

Abstract
In 2003 Rubenstein and Merzenich hypothesized that some forms of Autism (ASD) might be caused by a reduction in signal-to-noise in key neural circuits, which could be the result of changes in excitatory-inhibitory (E-I) balance. Here, we have clarified the concept of E-I balance, and updated the original hypothesis in light of the field’s increasingly sophisticated understanding of neuronal circuits. We discuss how specific developmental mechanisms, which reduce inhibition, affect cortical and hippocampal functions. After describing how mutations of some ASD genes disrupt inhibition in mice, we close by suggesting that E-I balance represents an organizing framework for understanding findings related to pathophysiology and for identifying appropriate treatments.
Introduction
In 2003 Rubenstein and Merzenich [1] hypothesized that some forms of Autism (ASD) might be caused by a reduction in signal-to-noise in key neural circuits, which could be caused by changing the circuit’s excitatory- inhibitory (E-I) balance. Reduced inhibition in the cortex and hippocampus, perhaps due to GABAergic interneuron defects would lead to “noisier” circuits and less efficient information processing. Generation of circuits that were too noisy, or too quiet (from too little excitation or too much inhibition) would be detrimental (Fig. 1). Since then, numerous reviews have presented updates related to this basic hypothesis, often by describing studies of mouse models of autism [2], or of neurobiological mechanisms, including neuronal homeostasis, synaptic autoregulatory feedback, and developmental disconnection, all of which could contribute to altered E-I balance [3–11].
Fig. 1.
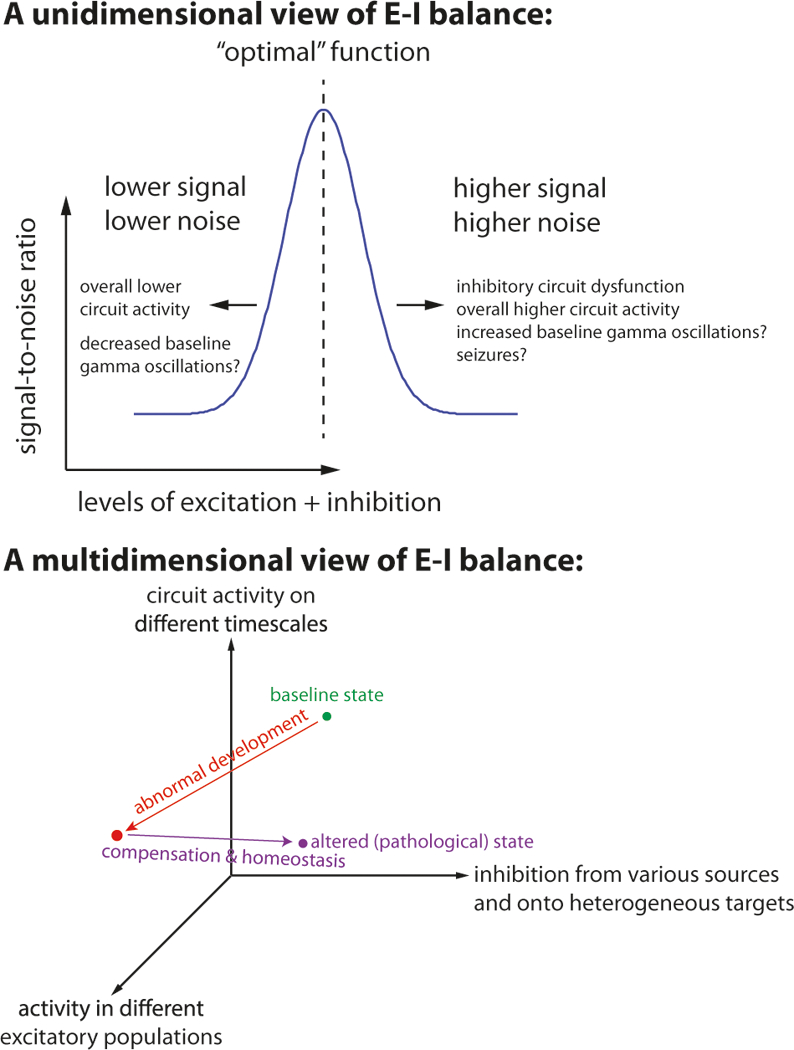
Top: in a unidimensional framework, E-I balance refers to the overall level of circuit activity. Dysfunction in inhibition (or excessive excitation) may cause balance to occur at an overall higher level of circuit activity. This may effectively decrease the signal to noise ratio, by increasing deleterious signals, i.e., “noise”, which render circuits less responsive to “signals”, i.e., behaviorally meaningful inputs. In this framework, “noise” might represent input that is not meaningful, or alternatively, an excessively high conductance state that produces low neuronal gain in response to signals. Conversely, excessive inhibitory feedback (or deficient excitation) may cause the circuit to achieve balance at an overall lower level of circuit activity, which may effectively reduce the signal to noise ratio by reducing the strength of signals. Bottom: In a more complex framework, E-I balance can be conceptualized as a multidimensional space. Different mechanisms, e.g., abnormal development due to altered gene expression or protein translation, compensatory or homeostatic processes, etc. perturb circuits along multiple dimensions in this space. In the future, biomarkers may be identified which indicate the state of the circuit along specific dimensions
Other reviews have discussed related concepts, e.g., that autism may reflect altered activity-dependent gene expression [12] and/or protein translation [13, 14].
Our goal here is not to supplant or duplicate these excellent reviews. Over the past 15 years, experimental studies have provided evidence that many mouse models of autism may involve alterations in the balance of excitation and inhibition, however, the nature of these changes are heterogeneous. Therefore, it is important to clarify the concept of E-I balance, provide a framework for organizing results, and update the original hypothesis in light of our increasingly sophisticated understanding of neuronal circuits. Furthermore, we will review recent work from our laboratories on specific developmental mechanisms which shed light on the effects of reducing inhibition on cortical and hippocampal functions. After describing how mutations of some ASD genes disrupt inhibition in mice, we close by suggesting that E-I balance represents an organizing framework for understanding findings related to pathophysiology and identifying appropriate treatments.
