

Abstract
Cortical progenitors begin to interpret leukemia inhibitory factor (LIF) and bone morphogenetic protein (BMP) as astrocyte-inducing signals during late embryonic cortical development, coincident with an increase in their expression of epidermal growth factor receptors (EGFRs). To determine whether the developmental change in EGFRs regulates the change in responsiveness to LIF and BMP, we analyzed cortical progenitors induced to express EGFRs prematurely and progenitors from late embryonic EGFR-null cortex. Premature elevation of EGFRs conferred premature competence to interpret LIF, but not BMP, as an astrocyte-inducing signal. EGFR-null progenitors from late embryonic cortex did not interpret LIF as an astrocyte-inducing signal but responded to BMP4. LIF responsiveness in EGFR-null cells was rescued by the addition of EGFRs but not by the stimulation of fibroblast growth factor receptors. Astrocyte differentiation induced by LIF depends on signal transducer and activator of transcription 3 (STAT3). We show that the level of STAT3 increases during late embryonic development in a subset of progenitors. EGFRs regulate this change in STAT3 and increase STAT3 phosphorylation in response to LIF. Increasing STAT3 prematurely with a retrovirus also increased the phosphorylation of STAT3 by LIF. In contrast to the finding with EGFRs, however, increasing STAT3 did not cause LIF to induce astrocytes, although it reduced expression of the neurogenic factor PAX6 (paired box gene 6 ). Our findings show that developmental changes in EGFRs regulate the competence of progenitors to interpret LIF as an astrocyte-inducing signal. EGFRs elevate STAT3 expression and increase its phosphorylation by LIF, but this is not sufficient to change LIF responsiveness to astrocyte induction, suggesting that EGFRs also regulate LIF responsiveness downstream of STAT3.
Keywords: EGF receptor, LIF, astrocyte, cortex, STAT3, stem cell, PAX6, BMP4
Introduction
During late embryonic development, there is a shift in cell-type specification from neurogenesis to gliogenesis (for review, see Bayer and Altman, 1991). Several intrinsic molecules that control the choice between neurogenesis and gliogenesis have been identified. Paired box gene 6 (PAX6) and the bHLH (basic helix-loop-helix) factors neurogenin 1 (ngn1) and ngn2 promote a neuronal fate and inhibit the development of astrocytes (Sun et al., 2001; Heins et al., 2002). ngns achieve this control of cell fate in part by antagonizing the action of signal transducers and activators of transcription (STATs) (Sun et al., 2001), which promote the development of astrocytes (Bonni et al., 1997; Rajan and McKay, 1998). Several extrinsic signals that induce the development of astrocytes have been identified. These include leukemia inhibitory factor (LIF)/ciliary neurotrophic factor (CNTF), bone morphogenetic proteins (BMPs), and epidermal growth factor (EGF)-family ligands (Gross et al., 1996; Johe et al., 1996; Burrows et al., 1997). The timing of astrocyte development is regulated by changes in ligand expression (Lillien et al., 1988; Stockli et al., 1991) and changes in the competence of progenitors to respond to these ligands in a specific manner (Mehler et al., 2000; Molne et al., 2000). Changes in competence are mediated at least in part by the ratio of ngn1 to STATs in progenitors (Sun et al., 2001).
The competence of progenitors to interpret LIF/CNTF, BMPs, and EGF-family ligands as astrocyte-inducing signals can first be observed at approximately embryonic day 14.5 (E14.5) in mice and E16 in rats (Burrows et al., 1997; Mehler et al., 2000; Molne et al., 2000). Our previous work demonstrated that the change in responsiveness to EGF-family ligands reflects an increase in the level of EGF receptors (EGFRs) expressed by progenitors between E13 and E16 in mice (E14.5–E16.5 in rats) (Eagleson et al., 1996; Burrows et al., 1997;Caric et al., 2001). The developmental change in EGFR expression is regulated by extrinsic signals, including fibroblast growth factor 2 (FGF2), which induces EGFR expression (Lillien and Raphael, 2000). The change in responsiveness to BMP and LIF does not reflect an absence of functional receptors and signal transduction machinery for these ligands in early cortical progenitors (Molne et al., 2000;Takizawa et al., 2001). In fact, early progenitors are responsive to BMP and LIF but generate neurons rather than astrocytes (Li et al., 1998; Molne et al., 2000).
Our previous work raised the possibility that EGFRs induce the development of astrocytes by regulating responses of progenitors to heterologous extrinsic signals, such as LIF/CNTF and BMPs (Burrows et al., 1997). To test this idea, we introduced high levels of EGFRs into cortical progenitors prematurely by retroviral transduction and asked whether this conferred competence to interpret LIF or BMP as astrocyte-inducing signals prematurely. We also assessed responses to LIF and BMP in EGFR-null progenitors after the change in responsiveness to LIF and BMP should have occurred. We noted that EGFRs regulated a developmental increase in the level of STAT3 and in the ability of LIF to induce STAT3 phosphorylation. To address the idea that EGFRs regulate responses to LIF by controlling the level of STAT3, we increased STAT3 in progenitors prematurely by retroviral transduction.
Materials and Methods
Animals. EGFR-null mice in a CD1 background (Threadgill et al., 1995) were purchased from The Jackson Laboratory (Bar Harbor, ME). Homozygous null and wild-type littermates were obtained by breeding heterozygotes. EGFR-null embryos were used at E16.5–E17. At this age, eyelids have fused in wild-type embryos but not in EGFR-null embryos. The genotype of embryos was confirmed by PCR. Only EGFR-null (−/−) and wild-type (+/+) embryos were used for analysis.
