α1Csubunit (red). The distribution of endogenous α1C protein in EGFP-transfected neurons was indistinguishable from untransfected neurons (data not shown). B, EGFP-VSNL exhibited a punctate expression pattern similar to the distribution of α1C in control cells. In neurons transfected with EGFP-VSNL, α1C labeling tended to be more diffuse, similar to staining in glial cells (arrowheads). C, EGFP-ITTL exhibited uniform fluorescence similar to EGFP, with no effect on α1C localization. D, EGFP-VSNL expression resulted in a significant (p < 0.001) reduction in the fluorescent clustering of α1C. The number of EGFP-VSNL puncta, when added to the number of residual α1C aggregations, was not significantly different (p > 0.05) from the total number of fluorescent clusters of α1C observed in control conditions.
Although EGFP was uniformly distributed throughout the cell, expression of EGFP-VSNL fluorescence was concentrated into puncta within the cell soma and dendrites (Fig. 1A,B). This expression pattern was similar to the localization of α1Cprotein under control conditions, suggesting that EGFP-VSNL competed with α1C for localization at PDZ binding sites. In keeping with this idea, the number of EGFP-VSNL puncta, when added to the number of residual α1C aggregations, was not significantly different (p > 0.05) from the total number of fluorescent α1C clusters observed under control conditions (Fig. 1D). The punctate distribution of EGFP-VSNL peaked ∼10–15 hr after transfection, becoming more difficult to resolve at 24 hr (see Fig. 4C), consistent with the idea that the PDZ binding sites normally taken up by L-type calcium channels were being fully occupied by EGFP-VSNL.
Fig. 4.

EGFP-VSNL does not affect global calcium signaling. A, Representative recordings of [Ca2+]i measurements during application of 20 mm K+ in EGFP- and EGFP-VSNL-expressing neurons. AP5 (25 μm) was present throughout the recording to block NMDA receptors. B, The stimulus-induced increase in somatic calcium concentrations did not differ between groups. C, D, Similar calcium photometry experiments in the presence of nifedipine. The rise in somatic intracellular calcium attributable to L-type and non-L-type calcium channels after depolarization is not different between EGFP- and EGFP-VSNL-expressing neurons.
To verify that mislocalization of α1C by EGFP-VSNL was specifically dependent on the VSNL sequence, EGFP was tethered to ITTL. ITTL are the last four amino acids of one splice variant of the calcium channel α1D(CaV1.3) subunit (Ihara et al., 1995; Safa et al., 2001) and were not predicted to interact with PDZ binding domains. As with EGFP, EGFP-ITTL exhibited a homogenous distribution throughout the cell, without altering α1C labeling (Fig.1C,D). Because most of the L-type calcium channels in brain are composed of either an α1C or an α1D subunit, the data suggest that these two populations of L-type calcium channels likely have different intracellular distributions and thus may interact with distinct second messenger systems (see Discussion).
Because the last four amino acids of receptors and ion channels promote specific interactions with particular PDZ domain proteins, and VSNL expression appeared to alter the subcellular distribution of endogenous α1C protein, we wanted to verify that EGFP-VSNL did indeed interact with PDZ domain proteins. To test this, COS-7 cells were transfected with cDNA encoding for NIL-16 (a PDZ domain protein hypothesized to interact with α1C) and either EGFP or EGFP-VSNL. In control experiments, EGFP expression was diffuse within the cytosol and nucleus (Fig.2A) and did not accumulate at the plasma membrane with NIL-16 (Fig.2B,C). Interestingly, when EGFP-VSNL was expressed without NIL-16, its cellular distribution was similar to EGFP (Fig.2D, bottom cell). However, when coexpressed with NIL-16, EGFP-VSNL was also observed as aggregations at the plasma membrane (Fig. 2D, top cell), colocalizing with NIL-16 (Fig. 2E,F). Coimmunoprecipitation experiments verified the interaction between EGFP-VSNL and NIL-16 (Fig.2G, lane 6). The interaction between these two proteins was dependent on VSNL, because EGFP did not coimmunoprecipitate with NIL-16 in control experiments (Fig. 2G, lane 3).
Fig. 2.

EGFP-VSNL interacts with the PDZ domain protein NIL-16. A–C, Confocal image of a COS cell expressing EGFP (green) and NIL-16 (red). The two proteins do not appreciably colocalize. D, Two cells transfected with EGFP-VSNL.E, The top cell also expressed NIL-16. F, By colocalizing with the fluorophore, NIL-16 promoted EGFP-VSNL expression at the plasma membrane (yellow). G, EGFP-VSNL (lane 6), but not EGFP (lane 3), coimmunoprecipitated with NIL-16, demonstrating that the interaction between the two proteins is dependent on the PDZ interaction sequence. EGFP and EGFP-VSNL are not detected when the antibody for NIL-16 is omitted from the immunoprecipitation step (lanes 1, 4). Direct probing of the cell lysate indicated comparable concentrations of EGFP and EGFP-VSNL protein (lanes 2, 5).
To verify that the alteration in neuronal α1Cimmunolabeling after EGFP-VSNL expression was not caused by a change in protein expression, L-type calcium channel currents were isolated using patch-clamp methods. Application of the L-type calcium channel blocker nifedipine (5 μm) resulted in a 34.1 ± 2.6% (mean ± SEM) reduction of the whole-cell calcium current in EGFP-VSNL-expressing neurons (n = 14). This percentage of L-type current was not significantly different from untransfected (35.6 ± 2.9%; n = 14) or EGFP transfected (37.8 ± 3.2%; n = 11) neurons (Fig.3). Furthermore, whole-cell calcium current densities remained unchanged, eliminating the possibility that EGFP-VSNL produced a general decrease in calcium channel expression (not transfected: 7.79 ± 0.98 pA/pF; EGFP: 9.62 ± 1.60 pA/pF; EGFP-VSNL: 9.32 ± 1.12 pA/pF).