Defining E-I balance
At the single neuron level, the numbers of excitatory and inhibitory synapses on individual cortical pyramidal neurons are highly-regulated by a process that reproducibly generates a relative invariant ratio of E/I synapses across dendritic segments [15].
Similarly, at the level of large scale cortical circuits, the ratio of excitatory to inhibitory cortical neurons is precisely controlled by multiple development process [16]. Thus, there appear to be homeostatic and developmental processes that maintain E-I balance at the level of single cells and cortical regions over long timescales. However, this does not exclude the possibility that the E/I ratio may vary between neurons and/or on more dynamic timescales.
From a global perspective, i.e., viewing excitation and inhibition as singular entities, if the level of excitation exceeds that of inhibition, then activity will increase, either until the ability of the circuit to generate activity is maximized, or until marginal increases in activity begin to recruit more inhibition than excitation, producing a state of “balance”. Conversely, if inhibition exceeds excitation, activity will decrease, either until the circuit is quiescent or until marginal decreases in activity cause larger drops in inhibition than excitation (also leading to overall balance). This is the simplest conceptualization of “excitation-inhibition (E-I) balance”. Of course, this is vastly oversimplified because circuit activity is not a singular (unidimensional) entity and both excitation and inhibition have dynamics on multiple timescales. However, before delving into these complications, we should note some useful aspects of the concept of E-I balance. First, the idea that activity will grow until a balanced state is achieved, does nicely describe what happens in isolated cortical circuits upon the optogenetic induction of activity. Stimulating some population of neurons causes activity to spread to other neurons and other layers until sufficient inhibition is recruited to produce balance [17, 18]. There is evidence that E-I balance in cortical layers 2/3 is important for optimal tuning of these circuits to respond to salient inputs and the generation of responses called neuronal avalanches [19].
Spontaneous UP states in brain slices and in vivo similarly begin in a small number of neurons, then spread to other neurons and layers where they recruit inhibition, producing an overall state of balance [20, 21]. Importantly, although circuits should be balanced, in a global sense, lest they succumb to runaway excitation or fall silent, transient imbalances, i.e., fluctuations in the level of activity, occur on fast timescales [22]. Furthermore, even balanced activity can be distributed across many configurations of neurons, and the particular population that is active at a given moment may be stable or change continuously over time. In particular, differences in local circuit connectivity, e.g., differential innervation of pyramidal neurons with different projection targets by GABAergic interneurons [23, 24], can thus lead to differences in levels of inhibition in different pathways, helping to shape the flow of activity through microcircuits. Thus, “E-I balance” usually refers a stable global level of activity within a particular circuit, even though individual groups of neurons may exhibit transient imbalances, and these groups of neurons can be dynamic over time.
In other words, even when a circuit is in E-I balance, the set of active neurons will likely evolve over time, giving rise to many possible configurations that all represent different forms of E-I balance. Changes in external input or short term synaptic plasticity may induce transitions between these configurations. The overall level of excitation and inhibition, as well as the detailed dynamics of excitatory and inhibitory synaptic conductances, will determine how much circuit activity changes in response to incoming input [25], and thus shape the signal-to-noise ratio as originally suggested by Rubenstein and Merzenich [1]. Fig. 1 illustrates a simple instantiation of this concept, although it goes without saying that changes in signal-to-noise ratio will depend on more than just the overall level of excitation and inhibition.
In this context, what does it mean for the E-I balance to be disturbed? This refers to a change in some form of excitation or inhibition such that the state or set of states for which E-I balance is achieved, is altered. In the simplified framework laid out above, this is often thought of as a change in the gain of either excitation or inhibition, causing a change in the overall level of activity at which balance is achieved. However, it is important to remember that excitation and inhibition are not unidimensional entities. Both excitation and inhibition originate from multiple sources and impinge onto different targets. For example, many studies have shown that various subtypes of inhibitory interneurons (e.g., PV vs. SOM expressing interneurons) differentially innervate different subtypes of pyramidal neurons (e.g., deep layer pyramidal neurons projecting callosally vs. projecting to subcortical targets) [23, 24, 26]. On the other hand, VIP-expressing inter-neurons innervate and inhibit other interneurons, such that activity in VIP+ interneurons can increase cortical excitation [27–29].
Thus, while the term “E-I balance” (or E/I ratio) suggests that excitation and inhibition are singular entities, in fact, both are multidimensional, and changes in E-I balance may comprise changes in the relative activity of different sub-types of excitatory or inhibitory neurons, rather than changes in the overall level of excitation or inhibition (Fig. 1, bottom). In summary, while we initially introduced a relatively simple concept of E-I balance, and provided evidence that this balance is maintained at both the single cell and global circuit levels, we then pointed out that E-I imbalance can occur on faster timescales, and that E-I balance is in fact highly multidimensional and complex. Having noted these complexities, let us now look at mechanisms which alter E-I balance, in order to show how the E-I balance concept can nonetheless aid in understanding how these mechanisms contribute to the pathophysiology of autism and related neuropsychiatric disorders.
Developmental mechanisms that can impact E-I balance in the cortex and hippocampus
The literature is replete with lessons about the developmental and genetic mechanisms that can lead to an increased E/I ratio and subsequent compensation in many regions of the neural axis. Here, we will focus the development of cortical and hippocampal inhibitory interneurons and the impact of defects in these processes on neural function [30–33].
