

Abstract
Synaptic and endogenous pacemaker properties have been hypothesized as principal cellular mechanisms for respiratory rhythm generation. This rhythmic activity is thought to originate in the pre-Bötzinger complex, an area that can generate fictive respiration when isolated in brainstem slice preparations of mice. In slice preparations, external potassium concentration ([K+]o) is typically elevated from 3 to 8 mm to induce rhythmic population activity. However, elevated [K+]o may not simply depolarize respiratory neurons but also change rhythm-generating mechanisms by inducing or altering pacemaker properties. To test this, we examined the membrane potential (Vm) of nonpacemaker neurons and endogenous bursting properties of pacemaker neurons before and after blockade of excitatory and inhibitory synaptic input in 3 mm [K+]o artificial CSF (aCSF). Most pacemaker neurons (82%) ceased to burst in 3 mm [K+]o aCSF after blockade of glutamatergic transmission. In all of these, endogenous bursting was restored on additional blockade of glycinergic and GABAergic inhibition. Thus, bursting properties are suppressed by endogenous synaptic inhibition, the level of which may determine whether network rhythmicity is generated in 3 mm(n = 12) or 8 mm (n= 40) [K+]o aCSF. In 3 mm[K+]o aCSF, synaptically isolated pacemaker neurons (n = 22) continued to burst over a wide range of imposed Vm. Furthermore, theVm of synaptically isolated pacemaker neurons was not significantly affected (p = 0.1; n = 10) when [K+]o was changed from 8 to 3 mm, whereas isolated nonpacemakers hyperpolarized (p < 0.001; n = 14). We conclude that respiratory pacemaker neurons possess membrane properties that stabilize their bursting against changes in [K+]o and imposed changes inVm.
Keywords: respiration, pacemaker properties, pattern generation, pre-Bötzinger complex, rhythm generation, potassium concentration
Introduction
Throughout the brain, intense neuronal activity can raise the extracellular potassium concentration ([K+]o) from 3 mm (resting) to up to 10–12 mm[K+]o, and this may alter rhythmogenesis for a variety of networks (Xiong and Stringer, 2000). Changes in [K+]o can have severe and even fatal consequences unless network or pacemaking mechanisms can compensate (Cohen et al., 1975; Johnson et al., 2001). Stabilization of this vital activity requires adjusting the excitability of neurons despite changes in [K+]o during sensory stimulation (Singer and Lux, 1975; Poolos et al., 1987), bursting during normoxia (Haller et al., 2001; Somjen, 2002), hypoxia (Hansen, 1985; Müller and Somjen, 2000), and epileptiform activity (Orkand, 1980; Jensen et al., 1994). The respiratory center within the brainstem must also face these challenges and indeed continues to be rhythmic over a range of [K+]o (Newstead et al., 1990; Peterson et al., 1992; Johnson et al., 2001). However, the mechanisms that allow the respiratory network to operate over a wide range of [K+]o are unknown. Here, we begin to examine this issue.
Increasing evidence suggests that the respiratory network is localized within the pre-Bötzinger complex (PBC). Lesioning of the PBC abolishes breathing in vivo (Ramirez et al., 1998; Gray et al., 2001), and isolated in a medullary brainstem slice, the PBC still generates respiratory rhythmic activity in vitro (Smith et al., 1991). However, experiments using this slice preparation are typically performed with [K+]o raised from 3 to 8 mm to induce rhythmic network activity at the population level within the PBC (Smith et al., 1991; Lieske et al., 2000). Elevating [K+]o may recruit neurons that are not endogenous pacemakers into a voltage range in which they express pacemaking properties (Richter and Spyer, 2001) similar to hyperkalemic induction of ectopic pacemakers in the heart (Cohen et al., 1975), granule cell bursting in the dentate gyrus (Pan and Stringer, 1997), and in hippocampal pyramidal neurons (Jensen et al., 1994). Indeed, computational models of inspiratory neurons suggest that they do not express pacemaking properties at physiological [K+]o (Rybak et al., 2001). This theoretical consideration has led to the hypothesis that the induction of pacemaker properties at nonphysiological [K+]o may significantly alter rhythm-generating mechanisms in the in vitro respiratory network (Rybak et al., 2001). If rhythm-generating mechanisms in vitro are fundamentally different from those in vivo, the usefulness of in vitro preparations for understanding the neuronal control of breathing becomes questionable. However, to date the effect of changing [K+]o on respiratory pacemaker neurons was only considered in modeling studies (Del Negro et al., 2001; Rybak et al., 2001); no experiments have tested whether pacemaker neurons are inactive at physiological [K+]o. It is also unknown how changes in [K+]o affect pacemaker activity. Here we show that pacemaker properties are present at physiological [K+]o, and these properties are surprisingly unaffected by changes in [K+]o, suggesting that respiratory pacemaker neurons may possess membrane properties that stabilize pacemaker activity. Such membrane properties may not only exist in respiratory neurons but may be a property of pacemaker neurons in general.
Materials and Methods
All experiments conformed to the guiding principles for the care and use of animals approved by the National Institutes of Health and the Animal Care and Use Committee at the University of Chicago. All efforts were made to minimize both the number of animals used and their suffering.
Medullary brain slice preparation. All experiments used the transverse, rhythmic medullary brain-slice preparation (Funk et al., 1994; Ramirez et al., 1996) (see Fig.1A,B). Mice (0- to 9-d-old CD-1 outbred mice; Charles River Laboratories, Wilmington, MA) were deeply anesthetized with ether (delivered by inhalation) and quickly decapitated at the C3/C4 spinal level (Ramirez et al., 1996). The brainstem was dissected in ice-cold artificial CSF (aCSF) that was equilibrated with carbogen (95% O2 and 5% CO2), pH 7.4. Rhythmic 650-μm-thick slices containing the PBC (Ramirez et al., 1996) were obtained by slicing the medulla using a microslicer (VT1000S; Leica, Nussloch, Germany). Slices were submerged in a recording chamber (6 ml) under circulating aCSF (30°C; flow rate, 17 ml/min; total volume, 200 ml) containing the following (in mm): 118 NaCl, 3 KCl, 1.5 CaCl2, 1 MgCl2 × 6 H2O, 25 NaHCO3, 1 NaH2PO4, and 30d-glucose, equilibrated with carbogen (95% O2 and 5% CO2), pH 7.4. KCl was elevated from 3 to 8 mm over a span of 30 min before commencing recordings. All aCSF chemicals were obtained from Sigma (St. Louis, MO). Bath temperature was monitored and automatically adjusted to 30 ± 0.7°C of the set temperature using a Warner Instruments (Hamden, CT) TC-344B temperature regulator with an in-line solution heater (SH-27B); bath temperature at various locations within the bath was routinely uniform.
Fig. 1.

Mouse medullary slice preparation containing the respiratory network. A, Extracellular suction electrodes are placed on the surface of the brain slice preparation to record population activity from the PBC. The hypoglossal motor nucleus is labeled (XII). B, PBC population activity (PBC trace) is integrated (∫PBC) and used as a marker for fictive eupneic inspiratory activity. Ci, Record of ∫PBC (top trace) and simultaneous intracellular recording from an inspiratory neuron in aCSF having 8 mm [K+]o.Cii, Both population (∫PBC, top trace) and neuron bursts cease in aCSF with 3 mm[K+]o. Di, Record from a different preparation than in Ci andCii shows fictive inspiratory bursts (∫PBC, top trace) and synchronized inspiratory neuron activity in aCSF having 8 mm [K+]o.Dii, Note that both the population and inspiratory neuron bursts persist in aCSF with 3 mm[K+]o. Calibration: (Cii) Ci–Dii, 15 mV, 10 sec.