Cultures. Timed pregnant CD1 or EGFR +/− females were killed with CO2, and embryos were collected in HBSS (Invitrogen, Gaithersburg, MD) at E11–E11.5, E13–E13.5, or E16–E17. Embryonic age was determined from crown–rump length and by examination of external features (Theiler, 1972). The dorsolateral cortex was dissected and cultured on nucleopore filters in serum-free medium in 35 mm dishes, as described previously (Burrows et al., 1997). Progenitors were infected with replication-incompetent retroviruses by placing a 30 μl drop of medium containing virus on the filters. These viruses provide a means of introducing extra wild-type EGFR or STAT3 (see below), in addition to serving as lineage markers. These viruses selectively infect dividing cells at the ventricular surface (Burrows et al., 1997). The frequency of infection was determined by counting the proportion of cells that express virally transduced genes 1 d after infection. For example, 0.002 ± 0.0002% of the cells in an E13 explant were infected by either control or EGFR viruses with titers of ∼1 × 107 colony-forming units (cfu)/ml. When viruses of lower (0.5 × 107) and higher (2 × 107) titers were compared, there was no significant difference in cell fate (data not shown). Viral stocks with titers of 0.5–2 × 107 cfu/ml were selected for infection of explants in this study. Growth factors were added as noted. They were added daily to the medium, beginning 1 d after explants were cultured. Growth factors included recombinant human FGF2 (10 ng/ml; R & D Systems, Minneapolis, MN), human BMP4 (10–100 ng/ml; R & D Systems); human TGFα (10 ng/ml; R & D Systems), and murine LIF [2000–10,000 U/ml; R & D Systems andChemicon (Temecula, CA)]. To quantify effects on cell-type specification among the infected cells after 4 din vitro, explants were dissociated [15 min in 0.1% trypsin (Sigma, St. Louis, MO)], and cells were allowed to attach to poly-d-lysine (PDL; Sigma)-coated slides for 1 hr and then processed for immunocytochemistry (see below). To assay the ability of LIF to induce STAT3 phosphorylation, explants were infected at E11 or E13, cultured without exogenous growth factors for 4 d, and then dissociated and plated as described above. Cells were allowed to recover from dissociation for 3 hr, stimulated with LIF for 20 min, and then fixed and stained as described below.
Viruses. Progenitors in explants were infected with control virus expressing either the histochemical marker β-geo (lacZ plus neomycin phosphatase) (Friedrich and Soriano, 1991) or enhanced green fluorescent protein (eGFP) (Clontech, Palo Alto, CA). The eGFP viral vector was made by subcloning the internal ribosome entry sequence (IRES)-eGFP sequence from IRES2-EGFP (Clontech) into the mouse mammary tumor virus vector pLIA (Bao and Cepko, 1997), after removing the fragment encoding IRES-alkaline phosphatase. The resulting vector (pLIE) was used to generate a viral vector that coexpresses STAT3 (rat cDNA) (Ripperger et al., 1995) and eGFP. Eighty-five to 90% of infected E11.5 cortical progenitors that express eGFP coexpressed a high level of STAT3 (both detected immunocytochemically) (see Fig.6A,B). The EGFR virus (Lillien, 1995) coexpresses β-geo (Friedrich and Soriano, 1991) and wild-type human EGFR (Velu et al., 1989). Virus stocks were made by transfecting ψ-2 cells with the viral vectors (Cepko et al., 1993). In the case of pLIE and pLIE-STAT3, cells were cotransfected with pSV2neo. Clones of transfected cells were selected in G418, screened for expression of the virally transduced genes, and titered with 3T3 cells (Cepko et al., 1993).
Fig. 6.

Premature expression of a high level of STAT3 reduces PAX6 but does not change responsiveness to LIF. A retrovirus that coexpresses eGFP (A) and wild-type STAT3 (B) was used to infect progenitors in E11.5 cortical explants. C, Four days after infection, STAT3 virus increases the proportion of cells that express the early astrocyte marker S-100β in the absence (−) of LIF but does not cause more cells to respond to LIF as an astrocyte-inducing signal. +, Addition of LIF. The proportion of infected E11.5 cells (D; arrow) that express the neurogenic factor PAX6 (E; arrow) is reduced by STAT3 virus compared with a control virus (F). *p = 0.02; **p = 0.003. GFP, Green fluorescent protein.
Immunocytochemistry. Cells on PDL slides were fixed in 4% PFA in either 0.1 m PO4, pH 7.4, or 3% PIPES buffer at room temperature for 10 min. Cells were rinsed and blocked in PBS containing 10% FBS and 0.1% Triton X-100 for 10 min. For phospho-STAT3 staining, cells were also fixed in methanol for 10 min at −20°C. Cells were incubated for 1 hr at room temperature in primary antibodies diluted in this block. Antibodies included rabbit anti-β-galactosidase (β-gal) (Cortex Biochem, San Leandro, CA), rabbit or mouse anti-eGFP (Molecular Probes, Eugene, OR), chick anti-eGFP (Chemicon), mouse anti-β-gal (Promega, Madison, WI), mouse anti-S-100β (Sigma), mouse anti-GFAP (Sigma), mouse anti-proliferating cell nuclear antigen (PCNA) (Sigma), sheep anti-EGFR (Upstate Biotechnology, Waltham, MA), mouse anti-β-tubulin (TuJ1; Covance Research Products, Berkeley, CA), rabbit anti-STAT3 (Cell Signaling Technology, Beverly, MA), rabbit anti-phospho-STAT3 (Tyr705; Cell Signaling Technology), mouse anti-PAX6 (Ericson et al., 1997) (Developmental Studies Hybridoma Bank, Iowa City, IA), mouse anti-RC2 (Misson et al., 1988) (Developmental Studies Hybridoma Bank), and rabbit anti-activated caspase 3 (Biovision, Mountain View, CA). For phospho-STAT3 staining, cells were incubated in primary antibodies overnight at 4°C. Cells were rinsed in PBS and incubated for 30 min in a mixture of secondary antibodies (Jackson ImmunoResearch, West Grove, PA): donkey anti-rabbit Cy3, donkey anti-sheep Cy3, donkey anti-rabbit Cy2, donkey anti-chick Cy2, or donkey anti-mouse IgG or IgM Cy2. To characterize the antigenic phenotype of E15 and E17 cells, dorsolateral cortex was dissected free of meninges, dissociated, plated on PDL slides, and stained as described for explants. Cells were rinsed and mounted in glycerol/PBS. Stained cells were analyzed with a Leica (Nussloch, Germany) DMR fluorescence microscope, and images were captured with a Sensys digital camera using IPLab and Photoshop software (Axon Instruments, Foster City, CA). One hundred cells per condition were counted per experiment. At least three experiments were performed for each condition. Data are presented as mean ± SEM. Statistical evaluation was performed using Student's t test and Statview software, with p < 0.05 considered significant.
Results
Premature elevation of EGFRs alters responses to LIF but not BMP
It has been reported that cortical progenitors from early embryonic rats and mice fail to differentiate into GFAP+ astrocytes when stimulated with either LIF/CNTF or BMPs in culture, although both factors induce astrocytes in cultures of older tissue (Gross et al., 1996; Johe et al., 1996; Li et al., 1998; Molne et al., 2000) (Fig.1A,B). Rather than becoming astrocytes, progenitor cells infected with a control virus at E11 or E13 and then exposed to LIF or BMP4 differentiated into neurons, their normal fate in vivo at these ages (Fig.1C).
Fig. 1.