Fig. 3.

EGFP-VSNL did not alter L-type calcium channel expression. A, A representative whole-cell patch-clamp recording taken from a neuron transfected with EGFP-VSNL (5 mm Ba2+ was used as the charge carrier). Application of nifedipine (5 μm) isolated the L-type current. The voltage waveform is shown above the current traces.B, The L-type calcium channel component of the whole-cell current was not significantly different between untransfected neurons and those transfected with either EGFP or EGFP-VSNL.
Although the above data demonstrate that the quantity of L-type calcium current remained unchanged in EGFP-VSNL-expressing neurons, we considered the possibility that the retention of current was caused by the replacement of α1C subunits with α1D. Because L-type calcium channels composed of α1C are more sensitive to DHP than those containing α1D (Xu and Lipscombe, 2001), the dose dependence of nifedipine block was determined. For each concentration of nifedipine examined (1 nm to 10 μm), block of L-type calcium current in EGFP-VSNL-expressing neurons (n = 7) did not differ significantly from control (n = 9) (supplemental Fig. 1; available at www.jneurosci.org). Furthermore, in both groups, a high-affinity (∼30 nm) and low-affinity (∼1 μm) DHP binding site was found, consistent with previous reports regarding the concentrations required to block α1C and α1D L-type calcium channels.
Global calcium dynamics remain unchanged after EGFP-VSNL expression
The next series of experiments determined that EGFP-VSNL did not affect global calcium influx. Neurons transfected with either EGFP or EGFP-VSNL were loaded with the calcium indicator Indo-1. Somatic calcium concentrations were measured during a 20 mmK+ (+25 μm AP5) stimulus protocol that mimicked the conditions used to activate CREB via calcium entry through L-type calcium channels (see below). Intracellular calcium concentrations in EGFP-VSNL-transfected neurons (n = 7) increased by 106.2 ± 20.5 nm, from a basal concentration of 46.7 ± 6.0 nm, which was not significantly different from their EGFP-transfected (n = 8) counterparts (110.3 ± 10.3 nm, 46.4 ± 4.4 nm) (Fig.4A,B). Furthermore, in the presence of 5 μm nifedipine, intracellular calcium concentrations increased by 40.2 ± 3.8 nm in EGFP-VSNL-transfected (n = 9) and 39.4 ± 5.6 nm in EGFP-transfected (n = 8) neurons (Fig. 4C,D). The approximate 60% block by nifedipine after 20 mmK+ depolarization was consistent with previous biophysical and imaging work suggesting that L-type calcium channels activate at more negative potentials than other voltage-gated calcium channels (Mermelstein et al., 2000, 2001).
EGFP-VSNL specifically disrupts L-type calcium channels from signaling to CREB
The effect of EGFP-VSNL on CREB activation was determined using a phospho-Ser133-specific CREB antibody (Ginty et al., 1993) with nuclear immunofluorescence quantified from confocal sections. To activate CREB via calcium entry through L-type calcium channels, neurons were depolarized for 3 min with 20 mm K+ (Mermelstein et al., 2001). To eliminate NMDA receptor-mediated CREB phosphorylation, AP5 was applied both 30 min before and during depolarization. In neurons expressing EGFP, 20 mm K+produced a significant increase (p < 0.001) in CREB phosphorylation, equal to the effect observed in neighboring untransfected cells (Fig. 5A). The 20 mm K+-induced increase in CREB phosphorylation was blocked by both nifedipine and KN-93 (2 μm) (p < 0.001) but not by the control drug KN-92 (10 μm) (Fig. 5B), demonstrating that under mild stimulation conditions, L-type channels mediate CREB phosphorylation via activation of a CaM-dependent protein kinase (Bito et al., 1996; Wu et al., 2001).
Fig. 5.
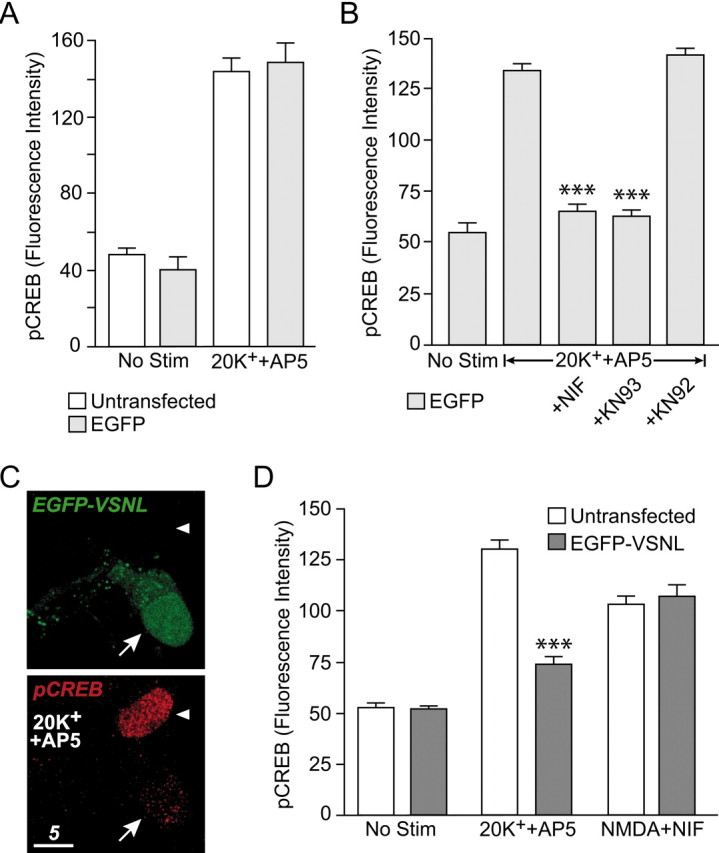
EGFP-VSNL specifically attenuated L-type calcium channel-mediated CREB phosphorylation. A, Immunolabeling studies demonstrated that expression of EGFP did not alter 20 mm K+- (+AP5) induced CREB phosphorylation. B, Nifedipine and the CaM kinase inhibitor KN-93 (2 μm) significantly (p < 0.001) attenuated CREB phosphorylation after depolarization by 20 mm K+. The negative control KN-92 (10 μm) had no effect.C, D, Expression of EGFP-VSNL significantly (p < 0.001) attenuated L-type calcium channel-mediated CREB phosphorylation. The arrow indicates a neuron expressing EGFP-VSNL. The arrowhead points to an untransfected neuron. The bar graph depicts summarized data. EGFP-VSNL attenuation of CREB phosphorylation was observed only following parameters designed to activate L-type calcium channels (i.e., 20 mmK+ + AP5). Conversely, NMDA receptor-mediated CREB phosphorylation (50 μm NMDA + nifedipine) was unaffected by EGFP-VSNL expression.
