

Abstract
Demyelinating diseases can be associated with painful sensory phenomena such as tactile allodynia and hyperalgesia. To study the mechanisms underlying demyelination-induced pain, we have characterized a novel model of demyelination of the sciatic or saphenous nerve. Topical lysolecithin application causes focal demyelination of afferent nerve A-fibers without axonal loss, as assessed either by electron and light microscopy or by immunohistochemical analysis of dorsal root ganglia (DRG) for a neuronal injury marker, activating transcription factor 3. Focal demyelination is accompanied by spontaneous action potentials in afferents and increased expression of neuropeptide Y and Nav1.3 sodium channels specifically in DRG neurons that coexpress a specific marker of myelinated afferents. In contrast, expression of tetrodotoxin-resistant, Nav1.8 sodium channels is specifically decreased in the same subgroup of DRG cells. Central sensitization of somatosensory processing is also induced, with increased behavioral reflex responsiveness to thermal and mechanical stimuli. These changes are reversed by intrathecal administration of an NMDA receptor antagonist or cannabinoid (CB) receptor agonist, but not by a μ-opioid receptor agonist. Recovery of behavioral reflexes occurred ∼3 weeks after lysolecithin treatment. This is the first time that demyelination of afferent A-fibers has been shown to specifically induce neuropathic pain and indicates that axonal damage is not a prerequisite for development of the pain state. The profile of phenotypic changes in DRG is distinct from other pain models and displays a sensitivity to NMDA and CB receptor agents that may be exploitable therapeutically.
Keywords: afferents, demyelination, neuropathic pain, cannabinoids, sodium channels, DRG
Introduction
Both the CNS and the peripheral nervous system (PNS) are susceptible to demyelinating disorders in humans. In the PNS, Charcot-Marie-Tooth disease types 1 and 4 (CMT1 and CMT4) and Guillain-Barré syndrome (GBS) are accompanied not only by segmental demyelination with reduced rates of nerve impulse transmission and impaired motor function, but also by abnormal sensory phenomena such as tactile allodynia (the perception of normally innocuous stimuli such as touching and brushing as painful), hyperalgesia (a heightened response to painful stimuli), and spontaneous pain (Carter et al., 1998). However, the mechanisms underlying neuropathic pain in demyelinating disease are poorly understood (Rasminsky, 1981).
Recently, we showed that mice lacking a functionalPeriaxin (Prx) gene develop a late-onset peripheral demyelination, which is associated with reflex behaviors corresponding to allodynia and hyperalgesia (Gillespie et al., 2000). These enhanced sensory responses are reversible by spinal administration of an NMDA receptor antagonist, implying a critical role for central sensitization in the pain behavior. Recently, mutations in the human PRX gene have been shown to underlie CMT4. Consistent with the mouse model, there is a particularly large sensory involvement in patients with CMT4F, who display various distal abnormalities including pain in both upper and lower extremities (Delague et al., 2000; Boerkoel et al., 2001; Guilbot et al., 2001;Takashima et al., 2002).
To determine how peripheral demyelination might contribute to the development of the ensuing neuropathic pain state, we have characterized a model of focal, reversible peripheral nerve demyelination of the sciatic or saphenous nerve using the selective demyelinating agent lysolecithin (lysophosphatidyl choline) (Hall and Gregson, 1971). Using this model we have studied the effects of demyelination on peripheral nerve function and on the phenotype of cells in the dorsal root ganglia (DRG), including changes in the expression of calcitonin gene-related peptide (CGRP), galanin, neuropeptide Y (NPY), and the Nav1.8 (sensory neuron-specific type 1) and Nav1.3 (brain type III) sodium channels. Furthermore, we have investigated key changes in somatosensory processing within the spinal cord. We show for the first time that transient demyelination causes a distinctive profile of phenotypic changes in DRG. These changes are accompanied by spontaneous activity in afferents and central sensitization of sensory processing, all in the apparent absence of damage to the afferent axons. The sensitivity of the behavioral reflex changes to NMDA receptor antagonists and cannabinoid (CB) receptor agonists suggests therapeutic strategies for pain induced by demyelination.
Materials and Methods
Animals and surgical methods. C57BL/6 mice over the age of 6 weeks were used for all experiments. Animals were anesthetized with 0.3 ml 25% Sagatal (Rhone Merieux, Hertfordshire, UK) in sterile physiological saline (intraperitoneal) and maintained on 1–2% halothane (Zeneca, Cheshire, UK) in O2. For all demyelination experiments, lysolecithin (Sigma, St. Louis, MO) was dissolved in physiological saline to give a final concentration of 15 mg/ml.
Saphenous and sciatic nerve demyelination. The saphenous branch of the femoral nerve was exposed at mid thigh level, and 5–10 μl lysolecithin solution was applied topically via a 25 gauge needle (Terumo) over a length of 5–6 mm, ensuring that the nerve remained coated with the solution for 10 min after which the reagent was washed off with sterile saline and the incision site was closed with 4.0 suture thread, marking the application site. For sciatic nerve demyelination experiments, once the nerve was exposed at mid thigh level, the two larger fascicles (tibial and peroneal) were separated by blunt dissection from the smallest sural fascicle. Lysolecithin was applied to the tibial and peroneal fascicles for 15 min. Sham control animals were prepared for both nerve preparations by omitting lysolecithin from the solution so that the nerve was exposed as above and sterile saline was applied topically for the relevant time.
Behavioral reflex testing. The threshold for hindpaw withdrawal in response to graded mechanical stimulation was measured in conscious animals with von Frey filaments (Stoelting, Wood Dale, IL), which provide a calibrated indentation pressure against the hairless skin of the hindpaws. The threshold response was defined by the filament that caused foot withdrawal at least 5 times in every 10 applications (Meyer et al., 1979; Chaplan et al., 1997). The time for hindpaw withdrawal in response to a quantified noxious thermal stimulus was measured using the Hargreaves-Plantar apparatus (Ugo Basile). The thermal stimulus was of infrared intensity setting 30 (>50°C) applied to the mid-plantar surface of each hindpaw (Hargreaves et al., 1988). The withdrawal was characterized as a brief paw flick, and the withdrawal latency was recorded in seconds.
Baseline measurements were obtained for all animals over the course of 1 week before surgery. Behavioral reflex tests were performed after surgery to identify the development of any reflex sensitivity in treated animals. The threshold value at each time point tested was calculated as the mean ± SEM. Any statistically significant differences in paw withdrawal threshold from mechanical stimulation between ipsilateral and contralateral paws were determined by a Mann–Whitney rank sum test. Likewise, any statistically significant differences in paw withdrawal latency from thermal stimulation were determined by a Student's paired t test.
Morphological investigations. Nerve morphology was examined in animals that previously had focal demyelination induced in either the saphenous or sciatic nerve. Sections of nerve from the region of lysolecithin application were dissected 13 d later, fixed for 4 hr in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1m sodium cacodylate buffer, pH 7.3, postfixed in OsO, and embedded in Araldite.
For light microscopy, 1 μm resin sections of the demyelinated section of the nerve (∼7–10 mm) were stained with Toluidine blue. Once the area of maximal demyelination was established by bright-field microscopy, ultrathin (80 nm) sections were stained with uranyl acetate and lead citrate and examined on a Phillips BioTwin electron microscope (FEI, UK Ltd., Cambridge, UK). Nerve counts were performed at the electron microscope (EM) level so that the amount of demyelination and integrity of large-diameter and small-diameter, unmyelinated axons could be determined. C-fibers were identified as small-diameter unmyelinated fibers present in bundles, surrounded by Schwann cell cytoplasm. The number of intact C-fibers was calculated from EM photographs of cross sections of the entire nerve. To minimize error, the images from all nerves (n = 4 per group) were randomly counted three times (while the source of each image was concealed), and the average for each nerve was calculated. To count the number of myelinated A-fibers, axons with all levels of myelination, including those that were completely demyelinated, must be considered. Therefore, A-fibers were identified as not only fibers with a myelin sheath, but also solitary fibers with a smaller overall diameter attributable to loss of myelin. This allowed distinction of demyelinated fibers from unmyelinated fibers, which usually occur in bundles. The statistical significance of any difference between treated and naive or sham nerves was determined using a Kruskal–Wallis one-way ANOVA on ranks with an all pair-wise multiple comparisons procedure (Dunn's method).
G-ratio. The g-ratio of an axon is a measure of myelin thickness calculated by dividing the axonal diameter by the total diameter of axon plus myelin sheath. This gives an indication of the level of demyelination in axons that are not completely demyelinated by lysolecithin treatment. The g-ratios of myelinated fibers were calculated from EM photographs of the whole cross-sectional area of each nerve using the IP Lab Spectrum P program (Scanalytics, Fairfax, VA). This also calculates the number of myelinated axons in the sample (292 μm2) and enables the proportion of completely demyelinated axons per nerve to be determined. The statistical significance of any difference among treated, naïve, or sham nerves was determined by Kruskal–Wallis one-way ANOVA on ranks with an all pair-wise multiple comparisons procedure (Dunn's method).
Electrophysiology. Electrophysiological recording of activity and function in afferent fiber units of peripheral nerve was performed on animals that previously had focal demyelination induced in the saphenous nerve. Recording experiments were performed on days 3 and 5 after surgery, times at which behavioral reflex sensitization had not developed, and also on days 11, 12, and 13 after surgery, corresponding to the peak time of development of behavioral reflex sensitization.
Mice were anesthetized with 5% urethane (1.25 gm/kg, i.p.) and maintained at 36–37°C with a radiant heat lamp. The saphenous nerve was exposed in the mid thigh and dissected from its associated vein and artery. Further dissection under liquid paraffin enabled the identification of afferent preparations comprising a small number of units. The mechanosensitivity of sensory receptors with low thresholds was measured using calibrated von Frey filaments, and the thermal threshold was measured with a calibrated radiant heat source. The mechanical threshold was defined as the pressure required to cause units to fire in response to direct application of von Frey filaments to the central zone of the identified receptive field, and the thermal threshold was defined as the temperature required to cause units to fire in response to direct application of the radiant heat source to the central zone of the identified receptive field. The conduction velocity of single identified afferent fibers was determined using bipolar electrodes and the peripheral stimulus technique (Iggo, 1958). The percentage of axons in each conduction velocity range, from 0–3 m/sec up to 18–21 m/sec, was calculated, and the statistical significance of any difference between each treatment group was determined by a Kruskal–Wallis one-way ANOVA on ranks with all pair-wise multiple comparisons procedure (Dunn's method). All recording traces of spike activity were obtained via a direct link of the oscilloscope to the Powerlab/MacLab Chart v3.6/s program (AD Instruments, Oxford, UK). Data were subsequently analyzed on the Claris Works 4.0 illustrator program (Claris Corporation, Smart Computing, Lincoln, NE).
Immunohistochemistry. For fixed-tissue immunohistochemistry, animals that 13 d previously had lysolecithin treatment of the right sciatic nerve or saphenous nerve were deeply anesthetized with halothane and perfused through the heart with heparinized saline followed by 4% paraformaldehyde (Sigma) in 0.1m phosphate buffer (Sigma). The DRG ipsilateral and contralateral to the lysolecithin-treated nerve or sham-operated nerve were removed, postfixed with the same fixative for 2 hr, and transferred through increasing concentrations of sucrose in 0.1 m phosphate buffer for 1 hr and left overnight in 25% sucrose in 0.1 m phosphate buffer. The DRG were then embedded in OCT mounting medium (Tissue Tek) and frozen over liquid nitrogen. For nonperfused tissue immunohistochemistry, animals were killed by CO2 inhalation 13 d after lysolecithin treatment. DRG ipsilateral and contralateral to the lysolecithin-treated nerve or sham-operated nerve were removed, snap frozen, and embedded in OCT mounting medium. Cryostat sections of all DRG (10 μm) were thaw-mounted on glass slides precoated with poly-L-lysine (Merck-BDH). For saphenous nerve-treated animals, L3, L4, and L5 DRG were analyzed, and for sciatic-treated animals, L4, L5, and L6 DRG were analyzed (all n = 4).