We restrict our discussion to the effects of mutations of the Dlx transcription factors on the generation and function of interneurons. Although allelic variation in the Dlx genes is not an established risk factor for neuropsychiatric dis-orders, the functions of these homeodomain transcription factors are illustrative of mechanisms spanning embryonic to adult stages that impacts E/I balance. There are six Dlx genes, all but Dlx3 and Dlx4 are expressed in forebrain GABAergic neurons [34]. Dlx1&2 and Dlx5&6 are bigene pairs; their expression begins in embryonic progenitors and neurons in the following order: Dlx2, Dlx1, Dlx5, and then Dlx6. There is functional redundancy between the Dlx genes, perhaps contributing to why mutations in Dlx genes are not observed in human brain disorders. However, there is an increasing evidence that targets of Dlx regulation that can cause human epilepsy and cognitive disorders (e.g., Arx, Gad1, Grin2b, Maf, and Zfhx1b) [35–37]. DLX2 expression and Dlx1&2 null mice provided the first evidence that the embryonic basal ganglia generate inter-neurons for telencephalon, including for the hippocampus (HIN), neocortex (CIN), and striatum (SIN) [38]. Thus, decreased inhibition can be the result of reduced subpallial interneuron production and their subsequent migration to the pallium (cortex and hippocampus).
In the absence of Dlx1&2, Dlx5&6 fail to induce, leading to many severe forebrain and craniofacial phenotypes and neonatal lethality [39]. However, in Dlx1&2 conditional knockouts (CKOs), one day of Dlx1&2 expression is sufficient to induce and maintain Dlx5&6 [37], and these mutants produce CINs that migrate and integrate into the cortex. CINs generated by Dlx1&2 CKOs using Dlxi12b-Cre (activity begins ~embryonic day (E) 11), or from Calretinin-Cre (activity begins -postnatal day (P) 1), have reduced mEPSCs. These mutant CINs receive reduced excitatory synapses and thus are less capable of generating inhibition.
Furthermore, Dlx1&2 drive expression of the genes encoding the enzymes that generate and package GABA (Gad1&2 and Vgat) [37, 40–42]. Probably as a result, Dlx1&2Dlxi12b-Cre CKOs also generate reduced mIPSC amplitude. In addition, Dlx1&2DM12b-Cre CKOs has increased CIN apoptosis in the first postnatal week, resulting in reduced numbers of PV, SST, and VIP CINs and HINs (~20–30%). Thus, Dlx1&2Dki12b-Cre CKOs reduce CINs numbers and function in at least three ways: (1) reducing mEPSCs and synapses onto CIN; (2) reducing pyramidal neuron mIPSC amplitude probably by reducing GABA release from CINs; and (3) reducing CIN numbers because of CIN death.
The first evidence for Dlx postnatal IN functions came from analysis of Dlx1 constitutive KOs [43]. These mutants show CIN and HIN death beginning at ~P30 with resultant decreases in mIPSC frequencies. In these Dlx1 single constitutive, and subsequently in Dlx1 CKOs, PV interneurons (MGE-derived) do not die, probably because Dlx1 expression is not detectable postnatally in PV interneurons [37, 43]. On the other hand, SST (MGE-derived) and VIP (CGE-derived) numbers decline by ~20–30%, thus reducing only dendrite-innervating interneurons.
It is important to note that many genes are important for the development, maturation, and function of specific classes of CINs. A brief summary of some recent findings include the following examples. The Prox1 transcription factor is required for the maturation of VIP, and other CGE-derived CINs [44]. CNTNAP4, a member of the neurexin superfamily of transmembrane molecules, promotes the function of cortical PV basket cells [45]. Finally, CCK and VGlut3 expressing basket cells lacking ErbB4, a tyrosine kinase receptor, have reduced inhibition onto pyramidal neurons [46].
Dlx1 mutants develop epilepsy and increased gamma oscillations around the same time when their interneurons die [43, 47]. Dlx1 mutant HINs have reduced excitatory postsynaptic current (EPSC) amplitude, which may contribute to reduced interneuron function [48]. Analysis of neocortical function in Dlx1 KOs showed that the primary visual cortex (V1) had reduced orientation selectivity [49]. In the primary auditory cortex (A1) Dlx1DlxI12b-Cre CKOs showed a rise in the baseline firing rates and reduction in tone-evoked responses [50]. Furthermore, A1 neurons had reduced receptive field size and shortened cortical responses, a phenotype which is opposite to that observed with acute reductions in inhibition. Seybold et al. [50]. proposed that the latter phenotypes arose secondary to a compensatory reduction in excitatory responses. A similar reduction is pyramidal neuron activity was also observed in V1 [49]. Thus, while some of the V1 and A1 phenotypes could arise directly from the loss of CIN-mediated inhibition, additional phenotypes may arise secondary to a compensatory reduction in excitation. Therefore, phenotypes are likely to change over time, as primary defects are combined with compensatory responses.
Compensatory changes in the Dlx1 KOs were more deeply investigated in the hippocampus (CA1) [47]. As in the neocortex, loss of HIN was associated with reduced synaptic inhibition, which then led to a compensatory reduction in miniature EPSCs on the CA1 pyramidal neurons. However, when a long-term potentiation (LTP) protocol was applied to hippocampal slices, the Dlx1 KO showed enhanced potentiation of activated synapses 1 h after induction compared with wild type. This finding was evident only after the death of interneurons beginning ~P30. These observations suggest that pyramidal neurons, in regions of the cortex/hippocampus that have reduced interneuron function, may compensate by reducing the numbers of excitatory synapses with detectable levels of AMPA containing plasma membrane glutamate receptors. At the same, there may be a greater potential for plasticity. This change in LTP may be a direct consequence of decreased inhibition lowering the LTP threshold, or a homeostatic increase in the proportion of silent synapses.
Thus, these experiments suggest the hypothesis that regions of the cortex/hippocampus with reduced interneuron function and excitatory circuit compensation could be susceptible to hyperexcitability if they are exposed to strong stimuli, which could lead to a pathological switch in circuit function and behavior [47]. Perhaps this could generate phenotypes such as an epileptic focus (as occurs in Dlx1 mutants), an emotional/anxiety hypersensitivity, or cognitive/perceptual defects (e.g., hallucinations).