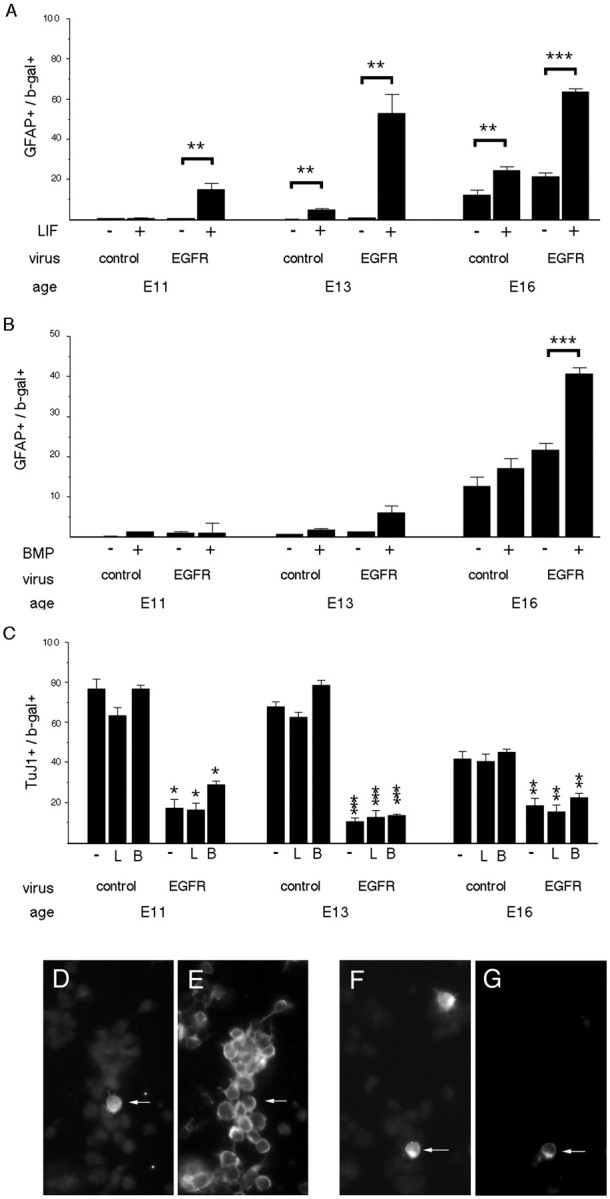
Premature expression of EGFRs induces a premature change in responsiveness to LIF. Progenitors in cortical explants were infected with either control virus or virus expressing EGFRs at E11, E13, or E16, and the proportion of infected cells (β-gal+) (D, F) that express the astrocyte marker GFAP (A, B, G) or the neuron marker TuJ1 (C, E) was determined 4 d later. Cultures were grown in the absence (−) or presence (+) of LIF (L) (A, C) or BMP4 (B) (B, C). EGFRs induce premature competence to interpret LIF as an astrocyte-inducing signal (A, F, G). Some EGFR-infected cells differentiate into neurons (C–E), but this is reduced compared with control-infected cells. Arrows point to double-labeled cells.D–G, E13 progenitors were infected with EGFR virus, grown without (D, E) or with (F, G) LIF, and then stained for β-gal (D) and TuJ1 (E) or β-gal (F) and GFAP (G). C, Comparisons are between EGFR virus and control virus. *p = 0.01; **p ≤ 0.006; ***p < 0.0001.
To determine whether the developmental increase in EGFR expression between E13 and E16 plays a role in the temporal change in responsiveness to LIF and/or BMP, we elevated EGFRs prematurely by infecting progenitors in explants with a retrovirus that transduces wild-type EGFRs and the histochemical marker β-gal. As a control, we used a virus transducing the marker alone. These viruses infect only dividing cells, thereby serving as a method of marking progenitors and following the fate of their progeny. The explants were grown in the absence or presence of LIF (2000 U/ml) or BMP4 (10 ng/ml) for 3 d, beginning 1 d after infection. The ability of the infected cells to respond to these factors by generating astrocytes rather than neurons was analyzed by double-labeling cells with a β-gal antibody to identify infected cells and with cell-type-specific antibodies for astrocytes (GFAP; S-100β) or neurons (TuJ1).
Premature elevation of EGFRs at E11 conferred premature competence to interpret LIF as an astrocyte-inducing signal in 15–20% of the infected cells (Fig. 1A,F,G). A larger proportion of the cells infected with EGFR virus generated astrocytes in response to LIF at later stages, E13 and E16 (Fig. 1A). In contrast, premature elevation of EGFRs did not confer competence to interpret BMP4 as an astrocyte-inducing signal prematurely, although it increased the proportion of cells that exhibited this response to BMP4 later, at E16 (Fig. 1B). In the absence of exogenous ligands, premature elevation of EGFRs reduced the differentiation of neurons at all ages (Fig. 1C). These observations suggest that increased expression of EGFRs is one of the molecular mechanisms used to regulate the timing of a change in responsiveness to LIF, but not to BMPs.
EGFRs are required for the developmental change in responsiveness to LIF but not BMP4
The results of viral transduction experiments indicated that elevated EGFR expression is sufficient for the change in responsiveness to LIF but did not address the issue of necessity. To determine whether EGFRs are required for the change in responsiveness to LIF or BMP4, explants of E16–E17 EGFR-null cortex were exposed to LIF or BMP4 (Fig.2). At this age, wild-type progenitors marked by infection with control virus respond to LIF by expressing S-100β, an early marker of astrocytes (Fig.2A,C,D), in addition to GFAP, a later marker of astrocytes (Fig. 1A). In contrast, EGFR-null progenitors did not express even the early astrocyte marker S-100β after stimulation with LIF (Fig. 2A) but differentiated into neurons instead (data not shown). If EGFRs were added back to EGFR-null progenitors, LIF could induce their differentiation into GFAP+ astrocytes, as observed in wild-type progenitors (Fig. 2B). In contrast to LIF, BMP4 could induce the late astrocyte marker GFAP in EGFR-null progenitors (Fig. 3). These findings provide additional support for the hypothesis that the developmental change in responsiveness to LIF is controlled by EGFRs.
Fig. 2.

The developmental change in responsiveness to LIF depends on EGFRs. Explants of E16–E17 cortex from EGFR-null mice (−/−) and wild-type littermates (+/+) were infected with control virus (A) or EGFR virus (B), and the proportion of infected cells (β-gal+) (C) that express the early astrocyte marker S-100β (A, D) or the late astrocyte marker GFAP (B) was determined after 3 d in the absence (−) or presence (+) of LIF. Competence to generate astrocytes in response to LIF is lost by EGFR-null cells (A) and restored by infecting progenitors with EGFR virus (B). Arrows point to a double-labeled cell expressing β-gal (C) and S-100β (D). *p = 0.03; **p = 0.001; ***p < 0.0001, comparing LIF with untreated cells.