For colocalization of Nav1.8 or the peptides CGRP or NPY with the A-fiber cell body marker neurofilament 200 kDa (NF-200) (Lawson and Waddell, 1991; Michael et al., 1999), or the C-fiber cell body marker peripherin (Goldstein et al., 1991), nonperfused DRG sections were immersion-fixed for 15 min in 4% paraformaldehyde in PBS. Sections were preincubated in buffer (0.1 mPBS, pH 7.4, containing 0.2% Triton X-100 and 2% gelatin) containing 10% normal goat serum or 10% normal donkey serum (for CGRP detection) for 1 hr at room temperature and incubated with primary antibodies diluted in buffer containing 4% goat or donkey serum overnight at 4°C. Perfused tissue was used for detecting colocalization of Nav1.3, galanin, or activating transcription factor 3 (ATF3) (Tsujino et al., 2000) with NF-200 or peripherin. The immunohistochemical procedure used was as for fresh tissue, omitting the immersion-fixation step.
Antisera were used at the following concentrations: rabbit anti-Nav1.8 [1:200; K107 supplied by S. Tate, GlaxoSmithKline; specificity of K107 was tested by preincubating primary antibody with dilutions of the relevant antigenic peptide (Amaya et al., 2000)]; rabbit anti-Nav1.3 sodium channel [1:200; Alomone Labs, Jerusalem, Israel (Black et al., 1999)]; sheep anti-CGRP [1:1500; Affiniti, Exeter, UK (Todd, 1997)]; rabbit anti-NPY [1:1000, Peninsula Laboratories, Belmont, CA (Polgar et al., 1999)]; rabbit anti-galanin (1:2000; Advanced Chemtech, Cambridgeshire, UK); rabbit anti-ATF3 (1:400; Santa Cruz Biotechnology, Santa Cruz, CA); mouse monoclonal anti-NF-200 kDa [1:2000; clone N52; Sigma (Bennett et al., 1998)]; and mouse monoclonal anti-peripherin [1:1000; Chemicon International, Harlow, UK (Amaya et al., 2000)]. Sections were then washed in buffer and incubated with the appropriate secondary antibodies linked to either tetramethyl rhodamine isothiocyanate (TRITC) (goat anti-mouse-TRITC, 1:200, to detect all mouse primary antisera; Southern Biotechology, Birmingham, AL) or fluorescein isothiocyanate (FITC) (goat anti-rabbit-FITC, 1:200, to detect all goat primary antisera; Cappell; donkey anti-sheep-FITC, 1:100, to detect anti-CGRP primary antiserum; Jackson ImmunoResearch Laboratories, West Grove, PA) for 2 hr at room temperature. Three final washes in 0.1 m PBS were conducted before coverslipping with Vecta-Shield (Vector Laboratories, Burlingame, CA) for analysis. Control sections were processed as above omitting the primary antisera. Observations were made and sections were photographed on an Olympus microscope equipped for epifluorescence. Counts of profiles labeled for immunopositive cells were performed on four to five randomly selected 10 μm sections of DRG (separation of 100 μm) from each of four animals in each group, and only neurons with clear nuclei were counted. Results were expressed either as a proportion of labeled profiles per section or the proportion per total number of single- or double-labeled profiles from all 16–20 sections. The statistical significance between groups was tested by Kruskal–Wallis one-way ANOVA on ranks with all pair-wise multiple comparison procedures (Dunn's method).
Intrathecal administration of drugs. Animals with focal demyelination induced in the saphenous nerve were used for all intrathecal injection experiments. Baseline measurements for mechanical allodynia and thermal hyperalgesia were recorded over a period of up to 2 hr before injection. The mice were briefly anesthetized with halothane and O2 and injected intrathecally at the level of the L4 spinal vertebra using a 25 gauge needle with the NMDA receptor antagonist 3-((R)-2-carboxypiperazine-4-yl)-propyl-1-phosphonic acid [(R)-CPP; Tocris Cookson, Bristol, UK; 100 pmol/10 μl saline], the selective μ opioid receptor agonist d-Ala2, MePhe4, Gly-ol enkephalin (DAMGO; Tocris Cookson; 10 pmol/10 μl saline), the mixed CB1/CB2cannabinoid receptor agonist mesylate (R)-(+)-(2,3-dihydro-5-methyl- 3-(4-morpholinylmethyl) pyrrolo (1,2,3-de)-1,4-benzoxazin-6-yl)-1-naphthalenylmethanone [WIN 55,212–2; Tocris; 60 pmol/10 μl saline with 0.02% dimethylformamide (DMF)], the selective CB1receptor antagonistN-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,3-dichlorophenyl)-4-methyl-1H- pyrazole-3-carboxamide (AM 251; Tocris; 100 pmol/10 μl saline with 0.3% DMF), or a combination of WIN 55,212–2 plus AM 251 (100 pmol/10 μl saline with 0.3% DMF). To determine the effects of each drug on both mechanical allodynia and thermal hyperalgesia, behavioral reflex testing commenced 15 min after injection to allow recovery from anesthesia and continued every 5 min thereafter until readings returned to baseline levels. For all drugs tested, any statistically significant difference of the postinjection paw withdrawal from baseline paw withdrawal thresholds was determined by a one-way repeated measures ANOVA with Dunnet's multiple comparisons versus control grouppost hoc analysis. Extensive control studies have shown that intrathecal injections of the vehicles, saline, or 0.1–0.5% DMF in saline have no effect on these behavioral reflex measures. Dye injections using Pontamine Sky Blue demonstrated that over various time periods up to 60 min after injection there was no spread of dye to the proximal spinal nerve or DRG.
Results
Lysolecithin treatment causes behavioral allodynia and hyperalgesia
Lysolecithin-treated mice displayed a markedly lowered threshold in reflex tests both of cutaneous mechanical sensitization using calibrated von Frey filaments and of thermal nociceptive sensitivity (Fig. 1). The heightened sensitivity to both tests was apparent from day 5 after lysolecithin treatment of saphenous or sciatic nerve, but reached peak values from day 9 until day 15 after surgery. Typically, after lysolecithin treatment of the saphenous nerve, von Frey thresholds were reduced from ∼700 to ∼300 mN/mm2, and thermal withdrawal latencies were reduced from ∼9 to 5 sec at peak behavioral change. Similarly, after lysolecithin treatment of the sciatic nerve, von Frey thresholds were reduced from ∼600 to ∼250 mN/mm2, and thermal withdrawal latencies were reduced from ∼11 to 5 sec at peak behavioral change. In each case the changes recovered to baseline levels by 23 d after treatment. There was no evidence for overt motor deficit after lysolecithin treatment of either nerve. Furthermore, mice that had undergone surgery and saline treatment of the saphenous or sciatic nerve showed no change from baseline von Frey threshold withdrawal values of between 600 and 700 mN/mm2 or baseline thermal paw withdrawal latencies of ∼10 sec (n = 4 in each case).
Fig. 1.
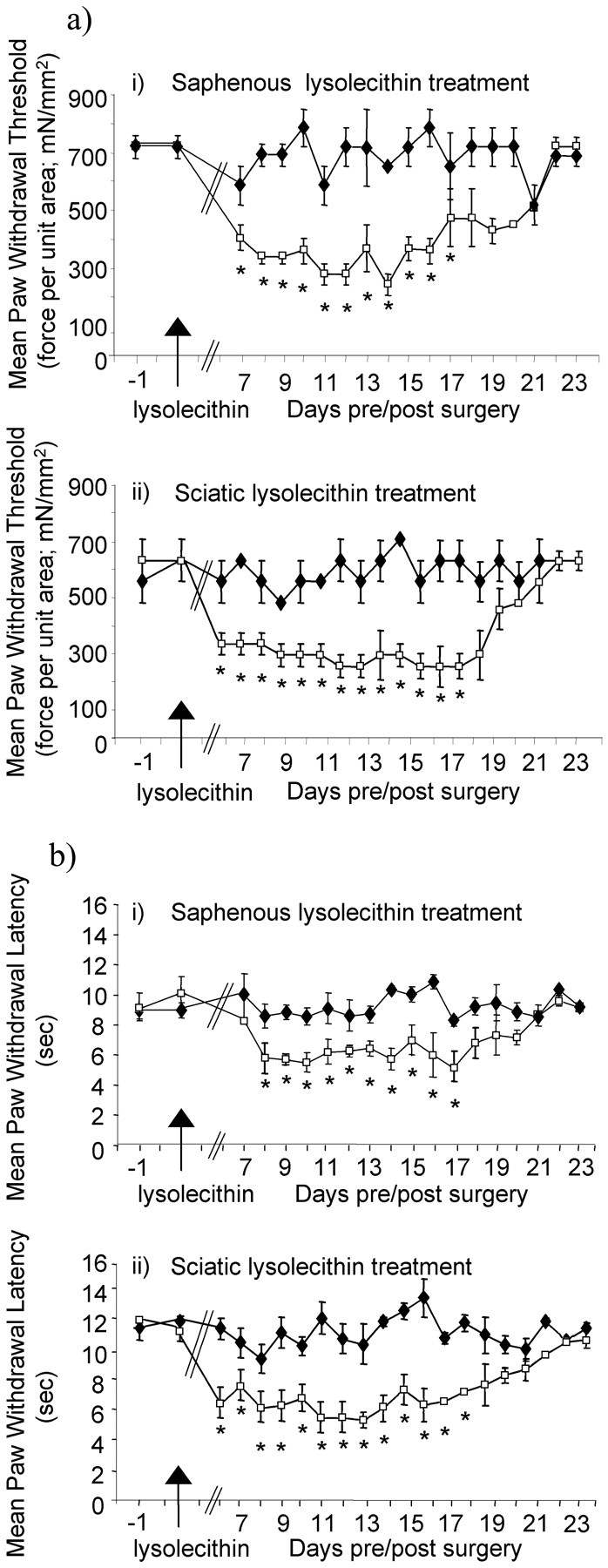
Development of mechanical allodynia and thermal hyperalgesia ipsilateral to lysolecithin treatment of the saphenous or sciatic nerve. a, Filament indentation pressure thresholds for paw withdrawal responses to von Frey filaments were measured after lysolecithin treatment (↑) to one saphenous (n = 6) (i) or one sciatic nerve (n = 6) (ii). Statistical significance of differences between ipsilateral (■) and contralateral (♦) paws (*p < 0.05) was determined by a Wilcoxon test. Each value is the mean ± SEM.b, The time taken for hindpaw withdrawal from a noxious thermal stimulus was measured after lysolecithin treatment (↑) to one saphenous (n = 6) (i) or one sciatic nerve (n = 6) (ii). Statistical significance between ipsilateral (■) and contralateral (♦) paws (*p < 0.05) was determined by a paired Student'st test. Each value is the mean ± SEM.
Lysolecithin treatment causes demyelination without overt axonal damage or sensory loss
To determine the extent of demyelination as a result of lysolecithin treatment, we examined demyelinated, sham, and naive peripheral nerves by light and electron microscopy at the time of peak behavioral change. A maximum of 40% of A-fibers in the saphenous nerve was completely demyelinated 11–13 d after lysolecithin treatment, and many remaining myelinated fibers had myelin of reduced thickness. This was observed by light microscopy (Fig.2a) and calculated from the number of myelinated fibers or formerly myelinated fibers present in each EM image (representing 292 μm2 of the nerve). Lysolecithin-treated saphenous nerve (n = 4) contained 4.9 ± 0.9 myelinated axonal profiles per area sampled (155 profiles over 33 images analyzed), and control nerve (n = 4) contained 8.2 ± 1.7 profiles per area sampled (261 myelinated axonal profiles over 32 images analyzed). After topical application of lysolecithin, a similar degree of demyelination was observed in the sciatic nerve as in the saphenous nerve (Fig.2a). The g-ratios were calculated for the remaining myelinated A-fibers in each nerve after treatment with lysolecithin. The g-ratio was significantly increased in the demyelinated saphenous nerve from 0.61 ± 0.01 in contralateral control nerves to 0.76 ± 0.01 in demyelinated nerves and similarly in the demyelinated sciatic nerve from 0.56 ± 0.02 in contralateral control nerves to 0.81 ± 0.03 in demyelinated nerves (Fig.2b), indicating a general decrease in myelin thickness. There was no significant difference in the level of myelination in sham-operated animals compared with control naive animals.
Fig. 2.