Electrophysiological recordings: integrated PBC population activity and current clamp recordings. Extracellular recordings were obtained with glass suction electrodes positioned on the slice surface in the PBC (see Fig. 1A). The signal collected was amplified and filtered (low-pass, 1.5 kHz; high-pass 250 Hz), rectified, and integrated using an electronic filter (time constant, 60 msec) (Ramirez et al., 1996) (see Fig.1B). The population activity is predominated by inspiratory neurons such that integrated PBC activity is in phase with integrated XII activity (Telgkamp and Ramirez, 1999) and PBC population bursts thus serve as a marker of fictive inspiration (see Fig. 1A,B).
Intracellular patch-clamp recordings were obtained from PBC neurons using the blind-patch technique (see Fig. 1Ci). Patch electrodes were manufactured from filamented borosilicate glass tubes (Clark G150F-4; Warner Instruments) and filled with a solution containing the following (in mm): 140 K-gluconic acid, 1 CaCl2 × 6 H2O, 10 EGTA, 2 MgCl2 × 6 H2O, 4 Na2ATP, and 10 HEPES. Only inspiratory neurons active in phase with population activity were considered in this study. The discharge pattern of each cell type was first identified in the cell-attached mode (in 8 mm[K+]o aCSF). Experiments were then performed in whole-cell patch-clamp mode. The membrane-potential values were corrected for liquid junction potentials as described by Neher (1992). In current-clamp, to isolate neurons from chemical synaptic input, we added glutamatergic, GABAergic, or glycinergic antagonists either individually or as a mixture. These drugs were bath applied at the following final concentrations (in μm): 40 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Tocris Cookson, Ballwin, MO), 10 μm(RS)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP; Tocris Cookson), 1 μmstrychnine (Sigma), and 20 μmbicuculline free-base (Sigma). Note that unlike bicuculline methiodide, the bicuculline free-base derivative is a specific GABA receptor antagonist and is not known to block apamin-sensitive Ca2+-activated K+ currents (Johnson and Seutin, 1997;Debarbieux et al., 1998).
Data analysis. All recordings were transferred to a personal computer using a Digidata (Axon Instruments, Foster City, CA) analog-to-digital conversion board. Data were stored using Axotape (version 2.0; Axon Instruments) and analyzed off-line. Only signals with good signal-to-noise ratios were quantitatively analyzed with software programs written using Igor Pro version 3.11 (WaveMetrics, Lake Oswego, OR). To make membrane potential (Vm) measurements (Table1) under various experimental conditions (e.g., 3 vs 8 mm[K+]o aCSF), theVm was averaged over a 100 sec period of time, and the resultant value was compared with similar data collected under a different condition from the same cell using a paired Student's t test. Because in certain situations (e.g., 8 mm[K+]o aCSF) neurons were firing action potentials and/or bursting, we eliminatedVm deflections attributable to spikes, bursts, and their afterhyperpolarizations from our averagedVm measurements using code written with Igor Pro so that the average accurately reflected the baselineVm.
Table 1.
Effect of changing aCSF [K+]ofrom 8 to 3 mm on Vm of synaptically isolated (in CNQX, CPP, bicuculline, and strychnine) pacemakers and nonpacemakers
| Vm (mV) | SD | Average change inVm (mV) | p | n | |
|---|---|---|---|---|---|
| Pacemakers | |||||
| 8 mm[K+]o | −57.21 | 2.81 | −1.23 | 0.106 | 10 |
| 3 mm[K+]o | −58.45 | 4.063 | −1.23 | 0.106 | 10 |
| Nonpacemakers | |||||
| 8 mm[K+]o | −61.78 | 6.56 | −5.25 | <0.001 | 14 |
| 3 mm[K+]o | −67.034 | 7.88 | −5.25 | <0.001 | 14 |
Results
Population activity in 3 mm[K+]o
The population activity recorded with [K+]o elevated to 8 mm (Fig.1B,Ci,Di, top traces) can be used to identify intracellularly recorded neuronal activity (Fig. 1Ci,Di, bottom traces). The neurons with activity that is in phase with the population bursts are termed inspiratory neurons (Fig. 1Ci,Di). After lowering the aCSF [K+]o from 8 to 3 mm, the majority (40 of 52) of slices ceased to generate population bursts (Fig. 1Cii). However, in some cases (12 of 52), rhythmic activity persisted in 3 mm[K+]o at the cellular and population levels (Fig. 1Dii); this has not been published previously. We next examined whether cessation of rhythmicity in most preparations was caused by effects of lower [K+]o on pacemakers and/or nonpacemakers by comparing their activity before and after isolation from chemical synaptic transmission.
Synaptically isolated inspiratory pacemaker neuron activity in elevated (8 mm) and physiological [K+]o
All of the pacemakers used for analysis met several criteria before being classified as inspiratory pacemaker neurons (n = 22). First, after isolation of the neuron from chemical synaptic input with bath applied CNQX, CPP, strychnine, and bicuculline, pacemaker neurons continued to burst in the absence of inspiratory population bursts (Fig.2A,B). Second, isolated pacemakers exhibited voltage-dependent bursting properties. That is, either brief depolarizing current injection could trigger a burst (data not shown), or hyperpolarizing current could terminate an ongoing burst; either of these reset the ongoing rhythm (Fig. 2B). Finally, current injection changed the bursting frequency of the neuron; depolarization increased (Fig. 2C, top trace) and hyperpolarization reduced (Fig.2C, bottom trace) the bursting frequency.
Fig. 2.
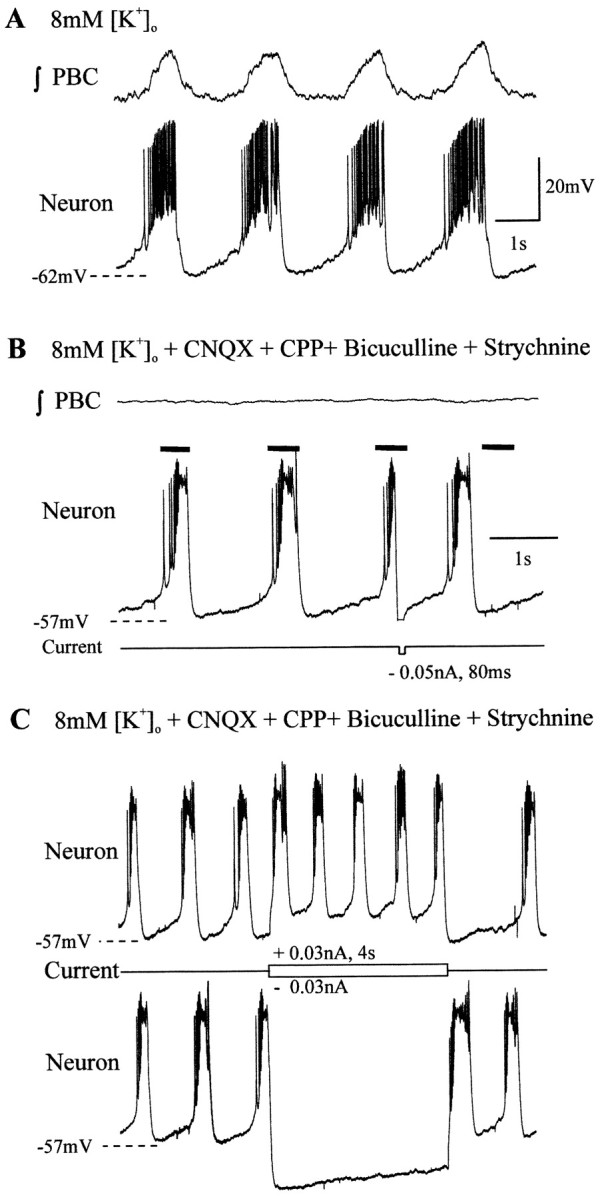
Criteria for identifying pacemakers.A, In 8 mm[K+]o aCSF, inspiratory pacemaker bursts are in phase with ∫PBC bursts. B, Bath application of CNQX, CPP, bicuculline, and strychnine eliminates ∫PBC bursts while pacemaker neurons continue bursting. Brief current injection reset the ongoing rhythm, the timing of which is demarcated by black bars above the neuron recording. C, Injecting depolarizing current into synaptically isolated pacemakers increased the bursting frequency (top neural trace), whereas hyperpolarizing current terminated bursting during current injection (bottom trace). The y-axis scale in A applies toB and C.