Although EGFP-transfected neurons exhibited a stimulation-induced increase in CREB phosphorylation identical to untransfected neurons, CREB phosphorylation was significantly reduced (p < 0.001) in neurons expressing EGFP-VSNL (Fig. 5C,D). The reduction in CREB phosphorylation occurred although the amount of calcium entering the cell through L-type calcium channels remained the same (Fig. 4). Intriguingly, the reduction in CREB phosphorylation by EGFP-VSNL was observed only with stimulation parameters designed to activate CREB specifically via calcium entry through L-type channels. EGFP-VSNL-transfected neurons treated for 3 min with NMDA (50 μm) in the presence of nifedipine did not exhibit decreased levels of CREB phosphorylation (Fig. 5D). Under these conditions, AP5 completely blocked stimulus-induced CREB phosphorylation, indicating an NMDA receptor-mediated event (data not shown). Furthermore, similar to L-type calcium channels, a significant component of CREB phosphorylation after brief NMDA receptor stimulation is dependent on activation of CaM-dependent protein kinases (Bito et al., 1996; Wu et al., 2001; Impey et al., 2002). Consistent with this finding, KN-93 significantly decreased CREB phosphorylation after depolarization in the presence of nifedipine (supplemental Fig. 2; available atwww.jneurosci.org). The effect of KN-93 was identical in both untransfected and EGFP-VSNL-transfected neurons, demonstrating that the influence of EGFP-VSNL on L-type calcium channel-mediated CREB signaling could not be attributed to a nonspecific effect on calcium, CaM, or CaM-dependent protein kinases.
We next sought to determine whether expression of EGFP-VSNL would impact CREB-dependent gene expression. In addition to either EGFP or EGFP-VSNL, neurons were transfected with a luciferase-based reporter of CRE-dependent transcription. A 3 min 20 mm K+ stimulus (+AP5) significantly (p < 0.05) increased CRE-dependent transcription [22,370 ± 9416 reflective light units (RLU)] in EGFP-transfected neurons (n = 9) in comparison with unstimulated cells (n = 10). Conversely, the increase in CRE-dependent transcription observed in EGFP-VSNL-transfected neurons (3213 ± 1697 RLU; n= 9) was not significantly different from their unstimulated controls (n = 10) (Fig.6A). Furthermore, when transfected with a constitutively active reporter, expression between groups was not significantly different (EGFP: 599,875 ± 59,744 RLU; EGFP-VSNL: 529,254 ± 25,171 RLU; n = 10 per group), eliminating the possibility that the reduction in CRE-dependent transcription observed in EGFP-VSNL transfected neurons was caused by a nonspecific decrease in gene expression.
Fig. 6.

EGFP-VSNL inhibited CRE-dependent transcription, whereas NFAT-dependent transcription was unaffected. A, A 3 min 20 mm K+ stimulus activated CRE-dependent transcription in EGFP (p < 0.05) but not in EGFP-VSNL-expressing neurons. Transcription was measured using a luciferase-based reporter construct. Similarly, after a 3 min depolarization with 90 mm K+, CRE-dependent transcription in EGFP-VSNL-transfected neurons was significantly attenuated (p < 0.02). AP5 was used to block NMDA receptors. B, After a 90 mm K+ stimulus, NFAT-dependent transcription was unaltered by EGFP-VSNL expression. The L-type calcium channel component of NFAT-dependent transcription triggered by endogenous synaptic activity was also unaffected by EGFP-VSNL.
K+ (90 mm) has also been used as a stimulus to activate L-type calcium channel-mediated gene expression (Bito et al., 1996). This stimulus will activate not only CREB, but other transcription factors as well, including NFATc4 (Graef et al., 1999). For CREB, the stronger depolarization results in increased calcium entry through L-type channels and greater CREB activation. Thus we tested whether expression of EGFP-VSNLwould again result in a diminution of CREB signaling. In EGFP-transfected neurons (n = 4), 90 mm K+ (+AP5) increased CRE-luciferase expression by 56,842 ± 18,978 RLU relative to unstimulated controls (n = 4). In contrast, EGFP-VSNL neurons (n = 5) exhibited an increase of only 6346 ± 1222 RLU when compared with unstimulated EGFP-VSNL neurons (n = 5). CRE-dependent transcription triggered by the brief stimulus was significantly less (p< 0.02) in EGFP-VSNL neurons. Consistent with the idea that attenuation of CRE-dependent transcription is through inhibition of CREB phosphorylation, EGFP-VSNL-expressing neurons also exhibited a significant (p < 0.05) decrease in 90 mm K+-induced CREB phosphorylation (data not shown).