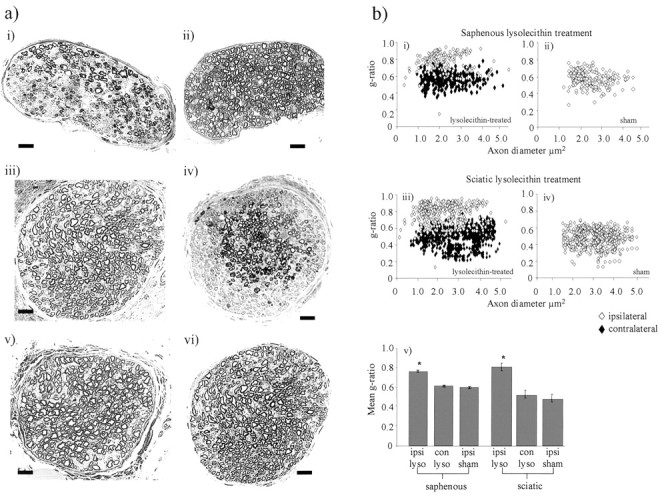
Effects of lysolecithin treatment on the myelination state of the saphenous and sciatic nerve.a, Toluidine blue-stained resin sections (1 μm) from the ipsilateral (i) and contralateral (ii) saphenous nerves of lysolecithin-treated animals on day 13 after treatment, ipsilateral saphenous nerve of sham-treated animals on day 13 after treatment (iii), ipsilateral (iv) and contralateral (v) sciatic nerves of lysolecithin-treated animals on day 13 after treatment, and ipsilateral sciatic nerve of sham-treated animals (vi) on day 13 after treatment were analyzed by light microscopy (n = 4 in each case). Contralateral nerves on day 13 after lysolecithin treatment and nerves ipsilateral to sham treatment all appeared to be morphologically normal with consistent levels of myelination across the nerve. Scale bar, 10 μm. b, Quantification of the degree of myelination of A-fibers after lysolecithin treatment to the saphenous nerve was determined as a g-ratio from electron microscope images of the ipsilateral and contralateral saphenous nerves of lysolecithin-treated (i) and sham animals (ii) on day 13 after, ipsilateral and contralateral sciatic nerve of lysolecithin-treated (iii) and sham animals (iv) on day 13 after treatment (n = 4 in each case). Results are shown as the mean g-ratio of each fiber as a function of the axon diameter (i–iv), and the data are summarized as bar charts below (v). The data show a general decrease in myelin thickness, i.e., increased g-ratio after lysolecithin treatment but not after sham surgery. Statistical significance (*p < 0.05) between conditions was determined by Kruskal–Wallis one-way ANOVA on ranks with an all pair-wise multiple comparisons procedure (Dunn's method).
Electron microscopy demonstrated that after lysolecithin treatment unmyelinated C-fiber bundles appeared to be morphologically normal, and fiber counts revealed no statistically significant change in the number of unmyelinated C-fibers per EM image of either saphenous or sciatic nerve (Fig. 3a,b, Table 1). Furthermore, the total number of larger A-fiber axons that were either myelinated or demyelinated was also unchanged in saphenous or sciatic nerve after lysolecithin (Fig.3a,b, Table 1). Hence, there was no evidence that treatment with lysolecithin under the conditions used in these experiments affects the integrity of axons.
Fig. 3.
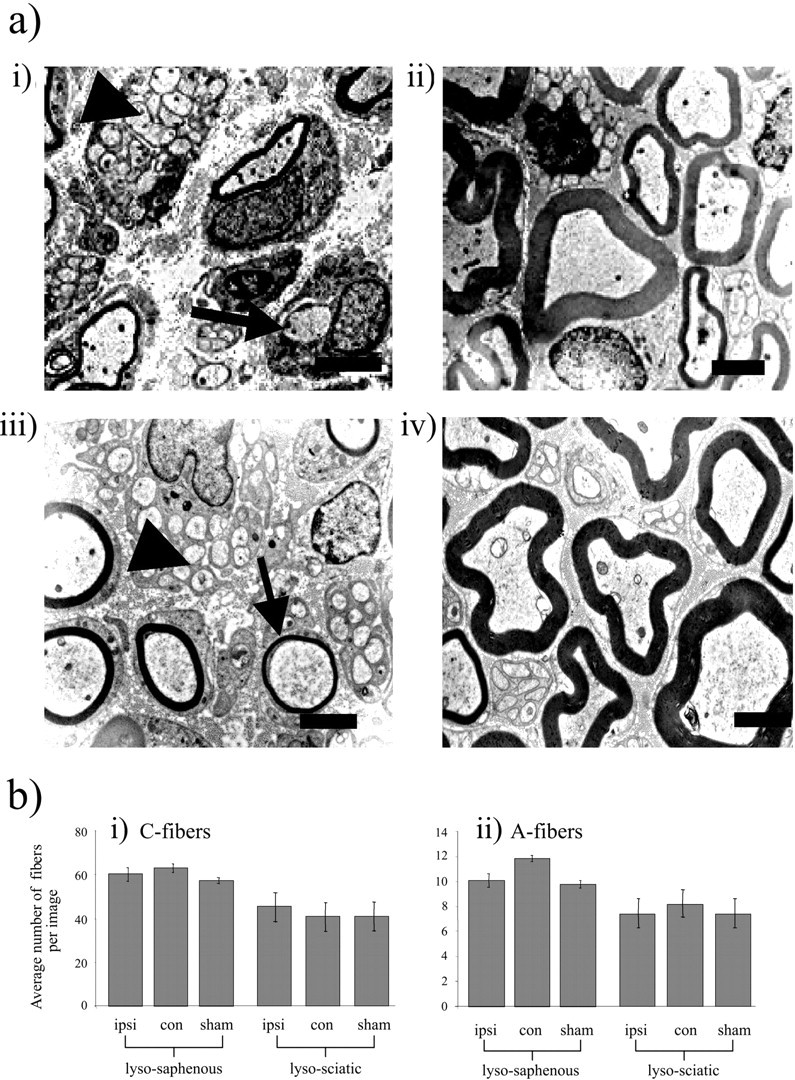
Effects of lysolecithin treatment on morphology of saphenous and sciatic nerve fibers at the EM level. a, Electron microscopy images of either the lysolecithin-treated saphenous (i) or sciatic (iii) nerves, compared with the equivalent sham-operated nerves [saphenous (ii) and sciatic (iv) nerves] on day 13 after treatment (n = 4 in each case). Thirteen days after lysolecithin treatment, ipsilateral nerves display complete demyelination in ∼40% of A-fibers with partial demyelination of many remaining myelinated fibers. The images demonstrate marked lysolecithin-induced reduction in the myelination of A-fibers (arrow), whereas C-fibers (arrowhead) remain intact (5000×). Scale bar, 2 μm. b, i andii show the mean number of C- and A-fibers recorded per image area in different conditions. Unmyelinated axons were identified as small-diameter fibers devoid of any myelin sheath (which were present in bundles, surrounded by Schwann cell cytoplasm) and were individually counted for all groups. A-fibers (with or without lysolecithin treatment) were identified as the larger-diameter axons with myelin sheaths or smaller-diameter thinly myelinated fibers or occasionally those with no myelin that were solitary. Data for both fiber types are presented as average numbers of fibers per 292 μm2 image ± SEM. A Kruskal-Wallis one-way ANOVA on ranks showed no statistically significant differences between treatment groups.
Table 1.
Effects of topical lysolecithin treatment on afferent fiber numbers
| Nerve | Treatment | Side | Average number of axons per 292 mm2 EM image | |
|---|---|---|---|---|
| A-fibers | C-fibers | |||
| Sciatic | Lysolecithin | Ipsi | 6.8 ± 1.5 | 45.1 ± 9.0 |
| Lysolecithin | Contra | 7.9 ± 2.2 | 42.9 ± 8.9 | |
| Sham | Ipsi | 6.7 ± 3.3 | 39.3 ± 7.3 | |
| Saphenous | Lysolecithin | Ipsi | 10.1 ± 0.5 | 60.1 ± 2.0 |
| Lysolecithin | Contra | 11.8 ± 0.2 | 62.9 ± 1.9 | |
| Sham | Ipsi | 9.7 ± 0.3 | 57.3 ± 1.3 | |
EM images were analyzed to count profiles characteristic of C-fiber axons or A-fiber axons that were either still myelinated or had been demyelinated to varying degrees. No differences were apparent in the number of A-fiber or C-fiber axons between lysolecithin-treated and control animals. Ipsi, Ipsilateral; Contra, contralateral.
Lysolecithin treatment induces ectopic firing in saphenous afferents in the absence of changes in conduction velocity or peripheral thresholds
At 3 and 5 d after lysolecithin treatment, reflex testing showed no detectable change in either mechanically or thermally evoked responses, indicating that a sensitized state had not yet developed. Neither at this time nor at the time of peak behavioral change (11–13 d after treatment) was there any significant difference among normal, sham, and demyelinated afferent fibers in the threshold for their firing response to von Frey filaments or to thermal nociceptive stimuli (Table 2). Thus there is no evidence that changes in peripheral transduction mechanisms might be responsible for the increased behavioral reflex sensitivity that was observed (Table 2). The focal nature of this demyelination together with the integrity of the axons was reflected in the normalrange of overall conduction velocities compared with sham and control nerves. The range included slow conduction velocities of <1 msec−1up to the fastest in the range of 20 msec–1. Furthermore, the number of units recorded for each conduction velocity did not differ significantly among the lysolecithin-treated, sham, and naive nerves (Fig.4a, Table 2). This provided further support for the view that there had been no significant axon damage.
Table 2.
Effects of topical lysolecithin treatment of the saphenous nerve on afferent fiber characteristics
| Treatment | Receptive field | Spontaneous activity (imp/sec) | von Frey filament threshold (mN/mm2) | Thermal threshold (oC) | Conduction velocity range (msec−1) |
|---|---|---|---|---|---|
| Naive | Normal | 0 | 8.41–16.70 | >48 | 1.4–20.0 |
| Sham (day 11–13) | Normal | 0 | 8.41–16.70 | >48 | 0.9–18.2 |
| Lysolecithin-treated (day 3- 5) | Normal | 0 | 8.41–16.70 | >48 | 0.5–16.6 |
| Lysolecithin-treated (day 11–13) | Normal | 0 | 8.41–16.70 | >48 | 1.3–16.0 |
Electrophysiological recordings of afferent fiber unit activity and cutaneous responsiveness were performed on animals 3–5 d (n = 4) and 11–13 d (n = 6) after lysolecithin treatment to the saphenous nerve, 11–13 d after sham treatment (n = 4), and on naive animals (n = 4). The total numbers of afferent units recorded in each condition were 21, 66, 25, and 49, respectively. Any sensitization of mechanical sensory receptors with high thresholds was measured using calibrated von Frey filaments, and the presence of any sensitization of thermally responsive receptors was measured with a calibrated radiant heat lamp. The conduction velocity range of afferent fiber units was determined using the peripheral stimulus technique via bipolar electrodes. There was no difference in threshold for unit firing in response to mechanical or thermal stimulation or conduction velocity range among any of the groups. Afferent fiber units of the lysolecithin-treated nerves (11–13 d after treatment) displayed spontaneous activity. imp/sec, Impulses per second.
Fig. 4.