To examine the activity of pacemaker neurons at physiological potassium concentrations, the aCSF containing 8 mm[K+]o was replaced with aCSF having 3 mm[K+]o (Fig.3A–C). Rhythmic bursting of most (n = 18 of 22) of the pacemakers in aCSF with 8 mm[K+]o was replaced with tonic activity on lowering the [K+]o to 3 mm[K+]o (Fig.3C) and remained tonic after blockade of glutamatergic synaptic transmission with CNQX and CPP (Fig. 3D). Blockade of inhibition restored endogenous bursting in all of the pacemakers that were not bursting after isolation from glutamatergic synaptic input in 3 mm[K+]o (Fig.3D,E). Because synaptic inhibition appeared to suppress pacemaker bursting in 3 mm[K+]o, we also tested whether (before blocking synaptic transmission) injecting depolarizing DC could bring them into a voltage range in which they burst. All examined inspiratory pacemakers that were tonic in 3 mm[K+]o (Fig.4A,B) burst on depolarizing constant current injection (n = 5) (Fig. 4C). These neurons generated rhythmic bursts without current injection after synaptic isolation (Fig.4D).
Fig. 3.
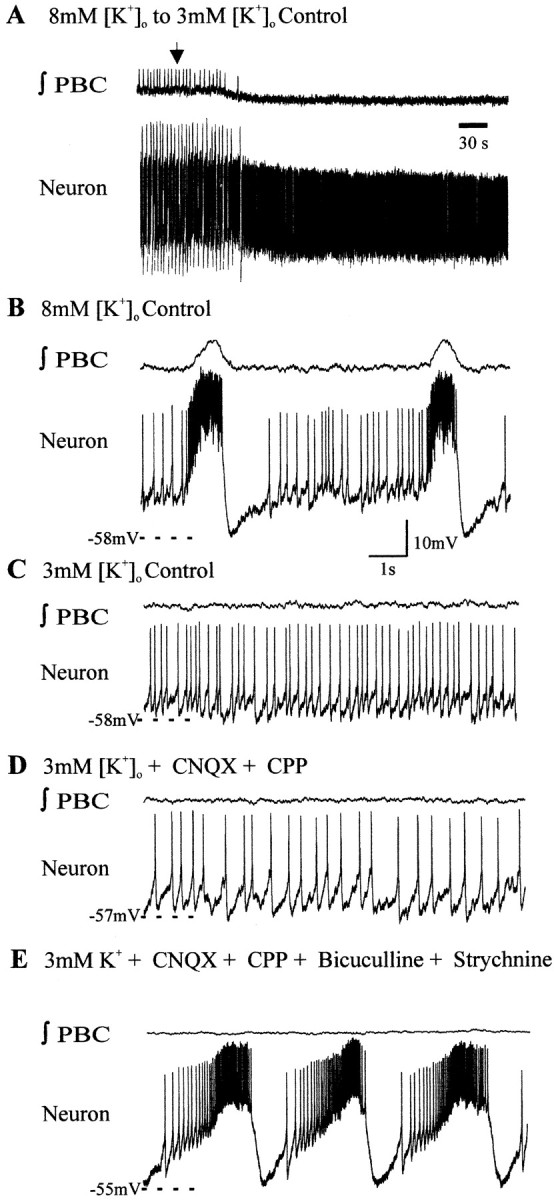
Synaptic inhibition can suppress pacemaker bursting. A, Population bursts (∫PBC) and some pacemakers cease bursting when 8 mm[K+]o aCSF is exchanged with 3 mm [K+]o aCSF (arrow indicates onset of exchange). B, Expanded record of neuron bursting in 8 mm[K+]o aCSF. C, Relatively tonic spiking in 3 mm[K+]o aCSF. D, The neuron continues to spike tonically in 3 mm[K+]o aCSF after blockade of glutamatergic transmission with CNQX and CPP. E, Endogenous bursting is restored after subsequent blockade of synaptic inhibition with strychnine and bicuculline. y-Axis scale in B applies to A–E.x-Axis scale in B applies toC–E.
Fig. 4.
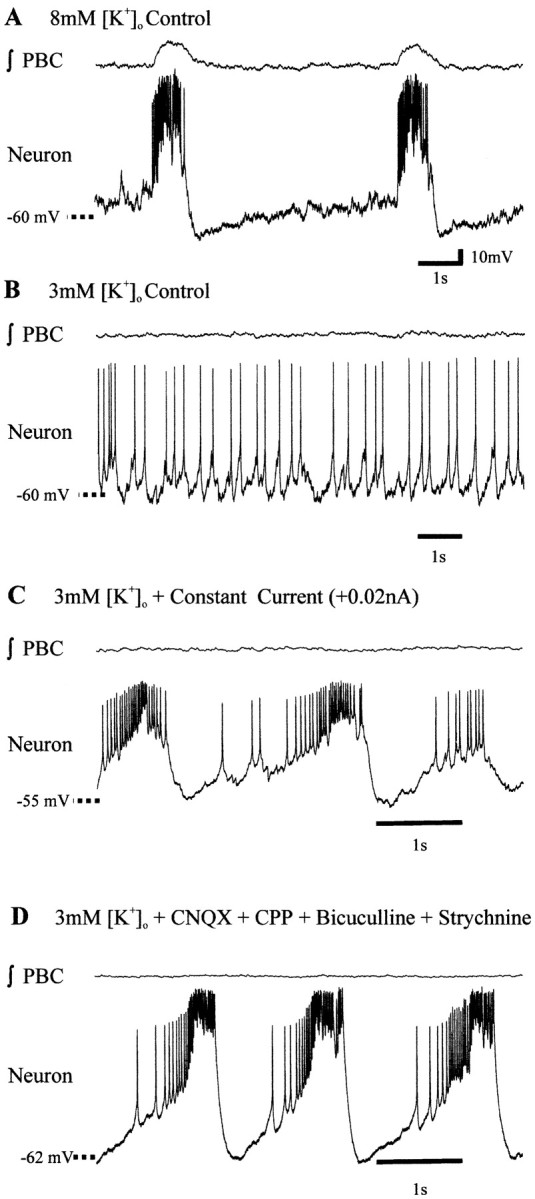
Pacemaker activity in 3 mm[K+]o aCSF is restored during depolarizing current injection. A, Under control conditions (8 mm[K+]o), pacemaker bursts are in phase with those of the population (∫PBC). B, The pacemaker is tonically active on lowering the aCSF [K+]o to 3 mm.C, Constant depolarizing current injection depolarized the neuron into a Vm in which bursting was restored. D, The pacemaker burst endogenously (i.e., without current injection) after synaptic isolation of the pacemaker in 3 mm [K+]o with bath-applied CNQX, CPP, bicuculline, and strychnine.y-Axis in A applies toA–D.