Because NFAT-dependent transcription has been shown to rely on calcium entry through L-type channels after a 90 mmK+ depolarization, we tested whether displacement of α1C L-type channels would also affect the activation of this transcription factor. Neurons were transfected with EGFP or EGFP-VSNL, along with a luciferase-based reporter of NFAT transcription. After a 3 min 90 mmK+ stimulus, EGFP-VSNL-transfected neurons (n = 10) exhibited an increase of 7217 ± 2409 RLU in comparison with unstimulated controls (n = 10), which was not significantly different from the stimulus-induced effect in EGFP neurons (8459 ± 1742 RLU; n = 10 per group) (Fig. 6B). AP5 was omitted from these experiments because NMDA receptors do not directly activate NFAT-dependent transcription under these conditions (Graef et al., 1999). In another test, nifedipine was used to block NFAT-dependent transcription mediated by endogenous synaptic activity. Again, EGFP-VSNL failed to block L-type channel-mediated NFAT-dependent transcription (EGFP: 21,437 ± 3982; EGFP-VSNL: 21,426 ± 2429 RLU; n = 10 per group), suggesting that a mechanism independent of α1C localization is responsible for L-type channel regulation of NFAT activity (see Discussion).
α1C subunits lacking the PDZ interaction sequence exhibit a significant impairment in the ability to signal to CREB
Recently, Dolmetsch et al. (2001) constructed an epitope-tagged α1C subunit containing a threonine to tyrosine point mutation at position 1039, resulting in the generation of a DHP-insensitive L-type calcium channel (DHP-LTC). Depolarizations in the presence of DHPs isolate CREB phosphorylation and CRE-dependent transcription arising from calcium entry specifically through these channels. Furthermore, DHP–LTCs are still sensitive to diltiazem (100 μm), a non-DHP blocker of L-type calcium channels. As such, we used this construct to examine the role of the PDZ interaction sequence on L-type calcium channel CREB responses.
Hippocampal neurons were transfected with DNA encoding for either the full-length DHP–LTC or a truncated version in which the PDZ interaction sequence was absent because of the insertion of a premature stop codon (DHP–LTCnoPDZ). Both constructs localized predominantly to the soma and proximal dendrites of neurons (Fig.7B,C), suggesting that removal of the terminal five amino acids did not have a dramatic effect on insertion of the subunit into the plasma membrane (see Discussion). Furthermore, after a 3 min stimulation with 20 mm K+ (+AP5 and nifedipine), increases in intracellular calcium attributable to both channels were not significantly different (DHP–LTC: 40.9 ± 6.4 nm, n = 7; DHP–LTCnoPDZ: 50.1 ± 5.4 nm, n = 6) (Fig.7A). Under these same stimulation conditions, neurons expressing DHP–LTCs exhibited a significant increase in CREB phosphorylation that could be blocked by diltiazem (Fig.7B). Conversely, neurons transfected with DHP–LTCnoPDZ did not exhibit the same stimulus-induced increase in CREB phosphorylation (Fig. 7C).
Fig. 7.

The PDZ interaction sequence on α1Cis required for L-type calcium channels to effectively signal to CREB.A, Diltiazem-sensitive (100 μm) increases in [Ca2+]i after a 3 min 20 mm K+ (+AP5 and nifedipine) depolarization are not significantly different between neurons transfected with the full-length, dihydropyridine-insensitive L-type calcium channel (DHP–LTC) and those expressing DHP–LTCnoPDZ.B, Neurons expressing DHP–LTCs (arrow) exhibit significant (p < 0.001) increases in CREB phosphorylation after the same stimulation conditions outlined above; untransfected neurons do not (arrowheads). The stimulus-induced increase in CREB phosphorylation was blocked by diltiazem.C, Unlike full-length channels, dihydropyridine-insensitive L-type calcium channels lacking the PDZ interaction sequence do not trigger CREB phosphorylation after a 3 min depolarization. D, In comparison with neurons transfected with DHP–LTC, neurons transfected with DHP–LTCnoPDZ exhibit significantly (p < 0.001) less diltiazem-sensitive CRE-dependent transcription after a 3 hr 20 mm K+ stimulus in the presence of AP5 and nifedipine.
In the final experiment, neurons were transfected with the CRE-luciferase reporter and either DHP–LTC or DHP–LTCnoPDZ. As reported originally by Dolmetsch et al. (2001), whereas DHP–LTCs are capable of activating CREB, they are not as proficient as endogenous L-type calcium channels. Thus, we were not surprised to find that neurons expressing DHP–LTCs did not consistently exhibit increases in luciferase expression after a 3 min 20 mmK+ stimulus (in the presence of AP5 and nifedipine). Consequently, neurons were stimulated with 20 mm K+ (+AP5 and nifedipine) for 3 hr in the presence or absence of diltiazem (Fig. 7D). Under these conditions, DHP–LTCs generated an increase in luciferase activity by 102,569 ± 9,899 RLU (n = 10 per group), significantly greater than CRE-dependent transcription attributable to DHP–LTCnoPDZs (42,752 ± 9077 RLU;n = 10 per group). Intriguingly, the results suggest that although DHP–LTCnoPDZs are capable of activating CREB, they are less efficient than those channels containing the PDZ interaction sequence (see Discussion).