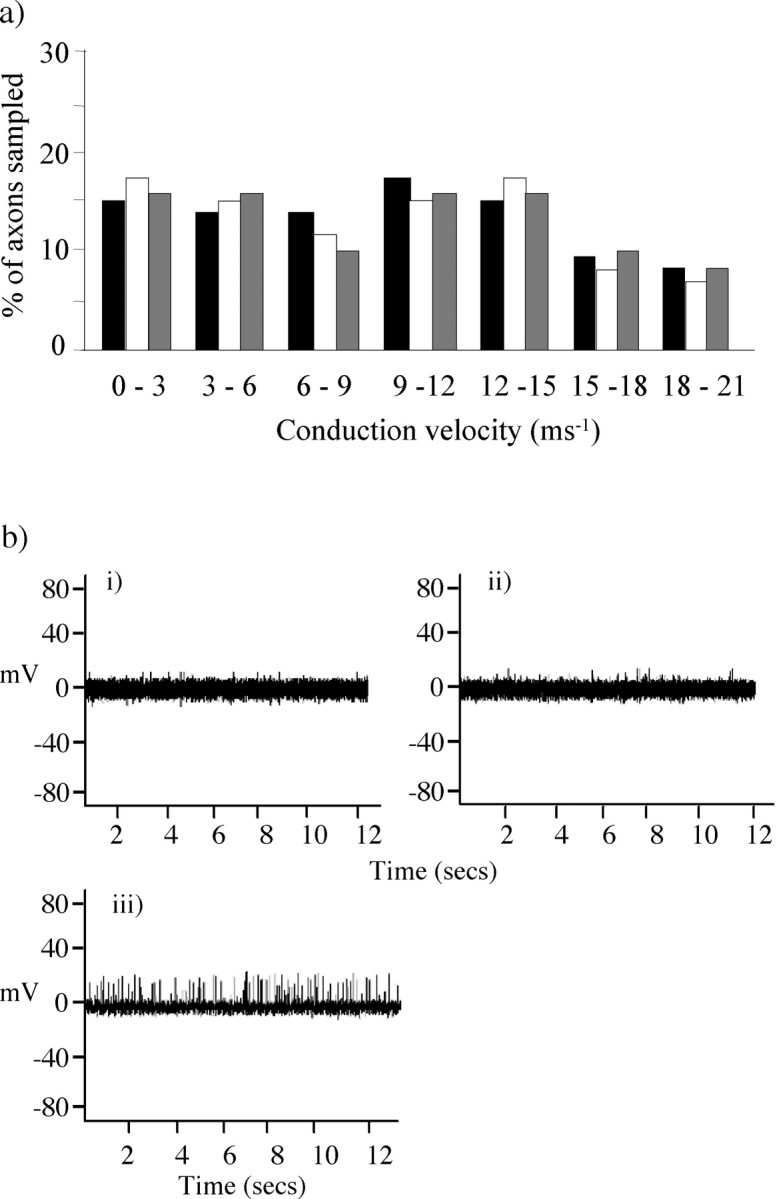
Effects of lysolecithin treatment on electrophysiological properties of saphenous afferents.a, Afferent fiber units from the lysolecithin-treated saphenous nerve of animals on days 11–13 after treatment (n = 6) displayed a range of conduction velocities that were not significantly different from those of fibers from naive nerves (n = 4). The range included slow conduction velocities of <1 msec−1 up to the fastest in the range of 20 msec–1. The number of units recorded for each conduction velocity did not differ significantly among the lysolecithin-treated (black column), naive (white column), and sham (gray column) nerves. b, Electrophysiological recordings of saphenous afferent fiber unit activity 11, 12, and 13 d after lysolecithin treatment to the saphenous nerve (n = 6), 11–13 d after sham treatment (n = 4), and in naive animals (n = 4). Spontaneous activity in saphenous afferents was recorded with a frequency of 2–3 Hz. Examples of electrophysiological recordings of naive (i), sham-treated (ii), and lysolecithin-treated (iii) nerves are displayed.
A common feature of lysolecithin-demyelinated nerves over a range of 11–13 d after application, however, has been the presence of a spontaneous low-frequency discharge (2–3 impulses per second), reminiscent of a similar discharge that we have observed in the sciatic nerve of Prx-null mice (Gillespie et al., 2000) (Fig.4b).
Expression of NF-200, peripherin, and the axonal damage marker ATF3 in DRG is unaffected by lysolecithin treatment
To assess the integrity of the cell bodies 13 d after lysolecithin treatment of the saphenous or sciatic nerve, we analyzed the expression of NF-200, which labels the larger-diameter, myelinated DRG cell population (Lawson and Waddell, 1991; Michael et al., 1999), and peripherin, a type III intermediate filament protein, which is normally expressed selectively by unmyelinated sensory neurons (Amaya et al., 2000). No significant difference was observed in the number of either NF-200 or peripherin-positive DRG neurons ipsilateral to lysolecithin compared with contralateral or sham treatment of the sciatic or saphenous nerve (Table 3).
Table 3.
Immunohistochemical assessment of structural markers and the neuropeptide CGRP in DRG cells after lysolecithin treatment
| Nerve | Treatment | Side | Number of positive cells per DRG | ||
|---|---|---|---|---|---|
| NF-200 | Peripherin | CGRP | |||
| Sciatic | Lysolecithin | Ipsi | 78.5 ± 3.2 | 129.7 ± 19.6 | 145.9 ± 19.7 |
| Lysolecithin | Contra | 77.3 ± 2.2 | 119.7 ± 4.6 | 154.5 ± 9.1 | |
| Sham | Ipsi | 68.1 ± 9.6 | 109.4 ± 9.6 | 149.6 ± 5.8 | |
| Sham | Contra | 73.4 ± 2.7 | 103.9 ± 1.9 | 129.9 ± 12.9 | |
| Saphenous | Lysolecithin | Ipsi | 67.1 ± 6.2 | 120.4 ± 11.6 | 152.8 ± 8.7 |
| Lysolecithin | Contra | 74.3 ± 13.9 | 124.7 ± 8.6 | 135.3 ± 7.4 | |
| Sham | Ipsi | 69.9 ± 9.7 | 119.5 ± 9.1 | 139.6 ± 5.8 | |
| Sham | Contra | 71.7 ± 9.6 | 121.7 ± 4.6 | 132.4 ± 9.6 | |
The expression of the neuronal structural markers peripherin and NF-200 and the peptide CGRP in DRG cells ipsilateral and contralateral to lysolecithin treatment of the saphenous or sciatic nerve on day 13 after treatment and from sham animals (n = 4 in each case). There was no difference in the number of cells expressing either structural marker after treatment with lysolecithin as determined by a Kruskal–Wallis one-way ANOVA on ranks or paired Student'st test. Data are presented as average number of immunopositive cells per DRG ± SEM. Ipsi, Ipsilateral; Contra, contralateral.
We also analyzed the expression of ATF3, a member of the ATF/cAMP response element-binding protein (ATF/CREB) family (Hai et al., 1999), which is induced in DRG neurons after peripheral nerve injury and thus considered to be a nerve injury marker, identifying axotomized neurons (Tsujino et al., 2000). As a positive control, we analyzed the expression of ATF3 in the DRG of mice, which were at the peak of neuropathic sensitization after chronic constriction injury (CCI) to the sciatic nerve. In CCI mice we found expression of ATF3 in 69 ± 16% of ipsilateral DRG neurons. Under control (sham) conditions we found a very small number of ATF3-positive cells, all of which were also positive for NF-200. However, there was no significant increase in the proportion of ATF3-immunopositive cells in the DRG ipsilateral to lysolecithin treatment of the sciatic nerve as compared with contralateral or sham-treated animals (Fig. 5a, Table4).
Fig. 5.
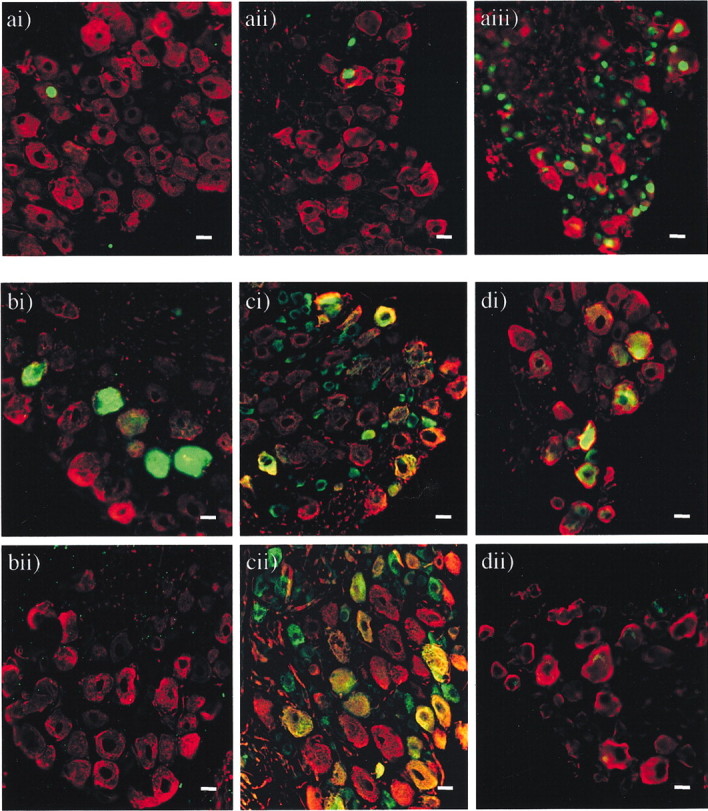
Immunohistochemical colocalization of neuronal subtype markers with ATF3, NPY, and the sodium channel subtypes Nav1.8 and Nav1.3 in DRG cells after lysolecithin treatment of the sciatic nerve. a, Expression of ATF3 in ipsilateral and contralateral DRG sections from lysolecithin-treated animals on day 13 after treatment (n = 4). There was no significant increase in expression of ATF3 from that seen in control DRG after lysolecithin treatment or sham treatment. The statistical significance (*p < 0.05) of any difference was determined by a Kruskal–Wallis one-way ANOVA on ranks with all pair-wise multiple comparison procedures (Dunn's method). Images show nuclei of ATF3-immunopositive cells labeled with FITC (green) and NF-200-immunopositive cells labeled with TRITC (red), in ipsilateral (i) and contralateral (ii) DRG from lysolecithin-treated animals and ipsilateral DRG (iii) from CCI animals. Scale bar, 20 μm.b, Expression of the neuropeptide NPY in ipsilateral and contralateral DRG sections from lysolecithin-treated animals at 13 d after treatment (n = 4). Images show NF-200-immunopositive cells labeled with TRITC (red) and NF-200/NPY-colocalized, immunopositive cells labeled with FITC (green) in DRG ipsilateral to lysolecithin treatment (i) as opposed to no positive cells in DRG contralateral to lysolecithin treatment (ii). Scale bar, 20 μm. c, The expression of the Nav1.8 channel in ipsilateral and contralateral DRG sections from lysolecithin-treated animals on day 13 after treatment (n = 4). After lysolecithin treatment of the sciatic nerve, there was a decrease in the number of cells expressing Nav1.8 in ipsilateral DRG compared with contralateral DRG, all of which were immunopositive for NF-200. There was no significant decrease in expression after sham treatment. The statistical significance (*p < 0.05) of any difference was determined by a Kruskal–Wallis one-way ANOVA on ranks with all pair-wise multiple comparison procedures (Dunn's method). Images show Nav1.8-immunopositive cells labeled with FITC (green) and NF-200-immunopositive cells labeled with TRITC (red), with coexpression appearing yellow in ipsilateral (i) and contralateral (ii) DRG from lysolecithin-treated animals. Scale bar, 20 μm. d, Expression of the Nav1.3 channel in ipsilateral and contralateral DRG sections from lysolecithin-treated animals 13 d after treatment and sham-treated animals on the same day after treatment (n = 4 in each case). Images show NF-200-immunopositive cells labeled with TRITC (red) and Nav1.3-immunopositive cells labeled with FITC (green) with coexpression appearing yellow in DRG ipsilateral to lysolecithin treatment (i) as opposed to no positive cells in DRG contralateral to lysolecithin treatment (ii). Scale bar, 20 μm.
Table 4.
Immunohistochemical assessment of ATF3, the neuropeptide NPY, and the sodium channel subtypes Nav1.8 and Nav1.3 in sciatic nerve DRG cells after lysolecithin treatment
| Treatment | Side | Positive cells as a proportion of NF-200/peripherin-positive cells (%) | ||||
|---|---|---|---|---|---|---|
| ATF3/ NF-200 | NPY/ NF-200 | Nav1.8/ NF-200 | Nav1.8/ peripherin | Nav1.3/ NF-200 | ||
| Lysolecithin | Ipsi | 7.3 ± 3.2 | 13.1 ± 2.2* | 15.7 ± 1.6* | 54.8 ± 3.7 | 18.5 ± 2.4* |
| Lysolecithin | Contra | 6.1 ± 1.4 | 0 | 24.0 ± 2.6 | 50.6 ± 6.3 | 0 |
| Sham | Ipsi | 8.2 ± 3.4 | 0 | 24.0 ± 2.9 | 51 ± 1.4 | 0 |
| Sham | Contra | 5.1 ± 3.2 | 0 | 22.5 ± 2.7 | 48.1 ± 2.4 | 0 |
The expression of NPY, ATF3, and the sodium channels Nav1.8 and Nav1.3 in DRG cells ipsilateral (Ipsi) and contralateral (Contra) to lysolecithin treatment of the sciatic nerve are shown for day 13 after treatment (n = 4) and from sham animals (n = 4). There was no significant increase in expression of ATF3 from that seen in contralateral DRG after lysolecithin treatment or sham treatment. All ATF3-expressing cells were NF-200 immunopositive, and none were peripherin immunopositive. After lysolecithin treatment of the sciatic nerve there was a significant increase in the number of cells expressing NPY, which were all positive for NF-200 and none of which were immunopositive for peripherin. There was no detectable expression of NPY in the DRG contralateral to lysolecithin treatment or in DRG cells from sham-treated animals. Ipsilateral to lysolecithin treatment, there was a decrease in the number of DRG cells expressing Nav1.8 together with NF-200 compared with contralateral DRG or sham controls. In contrast, there was no significant decrease in the proportion of cells coexpressing Nav1.8 and peripherin. Expression of the Nav1.3 channel became detectable in a subpopulation of NF-200- immunopositive cells after lysolecithin treatment, whereas there was no detectable expression in the contralateral DRG or in DRG from sham-treated animals. No coexpression of Nav1.3 with peripherin was detected. The statistical significance (* p < 0.05) of any difference between each group was determined by a Kruskal–Wallis one-way ANOVA on ranks with all pair-wise multiple comparison procedures (Dunn's method). Data are presented as the mean ± SEM percentage of total NF-200-positive cells that were positive for the relevant marker.