Interneuron transplantation experiments provided evidence that the Dlx1 mutant phenotypes arose secondary to the postnatal dysfunction and/or death of HINs [47]. Neonatal transplantation of wild type MGE-derived inter-neurons into the Dlx1 mutant hippocampus prevented or reversed the homeostatic changes to excitation and excitability in the CA1 pyramidal cells: miniature excitatory postsynaptic currents, input resistance, and LTP. Furthermore, interneuron transplantation rescued circuit-levels defects, as gamma oscillations become normalized. Thus, phenotypes caused by a HIN deficit, due to a mutation, can be rescued by wild type HINs.
Less is known about Dlx5&6 developmental functions. Dlx5, Dlx6, and Dlx5/6 constitutive mutants are developmental lethal [51]. Dlx5/6 mutations have exencephaly complicating analysis of forebrain development, postnatal analysis of development and neuronal function. These problems have been circumvented using MGE transplantation of the Dlx5 and Dlx5/6 mutants; the experiments provided evidence for a selective relative reduction of PV+ CIN numbers and associated dendritic morphological defects [51]. These results contrast with the Dlx1 mutants which has a selective sparing of PV CINs [43]. Below we will discuss Dlx5/6 heterozygotes and Dlx5 CKO states, where CIN functional defects have been identified.
Altered E-I balance in mouse models of autism (ASD)
Having described these developmental genetic mechanisms that can impact E-I balance, we will not summarize some recent studies that have examined whether mouse models of autism can be understood as alterations of E-I balance. This section is not intended to be comprehensive or assess the validity of these models of autism. Rather we will focus on some examples that illustrate how the E-I framework can be used. We begin with a highly influential study that used novel optogenetic tools to bidirectionally modulate E-I balance by nonspecifically exciting neurons and/or specifically exciting PV interneurons in the medial prefrontal cortex (mPFC) of wild-type (PV-Cre) mice [18]. Non-specifically exciting mPFC neurons was sufficient to disrupt normal social behavior. By contrast, specifically exciting PV interneurons in mPFC did not disrupt normal social behavior, and actually ameliorated social deficits caused by nonspecific excitation of the mPFC. The authors interpreted these results as broadly consistent with the original E-I imbalance hypothesis. Interestingly, the authors found that nonspecific excitation of mPFC neurons increased power across the gamma-frequency range (~50–100Hz). This change in gamma power parallels results in genetic models and will be discussed further below. More recently the same laboratory showed that optogenetic stimulation of PV interneurons is sufficient to rescue social deficits in CNTNAP2 knockout (KO) mice, a genetic model of autism and other neurodevelopmental disorders [52]. The authors proposed that CNTNAP2 mutant mice have deficiencies in PV interneuron recruitment during social interaction, such that optogenetically stimulating these interneurons restores the E-I balance to its wild-type state. These two studies suggest that a framework, based on a unidimensional conception of E-I balance, is sufficient to predict how pathological and/or therapeutic manipulations will impact social deficits in at least some cases—increasing the ratio of E/I (either by increasing excitation or by impairing inhibition) within mPFC tend to cause social deficits, whereas increasing inhibition decreases the E/I ratio and can ameliorate some social deficits. That being said, it is notable that in these two studies, restoration of E/I balance was achieved by targeting a specific class of GABAergic interneurons (PV interneurons)—this raises the question of whether any manipulations that target circuit inhibition would be similarly effective vs. whether specifically enhancing perisomatic inhibition is required.
Several other studies are broadly consistent with this view. Another group showed evidence for inhibitory dys-function in both a model of Dravet syndrome (Scn1a+/− mice), which includes epilepsy and autism, and the BTBR inbred mouse line, which exhibits social deficits and repetitive behaviors that model behavioral features of autism [53, 54]. Specifically, in both cases, they found a decrease in the spontaneous IPSC frequency. Furthermore, in both cases, nonspecifically enhancing inhibition via systemic injection of a benzodiazepine at sub-anxiolytic doses was sufficient to improve social behavior. Similar to patient populations, multisensory integration is deficient in multiple mouse models of autism, and other work in the BTBR inbred mouse strain suggests that this may reflect weakened GABAergic inhibition during an early sensitive period that can be similarly rescued by therapeutic benzodiazepine administration [55]. Again, this group described reduced spontaneous IPSC frequency in BTBR mice. Another group showed similar findings in mice with haploinsufficiency of Arid1b, a gene that has been strongly linked to autism by de novo mutations [56]. Arid1b+/− mice have decreased numbers of cortical GABAergic interneurons, a reduction in the frequency of miniature IPSCs, and social deficits that are also rescued by benzodiazepine treatment. More recently, a group showed that transplanting stem cells which differentiate into cortical interneurons, into ventral hippo-campus rescues social deficits in prenatal-methylazoxymethanol-treated rats [57]. Other work has shown that the TSC1-mTOR pathway, which has been implicated in autism, normally represses inhibitory synapses onto excitatory neurons, such that deficiency of Tsc1 leads to decreased inhibition and network hyperexcitability [58]. Mice lacking CHD2, a chromatin remodeling and autism risk gene, had fewer cortical and hippocampal interneurons, and a reduction in miniature IPSC frequency; the mutants’ cognitive phenotype was partially rescued by interneuron transplantation [59]. Finally, a large body of work has shown that Rett syndrome, which involves some autism-like features, includes deficits in inhibitory interneuron development and function [60], and that many abnormal behaviors in mice are driven by the loss of MeCP2 specifically in interneurons [61].