Discussion
The subcellular localization of α1C-comprised L-type calcium channels appears critical for CREB-dependent gene expression. EGFP-VSNL, which can interact with the PDZ domain protein NIL-16, reduced L-type calcium channel aggregations when expressed in neurons. After expression of EGFP-VSNL, CREB phosphorylation and CRE-dependent transcription mediated by calcium entry through L-type calcium channels was significantly attenuated, although increases in somatic calcium concentrations after stimulation remained unchanged. Furthermore, the ability of L-type calcium channels to activate CREB and CRE-dependent transcription after the removal of the PDZ interaction sequence was significantly compromised. Collectively, our results suggest that local calcium signaling plays a critical role in L-type calcium channel-mediated activation of CREB-dependent gene expression.
Over the last several years, PDZ domain-containing scaffold proteins have been shown to link ion channels with both cytoskeletal and signaling proteins. Because the highly polarized nature of neurons requires different cellular locales to support distinct functions, the active assembly of signal transduction pathways within particular regions of the plasma membrane provides a mechanism by which efficient signaling can occur. The most frequently studied of these structures are the postsynaptic densities, in which PDZ domain protein interactions regulate glutamate receptor activity and glutamate-induced plasticity. Here we provide evidence that L-type calcium channels may also be targeted to distinct intracellular scaffolding and signaling proteins to activate gene expression. Considering that different classes of voltage-gated calcium channels support unique cellular functions, the utilization of linking proteins is not surprising. In fact, compartmentalization of calcium channels may be a common theme, because recent data have shown that the N-type calcium channel subunit α1B-1 (CaV2.2a) contains targeting sequences to promote its interaction with the adaptor proteins Mint/x11-like protein 1 (Mint1) and calcium/calmodulin-dependent serine protein kinase (CASK) (Maximov et al., 1999; Kaneko et al., 2002; Maximov and Bezprozvanny, 2002). The resulting interactions target N-type calcium channels to the presynaptic terminal where they play a critical role in neurotransmitter release.
By manipulating various regions of the α1Csubunit, a clearer understanding of how L-type calcium channels are preferentially linked to CREB and CRE-dependent transcription is emerging. Dolmetsch et al. (2001) found that removal of the CaM-binding “IQ motif” from α1C produces L-type calcium channels incapable of signaling to CREB, demonstrating an inherent prerequisite within the channel for activation of CRE-dependent transcription. Yet various calcium channel α1 subunits, not just α1C, contain similar (or identical) IQ motifs and interact with CaM (Lee et al., 1999; Peterson et al., 1999). Thus, another mechanism must provide L-type calcium channels with a “private line” to the nucleus, allowing only these voltage-gated calcium channels to trigger CREB phosphorylation. We believe that this second requirement is fulfilled by interactions between α1C and PDZ domain proteins. Anchoring α1C to regions where CREB signaling is initiated would not only promote the ability of these particular L-type calcium channels to activate gene expression but also help to exclude other calcium channels from localizing to these same regions and thus signaling themselves.
The studies with DHP–LTC and DHP–LTCnoPDZ provide support for this hypothesis. After a brief and mild stimulation (3 min with 20 mm K+), activation of DHP–LTCs, but not DHP–LTCnoPDZs, results in the phosphorylation of CREB (Fig. 7). Interestingly, with longer stimulation conditions (e.g., 3 hr), L-type calcium channels lacking the PDZ interaction sequence can partially overcome their disadvantage in signaling to CREB, perhaps by overwhelming endogenous calcium buffering. Consistent with this idea, after a prolonged and robust depolarization (resulting in micromolar increases in intracellular calcium), DHP–LTCs containing the IQ motif but lacking the final 502 amino acids from its C terminus (including VSNL) exhibit CREB phosphorylation and CRE-dependent transcription to a lesser extent than the full-length channel (Dolmetsch et al., 2001).
In our experiments, we used two distinct strategies in examining the importance of the α1C PDZ interaction sequence on L-type calcium channel-mediated CREB responses. With EGFP-VSNL, we took a freely diffusible protein (EGFP) and promoted its targeting to the membrane surface (Fig. 1B). As a result, 10–15 hr after transfection, a clear difference in the distribution between EGFP and EGFP-VSNL was observed. At later time points (Fig. 5C), differences between the two fluorophores became more difficult to resolve. We attribute this phenomenon to EGFP-VSNL saturation of the PDZ protein binding sites, with excess EGFP-VSNL distributed similarly to EGFP. When comparing the localization of DHP–LTC with DHP–LTCnoPDZ, no striking differences were observed. Both proteins contain 24 transmembrane-spanning regions and the intracellular domains that promote interactions with the calcium channel accessory subunits necessary for channel insertion into the plasma membrane. As such, both DHP–LTC and DHP–LTCnoPDZ were targeted to the cell soma and dendrites (Fig. 7B,C). Presumably, at the subcellular level, differences in their localization exist.
Currently, only two PDZ domain proteins are known to interact with VSNL: CIPP and NIL-16 (Kurschner et al., 1998; Kurschner and Yuzaki, 1999). CIPP is expressed within the cerebellum; NIL-16 is expressed within the cerebellum and hippocampus. Thus, our working hypothesis is that in hippocampal neurons, interactions between L-type calcium channels and NIL-16 are necessary for efficient signaling to CREB. However, coimmunoprecipitation experiments examining potential interactions between α1C and NIL-16 in neurons have thus far been unsuccessful (Kurschner and Yuzaki, 1999; J. P. Weick and P. G. Mermelstein, unpublished observations). This is most likely attributable to various factors, including the relative low abundance of NIL-16 in neurons and the poor sensitivity of the NIL-16 antibody (Kurschner and Yuzaki, 1999). Future experiments will need to overcome these technical limitations to definitively reveal which PDZ domain protein(s) interacts with L-type calcium channels.