Lysolecithin treatment causes a distinctive pattern of neuropeptide expression in sensory neurons
After traumatic injury to the peripheral nerve, there are a number of neurochemical and morphological changes, both in peripheral nerve fibers and centrally in the spinal cord (Dray et al., 1994; Hokfelt et al., 1994), that can contribute to the altered sensory transmission associated with chronic pain states. Phenotypic changes occur in many primary afferent DRG neurons, resulting in the altered expression of neuropeptides including CGRP, galanin, and NPY (Villar et al., 1991;Hokfelt et al., 1994). We therefore investigated whether such changes occur in the demyelination-induced pain state to compare the changes with those occurring in other models of neuropathic pain in which axonal damage is a major factor. CGRP is normally expressed by approximately one-half of unmyelinated DRG neurons as well as one-fifth of those with Aδ myelinated neurons (Rosenfeld et al., 1983; Gibson et al., 1984; McCarthy and Lawson, 1990; Hokfelt et al., 1994) and is downregulated after peripheral nerve injury such as axotomy and CCI (Noguchi et al., 1990; Dumoulin et al., 1992). However, there was no significant difference in the numbers of ipsilateral versus contralateral DRG cells expressing immunoreactive CGRP after lysolecithin treatment or sham treatment of one sciatic nerve or of one saphenous nerve (Table 3), suggesting that lysolecithin treatment caused no extensive damage to the axons of CGRP-expressing cells.
Further immunohistochemical investigations were restricted to the sciatic model of lysolecithin-induced demyelination, because it is a larger nerve and would contribute a larger proportion of cell bodies in relevant DRGs, thereby facilitating detection of changes.
Galanin is normally expressed at very low levels in sensory and sympathetic neurons. However, after peripheral nerve lesions, it is strongly upregulated mainly in small and medium-sized neurons (Hokfelt et al., 1987; Villar et al., 1989), especially those that normally contain substance P and CGRP (Xu et al., 1990; Doughty et al., 1991;Kashiba et al., 1992), the majority of which are unmyelinated. However, we observed no significant galanin immunoreactivity in DRG cells ipsilateral or contralateral to focal demyelination of one sciatic nerve. As a positive control to test the specificity of the antibody, we used DRG from mice at peak neuropathic sensitization after sciatic CCI (Garry et al., 2003). These sections showed a significant proportion of mainly small diameter cells positive for galanin (ipsilateral 9.3 ± 2.4 cells per section; contralateral 0), verifying that the antibody could successfully detect the presence of galanin when expressed after traumatic nerve injury and that there is, in contrast, no significant upregulation after lysolecithin treatment.
NPY is not normally expressed in DRG cells (Lundberg et al., 1983). However, the expression of NPY is strongly upregulated in DRG neurons after axotomy (Wakisaka et al., 1991, 1992; Noguchi et al., 1993; Kashiba et al., 1994), partial sciatic injury (Ma and Bisby, 1998), CCI of the sciatic nerve (Nahin et al., 1994; Munglani et al., 1995), and streptozotocin-induced diabetic neuropathy (Rittenhouse et al., 1996). This upregulation occurs mainly in large primary sensory neurons containing NF-200 (Kashiba et al., 1994; Marchand et al., 1999). We also observed significant NPY immunoreactivity in ∼16% cells after demyelination of one sciatic nerve (185 NPY-positive cells from 1164 NF-200-positive cells counted over 26 sections;n = 4) as opposed to no detectable immunoreactivity in contralateral or sham nerves (Table 4). All NPY-immunopositive cells were also immunoreactive for NF-200, indicating that they were cells with myelinated fibers (Fig. 5b).
Afferent expression of NaV1.8 and NaV1.3 is altered by lysolecithin treatment
Normally, DRG neurons express a complex repertoire of sodium channel transcripts (Waxman et al., 1999) distinguished by their differential sensitivity to tetrodotoxin (TTX) (Black and Waxman, 1996). The TTX-resistant sodium channel NaV1.8 is expressed in ∼50% of small-diameter, unmyelinated DRG cells (Amaya et al., 2000) and in ∼20% of medium- to large-diameter, myelinated DRG cells. Our results from the DRG of contralateral untreated nerve confirm that result, showing expression of NaV1.8 in 1078 of the 2130 (51%) peripherin-immunopositive cells (C-fibers) and 500 of the 2086 (24%) NF-200-immunopositive cells (myelinated fibers). Likewise, in sham controls we saw Nav1.8 expressed in 51% of peripherin-immunopositive cells ipsilaterally and 48% contralaterally and in 23% of NF-200-immunopositive cells ipsilaterally and 22% contralaterally. It has been shown previously that after axotomy, CCI, or spinal nerve ligation, expression of Nav1.8 is downregulated in neurons in the DRG, a change that has been proposed to contribute to the production of a neuropathic pain state (Dib-Hajj et al., 1999; Waxman, 1999; Decosterd et al., 2002). In the L4, L5, and L6 DRG taken from animals 13 d after lysolecithin treatment to the sciatic nerve, there was a 34.1 ± 4.2% reduction of Nav1.8 expression in the ipsilateral DRG (62.4 ± 3.4 NaV1.8-immunopositive cells per section) versus the contralateral and sham DRG (with corresponding values of 94.7 ± 4.7 NaV1.8 and 95.7 ± 6.0, respectively). To address the question of whether these changes in sodium channel expression were restricted to a subset of neurons, we performed dual labeling experiments to reveal the colocalization of Nav1.8 with NF-200/peripherin. The reduction in Nav1.8 expression appeared to be restricted to NF-200-immunopositive cells, i.e., those presumed to have formerly myelinated axons (Fig. 5c, Table 4). No change in the number of cells coexpressing Nav1.8 and NF-200 was observed between the ipsilateral or contralateral DRG of sham control animals, and we found no decrease in the proportion of peripherin-immunoreactive cells expressing Nav1.8 after lysolecithin treatment (Table 4).
The Nav1.3 channel is normally found in DRG only during development, and consistent with this we could not detect expression of this channel in the DRG before treatment (Fig.5d, Table 4). However, expression of this channel has been reported to increase in sensory neurons after axotomy (Black et al., 1999), CCI, and spinal nerve ligation (Kim et al., 2001). After lysolecithin-induced demyelination of the sciatic nerve, we found a significant increase in the number of NF-200-immunoreactive cells per ipsilateral DRG that now expressed Nav1.3 (18.5 ± 2.4%) (Fig. 5d, Table 4). No Nav1.3 positive cells were also immunopositive for peripherin, showing that expression of the channel was not increased in C-fibers, and no Nav1.3 immunoreactivity was detected in contralateral or sham tissue.
Lysolecithin treatment causes NMDA receptor-dependent central sensitization that can be reversed by cannabinoids
Sensitization of cells in the dorsal horn of the spinal cord is a key change in other neuropathic pain states that is thought to involve activation of the NMDA receptor (Wilcox, 1991; Codderre and Melzack, 1992; Chaplan et al., 1997). We found that spinal NMDA receptors also play a role in the mechanical allodynia seen in animals with focal demyelination of the saphenous nerve, because behavioral sensitization was reversed after spinal administration of the selective NMDA receptor antagonist (R)-CPP (Lehmann et al., 1987) (Fig.6a). This parallels observations that we made previously in a demyelination model based on the periaxin-null mouse (Gillespie et al., 2000). Similarly, the reduction in thermal nociceptive response latency ipsilateral to lysolecithin treatment was reversed by (R)-CPP (Fig. 6e). There was no effect of (R)-CPP on thermal or mechanical responses contralateral to demyelination. Equivalent injections of the saline vehicle had no effect on mechanical or thermal reflex responses either ipsilateral or contralateral to lysolecithin (data not shown;n = 4).
Fig. 6.

Effects of intrathecal administration of NMDA and cannabinoid receptor-targeting agents on sensitized behavioral reflex responses after lysolecithin treatment. Paw withdrawal thresholds in response to cutaneous mechanical stimulation with von Frey filaments (a–d) and paw withdrawal latencies in response to cutaneous noxious thermal stimulation (e–h) were measured in animals at peak behavioral reflex sensitivity after topical application of lysolecithin to one saphenous nerve. Ipsilateral (■) and contralateral (♦) values are displayed before and after intrathecal administration of each pharmacological agent as paw withdrawal threshold in milliNewtons per millimeter squared (mN/mm2) for mechanical stimulation or paw withdrawal latency (seconds) for thermal stimulation. For all measurements, each value is the mean ± SEM, and statistical significance (*p < 0.05) of any differences between postinjection threshold values and preinjection baseline values was determined by a one-way repeated measures ANOVA with Dunnet's post hoc analysis. a,e, The NMDA receptor antagonist (R)-CPP (100 pmol) reversed the behavioral reflex sensitivity to mechanical stimulation (a) and thermal stimulation (e) in lysolecithin-treated animals for up to 65–70 min after intrathecal application (n = 8). b, f, The mixed CB1/CB2 cannabinoid receptor agonist WIN 55,212–2 (60 pmol) significantly reversed the behavioral reflex sensitivity to mechanical stimulation (b) and thermal stimulation (f) in lysolecithin-treated animals (n = 7) for up to 55–60 min after intrathecal application. c,g, When injected in combination, WIN 55,212–2 and AM 251 produced no significant effect on the behavioral reflex sensitivity to mechanical stimulation (c) and thermal stimulation (g) in lysolecithin-treated animals (n = 8). d, h, The CB1 receptor antagonist AM 251 (100 pmol) resulted in no change in the behavioral reflex sensitivity to mechanical stimulation (d) and thermal stimulation (h) in lysolecithin-treated animals (n = 7).
Because neuropathic pain states often show a relative insensitivity to spinal opioid analgesia compared with acute or peripheral inflammatory pain states (Arner and Meyerson, 1988;Fleetwood-Walker et al., 1988; Yaksh and Harty, 1988), we assessed the effect of the potent and selective μ-opioid receptor agonist DAMGO in the lysolecithin model. Spinal μ-opioid receptors exert powerful analgesic effects in inflammatory models of chronic pain (Malmberg and Yaksh, 1993; Chaplan et al., 1997). At the highest dose of intrathecal DAMGO that could be administered before overt psychomotor incoordination was seen (10 pmol/10 μl), there was no specific reversal of the mechanical allodynia or thermal hyperalgesia seen ipsilateral to lysolecithin treatment (data not shown; n = 4). The same intrathecal dose of DAMGO produces significant analgesia in tail pressure and thermal paw withdrawal tests in mice (Sakurada et al., 1999, 2001).
An alternative spinal modulatory system that can exert selective antinociceptive effects in both neuropathic and inflammatory hyperalgesia is the endogenous cannabinoid system (Herzberg et al., 1997; Richardson et al., 1998a; Fox et al., 2001). This is of particular interest because there is evidence that cannabinoids may exert analgesic and additional beneficial effects in demyelinating diseases such as multiple sclerosis (Consroe et al., 1997; Williamson and Evans, 2000; Robson, 2001). In this animal model of peripheral nerve demyelination, spinal administration of the cannabinoid receptor agonist WIN 55,212–2 attenuated the sensitization [that is the reduction in mechanical nociceptive thresholds (Fig. 6b) and thermal reflex latencies (Fig. 6f)] that was displayed ipsilateral to lysolecithin. In both cases, changes were limited to the lysolecithin-treated side and were statistically significant up to ∼1 hr after injection. The CB1 receptor in particular is implicated, because spinal administration of the selective CB1receptor antagonist AM 251 reversed the effects of WIN 55,212–2 on mechanical sensitization (Fig. 6c) and thermal sensitization (Fig. 6g). However, when administered alone, AM 251 had no significant effect, indicating that endogenous spinal CB1 receptors had not become tonically activated as a result of the lysolecithin treatment (Fig.6d,h).