These studies do not claim that excitation or inhibition are unidimensional entities. But they do suggest that for the purpose of correcting pathophysiological mechanisms that cause social deficits (including in mouse models of autism), focusing on GABAergic inhibition generally, or thinking in terms of a unidimensional ratio of E/I may be sufficient. By contrast, other studies suggest that mouse models of autism or mutations linked to autism may be associated with derangements of inhibition that cannot be understood simply in terms of overall E-I balance, and that correcting social behavior may require an explicit recognition of excitation and inhibition as multidimensional entities. In particular, two recent studies from our labs have shown that mutations in autism genes may differentially affect inhibition from different sources, e.g., from different subtypes of cortical interneurons. One study found that deficiency of Pten in MGE-derived neurons leads to a loss of cortical interneurons, and a preferential loss of SOM interneurons, such that the ratio of PV to SOM interneurons increases [62]. Furthermore, in this context, mutant PV interneurons send ectopic projections to superficial cortical layers, which are normally the targets of SOM interneurons, and levels of spontaneous inhibition in superficial pyramidal neurons were actually increased.
Interestingly, these mice—which had an increased PV: SOM ratio, ectopic PV+ projections to superficial layers, and increased spontaneous inhibition in superficial pyramidal neurons—also exhibited a broadband reduction in spontaneous EEG activity in the gamma band. This is notable, as the previous study by Yizhar et al. [18], showed that nonspecific excitation of the mPFC produced a broadband increase in spontaneous gamma power. Taken together, these findings suggest that overall increased activity in cortical circuits may be detectable via broadband increases in spontaneous gamma power. Conversely, excessive PV interneuron output may be associated with decreased cortical activity, resulting in broadband decreases in spontaneous gamma power. This may seem paradoxical at first given the large body of work suggesting that PV interneurons generate gamma oscillations evoked by sensory stimulation or behavioral tasks. But there is an emerging view that PV interneuron-generated gamma synchrony may be fundamentally distinct from the broadband “gamma oscillations” that are observed at baseline and during spontaneous behavior. Specifically, “gamma oscillations” that are measured via a broadband increase in power across a wide frequency range may simply reflect increases in overall levels of cortical activity that are not composed of rhythmic neural firing and are not well-synchronized across local circuits. By contrast, increases in gamma power that occur in more narrow frequency bands, e.g., in response to sensory stimulation or during tasks, may be indicative of the sort of rhythmic neural activity that is synchronized across cells and depends on feedback inhibition from PV interneurons.
Indeed, in previous work, we showed that Dlx5/6+/− mice, which have impaired PV interneuron function, exhibit an broadband increase in spontaneous gamma power (observed at baseline) but a decrease in task-evoked gamma oscillations [63]. Patient populations, specifically individuals with schizophrenia, seem to show a similar pattern: an increase in spontaneous, broadband gamma power measured under baseline conditions, and a decrease in gamma power evoked by tasks or stimuli [64]. All of these observations are consistent with the emerging view, described above, that spontaneous, broadband gamma power, measured under task-free conditions, may simply reflect the overall level of circuit activity, rather than activity which exhibits rhythmicity and/or synchrony at a specific frequency. Reductions in PV interneuron function may lead to increase in overall levels of circuit activity, causing a broadband increase in power across a wide range of frequencies including the gamma band. This is in contrast to task or stimulus evoked-gamma oscillations, which seem to depend on the integrity of PV interneuron output and exhibit peaks around specific frequencies, as well as synchronization between brain regions.
The case of the Dlx5/6+/− mice extends the earlier discussion of how genes involved in interneuron development can influence E-I balance. Constitutive knockout of Dlx5/6 leads to neonatal death. However, mice with either the heterozygous loss of Dlx5/6, or the conditional knockout of Dlx5/6 using DlxI12b-Cre, live into adulthood and are grossly normal. We carried out an extensive characterization of interneuron physiology, circuit physiology (measured via intracranial EEG), and behavior in these mice [63]. We found that in Dlx5/6 heterozygous mice, PV interneuron intrinsic properties, gamma oscillations (both at baseline and evoked by tasks), and cognitive flexibility are all abnormal, but these abnormalities only appear ~9–11 weeks of age. This suggests that Dlx5/6 play yet-to-be-defined roles in the post-pubertal maturation of PV interneurons, at least in prefrontal regions, in addition to their roles in early postnatal developmental and survival.
Another recent study from our laboratories showed that deficiency of the previously mentioned autism-associated gene CNTNAP2 in MGE-derived neurons causes a selective disruption in the physiology of PV, but not SOM, interneurons [65]. This raises the possibility that perhaps the aforementioned study from the Deisseroth laboratory, which ameliorated social deficits in CNTNAP2 KO mice by opto-genetically stimulating PV interneurons, was successful because it specifically enhanced PV interneuron function, in mice with preferential deficits in PV interneurons. Finally, we recently found that in mice exposed to valproic acid in utero, selective inhibition of dopamine D2 receptor-expressing (D2R+) neurons in mPFC can ameliorate social deficits, whereas nonspecific inhibition of the entire mPFC fails to do so (and actually makes social deficits worse) [66]. Thus in this last example, nonspecifically targeting excitation or inhibition may be counterproductive, and it is necessary to target excitatory activity within a specific neuronal class (D2R+ mPFC neurons) to achieve a therapeutic effect.
Notably, the original hypothesis relating E-I imbalance to autism was motivated in part by the high comorbidity between autism and seizure disorders. But even in the context of seizures, the idea that cortical inhibition is not a monolithic entity is now apparent. For example, we recently found that various classes of cortical GABAergic neurons are differentially recruited by and make distinct contributions to seizures [67]. In fact, the role of a single class of interneurons could evolve over the course of a seizure, such that interneurons which appear to suppress seizure initiation can also prolong the duration of seizures. This suggests that in the context of seizures, therapeutic efficacy may be maximized by targeting specific forms of inhibition, rather than inhibition generally.