Fig. 3.

The developmental change in responsiveness to BMP4 does not depend on EGFRs. Explants of E16–E17 cortex from EGFR-null mice (−/−) and wild-type littermates (+/+) were infected with a control virus, and the proportion of cells that expressed the late astrocyte marker GFAP was determined after 3 d in the absence (−) or presence (+) of BMP4. *p = 0.009, comparing BMP4 with untreated −/− cells.
Effects of FGF2 on LIF responsiveness require EGFRs
It has been suggested that FGF2 induces responsiveness to CNTF/LIF (Molne et al., 2000; Morrow et al., 2001). We have reported that FGF2 can induce elevated expression of EGFRs (Lillien and Raphael, 2000). Our results showing that EGFRs are required for LIF to induce astrocytes raised the question of whether EGFRs were also required for FGF2-dependent changes in LIF responsiveness. In wild-type E16 progenitors, FGF2 enhanced the proportion of GFAP+ astrocytes, and the combination of FGF2 and LIF also increased the proportion of astrocytes (Fig.4), as reported previously (Molne et al., 2000). In EGFR-null progenitors, FGF2 also promoted GFAP+ astrocyte differentiation to levels that were comparable with those of wild-type progenitors; however, LIF failed to induce additional GFAP+astrocytes when combined with FGF2 (Fig. 4). These observations indicate that FGFR activation cannot substitute for EGFRs in modulating responsiveness to LIF. Moreover, they suggest that the effect of FGF2 on LIF responsiveness requires EGFRs.
Fig. 4.

FGF2-dependent change in responsiveness to LIF requires EGFRs. Explants of E16–E17 cortex from EGFR-null mice (−/−) and wild-type littermates (+/+) were infected with a control virus, and the proportion of infected cells that express the astrocyte marker GFAP was determined after 3 d in the absence (−) or presence (+) of FGF2 or the combination of FGF2 and LIF. FGF2 alone increases astrocyte development in wild-type and EGFR-null cells but does not enhance astrocyte development in response to LIF in EGFR-null cells. *p ≤ 0.0004.
STAT3 levels in progenitors increase during development
Responses to LIF are mediated by STAT1 and STAT3 (Bonni et al., 1997). It has been proposed that the relative concentration of STATs and neurogenin determines whether progenitors become neurons or glia, with a high ngn/STAT ratio promoting a neuronal fate (Sun et al., 2001). STAT3 is expressed in the embryonic cortex before the change in LIF responsiveness occurs (Molne et al., 2000). At E17, however, there is cell-to-cell variability in the level of STAT3 expression, on the basis of fluorescence intensity measurements taken after staining with STAT3 antibody (Fig. 5A). Cells (n = 210) fell into three general categories: STAT3-negative/low (8% of cells; fluorescence intensity ≤10 arbitrary units above background), STAT3-moderate (82% of cells; fluorescence intensity 10–60 arbitrary units above background), and STAT3-high (10% of cells; fluorescence intensity >80 arbitrary units above background). At E17, ∼7% of the progenitor population defined by PCNA expression and 32% of the RC2+population (radial glia, now thought to be progenitors) (Malatesta et al., 2000; Noctor et al., 2001) expressed a high level of STAT3 (Fig.5B,C). In contrast, at earlier embryonic stages (E11, E13, and E15), there were relatively few PCNA+or RC2+ cells that expressed a high level of STAT3 immunoreactivity (Fig. 5B,C).
Fig. 5.

STAT3 expression changes during development and is regulated by EGFRs. A, E17 dorsolateral cortex was stained for STAT3. Note that four cells (arrows) express a high level of STAT3 immunoreactivity, whereas the majority express intermediate or low levels (see Results for quantification of the populations). The population of cells that expresses a high level of STAT3 appears at E15 and increases in size over 2 d, coexpressing the progenitor markers PCNA (B) or RC2 (C). D, Premature elevation of EGFRs induces a high level of STAT3 prematurely. Cells infected with the EGFR virus at E13 express the viral marker β-gal (E; arrows) and a high level of STAT3 (F; arrows). G, The size of the population of cells that expresses a high level of STAT3 is reduced in EGFR-null cortical explants (−/−), compared with wild type. *p ≤ 0.02; **p < 0.0001.
At E17, approximately one-half of the cells that expressed a high level of STAT3 also expressed a high level of EGFRs (Table1). Approximately one-half of the STAT3high cells expressed the early astrocyte marker S-100β, but none expressed the later marker GFAP. Few STAT3high cells expressed the neuronal marker TuJ1, but most were PCNA+. At E15, 40–50% of the STAT3high cells coexpressed PAX6, but this fell to <5% by E17. PAX6 is associated with competence to generate neurons (Heins et al., 2002). The data in Table 1 and Figure 5 indicate that the population of cells that expresses a high level of STAT3 is heterogeneous, increases in size between E15 and E17, and changes in a manner suggesting that the potential of these cells to generate neurons declines.
Table 1.
Antigenic phenotype of STAT3high cells
| Phenotype | E15 | E17 |
|---|---|---|
| EGFRhigh/STAT3high | ND | 49.3 ± 5.0 |
| S-100β/STAT3high | ND | 45.7 ± 0.7 |
| GFAP/STAT3high | ND | 0 ± 0 |
| TuJ1/STAT3high | ND | 2 ± 0.6 |
| PCNA/STAT3high | 78.3 ± 3.2 | 78.7 ± 2.9 |
| PAX6/STAT3high | 42.3 ± 7.8 | 3.2 ± 0.8 |
For each value, at least 100 cells were counted per cortex, and at least three different embryos were analyzed. Data are expressed as mean ± SEM. ND, Not determined.
EGFRs regulate STAT3 level
Although the subpopulations of embryonic cells that express high levels of EGFRs or STAT3 do not overlap completely, the timing of the changes in EGFR and STAT3 expression suggested that an increase in EGFRs might induce elevated expression of STAT3, and that this might underlie EGFR-dependent changes in LIF responsiveness. To determine whether EGFRs regulate the level of STAT3 expression, progenitors were infected with EGFR virus at E11, E13, and E16. At all ages, EGFRs increased STAT3 expression in approximately one-half of the infected cells (Fig. 5D–F). To determine whether the loss of EGFRs results in a failure of late embryonic cells to express a high level of STAT3, we examined EGFR-null cells marked by control virus at E16.5 and cultured as explants for 4 d. The proportion of control-infected cells that expressed a high level of STAT3 was reduced in EGFR-null cells (Fig. 5G). These observations suggest that EGFRs regulate the level of STAT3 in embryonic cortical progenitors.