Some of the pacemakers (n = 4 of 22) continued to generate endogenous bursting when aCSF [K+]o was lowered from 8 to 3 mm before blocking chemical synaptic transmission but in the absence of population activity (Fig.5A–C). These neurons continued bursting in 3 mm[K+]o after blockade of synaptic input with CNQX, CPP, bicuculline, and strychnine (Fig. 5D,E).
Fig. 5.

Pacemakers can burst in aCSF with 3 and 8 mm [K+]o without blocking synaptic transmission. A, Lowering the aCSF [K+]o from 8 to 3 mm(starting at downward arrow) typically results in a cessation of ∫PBC bursts (top trace), but some inspiratory neurons continue to burst rhythmically (Neuron). B, C, Expanded record of population (∫PBC) and cell (Neuron) in 8 mm[K+]o (B) and when aCSF [K+]o is lowered to 3 mm (C). Note that the neuron continues to burst rhythmically in 3 mm[K+]o, despite cessation of ∫PBC bursts. D, For pacemakers, raising aCSF [K+]o from 3 to 8 mm while chemical synaptic transmission is blocked does not result in a marked shift in the baseline Vm (solutions exchanged starting at downward arrow). E,F, Expanded record of synaptically isolated pacemaker activity in 3 mm [K+]oaCSF (E) and when the [K+]o is increased to 8 mm(F). y-Axis in Bapplies to B, C, and F.x-Axis in B and E applies to F.
Intrinsic membrane properties of pacemaker neurons anchorVm
As indicated in Table 1, the baselineVm of synaptically isolated pacemaker neurons did not significantly change on lowering the aCSF [K+]o from 8 to 3 mm (n = 10). Bursting in synaptically isolated pacemakers also continued with little apparent change in baseline Vm when [K+]o was increased from 3 to 8 mm (n = 3 tested) (Fig. 5D–F). To examine the bursting properties of synaptically isolated pacemakers over a range of imposedVm, we injected hyperpolarizing current pulses of several seconds in duration (n = 22). Stepping to various hyperpolarized Vmwith 10 sec current injections revealed typical voltage-dependent bursting properties (Fig.6A–C). That is, the frequency of bursting decreased at hyperpolarized levels, and endogenous bursting could be suppressed by strongly hyperpolarizing current injections (n = 22) (Fig.6A–C). However, the hyperpolarization evoked a slow depolarization in pacemaker neurons whereby the neuron depolarized throughout the constant current injection (n = 22) (Figs. 2C, 6C). In 13 of these pacemakers, we injected hyperpolarizing currents of varying intensity for >10 sec, and in each case, the Vm depolarized to a range in which endogenous bursting resumed (Fig.6D). The maximum hyperpolarization resulting from a current injection was plotted versusVm just before the first burst that occurred while the current injection was maintained. This plot revealed that pacemaker neurons have intrinsic membrane properties that depolarize them during the hyperpolarization (n = 13) (Fig. 7A). In four cases, pacemakers were hyperpolarized with constant current injected for the same duration (3.7 min). Measurement of theirVm at the onset of the current injection (−77.8 mV ± 1.9 SD) and just before terminating the hyperpolarizing pulse (−68.9 mV ± 2.3 SD) revealed that pacemakers depolarized during the course of hyperpolarizing current injection (p = 0.01; paired t test;n = 4) (Fig. 6D). The bursting frequency also increased throughout the maintained hyperpolarization (p < 0.001; one-way ANOVA), but the bursting frequency during the last 30 sec of current injection (0.21 ± 0.08, mean ± SD) did not reach control frequency (30 sec before current application; 0.36 ± 0.18 SD; p = 0.014; one sample t test) (Fig. 7B). Bursting frequency over the 30 sec after termination of current injection (0.37 ± 0.22 SD) was similar to baseline frequency measured 30 sec before hyperpolarizing current injection (p = 0.841; one-sample t test) (Fig. 7B).
Fig. 6.

Hyperpolarizing current injections into synaptically isolated pacemaker in 3 mm[K+]o aCSF. Endogenous bursting frequency is reduced or bursting is terminated during hyperpolarization to −65 mV with a 10 sec −0.06 nA current injection (A), −70 mV with a 10 sec −0.09 nA current injection (B), and −75 mV with a 10 sec −0.13 nA current injection (C).D, Hyperpolarization to −80 mV using a −0.15 nA constant current injection for a longer (3.7 min) duration does not terminate bursting. Calibration: (in A)A–D, 10 mV.
Fig. 7.
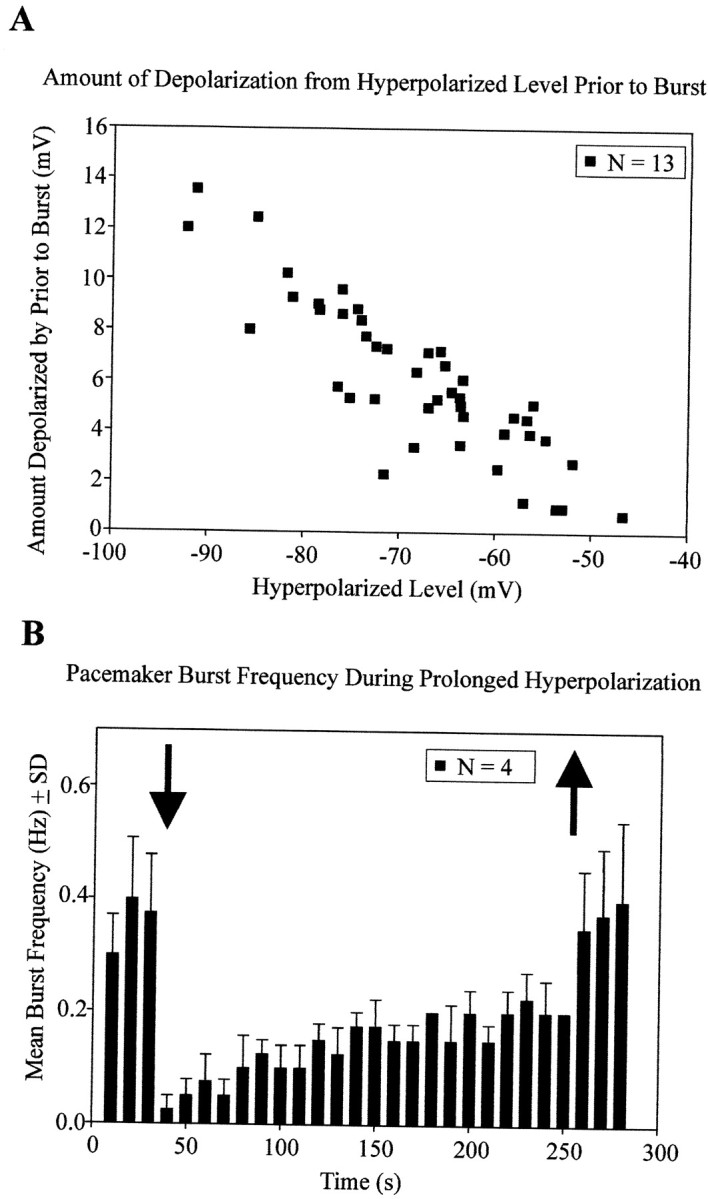
Pacemaker neurons depolarize during hyperpolarization. A, Vm of synaptically isolated pacemakers (in 3 mm[K+]o aCSF containing CNQX plus CPP, bicuculline, and strychnine) measured immediately after onset of current injection (x-axis) and immediately before the first burst (y-axis). Each square represents one current injection, and data are pooled from n = 13 neurons. B, Mean burst frequency histogram (10 sec bins:n = 4 mean ± SD) before, during, and after hyperpolarizing current injection for 3.7 min. The pacemakers in 3 mm [K+]o aCSF were synaptically isolated in CNQX, CPP, bicuculline, and strychnine. Onset and offset of current injection are indicated by arrows.