Summary and outlook
The concept of E/I balance is of course, simplified, because it suggests that excitation and inhibition are unidimensional entities and further simplifies the picture by considering their ratio. But we would argue that despite this high degree of simplification, several studies do suggest that in many cases, this is a useful first step to understanding pathophysiological mechanisms related to abnormal social behavior in mouse models of autism. Does this mean that the converse is true, that simply showing an alteration in some form of excitation or inhibition present is sufficient to explain pathophysiology, e.g., in a mouse with abnormal behavior? Of course not. Even though there may be many conditions involving deficient inhibition, for which behavior can be normalized by non-specifically increasing inhibition, in other cases, excessive or altered inhibition might be at fault, or it may be necessary to specifically enhance inhibition from particular sources (PV interneurons) or onto defined targets (e.g., D2R+ mPFC neurons). These scenarios represent fundamentally different kinds of circuit derangements that will likely require different types of therapeutic interventions. We argue that it is critical to understand what sort of circuit derangement is present, and that E-I balance can be a useful framework through which this issue could be explored.
Over the past 15 years, the E-I imbalance hypothesis has, in our mind, served an extremely important purpose, by stimulating many investigations into the consequences of abnormal development of inhibitory circuits, the state of excitation and inhibition in mouse models of autism, and the behavioral consequences of specifically perturbing excitatory vs. inhibitory systems. However, we would propose that the next phase of research should focus on (1) classifying the many possible “E-I imbalances” into some tractable categories of circuit derangements, (2) identifying biomarkers that might indicate which type of circuit derangement is present in a particular individual, and (3) testing whether determining what type of E-I imbalance/ circuit derangement is present in a given individual is sufficient to predict what sorts of behavioral abnormalities might be present, and/or which therapies might be effective. As outlined above, baseline gamma power may represent an example of one such biomarker, which might be useful for separating cases of increased vs. decreased PV interneuron function. Indeed, numerous studies have examined both resting state and evoked gamma oscillations in subjects with autism using EEG and MEG, although sample sizes for these studies tend to be small, use heterogeneous methods for quantifying gamma oscillations, and have some inconsistent findings [68–73]. In spite of this heterogeneity, quantifying specific aspects of rhythmic activity is a promising future direction because there is a great deal of evidence tying specific cellular deficits to deficit in specific aspects of oscillations, e.g., power or synchrony at a particular frequency and/or cross-frequency coupling [74–76].
Another potentially useful direction will be for computational modeling studies to explore how different sorts of derangements in inhibition can produce distinguishable changes in EEG biomarkers, along the lines of what we outlined above for baseline broadband gamma power vs. task-evoked increases in gamma power. No doubt, bio-markers like these are currently of limited translational utility on their own. However, it may be possible to identify a set of biomarkers which together could define the major circuit derangements present in a particular individual: nonspecific increases in activity, specific increases or decreases in output from a particular interneuron class, etc. The goal of these recommendations is to eventually transform the concept of E-I imbalance into a multidimensional framework for classifying circuit pathologies (Fig. 1, bottom), rather than a singular explanation for an entire class of what are actually heterogeneous neurodevelopmental dis-orders. A key test of this framework will then be to determine whether biomarkers that correlate with the presence of specific alterations in E-I balance, will then provide useful information about which therapeutic interventions will (or will not) normalize behavior in those cases.
We are intrigued by data in humans with autism that provides evidence for an alteration in the E/I balance [77, 78]. One study used an experimental paradigm called the binocular rivalry test of visual perceptual suppression [77]. Here, the left and right eyes are shown different objects, and the individual reports what they perceive. During the test, perception oscillates between awareness and suppression. Normal individuals more efficiently suppress one image than autistic individuals. MRI spectroscopy was used to assess the concentrations of GABA and glutamate in the visual cortex; GABA and glutamate concentrations were both linked with binocular rivalry dynamics in control individuals, whereas the link to GABA was not detected in autistic individuals, providing evidence for an increase in E/I signaling in the autistic visual cortex.
Another study, using magnetic resonance imaging, found evidence in the striatum of ASD individuals that glutamate concentration was reduced but GABA levels were normal. The altered glutamate/GABA ratio correlated with the severity of social symptoms [78]. This study raises an important point that one should not solely focus on cortical dysfunction in ASD. Going forward it will be important to learn whether modifying E/I balance in autistic individuals can improve some of their symptoms. In that regard Ajram et al. [79], found that riluzole, a sodium channel blocker, increased the prefrontal cortex inhibitory index in ASD but decreased it in controls (measured with magnetic resonance spectroscopy). Riluzole normalized prefrontal cortex functional connectivity in the ASD group. This encouraging study points to the possibility of pharmacological approaches for ASD therapies.
Acknowledgements
We thank Mackenzie Howard for discussions. VSS is supported by NIMH (R01 MH100292 and R01 MH106507).
JLRR is supported by Nina Ireland, the Simons Foundation (SFARI #309279), NINDS R01 NS34661, NIMH R01 MH081880, and NIMH R37 MH049428.