Future research will also need to identify other proteins localized to these signaling complexes. Because of its multiple roles in L-type calcium channel function, CaM may be concentrated within these subcellular regions. Interactions between α1C-comprised L-type calcium channels and CaM are required for channel inactivation and facilitation (Peterson et al., 1999; Zühlke et al., 1999, 2000; Erickson et al., 2001; Pitt et al., 2001). Therefore, by modulating calcium entry through L-type calcium channels, CaM may indirectly influence CREB phosphorylation. Furthermore, after brief stimulation, L-type calcium channel-induced CREB phosphorylation occurs via CaM-dependent protein kinase kinase and CaM-dependent protein kinase IV (CaMKIV), indicating that CaM plays a more direct role in CREB activation (Enslen et al., 1995; Tokumitsu et al., 1995; Bito et al., 1996; Ahn et al., 1999; Mermelstein et al., 2001). A necessary step in this process is the translocation of CaM from the cytosol into the nucleus (Deisseroth et al., 1998; Mermelstein et al., 2001; Wei et al., 2002). With longer depolarizations, L-type calcium channels mediate CREB phosphorylation through MAPK (mitogen-activated protein kinase) (Xing et al., 1996; Impey et al., 1998), a process also requiring CaM (Dolmetsch et al., 2001). Consequently, interactions within a clustered signaling complex, in which PDZ domain proteins play a critical role, may ensure that L-type calcium channels have access to sufficient concentrations of CaM to carry out multiple cellular functions.
Outside the hippocampus, the privileged role of L-type calcium channels in CREB signaling has been observed in the cerebral cortex, cerebellum, neostriatum, olfactory bulb, and retina (Murphy et al., 1991; Yoshida et al., 1995; Liu and Graybiel, 1996; Cigola et al., 1998). Calcium entry through NMDA receptors also activates CREB via a mechanism that uses local calcium microdomains (Hardingham et al., 2001, 2002). Cooperation between NMDA receptors and L-type calcium channels can occur to enhance CREB signaling (Nakazawa and Murphy, 1999;Rajadhyaksha et al., 1999), but depending on the stimulus conditions, activation of a single pathway can also take place (Hardingham et al., 2002). Our work has focused on studying how mild depolarizations lead to CREB phosphorylation and CRE-dependent transcription via CaM/CaMKIV activation, because this is the signaling pathway responsible for CREB regulation after synaptic activity at multiple stimulus frequencies (Deisseroth et al., 1996) and is a critical mediator of plasticityin vivo (Ho et al., 2000; Ribar et al., 2000).
With the expression of EGFP-VSNL, we were able to differentiate CREB phosphorylation caused by calcium entry through L-type calcium channels from NMDA receptors. But what is the purpose of having both L-type calcium channels and NMDA receptors signal to CREB? One possibility is that along with CREB phosphorylation, each pathway also activates other signaling molecules specific to that particular cascade, resulting in differences between L-type calcium channel- and NMDA receptor-mediated plasticity. For example, calcium entry through L-type calcium channels activates NFATc4, whereas calcium through NMDA receptors will not (Graef et al., 1999). Recently, L-type calcium channel-mediated, but not NMDA receptor-mediated, long-term potentiation and long-term depression were altered after disruption of the gene encoding extracellular matrix glycoprotein tenascin-C (Evers et al., 2002). These data further suggest that activation of L-type calcium channels and NMDA receptors lead to distinct aspects of plasticity.
In addition to CREB and NFATc4, L-type calcium channels have been shown to activate the transcription factors serum response factor and myocyte enhancer factor-2 (Misra et al., 1994; Mao et al., 1999). Because NFAT-dependent transcription was not affected by EGFP-VSNL, interactions between PDZ domain proteins and α1C are apparently not required for linking L-type calcium channels to the second messenger systems that trigger NFATc4 activation. Thus, multiple mechanisms seem to exist by which L-type calcium channels signal changes in gene expression. This adds another layer of cellular complexity to stimulus-induced signaling, because these separate gene activation pathways could be differentially regulated.
Notably, other discrepancies exist between activation of CREB and NFATc4 after the opening of L-type calcium channels. For example, NFATc4 requires dephosphorylation by calcineurin (Rao et al., 1997), previously shown to limit CREB phosphorylation (Bito et al., 1996). CREB and NFATc4 also exhibit differential sensitivities to the patterns of synaptic stimulation used to trigger their responses (P. G. Mermelstein and R. W. Tsien, unpublished observations). Furthermore, calcium entry through NMDA receptors, but not release from intracellular stores, will lead to rapid CREB activation, whereas the opposite is true for NFATc4. What can account for these differences between CREB and NFATc4 activation? One possibility is that NFAT-dependent transcription is being regulated by calcium entry through α1D-comprised L-type calcium channels. Experiments are under way to address this issue, as well as to determine whether interactions with PDZ domain proteins are required for L-type calcium channels to signal to transcription factors other than CREB.