Nonpacemaker inspiratory neuron activity in 3 and 8 mm [K+]o
Are intrinsic membrane properties that anchorVm against changes in [K+]o or hyperpolarization also present in inspiratory nonpacemakers? When the aCSF [K+]o is lowered from 8 to 3 mm, activity in nonpacemaker neurons behaved much like the population activity (Fig.8A) and hyperpolarized (Table 1). Synaptically driven rhythmic bursting that was present in 8 mm[K+]o ceased at 3 mm[K+]o aCSF (Fig.8A,B). The nonpacemaker neurons became inactive and did not generate spontaneous action potentials when the [K+]o was at 3 mm (Fig. 8A) or when they were isolated from glutamatergic synaptic input with CNQX and CPP (n = 25) (Fig. 8C). With glutamatergic input blocked, we also examined the effects of blocking inhibition in 10 nonpacemakers. These nonpacemaker neurons were either tonically active (n = 7) or silent (n = 3) (Fig.9A–C) after additional blockade of synaptic inhibition with bicuculline and strychnine. Unlike pacemakers (Fig. 4C), nonpacemaker inspiratory neurons (Fig.9A) generated tonic activity in response to depolarizing current injection in 3 mm[K+]o aCSF before (Fig. 9B) and after (Fig. 9C) isolation from chemical synaptic transmission (n = 5). In addition, these neurons did not burst when the [K+]o was raised from 3 to 8 mm (n = 5) (Fig.9D). The Vm of nonpacemakers did not change during prolonged (3.7 min) hyperpolarizing current injection to −81.3 mV ± 2.3 SD. The membrane potential just before terminating the current injection was −80.8 mV ± 2.9 SD (n = 5; p = 0.328; pairedt test) (Fig. 9E).
Fig. 8.
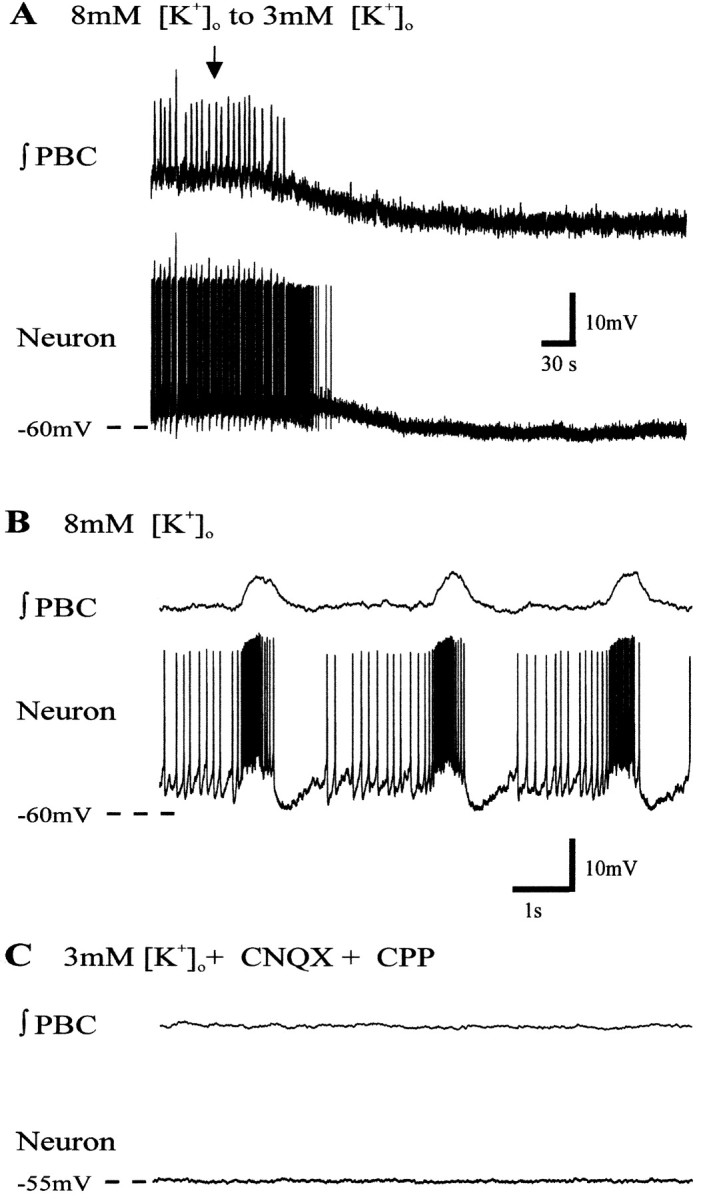
Nonpacemaker inspiratory neuron bursting ceases in 3 mm [K+]o aCSF.A, ∫PBC (top trace) and inspiratory neuron activity during replacement of 8 mm[K+]o aCSF with aCSF having 3 mm [K+]o (arrow indicates onset of aCSF exchange). Note that the neuron hyperpolarizes in 3 mm [K+]o aCSF.B, Expanded record of inspiratory neuron activity in 8 mm [K+]o.C, Both population and neuron activity remain silent in 3 mm [K+]o aCSF with blockade of glutamatergic input with CNQX and CPP. Calibration: (B) B, C, 10 mV, 1 sec.
Fig. 9.
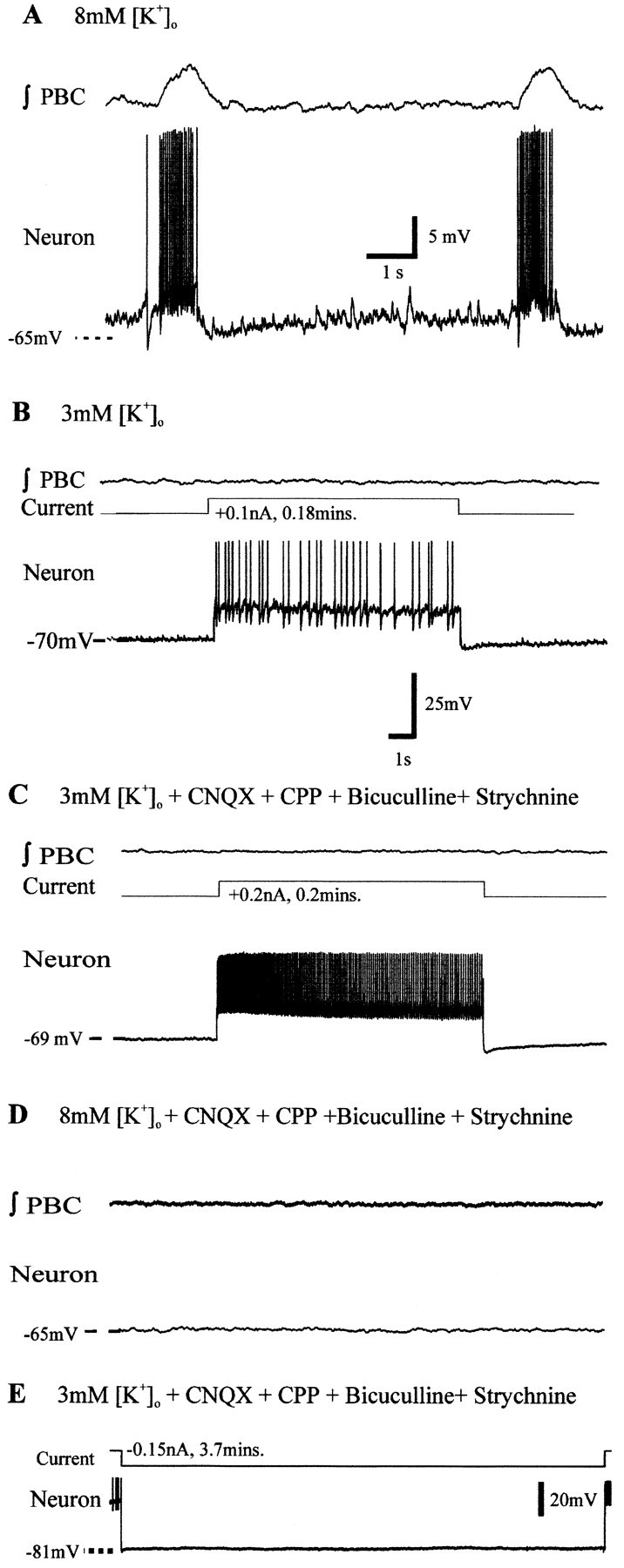
Nonpacemaker neurons are tonic in 3 mmaCSF on super-rheobase current injection. A, ∫PBC (top trace) and nonpacemaker inspiratory neuron activity in 8 mm[K+]o (bottom trace).B, In 3 mm aCSF, ∫PBC population bursts and nonpacemaker activity cease and depolarizing current injection evokes tonic activity (current). C, After blockade of chemical transmission with CNQX, CPP, bicuculline, and strychnine, ∫PBC population activity and nonpacemakers remain silent. The cell discharges tonically on depolarization in 3 mm[K+]o aCSF. D, Synaptically isolated nonpacemaker remains silent when aCSF [K+]o is raised from 3 to 8 mm. Calibration: (B)C–E, 25 mV, 1 sec. Nonpacemakers do not depolarize during prolonged hyperpolarizing current injection.