Increased STAT3 expression is not sufficient to change LIF responsiveness
STAT3 has been shown to be required for astrocyte generation in response to LIF/CNTF (Bonni et al., 1997). Our data raised the possibility that EGFRs mediate changes in LIF responsiveness by increasing the level of STAT3. To determine whether a high level of STAT3 is sufficient to induce a change in responsiveness to LIF, E11.5 progenitors were infected with a virus that expresses STAT3 and the reporter eGFP (Fig.6A,B) or a virus that expresses eGFP alone. Infection with STAT3 virus did not confer competence to interpret LIF as an astrocyte-inducing signal in E11.5 progenitors (Fig. 6C) or E13.5 progenitors (11 ± 1% for control virus vs 8.5 ± 0.5% for STAT3 virus). Instead, these cells differentiated into neurons (data not shown). STAT3 did increase the proportion of cells that expressed the early astrocyte marker S-100β in the absence of LIF (Fig. 6C), but these cells did not express the later marker GFAP (data not shown). LIF has been reported to act synergistically with BMP to induce GFAP+ astrocytes (Nakashima et al., 1999); however, premature elevation of STAT3 did not confer competence to interpret the combination of BMP4 and LIF, or BMP alone, as astrocyte-inducing signals (data not shown).
Although increasing STAT3 did not change the way progenitors interpreted LIF, it did reduce the proportion of PAX6+ cells (Fig.6D–F). STAT3 virus did not alter PCNA expression significantly (data not shown), suggesting that the reduction in PAX6 did not simply reflect a reduction in dividing progenitors. A significant reduction in the proportion of PAX6+ cells was seen as early as 2 d after infection with STAT3 virus: 6.9 ± 0.9% of STAT3-infected E12 cells were PAX6+, compared with 11.3 ± 1.3% of control-infected cells (n = 3;p = 0.04). As noted in Table 1, coexpression of PAX6 and STAT3 declines between E15 and E17. Our findings suggest that elevated STAT3 may induce the decline in PAX6 expression. This could facilitate the transition in cell fate specification from neurons to astrocytes, although expression of a high level of STAT3 is not sufficient on its own to alter responsiveness to LIF or BMP4.
LIF-induced STAT3 phosphorylation is enhanced by EGFRs and elevated STAT3
The induction of astrocytes by LIF is associated with the tyrosine phosphorylation and nuclear translocation of STAT3 (Bonni et al., 1997). We wondered whether increased STAT3 expression failed to confer astrocyte induction in response to LIF because of a limitation in STAT3 phosphorylation. To address this, we infected progenitors at E11 and E13 with control, EGFR, or STAT3 viruses and assessed the expression and nuclear localization of phospho-STAT3 (Tyr705) 4 d later, after stimulation with LIF for 20 min.
Cells infected with EGFR virus exhibited nuclear phospho-STAT3 staining after stimulation with LIF (Fig.7A,B), as did cells infected with STAT3 virus (Fig. 7C,D). The proportion of phospho-STAT3+ cells observed after stimulation with LIF was greater among EGFR-infected cells than control-infected cells at either E11 or E13 (Fig. 7E). The increase in phospho-STAT3 was greater at E13, although the proportion of EGFR-infected cells that express a high level of STAT3 is comparable at E11 and E13 (Fig. 5D). The difference in LIF-induced STAT3 phosphorylation at E11 and E13 is consistent with the age-related difference in LIF-induced astrocyte development noted in Figure 1. The ability of LIF to induce STAT3 phosphorylation appears to be developmentally regulated by an EGFR-independent mechanism that limits astrocyte development. Surprisingly, the proportion of STAT3 virus-infected cells that were phospho-STAT3+ after stimulation with LIF was comparable with that observed among EGFR-infected cells (Fig.7E). This suggests that the failure of STAT3 elevation to confer astrocyte induction in response to LIF does not reflect a limitation of its phosphorylation. Instead, astrocyte induction by LIF appears to be limited downstream of STAT3 phosphorylation. Elevated expression of EGFRs can overcome this limitation to turn LIF into an astrocyte-inducing signal.
Fig. 7.
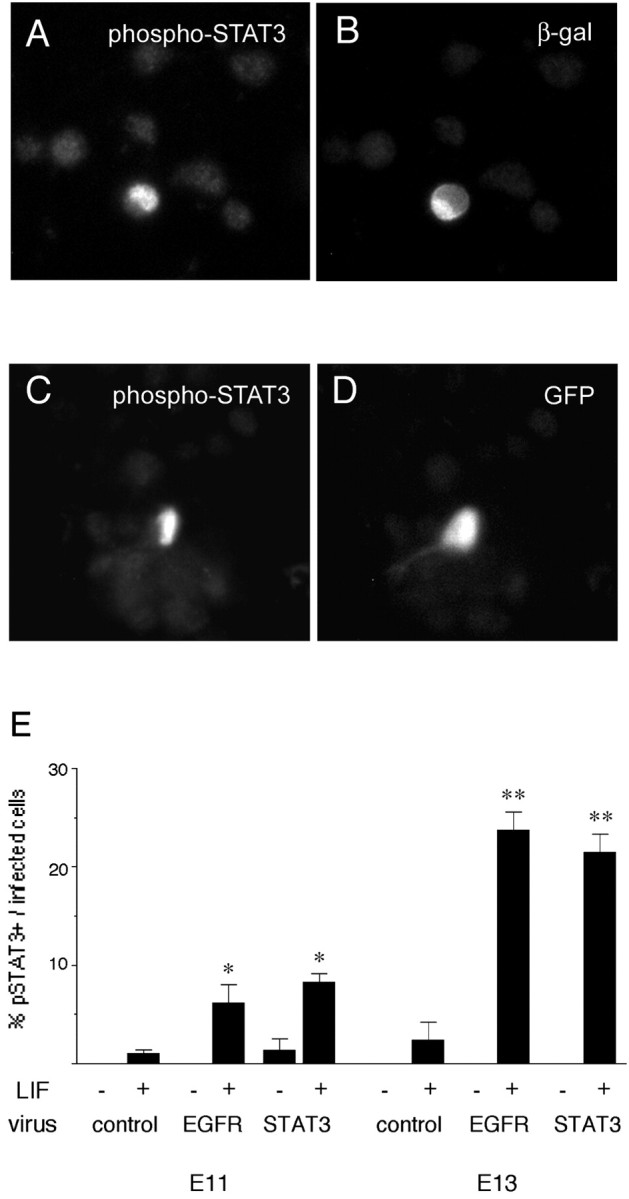
LIF induces STAT3 phosphorylation. E13 explants were infected with EGFR virus (A, B) or STAT3 virus (C, D), dissociated 4 d later, stimulated with LIF for 20 min, and then stained with antibodies to phospho-STAT3 (A, C) and viral markers (B, D).E, The proportion of cells that respond to LIF by phosphorylating STAT3 (pSTAT3) is greater among EGFR-infected and STAT3-infected cells compared with control-infected cells at E11 and E13. −, No LIF; +, addition of LIF. *p ≤ 0.01; **p < 0.0001, comparing LIF with no LIF. More EGFR-infected and STAT3-infected cells responded to LIF at E13 than at E11 (p < 0.0025).