Footnotes
Compliance with ethical standards
Conflict of interest JLRR is cofounder, stockholder, and currently on the scientific board of Neurona, a company studying the potential therapeutic use of interneuron transplantation. VSS receives research funding from Neurona.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee E, Lee J, Kim E. Excitation/Inhibition imbalance in animal models of autism spectrum disorders. Biol Psychiatry. 2017;81: 838–47. [DOI] [PubMed] [Google Scholar]
- 3.Bourgeron T From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–63. [DOI] [PubMed] [Google Scholar]
- 4.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17: 103–11. [DOI] [PubMed] [Google Scholar]
- 5.Mullins C, Fishell G, Tsien RW. Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron. 2016;89:1131–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. 2015;87: 684–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455: 912–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubenstein JLR. Three hypotheses for developmental defects that may underlie some forms of autism. Curr Opin Neurol. 2010;23:118–23. [DOI] [PubMed] [Google Scholar]
- 9.Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F, et al. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26: 363–72. [DOI] [PubMed] [Google Scholar]
- 10.Wondolowski J, Dickman D. Emerging links between homeostatic synaptic plasticity and neurological disease. Front Cell Neurosci. 2016;7:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canitano R, Pallagrosi M. Autism spectrum disorders and schi-zophrenia spectrum disorders: excitation/inhibition imbalance and developmental trajectories. Front Psychiatry. 2017;8:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci. 2013;16: 1530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelleher RJ 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–6. [DOI] [PubMed] [Google Scholar]
- 15.Iascone DM, Li Y, Sümbül U, Doron M, Chen H, Andreu V, et al. Principles of excitatory and inhibitory synaptic organization constrain dendritic spiking in pyramidal neurons. BioRxiv. 2018. 10.1101/395384. [DOI] [Google Scholar]
- 16.Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG. Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron. 2013;80:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adesnik H, Scanziani M. Lateral competition for cortical space by layer-specific horizontal circuits. Nature. 2010;464:1155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477: 171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shew WL, Yang H, Yu S, Roy R, Plenz D. Information capacity and transmission are maximized in balanced cortical networks with neuronal avalanches. J Neurosci. 2011;31:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26: 4535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–34. [DOI] [PubMed] [Google Scholar]
- 22.Litwin-Kumar A, Doiron B. Slow dynamics and high variability in balanced cortical networks with clustered connections. Nat Neurosci. 2012;15:1498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee AT, Gee SM, Vogt D, Patel T, Rubenstein JL, Sohal VS. Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron. 2014a;81:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SH, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, et al. Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron. 2014b;82:1129–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasenstaub A, Sachdev RN, McCormick DA. State changes rapidly modulate cortical neuronal responsiveness. J Neurosci. 2007;27:9607–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J, Tucciarone J, Padilla-Coreano N, He M, Gordon JA, Huang ZJ. Selective inhibitory control of pyramidal neuron ensembles and cortical subnetworks by chandelier cells. Nat Neurosci. 2017;20:1377–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci. 2013;16:1662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci. 2013;16: 1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu J, Anderson SA. Development of cortical interneurons. Neuropsychopharmacology. 2015;40:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wamsley B, Fishell G. Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat Rev Neurosci. 2017; 18:299–309. [DOI] [PubMed] [Google Scholar]
- 32.Lim L, Mi D, Llorca A, Marin O. Development and functional diversification of cortical interneurons. Neuron. 2018;100: 294–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu JS, Vogt D, Sandberg M, Rubenstein JL. Cortical interneuron development: a tale of time and space. Development. 2017;144: 3867–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panganiban G, Rubenstein JLR. Developmental functions of the Distal-less (Dlx) homeobox genes. Development. 2002;129: 4371–86. [DOI] [PubMed] [Google Scholar]
- 35.Colasante G, Patrick Collombat P, Raimondi V, Bonanomi D, Ferrai C, Maira M, et al. Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic inter-neurons. J Neurosci. 2008;28:10674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKinsey GL, Lindtner S, Trzcinski B, Visel A, Pennacchio L, Huylebroeck D, et al. Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron. 2013;77:83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pla R, Stanco A, Howard MA, Vogt D, Rubin A, Mortimer N, et al. Dlx1/2 promote interneuron GABA synthesis, synaptogenesis, and dendritogenesis through Grin2b. Cereb Cortex. 2017;28:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson SA, Eisenstat D, Shi L, Rubenstein JLR. Interneuron migration from basal forebrain:dependence on Dlx genes. Science. 1997a;278:474–6. [DOI] [PubMed] [Google Scholar]
- 39.Anderson S, Qiu M, Bulfone A, Eisenstat D, Meneses JJ, Pedersen RA, et al. Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late-born striatal cells. Neuron. 1997b;19:27–37. [DOI] [PubMed] [Google Scholar]
- 40.Long JE, Garel S, Alvarez-Dolado M, Yoshikawa K, Osumi N, Alvarez-Buylla A, et al. Dlx-dependent and independent regulation of olfactory bulb interneuron differentiation. J Neurosci. 2007;27:3230–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Long JE, Swan C, Liang WS, Cobos I, Potter GB, Rubenstein JLR. Dlx1&2 and Mash1 transcription factors control striatal patterning and differentiation through parallel and overlapping pathways. J Comp Neurol. 2009;512:556–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le TN, Zhou QP, Cobos I, Zhang S, Zagozewski J, Japoni S, et al. GABAergic interneuron differentiation in the basal forebrain is mediated through direct regulation of glutamic acid decarboxylase isoforms by Dlx homeobox transcription factors. J Neurosci. 2017;37:8816–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cobos I, Calcagnotto ME, Vilaythong AJ, Noebels J, Baraban SC, Rubenstein JLR. Mice lacking the Dlx1 transcription factor exhibit subtype-specific loss of interneurons, reduced synaptic inhibition and epilepsy. Nature Neurosci. 2005;8:1059–68. [DOI] [PubMed] [Google Scholar]