Discussion
The results of this study indicate that in the brainstem slice preparation, the respiratory network (PBC) and pacemaker neurons can produce fictive respiratory activity at physiological extracellular potassium concentrations ([K+]o). Thus, pacemakers may play a key role in respiratory rhythmogenesis under physiological conditions. Data obtained by Johnson et al. (2001)revealed that in vitro the presence of endogenous synaptic inhibition can suppress PBC population bursts, and we add here that endogenous bursting properties of pacemakers can also be suppressed by synaptic inhibition in aCSF with 3 mm[K+]o. Intrinsic membrane properties of pacemakers, but not nonpacemaker neurons, can anchor their membrane potential within a range in which they burst endogenously, despite imposed hyperpolarization or changes in [K+]o.
A central issue to understanding breathing in mammals is to identify the neural mechanisms underlying rhythm generation for a variety of breathing patterns (e.g., eupnea, sighs, and gasps). Respiratory pacemakers have been hypothesized to play a key role in rhythmogenesis, but those isolated in various in vitro preparations have been described in aCSF containing elevated [K+]o(Thoby-Brisson and Ramirez, 2000; for review, see Richter and Spyer, 2001). This paradigm has fostered controversy over whether endogenous pacemakers contribute to the respiratory rhythm, because in other systems, neurons that do not express pacemaking properties at 3 mm[K+]o burst endogenously when [K+]o is raised (Jensen et al., 1994; Jensen and Yaari, 1997; Su et al., 2001). Furthermore, modeling of respiratory neurons predicted, on the basis of theoretical considerations, that potassium concentrations must be >6 mm for endogenous bursting to occur and suggested that endogenous pacemakers cannot be responsible for, or even contribute to, respiratory rhythmogenesis at physiological potassium concentrations (Rybak et al., 2001). For these reasons, several authors have proposed that respiratory rhythmogenesis is an emergent network property that depends on synaptic interactions (for review, see Richter and Spyer, 2001). However, the results presented here argue against excluding pacemakers as playing a role in rhythmogenesis, because both the population and endogenous pacemakers can generate respiratory activity in 3 mm[K+]o aCSF (Figs.1Dii, 5A).
The possibility that pacemaker neurons are essential for respiratory rhythm generation has been questioned recently on the basis of pharmacological experiments (Del Negro et al., 2002). Although strain and species differences are possible, these studies have demonstrated that riluzole, a blocker of the persistent sodium current that is found in some respiratory pacemakers, did not abolish respiratory rhythm generation in either mice or rats. This conclusion is based on the assumption that all pacemaker neurons have similar bursting properties and are sensitive to riluzole. However, this is not the case. The respiratory network not only contains the pacemaker neurons whose bursting depends on the persistent sodium current but also pacemaker neurons with bursting properties that depend on calcium currents (Thoby-Brisson and Ramirez, 2001). The majority of neurons in the study by Del Negro et al. (2002) were examined in zero extracellular calcium concentrations that abolish bursting in the Ca2+-dependent pacemaker population. Thus, the riluzole sensitivity of these pacemaker neurons remains unexamined. Indeed, our studies suggest that not all pacemaker neurons are riluzole sensitive, and some of these continue to burst after blockade of voltage-dependent Ca2+ channels with cadmium (Parkis et al., 2002). Although it remains unclear whether pacemaker neurons are essential for respiratory rhythm generation, this study demonstrates that pacemaker neurons could contribute to respiratory rhythm generation, not only at high but also physiological [K+]o. Clearly, many properties of the respiratory network are consistent with the concept that pacemaker neurons contribute to respiratory rhythm generation. The frequency of pacemaker bursts mimics the biphasic hypoxic response (Thoby-Brisson and Ramirez, 2000), and modeling studies demonstrate that many characteristics of the respiratory network can be explained by the activation and inactivation properties of endogenously bursting neurons (Butera et al., 1999a,b).
Our study also contributed to a better understanding of the role of synaptic inhibition in the respiratory network. Previous studies have demonstrated that synaptic inhibition plays a critical role in the generation of the respiratory pattern that underlies eupneic activity (Lieske et al., 2000; Büsselberg et al., 2001). However, rhythmogenesis persists after the blockade of synaptic inhibition (Figs. 2B, 3E, 4D,5D, 6A) (Smith et al., 1991, 2000; Ramirez et al., 1997; Shao and Feldman, 1997; Brockhaus and Ballanyi, 1998;Rekling and Feldman, 1998; Butera et al., 1999; Koshiya and Smith, 1999). Here, we demonstrate that inhibition plays a critical role in modulating pacemaker activity, as occurs in other systems. For instance, inhibition regulates endogenous bursting in the stomatogastric system of crustacea (Cazalets et al., 1987), the spinal cord (Su, 2001), and the substantia nigra (Silva and Bunney, 1988). Our data show that inhibition can suppress endogenous bursting of some (Fig. 3E) but not all (Fig. 5C) pacemakersin vitro, which could explain why respiratory network activity ceases at physiological potassium concentrations (Fig.1Cii) (Johnson et al., 2001). Although this finding may suggest that network and inspiratory pacemaker activity primarily depends on modulation of inhibition (i.e., conditional pacemaking), the same level of inhibition may not be present in vivo. Along these lines, whereas elevating [K+]o raised excitability of nonpacemaker neurons (Table 1), it may also suppress chloride-mediated GABAergic inhibition (Traynelis and Dingledine, 1989;Jensen et al., 1994; Pan and Stringer, 1997); both of these factors may enhance the likelihood of evoking population activity in vitro. That said, inhibition does not interfere with the mechanism of pacemaker bursting, because depolarizing pacemakers that are tonically firing in 3 mm[K+]o can bring them into a Vm range in which bursting occurs (Fig. 4C).