Discussion
Our previous work suggested that a developmental change in cortical progenitor expression of EGFRs contributed to the transition from neurogenesis to gliogenesis (Burrows et al., 1997). Others have shown that progenitor competence to interpret LIF/CNTF and BMP as astrocyte-inducing signals is acquired during late embryonic development in rat and mouse cortex (Li et al., 1998; Mehler et al., 2000; Molne et al., 2000). In the present study, we examined the relationship between the increase in EGFR expression and the acquisition of competence to interpret LIF and BMP as astrocyte-inducing signals. We demonstrated that EGFRs regulate the change in responsiveness to LIF but not to BMPs (Fig.8). To address the molecular mechanism underlying this effect of EGFRs, we tested the idea that the expression of STAT3, which mediates astrocyte differentiation, is regulated by EGFRs and is sufficient for the effect of EGFRs on LIF responsiveness. We show that EGFRs upregulate levels of STAT3 and its phosphorylation in response to LIF, and that STAT3 misexpression downregulates the neurogenic factor PAX6. Increased STAT3 expression also enhances LIF-induced phosphorylation of STAT3, but this does not induce the change in LIF responsiveness to astrocyte induction. Our findings suggest that EGFRs overcome a limitation downstream of STAT3 elevation and phosphorylation that allows LIF to induce astrocytes.
Fig. 8.

Relationship between EGFR expression and responsiveness to LIF. Progenitors that express low (A) or no (D) EGFRs generate neurons when stimulated with LIF, whereas progenitors that express a high level of EGFRs (B, C) generate astrocytes in response to LIF.
How does increased EGFR expression alter LIF responsiveness?
Our observation that EGFR-null progenitors fail to undergo the normal developmental change in responsiveness to LIF suggests that the EGFR is required for this change to occur. This contradicts a previous study suggesting that EGFRs were not required. This interpretation was based on the observation that an inhibitor of the EGFR tyrosine kinase did not prevent the developmental change in responsiveness to CNTF/LIF (Zhu et al., 2000). Mutant forms of the EGFR that lack intrinsic tyrosine kinase activity have been shown to mediate signal transduction, as least in part by serving as a substrate for other kinases and a scaffold for the assembly of signal transduction complexes (Wright et al., 1995; Yamauchi et al., 1997; Deb et al., 2001). In cortical progenitors, EGFRs might therefore regulate responsiveness to LIF by such a kinase-independent mechanism.
It has been suggested that the process of gliogenesis can be separated into two steps that are regulated by distinct extrinsic signals. Restriction to a glial fate could first be specified by a signal such as FGF2, followed by terminal differentiation into astrocytes in response to CNTF/LIF (Morrow et al., 2001). Where does the EGFR fit in? EGFR activation per se does not restrict fate to gliogenesis (Reynolds and Weiss, 1992; Ferri and Levitt, 1995; Burrows et al., 1997; Morrow et al., 2001). Instead, EGFR stimulation appears to block restriction to a neuronal fate (Fig. 1C), maintaining progenitors in a multipotent state that allows the generation of both neuronal and glial progeny (Burrows et al., 1997;Morrow et al., 2001). We reported previously that EGFRs enhance proliferation in addition to promoting astrocyte development (Burrows et al., 1997). The impact of EGFRs on LIF responsiveness could reflect mitotic expansion of multipotent cells that are competent to interpret LIF as an astrocyte-inducing signal. We did not observe a selective effect of EGFR virus on survival; for example, after 4 d in vitro, 1.4 ± 0.5% of control-infected E13 cells expressed activated caspase 3, a marker of dying cells, compared with 1 ± 0.7% of EGFR-infected cells (n = 4). It is therefore not likely that selective survival of a progenitor subpopulation underlies the impact of EGFRs on LIF responsiveness.
Previous work has shown that FGF2 promotes astrocyte differentiation in response to LIF (Molne et al., 2000; Morrow et al., 2001). The present study shows that this effect of FGF2 requires EGFRs. We have shown previously that FGF2 induces expression of a high level of EGFRs prematurely (Lillien and Raphael, 2000). Normally, during development, these signals may act sequentially, with FGF2 inducing an increase in EGFR expression, which then alters responsiveness to LIF.
Regulation of STAT3 expression and phosphorylation
Early progenitors express STAT3 as well as functional LIF receptors (Molne et al., 2000), but we noticed an increase in the level of STAT3 expression among a subpopulation of progenitors, beginning at E15. Over 2 d, the size of the population that expressed a high level of STAT3 increased. At E17, approximately one-half of the STAT3high cells coexpressed a high level of EGFRs. These observations suggested a causal relationship between EGFRs and the level of STAT3. Several lines of evidence support the idea that EGFRs contribute to the developmental increase in STAT3 expression. First, a premature elevation of EGFRs induced a premature increase in STAT3 expression. Second, the proportion of STAT3high cells was reduced in late embryonic EGFR-null cortex. Not all STAT3high cells expressed a high level of EGFRs, suggesting that additional mechanisms contribute to the regulation of STAT3 expression.