Discussion
Abnormal sensory phenomena including spontaneous pain, hyperalgesia, and allodynia are associated with human peripheral demyelinating neuropathies such as types of CMT and GBS. In CMT, although sensory deficits are usually less severe than motor problems, pain is a problem in many sufferers and is a clear aspect of the clinical picture in CMT4F (Carter et al., 1998; Boerkoel et al., 2001;Guilbot et al., 2001; Takashima et al., 2002). Similarly, GBS or acute inflammatory polyneuropathy may involve pain in 54–84% of cases (Asbury, 1990; Moulin, 1998). Because the mechanisms underlying neuropathic pain in demyelinating disease are poorly understood (Rasminsky, 1981), we developed and characterized an animal model of focal peripheral nerve demyelination.
Morphology of the demyelinated nerve
To understand the mechanisms underlying demyelination-induced neuropathic pain, it is important to establish whether axonopathic changes may contribute. The protocol used here results in demyelination of rapid onset confined to the immediate vicinity of the site of injection, causing little damage to the Schwann cell cytoplasm (Love et al., 1986) or the axon (Figs. 2, 3). However, because small-diameter fibers may be damaged by lysolecithin injection into the nerve (Mitchell and Caren, 1982), we applied lysolecithin topically to the saphenous or sciatic nerve, producing a focal demyelination without detectable axon damage or loss as assessed by light and electron microscopy. Correspondingly, in immunohistochemical studies we found neither loss of neuronal somata in the DRG, which were positive for NF-200 or peripherin, nor increased expression of the neuronal injury marker ATF3. This model of peripheral nerve demyelination appears to involve neither axonopathy nor cell loss and in that sense is less severe than models of neuropathic pain such as axotomy, CCI, and spinal nerve ligation (SNL), which involve axonal injury.
Demyelination-induced allodynia and hyperalgesia: the role of functional changes in peripheral nerve
Damage to sensory nerves as a result of human peripheral demyelinating disease has been linked to pain and heightened sensitivity to touch (Pentland and Donald, 1994; Devor and Seltzer, 1999). After lysolecithin treatment of either the mixed sciatic nerve or the sensory saphenous nerve, we have demonstrated, for the first time, the development of mechanical allodynia and thermal hyperalgesia. Peripheral sensitization may contribute to neuropathic pain behavior in other models (Koltzenburg, 1998; Woolf and Costigan, 1999), but in the demyelination model we could find no significant changes in the cutaneous threshold for action potential generation by mechanical or thermal nociceptive stimuli in either A- or C-fibers. Some forms of mechanical allodynia are considered to be mediated by myelinated A-fibers providing inputs that are inappropriately processed by sensitized dorsal horn neurons (Woolf, 1997; Koltzenburg, 1998). Indeed, in the conditions that occur after section of peripheral or spinal nerves (for example, with phenotypic changes in DRG cells), spontaneous, ectopic activity in A, but not C afferents may play a role in the induction of neuropathic pain behaviors (C. N. Liu et al., 2000; X. Liu et al., 2000). Such activity may be important in the development of neuropathic pain here because repetitive ectopic firing may underlie abnormal sensation in diseases involving segmental demyelination, particularly where axonopathic changes are lacking (Rasminsky, 1981). The presence of low-frequency spontaneous discharge in demyelinated saphenous nerve corresponded to times of peak behavioral reflex sensitivity and was similar to observations in thePrx-null mouse (Gillespie et al., 2000). Because inflammatory neuritis can lead to a sensitized neuropathic pain state (Eliav et al., 1999), the possibility should be considered that a neuroimmune response could contribute to generating the allodynia and hyperalgesia observed here. However, in the Prx-null mouse, an alternative model of demyelination-induced neuropathic pain, minimal evidence could be found for macrophage infiltration of the degenerating structures (Gillespie et al., 2000), suggesting that demyelination per se is a critical factor in generating the neuropathic pain state.
Phenotypic changes in the DRG
After peripheral nerve lesions, changes in the expression of peptides occur in primary sensory neurons that may contribute to mediating central sensitization (Hokfelt et al., 1994). We assessed whether changes in the lysolecithin model are similar to those in other models of neuropathic pain, such as the downregulation of CGRP and the upregulation of galanin and NPY. Because this model appears to cause morphological and functional changes only in myelinated afferents, we asked whether changes in peptide expression are similarly restricted. Alterations in CGRP and galanin expression (generally in small-diameter afferents) have been implicated in the development of neuropathic pain (Hokfelt et al., 1987; Villar et al., 1989; Dumoulin et al., 1992; Kerr et al., 2000; Wynick et al., 2001), but we saw no change in these peptides after peripheral nerve demyelination.
We did find, however, upregulated expression of NPY, restricted to large NF-200-positive cells ipsilateral to focal demyelination. This parallels changes seen in other models of neuropathic pain. Notably, the upregulation of NPY occurred without any alteration in the expression of ATF3, indicating that direct neuronal injury was not responsible for the effect. After axotomy (Waxman et al., 1994), CCI (Dib-Hajj et al., 1999), and SNL (Kim et al., 2001), changes are seen in the expression of sodium channel isoforms, leading to functional changes in afferent sodium currents. For example, Nav1.8 [expressed preferentially in small- and medium-diameter DRG and implicated in pathological pain states (Akopian et al., 1996; Sangameswaran et al., 1996; Novakovic et al., 1998)], is downregulated (Dib-Hajj et al., 1996; Waxman, 1999), whereas Nav1.3 (normally found only in the DRG during development) is upregulated (Waxman et al., 1994; Waxman, 1999). We found a similar decrease in the expression of Nav1.8 channels in the DRG after focal demyelination that was restricted to formerly myelinated cells. In contrast, Nav1.3 was upregulated in corresponding cells. This further supports the specificity of A-fiber changes in evoking neuropathic pain after demyelination. Changes in channel type and distribution could contribute to hyperexcitability and ectopic pacemaker activity (Matzner and Devor, 1992, 1994; Devor et al., 1994;Cummins and Waxman, 1997; Omana-Zapata et al., 1997; Waxman, 1999). Na+ channel-blocking agents may be effective analgesics not only in nerve injury-induced neuropathic pain (Chabal et al., 1992; Devor et al., 1992; Omana-Zapata et al., 1997;Rizzo, 1997), but also in demyelination-induced pain.
Central changes after afferent demyelination and central modulation of sensitization
Afferent hyperexcitability and ectopic firing after lysolecithin treatment provide a basis for the ongoing input that brings about central sensitization. Because a spinal NMDA antagonist reverses sensitization in lysolecithin-treated mice, central changes play a critical role in the phenotype. NMDA receptors are similarly important in the mechanical allodynia and thermal hyperalgesia of other chronic pain models (Dickenson and Sullivan, 1987; Mao et al., 1993; Bennett, 1994; Chaplan et al., 1997; Woolf and Costigan, 1999). The relative insensitivity of neuropathic pain models to spinally administered opioid analgesics (Arner and Meyerson, 1988; Yaksh and Harty, 1988; Lee et al., 1995) also corresponds to our findings in the lysolecithin demyelination model.
Cannabinoid receptor drugs may represent an alternative strategy for the relief of chronic pain. Spinal cannabinoids can effectively modulate nociception (Herzberg et al., 1997; Richardson et al., 1998a;Martin et al., 1999; Bridges et al., 2001; Fox et al., 2001; Kelly and Chaplan, 2001), and the majority of these analgesic effects appear to be CB1 receptor-mediated (Richardson et al., 1998b). In line with experiments on nerve-injury pain (Herzberg et al., 1997; Bridges et al., 2001; Fox et al., 2001), we found that intrathecal administration of the mixed CB receptor agonist WIN 55,212–2 attenuated mechanical allodynia and thermal hyperalgesia in lysolecithin-treated mice. This effect was reversed by the CB1 antagonist AM 251, although AM 251 had little effect alone. It is possible that additional, noncannabinoid side effects of the compounds may occur in the immediate vicinity of the intrathecal injections where the local concentrations are higher, but the most likely explanation is that the WIN 55,212–2-induced, AM-251-reversed analgesia is mediated by CB1receptors. The efficacy of cannabinoids in the treatment of neuropathic pain may be linked to their actions on myelinated A-fibers. Under normal conditions, spinal cannabinoid receptors may predominantly regulate C-fiber input to the spinal cord, but after nerve injury these effects may be lost, whereas inhibitory influences on Aδ-fiber inputs are retained (Chapman, 2001). This might result in cannabinoid agonists causing a reduction of mechanical allodynia, with more limited effect on thermal hyperalgesia, consistent with evidence from a number of studies (Herzberg et al., 1997; Bridges et al., 2001; Fox et al., 2001). In the present model, in which C-fiber primary afferents appear to be unaffected, both mechanical (A-fiber) and thermal (C-fiber) inputs should be targeted effectively by cannabinoids, as observed experimentally. These findings point further to the potential use of cannabinoids as therapeutic agents in human demyelinating neuropathies.
Footnotes
This work was supported by the Wellcome Trust (S.F.W., P.J.B.). We thank the Medical Research Council for the award of a Studentship (V.C.J.W.). We thank staff at the Wellcome Animal Research Unit for animal husbandry and R. Mitchell and M. Koltzenburg for helpful suggestions.
Correspondence should be addressed to Dr. S. M. Fleetwood-Walker, Division of Preclinical Veterinary Sciences, University of Edinburgh, Summerhall, Edinburgh EH9 1QH, UK. E-mails.m.fleetwood-walker@ed.ac.uk.