Another important finding of our study was that lowering the [K+]o from 8 to 3 mm did not significantly change theVm of synaptically isolated pacemaker neurons (Table 1). Thus, future neural models of the respiratory network should consider that the excitability of pacemaker neurons is not primarily determined by EK+ (Del Negro et al., 2001). More importantly, unlike nonpacemaker neurons, intrinsic membrane properties of pacemakers seem to anchor theirVm in a range in which they express pacemaking properties. After hyperpolarizing current injection, pacemaker neurons had intrinsic membrane properties that depolarized them into a Vm in which they express pacemaker bursting. This may serve to enhance the stability of rhythmogenesis during variations in [K+]o, as occurs for bursting activity during normoxia (Haller et al., 2001; Somjen, 2002) and hypoxic conditions (Hansen, 1985; Müller and Somjen, 2000). Future studies will be necessary to determine the type of ion channel that is responsible for this depolarization. One obvious candidate is Ih current. However, this ionic current is only expressed in some, but not all, respiratory pacemaker neurons (Thoby-Brisson et al., 2000). Furthermore, the putative ion channel appears to have a much slower time constant, because the depolarization developed in tens of seconds (Figs.6D, 7B) rather than within 1.5 sec, as is typical for the Ih current (Thoby-Brisson et al., 2000). Whatever the underlying mechanism, intrinsic pacemaker properties could contribute to stabilization of the Vm of pacemakers to promote bursting despite changes in [K+]o (Table 1, Fig. 6D) (Kramer and Zucker, 1985; Andrew, 1987). These intrinsic mechanisms may be widely responsible for stabilizing rhythmic bursting of pacemakers in a variety of systems including Aplysia (Kramer and Zucker, 1985), supraoptic magnocellular neurons (Andrew, 1987), and nucleus principalis trigeminal neurons (Sandler et al., 1998).
The results support the hypothesis that PBC pacemakers may contribute to respiratory rhythmogenesis. These data suggest that it is not necessary to assume that the rhythm relies on network properties. In addition, it would be useful to integrate these findings into new models of respiratory rhythm generation, so the models more accurately reflect results from experimental manipulation. Finally, the degree of synaptic inhibition may permit flexibility in terms of control of pacemakers so that the respiratory pattern can be adjusted to meet the respiratory needs of the animal.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant 1F32HL67659 (A.K.T.), a Pew Charitable Trust Fellowship (F.P.), and NIH Grant RO1-HL 60120 (J.M.R.). We thank Drs. Steve P. Lieske for technical support in data analysis and editing and Marjorie Parkis for editorial comments.
Correspondence should be addressed to Dr. Andrew K. Tryba, The University of Chicago, Department of Organismal Biology and Anatomy, 1027 East 57th Street, Chicago, IL 60637-1508. E-mail:Tech10s@techsan.org.
References
- 1.Andrew RD. Endogenous bursting by rat suproptic neuroendocrine cells is calcium dependent. J Physiol (Lond) 1987;384:451–465. doi: 10.1113/jphysiol.1987.sp016463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brockhaus J, Ballanyi K. Synaptic inhibition in the respiratory network of neonatal rats. Eur J Neurosci. 1998;10:3823–3839. doi: 10.1046/j.1460-9568.1998.00396.x. [DOI] [PubMed] [Google Scholar]
- 3.Busselberg D, Bischoff AM, Paton JF, Richter DW. Reorganization of respiratory network activity after loss of glycinergic inhibition. Pflügers Arch. 2001;441:444–449. doi: 10.1007/s004240000453. [DOI] [PubMed] [Google Scholar]
- 4.Butera RJ, Jr, Rinzel J, Smith JC. Models of respiratory rhythm generation in the pre-Botzinger complex. I. Bursting pacemaker neurons. J Neurophysiol. 1999a;81:382–397. doi: 10.1152/jn.1999.82.1.382. [DOI] [PubMed] [Google Scholar]
- 5.Butera RJ, Jr, Rinzel J, Smith JC. Models of respiratory rhythm generation in the pre-Botzinger complex. II. Populations of coupled pacemaker neurons. J Neurophysiol. 1999b;81:398–415. doi: 10.1152/jn.1999.82.1.398. [DOI] [PubMed] [Google Scholar]
- 6.Cazalets JR, Cournil I, Geffard M, Moulins M. Suppression of oscillatory activity in crustacean pyloric neurons: implication of GABAergic inputs. J Neurosci. 1987;7:2884–2893. doi: 10.1523/JNEUROSCI.07-09-02884.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen HC, Rosen KM, Pick A. Disorders of impulse conduction and impulse formation caused by hyperkalemia in man. Am Heart J. 1975;89:501–509. doi: 10.1016/0002-8703(75)90158-1. [DOI] [PubMed] [Google Scholar]
- 8.Debarbieux F, Brunton J, Charpak S. Effect of bicuculline on thalamic activity: a direct blockade of IAHP in reticularis neurons. J Neurophysiol. 1998;79:2911–2918. doi: 10.1152/jn.1998.79.6.2911. [DOI] [PubMed] [Google Scholar]
- 9.Del Negro CA, Johnson SM, Buter RJ, Smith JC. Models of respiratory rhythm generation in the pre-Botzinger complex. III. Experimental tests of model predictions. J Neurophysiol. 2001;86:59–74. doi: 10.1152/jn.2001.86.1.59. [DOI] [PubMed] [Google Scholar]
- 10.Del Negro CA, Koshiya N, Butera RJ, Jr, Smith JC. Persistent sodium current, membrane properties and bursting behavior of pre- Bötzinger complex inspiratory neurons in vitro. J Neurophysiol. 2002;88:2242–2250. doi: 10.1152/jn.00081.2002. [DOI] [PubMed] [Google Scholar]
- 11.Funk G, Smith JC, Feldman JL. Development of thyrotropin-releasing hormone and norepinephrine potentiation of inspiratory-related hypoglossal motorneuron discharge in neonatal and juvenile mice in vitro. J Neurophysiol. 1994;72:2538–2541. doi: 10.1152/jn.1994.72.5.2538. [DOI] [PubMed] [Google Scholar]
- 12.Gray PA, Janczewski WA, Mellen N, McCrimmon DR, Feldman JL. Normal breathing requires preBotzinger complex neurokinin-1 receptor-expressing neurons. Nat Neurosci. 2001;4:927–930. doi: 10.1038/nn0901-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haller M, Mironov SL, Karschin A, Richter DW. Dynamic activation of Katp channels in rhythmically active neurons. J Physiol (Lond) 2001;537:69–81. doi: 10.1111/j.1469-7793.2001.0069k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen AJ. Effects of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- 15.Jensen MS, Yaari Y. Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J Neurophysiol. 1997;77:1224–1233. doi: 10.1152/jn.1997.77.3.1224. [DOI] [PubMed] [Google Scholar]
- 16.Jensen MS, Azouz Y, Yaari Y. Variant firing patterns in rate hippocampal pyramidal cells modulated by extracellular potassium. J Neurophysiol. 1994;71:831–839. doi: 10.1152/jn.1994.71.3.831. [DOI] [PubMed] [Google Scholar]