It has been suggested that the choice between neurogenesis and gliogenesis depends at least in part on a competitive relationship between the neurogenic factor ngn1 and STAT3 for limiting concentrations of CREB binding protein/p300 (Sun et al., 2001). This hypothesis implies that increasing the ratio of STAT3 to neurogenic factors might shift the balance and promote gliogenesis. Several observations support the idea that increased STAT3 expression contributes to neuron–astrocyte choice. The developmental increase in STAT3 level occurs during the transition from neurogenesis to gliogenesis. At E15, 40–50% of the STAT3high cells coexpressed PAX6, which has been shown to promote a neuronal fate (Heins et al., 2002). Over 2 d, however, the proportion of STAT3high cells that coexpressed PAX6 declined. When we bypassed the EGFR and increased STAT3 prematurely with a retrovirus, PAX6 expression declined. The increase in STAT3 expression might therefore contribute to the transition from neurogenesis to gliogenesis by regulating PAX6. PAX6 is a positive regulator of ngn2 (Scardigli et al., 2001; Heins et al., 2002), so it is possible that STAT3 upregulation could indirectly reduce ngn2 expression by negatively regulating PAX6.
In contrast to increased EGFR expression, increasing STAT3 with a retrovirus was not enough to change responsiveness to LIF, BMPs, or the combination of these factors. Activation of threshold levels of EGFRs therefore changes LIF responsiveness by additional mechanisms, although elevated STAT3 could still be required for the change in LIF responsiveness. Increasing STAT3 in the absence of exogenous LIF induced a small increase in the early astrocyte marker S-100β that was comparable with the response of control-infected cells to LIF. Increased STAT3 expression might therefore sensitize cells to endogenous signal(s) that initiate astrocyte differentiation, but it does not appear to be sufficient to drive cells to a GFAP+ state. The endogenous signals responsible for this effect on S-100β expression have not been identified but could include LIF.
LIF activation of STAT3 signaling involves phosphorylation of STAT3 on tyrosine 705 (Bonni et al., 1997). We found that the ability of LIF to induce STAT3 phosphorylation was enhanced by elevating EGFRs. Moreover, when we bypassed EGFRs and increased the level of STAT3 directly, the ability of LIF to induce STAT3 phosphorylation was also enhanced. Because this was not enough to cause LIF to induce astrocytes, something downstream of STAT3 phosphorylation appears to be limiting. It has been noted that methylation of the GFAP promoter is developmentally regulated and also serves to limit astrocyte development (Takizawa et al., 2001). Elevated EGFR expression is able to overcome limitations downstream of STAT3 phosphorylation to confer astrocyte induction by LIF. It remains to be determined whether EGFRs achieve this by altering methylation or antagonizing one of the inhibitors of LIF signaling downstream of phospho-STAT3, such as PIAS3 (protein inhibitor of activated STAT3) (for review, see O'Shea et al., 2002).
Conclusions
Several lines of evidence implicate EGFRs in the generation of astrocytes. For example, astrocyte development is abnormal in EGFR-null mice (Kornblum et al., 1998), and misexpression of EGFRs in vivo promoted astrocyte development prematurely (Burrows et al., 1997). Results from the present study demonstrate that EGFRs can regulate the development of astrocytes by modulating responsiveness to heterologous signals such as LIF. Although EGFRs contribute to astrocyte generation, astrocytes can also be induced by EGFR-independent mechanisms. For example, FGF2 and BMP4 induced astrocyte development in EGFR-null cells.
We observed developmental changes in the effectiveness of EGFR stimulation in eliciting changes in STAT3 phosphorylation and LIF responsiveness (Figs. 1A, 7E). Thus, additional developmentally regulated events appear to restrict astrocyte generation in the early embryonic cortex. Astrocyte induction by LIF/CNTF is inhibited by a number of extrinsic and intrinsic molecules. These include PDGF (Park et al., 1999), C/EBP (CCAAT/enhancer-binding protein) (Menard et al., 2002), and SOCS (suppressor of cytokine signaling) family members (for review, see Turnley and Bartlett, 2000). The ability of EGFRs to change LIF responsiveness in early progenitors could be limited by these molecules. EGFRs did not regulate changes in responsiveness to BMPs, indicating that astrocyte induction by these molecules is controlled by distinct mechanisms that remain to be identified.
Footnotes
This work was supported by National Institutes of Health Grant RO1 NS38306. We thank Amy Sinor and Alexandra Gulacsi for their comments on this manuscript.
Correspondence should be addressed to Dr. Laura Lillien, Department of Neurobiology and Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, W1454 Biomedical Science Tower, Pittsburgh, PA 15261. E-mail: lillien+@pitt.edu.
References
- 1.Bao ZZ, Cepko CL. The expression and function of Notch pathway genes in the developing rat eye. J Neurosci. 1997;17:1425–1434. doi: 10.1523/JNEUROSCI.17-04-01425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayer SA, Altman J. Neocortical development. Raven; New York: 1991. [Google Scholar]
- 3.Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 4.Burrows RC, Wancio D, Levitt P, Lillien L. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron. 1997;19:251–267. doi: 10.1016/s0896-6273(00)80937-x. [DOI] [PubMed] [Google Scholar]
- 5.Caric D, Raphael H, Viti J, Feathers A, Wancio D, Lillien L. EGFRs mediate chemotactic migration in the developing telencephalon. Development. 2001;128:4203–4216. doi: 10.1242/dev.128.21.4203. [DOI] [PubMed] [Google Scholar]
- 6.Cepko CL, Ryder EF, Austin CP, Walsh C, Fekete DM. Lineage analysis using retrovirus vectors. Methods Enzymol. 1993;225:933–960. doi: 10.1016/0076-6879(93)25059-b. [DOI] [PubMed] [Google Scholar]
- 7.Deb TB, Su L, Wong L, Bonvini E, Wells A, David M, Johnson GR. Epidermal growth factor (EGF) receptor kinase-independent signaling by EGF. J Biol Chem. 2001;276:15554–15560. doi: 10.1074/jbc.M100928200. [DOI] [PubMed] [Google Scholar]
- 8.Eagleson KL, Ferri RT, Levitt P. Complementary distribution of collagen type IV and the epidermal growth factor receptor in the embryonic rat telencephalon. Cereb Cortex. 1996;6:540–549. doi: 10.1093/cercor/6.3.540. [DOI] [PubMed] [Google Scholar]
- 9.Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- 10.Ferri RT, Levitt P. Regulation of regional differences in the fate of cerebral cortical neurons by EGF family-matrix interactions. Development. 1995;12:1151–1160. doi: 10.1242/dev.121.4.1151. [DOI] [PubMed] [Google Scholar]