- 44.Miyoshi G, Young A, Petros T, Karayannis T, McKenzie Chang M, Lavado A, et al. Prox1 regulates the subtype-specific development of caudal ganglionic eminence-derived GABAergic cortical interneurons. J Neurosci. 2015;35:12869–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karayannis T, Au E, Patel JC, Kruglikov I, Markx S, Delorme R, et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature. 2014;511:236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Pino I, Brotons-Mas JR, Marques-Smith A, Marighetto A, Frick A, Marin O, et al. Abnormal wiring of CCK+ basket cells disrupts spatial information coding. Nat Neurosci. 2017;20: 784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Howard MA, Rubenstein JL, Baraban SC. Bidirectional homeostatic plasticity induced by interneuron cell death and transplantation in vivo. Proc Natl Acad Sci USA. 2014;111:492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones D, Howard M, Stanco A, Rubenstein JLR, Baraban S. Deletion of Dlx1 results in reduced glutamatergic input to hippocampal interneurons. J Neurophys. 2011;105:1984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mao R, Schummers J, Knoblich U, Lacey CJ, Van Wart A, Cobos I, et al. Influence of a subtype of inhibitory interneuron on stimulus-specific responses in visual cortex. Cereb Cortex. 2012;22:493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seybold BA, Stanco A, Cho K, Potter G, Kim C, Sohal V, et al. Chronic reduction in inhibition reduces receptive field size in mouse auditory cortex. PNAS. 2012;109:13829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Dye C, Sohal V, Long J, Estrada R, Roztocil T, et al. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J Neurosci. 2010;30: 5334–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, et al. Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci Transl Med. 2017;9:eaah6733. pii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489: 385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han S, Tai C, Jones CJ, Scheuer T, Catterall WA. Enhancement of inhibitory neurotransmission by GABAA receptors havingalpha2,3-subunits ameliorates behavioral deficits in a mouse model of autism. Neuron. 2014;81:1282–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gogolla N, Takesian AE, Feng G, Fagiolini M, Hensch TK. Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron. 2014;83:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jung EM, Moffat JJ, Liu J, Dravid SM, Gurumurthy CB, Kim WY. Aridlb haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat Neurosci. 2017;20: 1694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donegan JJ, Tyson JA, Branch SY, Beckstead MJ, Anderson SA, Lodge DJ. Stem cell-derived interneuron transplants as a treatment for schizophrenia: preclinical validation in a rodent model. Mol Psychiatry. 2017;22:1492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron. 2013;78:510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim YJ, Khoshkhoo S, Frankowski JC, Zhu B, Abbasi S, Lee S, et al. Chd2 is necessary for neural circuit development and long-term memory. Neuron. 2018;100:1180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mierau SB, Patrizi A, Hensch TK, Fagiolini M. Cell-specific regulation of N-Methyl-D-aspartate receptor maturation by Mecp2 in cortical circuits. Biol Psychiatry. 2016;79:746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereo-typies and Rett syndrome phenotypes. Nature. 2010;468: 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vogt D, Cho KK, Lee AT, Sohal VS, Rubenstein JL. The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep. 2015;11:944–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, Sohal VS. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/−) mice. Neuron. 2015;85: 1332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mathalon DH, Sohal VS. Neural oscillations and synchrony in brain dysfunction and neuropsychiatric disorders: it’s about time. JAMA Psychiatry. 2015;72:840–4. [DOI] [PubMed] [Google Scholar]
- 65.Vogt D, Cho KKA, Shelton SM, Paul A, Huang ZJ, Sohal VS, et al. Mouse Cntnap2 and human CNTNAP2 ASD alleles cell autonomously regulate PV+cortical interneurons. Cereb Cortex. 2017;28:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brumback AC, Ellwood IT, Kjaerby C, Iafrati J, Robinson S, Lee AT, et al. Identifying specific prefrontal neurons that contribute to autism-associated abnormalities in physiology and social behavior. Mol Psychiatry. 2018;23:2078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khoshkhoo S, Vogt D, Sohal VS. Dynamic, cell-type-specific roles for GABAergic interneurons in a mouse model of opto-genetically inducible seizures. Neuron. 2017;93:291–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown C, Gruber T, Boucher J, Rippon G, Brock J. Gamma abnormalities during perception of illusory figures in autism. Cortex. 2005;41:364–76. [DOI] [PubMed] [Google Scholar]
- 69.Orekhova EV, Stroganova TA, Nygren G, Tsetlin MM, Posikera IN, Gillberg C, et al. Excess of high frequency electro-encephalogram oscillations in boys with autism. Biol Psychiatry. 2007;62:1022–9. [DOI] [PubMed] [Google Scholar]
- 70.Rojas DC, Maharajh K, Teale P, Rogers SJ. Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism. BMC Psychiatry. 2008;8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun L, Grutzner C, Bolte S, Wibral M, Tozman T, Schlitt S, et al. Impaired gamma-band activity during perceptual organization in adults with autism spectrum disorders: evidence for dysfunctional network activity in frontal-posterior cortices. J Neurosci. 2012;32:9563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Port RG, Gaetz W, Bloy L, Wang D-J, Blaskey L, Kuschner ES, et al. Exploring the relationship between cortical GABA concentrations, auditory gamma-band responses and development in ASD: evidence for an altered maturational trajectory in ASD. Autism Res. 2017;10:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lajiness-O’Neill R, Brennan JR, Moran JE, Richard AE, Flores AM, Swick C, et al. Patterns of altered neural synchrony in the default mode network in autism spectrum disorder revealed with magnetoencephalography (MEG): Relationship to clinical symptomatology. Autism Res. 2018;11:434–49. [DOI] [PubMed] [Google Scholar]
- 74.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Veit J, Hakim R, Jadi MP, Sejnowski TJ, Adesnik H. Cortical gamma band synchronization through somatostatin interneurons. Nat Neurosci. 2017;20:951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Port RG, Berman J, Liu S, Featherstone RE, Roberts TPL, Siegel SJ. Parvalbumin cell ablation of NMDA-R1 leads to altered phase, but not amplitude, of gamma-band cross frequency coupling. Brain Connect. 2018. 10.1089/brain.2018.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robertson CE, Ratai EM, Kanwisher N. Reduced GABAerGic action in the autistic brain. Curr Biol. 2016;26:80–85. [DOI] [PubMed] [Google Scholar]
- 78.Horder J, Petrinovic MM, Mendez MA, Bruns A, Takumi T, Spooren W, et al. Glutamate and GABA in autism spectrum disorder—a translational magnetic resonance spectroscopy study in man and rodent models. Transl Psychiatry. 2018;8: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ajram LA, Horder J, Mendez MA, Galanopoulos A, Brennan LP, Wichers RH, et al. Shifting brain inhibitory balance and connectivity of the prefrontal cortex of adults with autism spectrum disorder. Transl Psychiatry. 2017;7:e1137. [DOI] [PMC free article] [PubMed] [Google Scholar]