Understanding how L-type calcium channels regulate different transcription factors is of significant interest, because their coordinated activities guide the expression of immediate-early genes, growth factors, signaling, and structural proteins. Furthermore, through changes in gene expression, L-type calcium channels affect cell fate and axonal and dendritic guidance and influence long-term potentiation and depression (for review, see Mermelstein et al., 2000). The concept of localized calcium signaling within subcellular microdomains has been suggested as a mechanism of signaling specificity for years. Here we provide some of the first evidence that L-type calcium channels signal to the transcription factor CREB using such a process. Further examination of this and other signaling pathways leading to variations in gene expression after L-type calcium channel activation will play an important role in understanding how cellular activity leads to long-term changes in cell structure and function.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS41302 and a Whitehall Foundation grant (P.G.M.). R.D.G. is supported by NIH Training Grant DA07234. We thank Drs. Linda Boland, Eric Newman, Harry Orr, and Kevin Wickman for their scientific input; Drs. Stan Thayer and Yuriy Usachev for their assistance with the photometry studies; Dr. Chris Gomez, Robert Raike, and Holly Kordasiewicz for providing COS cells; and Bryan Becklund, Marissa Iden, and Kathryn Klammer for their technical support. We thank Drs. Michael Greenberg and Ricardo Dolmetsch for the DHP–LTC construct and Dr. Cornelia Kurschner for the NIL-16 plasmid.
Correspondence should be addressed to Paul G. Mermelstein, Department of Neuroscience, University of Minnesota, 6 145 Jackson Hall, 321 Church Street Southeast, Minneapolis, MN 55455. E-mail:pmerm@umn.edu.
References
- 1.Agrawal SK, Nashmi R, Fehlings MG. Role of L- and N-type calcium channels in the pathophysiology of traumatic spinal cord white matter injury. Neuroscience. 2000;99:179–188. doi: 10.1016/s0306-4522(00)00165-2. [DOI] [PubMed] [Google Scholar]
- 2.Ahn S, Ginty DD, Linden DJ. A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron. 1999;23:559–568. doi: 10.1016/s0896-6273(00)80808-9. [DOI] [PubMed] [Google Scholar]
- 3.Anderson LE, Seybold VS. Phosphorylated cAMP response element binding protein increases in neurokinin-1 receptor-immunoreactive neurons in rat spinal cord in response to formalin-induced nociception. Neurosci Lett. 2000;283:29–32. doi: 10.1016/s0304-3940(00)00908-3. [DOI] [PubMed] [Google Scholar]
- 4.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 5.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 6.Bonni A, Greenberg ME. Neurotrophin regulation of gene expression. Can J Neurol Sci. 1997;24:272–283. doi: 10.1017/s0317167100032935. [DOI] [PubMed] [Google Scholar]
- 7.Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 8.Chung YH, Shin CM, Kim MJ, Cha CI. Enhanced expression of L-type Ca2+ channels in reactive astrocytes after ischemic injury in rats. Neurosci Lett. 2001;302:93–96. doi: 10.1016/s0304-3940(01)01683-4. [DOI] [PubMed] [Google Scholar]
- 9.Cigola E, Volpe BT, Lee JW, Franzen L, Baker H. Tyrosine hydroxylase expression in primary cultures of olfactory bulb: role of L-type calcium channels. J Neurosci. 1998;18:7638–7649. doi: 10.1523/JNEUROSCI.18-19-07638.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craven SE, Bredt DS. PDZ proteins organize synaptic signaling pathways. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 11.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 12.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 13.Dolmetsch RE, Pajvani V, Fife K, Spotts J, Greenberg ME. Signaling to the nucleus by and L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 14.Enslen H, Tokumitsu H, Soderling TR. Phosphorylation of CREB by CaM-kinase IV activated by CaM-kinase IV kinase. Biochem Biophys Res Commun. 1995;207:1038–1043. doi: 10.1006/bbrc.1995.1289. [DOI] [PubMed] [Google Scholar]
- 15.Erickson MG, Alseikhan BA, Perterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- 16.Evers MR, Salmen B, Bukalo O, Rollenhagen A, Bosl MR, Morellini F, Bartsch U, Dityatev A, Schachner M. Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J Neurosci. 2002;22:7177–7194. doi: 10.1523/JNEUROSCI.22-16-07177.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- 18.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 19.Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- 20.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 21.Hardingham GE, Arnold FJ, Bading H. A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci. 2001;4:565–566. doi: 10.1038/88380. [DOI] [PubMed] [Google Scholar]
- 22.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 23.Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1988. [Google Scholar]
- 24.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20:6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ihara Y, Yamada Y, Fujii Y, Gonoi T, Yano H, Yasuda K, Inagaki N, Seino Y, Seino S. Molecular diversity and functional characterization of voltage- dependent calcium channels (CACN4) expressed in pancreatic beta-cells. Mol Endocrinol. 1995;9:121–130. doi: 10.1210/mend.9.1.7760845. [DOI] [PubMed] [Google Scholar]
- 27.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 28.Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 29.Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci. 1997;17:1776–1785. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaneko S, Cooper CB, Nishioka N, Yamasaki H, Suzuki A, Jarvis SE, Akaike A, Satoh M, Zamponi GW. Identification and characterization of novel human Ca(v)2.2 (α1B) calcium channel variants lacking the synaptic protein interaction site. J Neurosci. 2002;22:82–92. doi: 10.1523/JNEUROSCI.22-01-00082.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang H, Sun LD, Atkins CM, Soderling TR, Wilson MA, Tonegawa S. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell. 2001;106:771–783. doi: 10.1016/s0092-8674(01)00497-4. [DOI] [PubMed] [Google Scholar]