References
- 1.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 2.Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- 3.Arner S, Meyerson BA. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11–23. doi: 10.1016/0304-3959(88)90198-4. [DOI] [PubMed] [Google Scholar]
- 4.Asbury AK. Guillain-Barre syndrome: historical aspects. Ann Neurol [Suppl] 1990;27:S2–S6. doi: 10.1002/ana.410270703. [DOI] [PubMed] [Google Scholar]
- 5.Bennett DL, Michael GJ, Ramachandran N, Munson JB, Averill S, Yan Q, McMahon SB, Priestley JV. A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. J Neurosci. 1998;18:3059–3072. doi: 10.1523/JNEUROSCI.18-08-03059.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett GJ. Animal models of neuropathic pain. In: Gebhart GF, Hammond DL, Jensen TS, editors. Proceedings of the 7th World Congress on Pain, Vol 2. IASP; Seattle: 1994. pp. 495–510. [Google Scholar]
- 7.Black JA, Waxman SG. Sodium channel expression: a dynamic process in neurons and non-neuronal cells. Dev Neurosci. 1996;18:139–152. doi: 10.1159/000111403. [DOI] [PubMed] [Google Scholar]
- 8.Black JA, Cummins TR, Plumpton C, Chen YH, Hormuzdiar W, Clare JJ, Waxman SG. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol. 1999;82:2776–2785. doi: 10.1152/jn.1999.82.5.2776. [DOI] [PubMed] [Google Scholar]
- 9.Boerkoel CF, Takashima H, Stankiewicz P, Garcia CA, Leber SM, Rhee-Morris L, Lupski JR. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet. 2001;68:325–333. doi: 10.1086/318208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bridges D, Ahmad K, Rice AS. The synthetic cannabinoid WIN55, 212–2 attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Br J Pharmacol. 2001;133:586–594. doi: 10.1038/sj.bjp.0704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter GT, Jensen MP, Galer BS, Kraft GH, Crabtree LD, Beardsley RM, Abresch RT, Bird TD. Neuropathic pain in Charcot-Marie-Tooth disease. Arch Phys Med Rehabil. 1998;79:1560–1564. doi: 10.1016/s0003-9993(98)90421-x. [DOI] [PubMed] [Google Scholar]
- 12.Chabal C, Jacobson L, Russell LC, Burchiel KJ. Pain response to perineuromal injection of normal saline, epinephrine, and lidocaine in humans. Pain. 1992;49:9–12. doi: 10.1016/0304-3959(92)90181-A. [DOI] [PubMed] [Google Scholar]
- 13.Chaplan SR, Malmberg AB, Yaksh TL. Efficacy of spinal NMDA receptor antagonism in formalin hyperalgesia and nerve injury evoked allodynia in the rat. J Pharmacol Exp Ther. 1997;280:829–838. [PubMed] [Google Scholar]
- 14.Chapman V. Functional changes in the inhibitory effect of spinal cannabinoid (CB) receptor activation in nerve injured rats. Neuropharmacology. 2001;41:870–877. doi: 10.1016/s0028-3908(01)00125-3. [DOI] [PubMed] [Google Scholar]
- 15.Codderre TJ, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Consroe P, Musty R, Rein J, Tillery W, Pertwee R. The perceived effects of smoked cannabis on patients with multiple sclerosis. Eur Neurol. 1997;38:44–48. doi: 10.1159/000112901. [DOI] [PubMed] [Google Scholar]
- 17.Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Decosterd I, Ji RR, Abdi S, Tate S, Woolf CJ. The pattern of expression of the voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain. 2002;96:269–277. doi: 10.1016/S0304-3959(01)00456-0. [DOI] [PubMed] [Google Scholar]
- 19.Delague V, Bareil C, Tuffery S, Bouvagnet P, Chouery E, Koussa S, Maisonobe T, Loiselet J, Megarbane A, Claustres M. Mapping of a new locus for autosomal recessive demyelinating Charcot-Marie-Tooth disease to 19q13.1–13.3 in a large consanguineous Lebanese family: exclusion of MAG as a candidate gene. Am J Hum Genet. 2000;67:236–243. doi: 10.1086/302980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devor M, Seltzer Z. Pathophysiology of damaged nerves in relation to chronic pain. In: Wall PD, Melzack R, editors. Textbook of pain. Churchill Livingstone; London: 1999. pp. 129–164. [Google Scholar]
- 21.Devor M, Wall PD, Catalan N. Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. Pain. 1992;48:261–268. doi: 10.1016/0304-3959(92)90067-L. [DOI] [PubMed] [Google Scholar]
- 22.Devor M, Janig W, Michaelis M. Modulation of activity in dorsal root ganglion neurons by sympathetic activation in nerve-injured rats. J Neurophysiol. 1994;71:38–47. doi: 10.1152/jn.1994.71.1.38. [DOI] [PubMed] [Google Scholar]
- 23.Dib-Hajj S, Black JA, Felts P, Waxman SG. Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci USA. 1996;93:14950–14954. doi: 10.1073/pnas.93.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dib-Hajj SD, Fjell J, Cummins TR, Zheng Z, Fried K, LaMotte R, Black JA, Waxman SG. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain. 1999;83:591–600. doi: 10.1016/S0304-3959(99)00169-4. [DOI] [PubMed] [Google Scholar]
- 25.Dickenson AH, Sullivan AF. Evidence for a role of the NMDA receptor in the frequency dependent potentiation of deep rat dorsal horn nociceptive neurones following C fiber stimulation. Neuropharmacology. 1987;26:1235–1238. doi: 10.1016/0028-3908(87)90275-9. [DOI] [PubMed] [Google Scholar]
- 26.Doughty SE, Atkinson ME, Shehab SA. A quantitative study of neuropeptide immunoreactive cell bodies of primary afferent sensory neurons following rat sciatic nerve peripheral axotomy. Regul Pept. 1991;35:59–72. doi: 10.1016/0167-0115(91)90254-e. [DOI] [PubMed] [Google Scholar]
- 27.Dray A, Urban L, Dickenson A. Pharmacology of chronic pain. Trends Pharmacol Sci. 1994;15:190–197. doi: 10.1016/0165-6147(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 28.Dumoulin FL, Raivich G, Haas CA, Lazar P, Reddington M, Streit WJ, Kreutzberg GW. Calcitonin gene-related peptide and peripheral nerve regeneration. Ann NY Acad Sci. 1992;657:351–360. doi: 10.1111/j.1749-6632.1992.tb22782.x. [DOI] [PubMed] [Google Scholar]
- 29.Eliav E, Herzberg U, Ruda MA, Bennett GJ. Neuropathic pain from an experimental neuritis of the rat sciatic nerve. Pain. 1999;83:169–182. doi: 10.1016/s0304-3959(99)00102-5. [DOI] [PubMed] [Google Scholar]
- 30.Fleetwood-Walker SM, Hope PJ, Mitchell R, el Yassir N, Molony V. The influence of opioid receptor subtypes on the processing of nociceptive inputs in the spinal dorsal horn of the cat. Brain Res. 1988;451:213–226. doi: 10.1016/0006-8993(88)90766-4. [DOI] [PubMed] [Google Scholar]
- 31.Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, James I. The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100. doi: 10.1016/s0304-3959(00)00474-7. [DOI] [PubMed] [Google Scholar]
- 32.Garry EM, Moss A, Delaney A, O'Neill F, Blakemore J, Bowen J, Husi M, Mitchell R, Grant SGN, Fleetwood-Walker SM. Neuropathic sensitisation of behavioral reflexes and spinal NMDA receptor/CaM kinase II interactions are disrupted in PSD-95 mutant mice. Curr Biol. 2003;13:321–328. doi: 10.1016/s0960-9822(03)00084-8. [DOI] [PubMed] [Google Scholar]
- 33.Gibson SJ, Polak JM, Bloom SR, Sabate IM, Mulderry PM, Ghatei MA, McGregor GP, Morrison JF, Kelly JS, Evans RM. Calcitonin gene-related peptide immunoreactivity in the spinal cord of man and of eight other species. J Neurosci. 1984;4:3101–3111. doi: 10.1523/JNEUROSCI.04-12-03101.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillespie CS, Sherman DL, Fleetwood-Walker SM, Cottrell DF, Tait S, Garry EM, Wallace VC, Ure J, Griffiths IR, Smith A, Brophy PJ. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron. 2000;26:523–531. doi: 10.1016/s0896-6273(00)81184-8. [DOI] [PubMed] [Google Scholar]
- 35.Goldstein ME, House SB, Gainer H. NF-L and peripherin immunoreactivities define distinct classes of rat sensory ganglion cells. J Neurosci Res. 1991;30:92–104. doi: 10.1002/jnr.490300111. [DOI] [PubMed] [Google Scholar]
- 36.Guilbot A, Williams A, Ravise N, Verny C, Brice A, Sherman DL, Brophy PJ, LeGuern E, Delague V, Bareil C, Megarbane A, Claustres M. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum Mol Genet. 2001;10:415–421. doi: 10.1093/hmg/10.4.415. [DOI] [PubMed] [Google Scholar]
- 37.Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Exp. 1999;7:321–335. [PMC free article] [PubMed] [Google Scholar]
- 38.Hall SM, Gregson NA. The in vivo and ultrastructural effects of injection of lysophosphatidyl choline into myelinated peripheral nerve fibers of the adult mouse. J Cell Sci. 1971;9:769–789. doi: 10.1242/jcs.9.3.769. [DOI] [PubMed] [Google Scholar]
- 39.Hargreaves K, Dubner R, Brown FFC, Jovis J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 40.Herzberg U, Eliav E, Bennett GJ, Kopin IJ. The analgesic effects of R(+)-WIN 55, 212–2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neurosci Lett. 1997;221:157–160. doi: 10.1016/s0304-3940(96)13308-5. [DOI] [PubMed] [Google Scholar]
- 41.Hokfelt T, Wiesenfeld-Hallin Z, Villar M, Melander T. Increase of galanin-like immunoreactivity in rat dorsal root ganglion cells after peripheral axotomy. Neurosci Lett. 1987;83:217–220. doi: 10.1016/0304-3940(87)90088-7. [DOI] [PubMed] [Google Scholar]
- 42.Hokfelt T, Zhang X, Wiesenfeld-Hallin Z. Messenger plasticity in primary sensory neurons following axotomy and its functional implications. Trends Neurosci. 1994;17:22–30. doi: 10.1016/0166-2236(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 43.Iggo A. The electrophysiological identification of single nerve fibers with particular reference to the slowly conducting vagal afferent fibers in the cat. J Physiol (Lond) 1958;142:110–126. doi: 10.1113/jphysiol.1958.sp006002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kashiba H, Senba E, Kawai Y, Ueda Y, Tohyama M. Axonal blockade induces the expression of vasoactive intestinal polypeptide and galanin in rat dorsal root ganglion neurons. Brain Res. 1992;577:19–28. doi: 10.1016/0006-8993(92)90532-e. [DOI] [PubMed] [Google Scholar]
- 45.Kashiba H, Noguchi K, Ueda Y, Senba E. Neuropeptide Y and galanin are coexpressed in rat large type A sensory neurons after peripheral transection. Peptides. 1994;15:411–416. doi: 10.1016/0196-9781(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 46.Kelly S, Chapman V. Selective cannabinoid CB1 receptor activation inhibits spinal nociceptive transmission in vivo. J Neurophysiol. 2001;86:3061–3064. doi: 10.1152/jn.2001.86.6.3061. [DOI] [PubMed] [Google Scholar]
- 47.Kerr BJ, Wynick D, Thompson SW, McMahon SB. The biological role of galanin in normal and neuropathic states. Prog Brain Res. 2000;129:219–230. doi: 10.1016/S0079-6123(00)29016-X. [DOI] [PubMed] [Google Scholar]
- 48.Kim CH, Oh Y, Chung JM, Chung K. The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res Mol Brain Res. 2001;95:153–161. doi: 10.1016/s0169-328x(01)00226-1. [DOI] [PubMed] [Google Scholar]
- 49.Koltzenburg M. Painful neuropathies. Curr Opin Neurol. 1998;11:515–521. doi: 10.1097/00019052-199810000-00014. [DOI] [PubMed] [Google Scholar]
- 50.Lawson SN, Waddell PJ. Soma neurofilament immunoreactivity is related to cell size and fiber conduction velocity in rat primary sensory neurons. J Physiol (Lond) 1991;435:41–63. doi: 10.1113/jphysiol.1991.sp018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee YW, Chaplan SR, Yaksh TL. Systemic and supraspinal, but not spinal, opiates suppress allodynia in a rat neuropathic pain model. Neurosci Lett. 1995;199:111–114. doi: 10.1016/0304-3940(95)12034-2. [DOI] [PubMed] [Google Scholar]
- 52.Lehmann J, Schneider J, McPherson S, Murphy DE, Bernard P, Tsai C, Bennett DA, Pastor G, Steel DJ, Boehm C. CPP, a selective N-methyl-d-aspartate (NMDA)-type receptor antagonist: characterization in vitro and in vivo. J Pharmacol Exp Ther. 1987;240:737–746. [PubMed] [Google Scholar]
- 53.Liu CN, Wall PD, Ben Dor E, Michaelis M, Amir R, Devor M. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain. 2000;85:503–521. doi: 10.1016/S0304-3959(00)00251-7. [DOI] [PubMed] [Google Scholar]
- 54.Liu X, Eschenfelder S, Blenk KH, Janig W, Habler H. Spontaneous activity of axotomized afferent neurons after L5 spinal nerve injury in rats. Pain. 2000;84:309–318. doi: 10.1016/s0304-3959(99)00211-0. [DOI] [PubMed] [Google Scholar]