- 11.Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 12.Gross RE, Mehler MF, Mabie PC, Zang Z, Santschi L, Kessler JA. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron. 1996;17:595–606. doi: 10.1016/s0896-6273(00)80193-2. [DOI] [PubMed] [Google Scholar]
- 13.Heins N, Malatesta P, Cecconi F, Nakafuku M, Tucker KL, Hack MA, Chapouton P, Barde YA, Gotz M. Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci. 2002;5:308–315. doi: 10.1038/nn828. [DOI] [PubMed] [Google Scholar]
- 14.Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 1996;10:3129–3140. doi: 10.1101/gad.10.24.3129. [DOI] [PubMed] [Google Scholar]
- 15.Kornblum HI, Hussain R, Wiesen J, Miettinen P, Zurcher SD, Chow K, Derynck R, Werb Z. Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J Neurosci Res. 1998;53:697–717. doi: 10.1002/(SICI)1097-4547(19980915)53:6<697::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Cogswell CA, LoTurco JJ. Neuronal differentiation of precursors in the neocortical ventricular zone is triggered by BMP. J Neurosci. 1998;18:8853–8862. doi: 10.1523/JNEUROSCI.18-21-08853.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lillien L. Changes in retinal cell fate induced by overexpression of EGF receptor. Nature. 1995;377:158–162. doi: 10.1038/377158a0. [DOI] [PubMed] [Google Scholar]
- 18.Lillien L, Raphael H. BMP and FGF regulate the development of EGF-responsive neural progenitor cells. Development. 2000;127:4993–5005. doi: 10.1242/dev.127.22.4993. [DOI] [PubMed] [Google Scholar]
- 19.Lillien LE, Sendtner M, Rohrer H, Hughes SM, Raff MC. Type-2 astrocyte development in rat brain cultures is initiated by a CNTF-like protein produced by type-1 astrocytes. Neuron. 1988;1:485–494. doi: 10.1016/0896-6273(88)90179-1. [DOI] [PubMed] [Google Scholar]
- 20.Malatesta P, Hartfuss E, Gotz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development. 2000;127:5253–5263. doi: 10.1242/dev.127.24.5253. [DOI] [PubMed] [Google Scholar]
- 21.Mehler MF, Mabie PC, Zhu G, Gokhan S, Kessler JA. Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Dev Neurosci. 2000;22:74–85. doi: 10.1159/000017429. [DOI] [PubMed] [Google Scholar]
- 22.Menard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabe-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36:597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- 23.Misson JP, Edwards MA, Yamamoto M, Caviness VS., Jr Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res Dev Brain Res. 1988;44:95–108. doi: 10.1016/0165-3806(88)90121-6. [DOI] [PubMed] [Google Scholar]
- 24.Molne M, Studer L, Tabar V, Ting YT, Eiden MV, McKay RD. Early cortical precursors do not undergo LIF-mediated astrocytic differentiation. J Neurosci Res. 2000;59:301–311. doi: 10.1002/(sici)1097-4547(20000201)59:3<301::aid-jnr3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 25.Morrow T, Song MR, Ghosh A. Sequential specification of neurons and glia by developmentally regulated extracellular factors. Development. 2001;128:3585–3594. doi: 10.1242/dev.128.18.3585. [DOI] [PubMed] [Google Scholar]
- 26.Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M, Miyazono K, Taga T. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 1999;284:479–482. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- 27.Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- 28.O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109:S121–S131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 29.Park JK, Williams BP, Alberta JA, Stiles CD. Bipotent cortical progenitor cells process conflicting cues for neurons and glia in a hierarchical manner. J Neurosci. 1999;19:10383–10389. doi: 10.1523/JNEUROSCI.19-23-10383.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajan P, McKay RD. Multiple routes to astrocytic differentiation in the CNS. J Neurosci. 1998;18:3620–3629. doi: 10.1523/JNEUROSCI.18-10-03620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 32.Ripperger JA, Fritz S, Richter K, Hocke GM, Lottspeich F, Fey GH. Transcription factors Stat3 and Stat5b are present in rat liver nuclei late in an acute phase response and bind interleukin-6 response elements. J Biol Chem. 1995;270:29998–30006. doi: 10.1074/jbc.270.50.29998. [DOI] [PubMed] [Google Scholar]
- 33.Scardigli R, Schuurmans C, Gradwohl G, Guillemot F. Crossregulation between Neurogenin2 and pathways specifying neuronal identity in the spinal cord. Neuron. 2001;31:203–217. doi: 10.1016/s0896-6273(01)00358-0. [DOI] [PubMed] [Google Scholar]
- 34.Stockli KA, Lillien LE, Naher-Noe M, Breitfeld G, Hughes RA, Raff MC, Thoenen H, Sendtner M. Regional distribution, developmental changes, and cellular localization of CNTF-mRNA and protein in the rat brain. J Cell Biol. 1991;115:447–459. doi: 10.1083/jcb.115.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell. 2001;104:365–376. doi: 10.1016/s0092-8674(01)00224-0. [DOI] [PubMed] [Google Scholar]
- 36.Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. 2001;1:749–758. doi: 10.1016/s1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- 37.Theiler K. The house mouse. Springer; New York: 1972. [Google Scholar]
- 38.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, Barnard JA, Yuspa SH, Coffey RJ, Magnuson T. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 39.Turnley AM, Bartlett PF. Cytokines that signal through the leukemia inhibitory factor receptor-beta complex in the nervous system. J Neurochem. 2000;74:889–899. doi: 10.1046/j.1471-4159.2000.0740889.x. [DOI] [PubMed] [Google Scholar]
- 40.Velu TJ, Beguinot L, Vass WC, Zhang K, Pastan I, Lowy DR. Retroviruses expressing different levels of the normal epidermal growth factor receptor: biological properties and new bioassay. J Cell Biochem. 1989;39:153–166. doi: 10.1002/jcb.240390207. [DOI] [PubMed] [Google Scholar]
- 41.Wright JD, Reuter CW, Weber MJ. An incomplete program of cellular tyrosine phosphorylations induced by kinase-defective epidermal growth factor receptors. J Biol Chem. 1995;270:12085–12093. doi: 10.1074/jbc.270.20.12085. [DOI] [PubMed] [Google Scholar]
- 42.Yamauchi T, Ueki K, Tobe K, Tamemoto H, Sekine N, Wada M, Honjo M, Takahashi M, Takahashi T, Hirai H, Tushima T, Akanuma Y, Fujita T, Komuro I, Yazaki Y, Kadowaki T. Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature. 1997;390:91–96. doi: 10.1038/36369. [DOI] [PubMed] [Google Scholar]
- 43.Zhu G, Mehler MF, Mabie PC, Kessler JA. Developmental changes in neural progenitor cell lineage commitment do not depend on epidermal growth factor receptor signaling. J Neurosci Res. 2000;59:312–320. [PubMed] [Google Scholar]