- 17.Johnson SM, Koshiya N, Smith JC. Isolation of the kernel for respiratory rhythm generation in a novel preparation: the pre-Bötzinger complex “island.”. J Neurophysiol. 2001;85:1772–1776. doi: 10.1152/jn.2001.85.4.1772. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SW, Seutin V. Bicuculline methiodide potentiates NMDA-dependent burst firing in rat dopamine neurons by blocking apamin-sensitive Ca2+-activated K+ currents. Neurosci Lett. 1997;231:13–16. doi: 10.1016/s0304-3940(97)00508-9. [DOI] [PubMed] [Google Scholar]
- 19.Koshiya N, Smith JC. Neuronal pacemaker for breathing visualized in vitro. Nature. 1999;400:360–363. doi: 10.1038/22540. [DOI] [PubMed] [Google Scholar]
- 20.Kramer RH, Zucker RS. Calcium-induced inactivation of calcium current causes inter-burst hyperpolarization of Aplysia bursting neurons. J Physiol (Lond) 1985;362:131–160. doi: 10.1113/jphysiol.1985.sp015667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieske SP, Thoby-Brisson M, Telgkamp P, Ramirez JM. Reconfiguration of the neural network controlling multiple breathing patterns: eupnea, sighs and gasps. Nat Neurosci. 2000;3:600–607. doi: 10.1038/75776. [DOI] [PubMed] [Google Scholar]
- 22.Müller M, Somjen GG. Na+ and K+ concentrations, extra- and intracellular voltages and the effect of TTX in hypoxic rat hippocampal slices. J Neurophysiol. 2000;83:735–745. doi: 10.1152/jn.2000.83.2.735. [DOI] [PubMed] [Google Scholar]
- 23.Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- 24.Newstead CG, Donaldson GC, Sneyd JR. Potassium as a respiratory signal in humans. J Appl Physiol. 1990;69:1799–1803. doi: 10.1152/jappl.1990.69.5.1799. [DOI] [PubMed] [Google Scholar]
- 25.Orkand RK. Extracellular potassium accumulation in the nervous system. FASEB J. 1980;39:1515–1518. [PubMed] [Google Scholar]
- 26.Pan E, Stringer JL. Role of calcium and potassium in generation of cellular bursts in the dentate gyrus. J Neurophysiol. 1997;77:2293–2299. doi: 10.1152/jn.1997.77.5.2293. [DOI] [PubMed] [Google Scholar]
- 27.Parkis MA, Peña F, Ramirez JM. The roles of pacemaker neurons in the generation of different respiratory rhythms. Soc Neurosci Abstr. 2002;28:362.5. [Google Scholar]
- 28.Paterson DJ, Dorrington KL, Bergel DH, Kerr G, Miall RC, Stein JF, Nye PC. Effect of potassium on ventilation in the rhesus monkey. Exp Physiol. 1992;77:217–220. doi: 10.1113/expphysiol.1992.sp003577. [DOI] [PubMed] [Google Scholar]
- 29.Poolos NP, Mauk MD, Kocsis JD. Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol. 1987;58:404–416. doi: 10.1152/jn.1987.58.2.404. [DOI] [PubMed] [Google Scholar]
- 30.Ramirez JM, Schwarzacher SW, Pierrefiche O, Olivera BM, Richter DW. Selective lesioning of the cat pre-Bötzinger complex in vivo eliminates breathing but not gasping. J Physiol (Lond) 1996;507:895–907. doi: 10.1111/j.1469-7793.1998.895bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez JM, Telgkamp P, Elsen FP, Quellmalz UJA, Richter DW. Respiratory rhythm generation in mammals: synaptic and membrane properties. Respir Physiol. 1997;110:71–85. doi: 10.1016/s0034-5687(97)00074-1. [DOI] [PubMed] [Google Scholar]
- 32.Ramirez JM, Quellmalz UJA, Wilken B, Richter DW. The hypoxic response of neurons within the in vitro mammalian respiratory network. J Physiol (Lond) 1998;507:571–582. doi: 10.1111/j.1469-7793.1998.571bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rekling JC, Feldman JL. PreBotzinger complex and pacemaker neurons: hypothesized site and kernel for respiratory rhythm generation. Annu Rev Physiol. 1998;60:385–405. doi: 10.1146/annurev.physiol.60.1.385. [DOI] [PubMed] [Google Scholar]
- 34.Richter DW, Spyer KM. Studying rhythmogenesis of breathing: comparison of in vivo and in vitro models. Trends Neurosci. 2001;24:464–472. doi: 10.1016/s0166-2236(00)01867-1. [DOI] [PubMed] [Google Scholar]
- 35.Rybak IA, St John WM, Paton JF. Models of neuronal bursting behavior: implications for in-vivo versus in-vitro respiratory rhythmogenesis. Adv Exp Med Biol. 2001;499:159–164. doi: 10.1007/978-1-4615-1375-9_25. [DOI] [PubMed] [Google Scholar]
- 36.Sandler VM, Puil E, Schwarz DWF. Intrinsic response properties of bursting neurons in the nucleus principalis trigemini of the gerbil. Neuroscience. 1998;83:891–904. doi: 10.1016/s0306-4522(97)00415-6. [DOI] [PubMed] [Google Scholar]
- 37.Shao YM, Feldman JL. Respiratory rhythm generation and synaptic inhibition of expiratory neurons in pre-Botzinger complex: differential roles of glycinergic and GABAergic neural transmission. J Neurophysiol. 1997;77:1853–1860. doi: 10.1152/jn.1997.77.4.1853. [DOI] [PubMed] [Google Scholar]
- 38.Silva NL, Bunney BS. Intracellular studies of dopamine neurons in vitro: pacemakers modulated by dopamine. Eur J Pharmacol. 1988;149:307–315. doi: 10.1016/0014-2999(88)90661-9. [DOI] [PubMed] [Google Scholar]
- 39.Singer W, Lux HD. Extracellular potassium gradients and visual receptive fields in the cat striate cortex. Brain Res. 1975;96:378–383. doi: 10.1016/0006-8993(75)90751-9. [DOI] [PubMed] [Google Scholar]
- 40.Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith JC, Butera RJ, Koshiya N, Del Negro C, Wilson CG, Johnson SM. Respiratory rhythm generation in neonatal and adult mammals: the hybrid pacemaker-network model. Respir Physiol. 2000;122:131–147. doi: 10.1016/s0034-5687(00)00155-9. [DOI] [PubMed] [Google Scholar]
- 42.Somjen GG. Ion regulation in the brain: implications for pathophysiology. The Neuroscientist. 2002;8:254–267. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- 43.Su CK. Intraspinal amino acid neurotransmitter activities are involved in the generation of rhythmic sympathetic nerve discharge in newborn rat spinal cord. Brain Res. 2001;904:112–125. doi: 10.1016/s0006-8993(01)02495-7. [DOI] [PubMed] [Google Scholar]
- 44.Su H, Alroy G, Kirson ED, Yaari Y. Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons . J Neurosci. 2001;21:4173–4182. doi: 10.1523/JNEUROSCI.21-12-04173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Telgkamp P, Ramirez JM. Differential response of respiratory nuclei to anoxia in rhythmic brainstem slices of mice. J Neurophysiol. 1999;82:2163–2170. doi: 10.1152/jn.1999.82.5.2163. [DOI] [PubMed] [Google Scholar]
- 46.Thoby-Brisson M, Ramirez J-M. Role of inspiratory pacemaker neurons in mediating the hypoxic response of the respiratory network in vitro. J Neurosci. 2000;20:5858–5866. doi: 10.1523/JNEUROSCI.20-15-05858.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thoby-Brisson M, Ramirez J-M. Identification of two types of inspiratory pacemaker neurons in the isolated respiratory neural network of mice. J Neurophysiol. 2001;86:104–112. doi: 10.1152/jn.2001.86.1.104. [DOI] [PubMed] [Google Scholar]
- 48.Traynelis SF, Dingledine R. Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol. 1989;61:927–938. doi: 10.1152/jn.1989.61.5.927. [DOI] [PubMed] [Google Scholar]
- 49.Xiong ZQ, Stringer JL. Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol. 2000;83:1443–1451. doi: 10.1152/jn.2000.83.3.1443. [DOI] [PubMed] [Google Scholar]