- 32.Koch WJ, Ellinor PT, Schwartz A. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. J Biol Chem. 1990;265:17786–17791. [PubMed] [Google Scholar]
- 33.Kurschner C, Yuzaki M. Neuronal interleukin-16 (NIL-16): a dual function PDZ domain protein. J Neurosci. 1999;19:7770–7780. doi: 10.1523/JNEUROSCI.19-18-07770.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurschner C, Mermelstein PG, Holden WT, Surmeier DJ. CIPP, a novel multivalent PDZ domain protein, selectively interacts with Kir4.0 family members, NMDA receptor subunits, neurexins, and neuroligins. Mol Cell Neurosci. 1998;11:161–172. doi: 10.1006/mcne.1998.0679. [DOI] [PubMed] [Google Scholar]
- 35.Lanahan A, Worley P. Immediate-early genes and synaptic function. Neurobiol Learn Mem. 1998;70:37–43. doi: 10.1006/nlme.1998.3836. [DOI] [PubMed] [Google Scholar]
- 36.Latour I, Gee CE, Robitaille R, Lacaille JC. Differential mechanisms of Ca2+ responses in glial cells evoked by exogenous and endogenous glutamate in rat hippocampus. Hippocampus. 2001;11:132–145. doi: 10.1002/hipo.1031. [DOI] [PubMed] [Google Scholar]
- 37.Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 38.Liu FC, Graybiel AM. Spatiotemporal dynamics of CREB phosphorylation: transient versus sustained phosphorylation in the developing striatum. Neuron. 1996;17:1133–1144. doi: 10.1016/s0896-6273(00)80245-7. [DOI] [PubMed] [Google Scholar]
- 39.Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- 40.Maximov A, Bezprozvanny I. Synaptic targeting of N-type calcium channels in hippocampal neurons. J Neurosci. 2002;22:6939–6952. doi: 10.1523/JNEUROSCI.22-16-06939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maximov A, Sudhof TC, Bezprozvanny I. Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem. 1999;274:24453–24456. doi: 10.1074/jbc.274.35.24453. [DOI] [PubMed] [Google Scholar]
- 42.Melzack R, Coderre TJ, Katz J, Vaccarino AL. Central neuroplasticity and pathological pain. Ann NY Acad Sci. 2001;933:157–174. doi: 10.1111/j.1749-6632.2001.tb05822.x. [DOI] [PubMed] [Google Scholar]
- 43.Mermelstein PG, Bito H, Deisseroth K, Tsien RW. Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci. 2000;20:266–273. doi: 10.1523/JNEUROSCI.20-01-00266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mermelstein PG, Deisseroth K, Dasgupta N, Isaksen AL, Tsien RW. Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci USA. 2001;98:15342–15347. doi: 10.1073/pnas.211563998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- 46.Misra RP, Bonni A, Miranti CK, Rivera VM, Sheng M, Greenberg ME. L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway. J Biol Chem. 1994;269:25483–25493. [PubMed] [Google Scholar]
- 47.Morgan JI, Curran T. Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annu Rev Neurosci. 1991;14:421–451. doi: 10.1146/annurev.ne.14.030191.002225. [DOI] [PubMed] [Google Scholar]
- 48.Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- 49.Nakazawa H, Murphy TH. Activation of nuclear calcium dynamics by synaptic stimulation in cultured cortical neurons. J Neurochem. 1999;73:1075–1083. doi: 10.1046/j.1471-4159.1999.0731075.x. [DOI] [PubMed] [Google Scholar]
- 50.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 51.Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 53.Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 54.Rajadhyaksha A, Barczak A, Macías W, Leveque JC, Lewis SE, Konradi C. L-Type Ca(2+) channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J Neurosci. 1999;19:6348–6359. doi: 10.1523/JNEUROSCI.19-15-06348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 56. Ribar TJ, Rodriguiz RM, Khiroug L, Wetsel WC, Augustine GJ, Means AR. Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J Neurosci 20 2000. RC107(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Safa P, Boulter J, Hales TG. Functional properties of Cav1.3 (alpha1D) L-type Ca2+ channel splice variants expressed by rat brain and neuroendocrine GH3 cells. J Biol Chem. 2001;276:38727–38737. doi: 10.1074/jbc.M103724200. [DOI] [PubMed] [Google Scholar]
- 58.Sheng M, Pak DTS. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2000;62:755–778. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- 59.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 60.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 61.Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM. Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron. 1991;7:45–57. doi: 10.1016/0896-6273(91)90073-9. [DOI] [PubMed] [Google Scholar]
- 62.Tokumitsu H, Enslen H, Soderling TR. Characterization of a Ca2+/calmodulin-dependent protein kinase cascade. Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. J Biol Chem. 1995;270:19320–19324. doi: 10.1074/jbc.270.33.19320. [DOI] [PubMed] [Google Scholar]
- 63.Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M. Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci. 2002;5:573–579. doi: 10.1038/nn0602-855. [DOI] [PubMed] [Google Scholar]
- 64.Werth JL, Usachev YM, Thayer SA. Modulation of calcium efflux from cultured rat dorsal root ganglion neurons. J Neurosci. 1996;16:1008–1015. doi: 10.1523/JNEUROSCI.16-03-01008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 66.Wu GY, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 2001;98:2808–2813. doi: 10.1073/pnas.051634198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 68.Xu W, Lipscombe D. Neuronal Ca(V)1.3α(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshida K, Imaki J, Matsuda H, Hagiwara M. Light-induced CREB phosphorylation and gene expression in rat retinal cells. J Neurochem. 1995;65:1499–1504. doi: 10.1046/j.1471-4159.1995.65041499.x. [DOI] [PubMed] [Google Scholar]
- 70.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 71.Zühlke RD, Pitt GS, Tsien RW, Reuter H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the(alpha)1C subunit. J Biol Chem. 2000;275:21121–21129. doi: 10.1074/jbc.M002986200. [DOI] [PubMed] [Google Scholar]