- 55.Love S, Jacobs JM, Myers R. Chronic demyelination in mouse peripheral nerve produced by lysophosphatidyl choline and X-irradiation: ultrastructural observations. J Neurocytol. 1986;15:155–167. doi: 10.1007/BF01611652. [DOI] [PubMed] [Google Scholar]
- 56.Lundberg JM, Terenius L, Hokfelt T, Goldstein M. High levels of neuropeptide Y in peripheral noradrenergic neurons in various mammals including man. Neurosci Lett. 1983;42:167–172. doi: 10.1016/0304-3940(83)90401-9. [DOI] [PubMed] [Google Scholar]
- 57.Ma W, Bisby MA. Partial and complete sciatic nerve injuries induce similar increases of neuropeptide Y and vasoactive intestinal peptide immunoreactivities in primary sensory neurons and their central projections. Neuroscience. 1998;86:1217–1234. doi: 10.1016/s0306-4522(98)00068-2. [DOI] [PubMed] [Google Scholar]
- 58.Malmberg AB, Yaksh TL. Pharmacology of the spinal action of ketorolac, morphine, ST-91, U50488H, and L-PIA on the formalin test and an isobolographic analysis of the NSAID interaction. Anesthesiology. 1993;79:270–281. doi: 10.1097/00000542-199308000-00012. [DOI] [PubMed] [Google Scholar]
- 59.Mao J, Price DD, Hayes RL, Lu J, Mayer DJ, Frenk H. Intrathecal treatment with dextrorphan or ketamine potently reduces pain-related behaviors in a rat model of peripheral mononeuropathy. Brain Res. 1993;605:164–168. doi: 10.1016/0006-8993(93)91368-3. [DOI] [PubMed] [Google Scholar]
- 60.Marchand JE, Cepeda MS, Carr DB, Wurm WH, Kream RM. Alterations in neuropeptide Y, tyrosine hydroxylase, and Y-receptor subtype distribution following spinal nerve injury to rats. Pain. 1999;79:187–200. doi: 10.1016/s0304-3959(98)00165-1. [DOI] [PubMed] [Google Scholar]
- 61.Martin WJ, Loo CM, Basbaum AI. Spinal cannabinoids are anti-allodynic in rats with persistent inflammation. Pain. 1999;82:199–205. doi: 10.1016/S0304-3959(99)00045-7. [DOI] [PubMed] [Google Scholar]
- 62.Matzner O, Devor M. Na+ conductance and the threshold for repetitive neuronal firing. Brain Res. 1992;597:92–98. doi: 10.1016/0006-8993(92)91509-d. [DOI] [PubMed] [Google Scholar]
- 63.Matzner O, Devor M. Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J Neurophysiol. 1994;72:349–359. doi: 10.1152/jn.1994.72.1.349. [DOI] [PubMed] [Google Scholar]
- 64.McCarthy PW, Lawson SN. Cell type and conduction velocity of rat primary sensory neurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience. 1990;34:623–632. doi: 10.1016/0306-4522(90)90169-5. [DOI] [PubMed] [Google Scholar]
- 65.Meyer OA, Tilson HA, Byrd WC, Riley MT. A method for the routine assessment of fore- and hindlimbs grip strength of rats and mice. Neurobehav Toxicol. 1979;1:233–236. [PubMed] [Google Scholar]
- 66.Michael GJ, Averill S, Shortland PJ, Yan Q, Priestley JV. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large trkB and trkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. Eur J Neurosci. 1999;11:3539–3551. doi: 10.1046/j.1460-9568.1999.00767.x. [DOI] [PubMed] [Google Scholar]
- 67.Mitchell J, Caren CA. Degeneration of non-myelinated axons in the rat sciatic nerve following lysolecithin injection. Acta Neuropathol (Berl) 1982;56:187–193. doi: 10.1007/BF00690634. [DOI] [PubMed] [Google Scholar]
- 68.Moulin DE. Pain in central and peripheral demyelinating disorders: multiple sclerosis and Guilain-Barre syndrome. Neurol Clin. 1998;16:889–897. doi: 10.1016/s0733-8619(05)70103-1. [DOI] [PubMed] [Google Scholar]
- 69.Munglani R, Bond A, Smith GD, Harrison SM, Elliot PJ, Birch PJ, Hunt SP. Changes in neuronal markers in a mononeuropathic rat model relationship between neuropeptide Y, pre-emptive drug treatment and long-term mechanical hyperalgesia. Pain. 1995;63:21–31. doi: 10.1016/0304-3959(95)00013-I. [DOI] [PubMed] [Google Scholar]
- 70.Nahin RL, Ren K, De Leon M, Ruda M. Primary sensory neurons exhibit altered gene expression in a rat model of neuropathic pain. Pain. 1994;58:95–108. doi: 10.1016/0304-3959(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 71.Noguchi K, Senba E, Morita Y, Sato M, Tohyama M. Alpha-CGRP and beta-CGRP mRNAs are differentially regulated in the rat spinal cord and dorsal root ganglion. Brain Res Mol Brain Res. 1990;7:299–304. doi: 10.1016/0169-328x(90)90080-w. [DOI] [PubMed] [Google Scholar]
- 72.Noguchi K, De Leon M, Nahin RL, Senba E, Ruda MA. Quantification of axotomy-induced alteration of neuropeptide mRNAs in dorsal root ganglion neurons with special reference to neuropeptide Y mRNA and the effects of neonatal capsaicin treatment. J Neurosci Res. 1993;35:54–66. doi: 10.1002/jnr.490350108. [DOI] [PubMed] [Google Scholar]
- 73.Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR, Eglen RM, Hunter JC. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18:2174–2187. doi: 10.1523/JNEUROSCI.18-06-02174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Omana-Zapata I, Khabbaz MA, Hunter JC, Clarke DE, Bley KR. Tetrodotoxin inhibits neuropathic ectopic activity in neuromas, dorsal root ganglia and dorsal horn neurons. Pain. 1997;72:41–49. doi: 10.1016/s0304-3959(97)00012-2. [DOI] [PubMed] [Google Scholar]
- 75.Pentland B, Donald SM. Pain in the Guillain-Barre syndrome: a clinical review. Pain. 1994;59:159–164. doi: 10.1016/0304-3959(94)90068-X. [DOI] [PubMed] [Google Scholar]
- 76.Polgar E, Shehab SA, Watt C, Todd AJ. GABAergic neurons that contain neuropeptide Y selectively target cells with the neurokinin 1 receptor in laminas III and IV of the rat spinal cord. J Neurosci. 1999;19:2637–2646. doi: 10.1523/JNEUROSCI.19-07-02637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rasminsky M. Hyperexcitability of pathologically myelinated axons and positive symptoms in multiple sclerosis. Adv Neurol. 1981;31:289–297. [PubMed] [Google Scholar]
- 78.Richardson JD, Aanonsen L, Hargreaves KM. Antihyperalgesic effects of spinal cannabinoids. Eur J Pharmacol. 1998a;345:145–153. doi: 10.1016/s0014-2999(97)01621-x. [DOI] [PubMed] [Google Scholar]
- 79.Richardson JD, Kilo S, Hargreaves KM. Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain. 1998b;75:111–119. doi: 10.1016/S0304-3959(97)00213-3. [DOI] [PubMed] [Google Scholar]
- 80.Rittenhouse PA, Marchand JE, Chen J, Kream RM, Leeman SE. Streptozotocin-induced diabetes is associated with altered expression of peptide-encoding mRNAs in rat sensory neurons. Peptides. 1996;17:1017–1022. doi: 10.1016/0196-9781(96)00129-5. [DOI] [PubMed] [Google Scholar]
- 81.Rizzo MA. Successful treatment of painful traumatic mononeuropathy with carbamazepine: insights into a possible molecular pain mechanism. J Neurol Sci. 1997;152:103–106. doi: 10.1016/s0022-510x(97)00143-3. [DOI] [PubMed] [Google Scholar]
- 82.Robson P. Therapeutic aspects of cannabis and cannabinoids. Br J Psychiatry. 2001;178:107–115. doi: 10.1192/bjp.178.2.107. [DOI] [PubMed] [Google Scholar]
- 83.Rosenfeld MG, Mermod JJ, Amara SG, Swanson LW, Sawchenko PE, Rivier J, Vale WW, Evans RM. Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific RNA processing. Nature. 1983;304:129–135. doi: 10.1038/304129a0. [DOI] [PubMed] [Google Scholar]
- 84.Sakurada S, Zadina JE, Kastin AJ, Katsuyama S, Fujimura T, Murayama K, Yuki M, Ueda H. Differential involvement of mu-opioid receptor subtypes in endomorphin-1- and –2-induced antinociception. Eur J Pharmacol. 1999;372:25–30. doi: 10.1016/s0014-2999(99)00181-8. [DOI] [PubMed] [Google Scholar]
- 85.Sakurada S, Hayashi T, Yuhki M, Orito T, Zadina JE, Kastin AJ, Fujimura T, Murayama K, Sakurada C, Sakurada T, Narita M, Suzuki T, Tan-no K, Tseng LF. Differential antinociceptive effects induced by intrathecally administered endomorphin-1 and endomorphin-2 in the mouse. Eur J Pharmacol. 2001;427:203–210. doi: 10.1016/s0014-2999(01)01238-9. [DOI] [PubMed] [Google Scholar]
- 86.Sangameswaran L, Delgado SG, Fish LM, Koch BD, Jakeman LB, Stewart GR, Sze P, Hunter JC, Eglen RM, Herman RC. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J Biol Chem. 1996;271:5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- 87.Takashima H, Boerkoel CF, De Jonghe P, Ceuterick C, Martin JJ, Voit T, Schroder JM, Williams A, Brophy PJ, Timmerman V, Lupski JR. Periaxin mutations cause a broad spectrum of demyelinating neuropathies. Ann Neurol. 2002;51:709–715. doi: 10.1002/ana.10213. [DOI] [PubMed] [Google Scholar]
- 88.Todd AJ. A method for combining confocal and electron microscopic examination of sections processed for double- or triple-labeling immunocytochemistry. J Neurosci Methods. 1997;73:149–157. doi: 10.1016/s0165-0270(97)02222-x. [DOI] [PubMed] [Google Scholar]
- 89.Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, Yonenobu K, Ochi T, Noguchi K. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol Cell Neurosci. 2000;15:170–182. doi: 10.1006/mcne.1999.0814. [DOI] [PubMed] [Google Scholar]
- 90.Villar MJ, Cortes R, Theodorsson E, Wiesenfeld-Hallin Z, Schalling M, Fahrenkrug J, Emson PC, Hokfelt T. Neuropeptide expression in rat dorsal root ganglion cells and spinal cord after peripheral nerve injury with special reference to galanin. Neuroscience. 1989;33:587–604. doi: 10.1016/0306-4522(89)90411-9. [DOI] [PubMed] [Google Scholar]
- 91.Villar MJ, Wiesenfeld-Hallin Z, Xu XJ, Theodorsson E, Emson PC, Hokfelt T. Further studies on galanin-, substance P-, and CGRP-like immunoreactivities in primary sensory neurons and spinal cord: effects of dorsal rhizotomies and sciatic nerve lesions. Exp Neurol. 1991;112:29–39. doi: 10.1016/0014-4886(91)90111-o. [DOI] [PubMed] [Google Scholar]
- 92.Wakisaka S, Kajander KC, Bennett GJ. Increased neuropeptide Y (NPY)-like immunoreactivity in rat sensory neurons following peripheral axotomy. Neurosci Lett. 1991;124:200–203. doi: 10.1016/0304-3940(91)90093-9. [DOI] [PubMed] [Google Scholar]
- 93.Wakisaka S, Kajander KC, Bennett GJ. Effects of peripheral nerve injuries and tissue inflammation on the levels of neuropeptide Y-like immunoreactivity in rat primary afferent neurons. Brain Res. 1992;598:349–352. doi: 10.1016/0006-8993(92)90206-o. [DOI] [PubMed] [Google Scholar]
- 94.Waxman SG. The molecular pathophysiology of pain: abnormal expression of sodium channel genes and its contributions to hyperexcitability of primary sensory neurons. Pain [Suppl] 1999;6:S133–S140. doi: 10.1016/S0304-3959(99)00147-5. [DOI] [PubMed] [Google Scholar]
- 95.Waxman SG, Kocsis JD, Black JA. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol. 1994;72:466–470. doi: 10.1152/jn.1994.72.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci USA. 1999;96:7635–7639. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wilcox GL. Excitatory neurotransmitters and pain. In: Bond MR, Charlton JE, Woolf CJ, editors. Proceedings of the 6th World Congress of Pain. Elsevier; Amsterdam: 1991. pp. 97–117. [Google Scholar]
- 98.Williamson EM, Evans FJ. Cannabinoids in clinical practice. Drugs. 2000;60:1303–1314. doi: 10.2165/00003495-200060060-00005. [DOI] [PubMed] [Google Scholar]
- 99.Woolf CJ. Molecular signals responsible for the reorganisation of the synaptic circuitry of the dorsal horn after peripheral nerve injury: the mechanisms of tactile allodynia. In: Borsook D, editor. Molecular neurobiology of pain. IASP; Seattle: 1997. pp. 171–200. [Google Scholar]
- 100.Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA. 1999;96:7723–7730. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wynick D, Thompson SW, McMahon SB. The role of galanin as a multi-functional neuropeptide in the nervous system. Curr Opin Pharmacol. 2001;1:73–77. doi: 10.1016/s1471-4892(01)00006-6. [DOI] [PubMed] [Google Scholar]
- 102.Xu XJ, Wiesenfeld-Hallin Z, Villar MJ, Fahrenkrug J, Hokfelt T. On the role of galanin, substance P and other neuropeptides in primary sensory neurons of the rat: studies on spinal reflex excitability and peripheral axotomy. Eur J Neurosci. 1990;2:733–743. doi: 10.1111/j.1460-9568.1990.tb00464.x. [DOI] [PubMed] [Google Scholar]
- 103.Yaksh TL, Harty GJ. Pharmacology of the allodynia in rats evoked by high dose intrathecal morphine. J Pharmacol Exp Ther. 1988;244:501–507. [PubMed] [Google Scholar]