

Abstract
Mutants of human Cellular Retinol Binding Protein II (hCRBPII) were engineered to bind a julolidine retinal analog for the purpose of developing a ratiometric pH sensor. The design relied on the electrostatic influence of a titratable amino acid side chain, affecting the absorption, and thus emission of the protein/fluorophore complex. The ratio of emissions obtained at two excitation wavelengths that correspond to the absorption of the two forms of the protein/fluorophore complex, leads to a concentration independent measure of pH.
Keywords: ratiometric, pH probe, hCRBPII, julolidine, protein engineering
Graphical Abstract
An engineered human Cellular Retinol Binding Protein II, capable of binding a julolidine retinal analog as an iminium, shows pH dependent wavelength regulation. The emission measured at two different excitation wavelengths leads to a ratiometric pH sensor, with working pH spanning 6–8.

Introduction
Intracellular pH regulation is critical for cellular function. Small changes in proton concentration can alter the activity of proteins and enzymes in the cellular milleau, affecting different processes such as metabolism and growth.[1] Cellular regulation of a number of pathways is also triggered by changes of the intracellular pH, and so are changes in cell signaling and communication.[2] There are numerous occasions, notwithstanding those cited above, that require a robust, sensitive, and accurate methodology to measure pH in biological settings.
Variants of Green Fluorescence Protein (GFP) have been developed and are currently utilized heavily as intracellular pH reporters.[3] Fluorescence of GFP depends on the rigidity of the encapsulated chromophore and also the formation of the phenolate anion, achieved through excited state proton transfer (ESPT).[4] The spectroscopic changes, going from phenol to phenolate is the foundation for GFP-based pH sensor development. pH-sensitive GFP variants can be subdivided into three groups.[5] Change in pH leads to changes in emission intensity for the first group. These GFP-based pH sensors function well as probes for monitoring pH fluctuations inside the cell. The second group has dual excitation and/or emission profiles that have been successfully implemented as ratiometric pH probes. The third group consists of protein fusion chimeras that gain the new acid/base sensitive character upon altering the protein construct. Nevertheless, all GFP-derived probes rely on the protonation/deprotonation of the hydroxyl group belonging to Tyr 66 (numbering refers to wildtype GFP).[6] The spectroscopic properties of the probe vary based on the chromophore’s structure (mutations that can change the nature of the mature chromophore) or its interactions with the protein environment.[7] Complimentary to the success of GFP as a pH probe, we sought to develop a ratiometric protein-based pH sensor that would require little to no time for maturity, have good brightness, and have a readout based on the titration of a particular amino acid side chain and not the actual chromophore itself. In this fashion, the fluorescence of the probe is not compromised at different pHs, instead the absorption of the peak is shifted based on the protonation state of the interacting residues. Herein, we report results of a single protein ratiometric pH sensor based on reengineering of the human Cellular Retinol Binding Protein II (hCRPBII) bound via an iminium to a julolidine retinal analog. The sensitivity of the bound chromophore to the electrostatic environment within the binding cavity leads to changes in the absorption profile of the system, which is exploited to generate a ratiometric pH sensor.
Results and Discussion
Our efforts in wavelength regulation of retinal, as inspired by the ability of rhodopsin to modulate absorption, and thus enable color vision, led us to the reengineering of the family of intracellular fatty acid binding proteins. During this campaign, we reported not only on the ability to alter wavelength of absorption based on electrostatic perturbations,[8] but also demonstrated the ability to use these systems as a reporter for measuring pH.[8c] Mutants of Cellular Retinoic Acid Binding Protein II (CRABPII) bound to all-trans-retinal, displayed a pH dependent color change as a result of altering the pKa of the iminium. Nonetheless, there were two drawbacks to our initial proof-of-principle system: 1. The chromophore was not fluorescent, and thus could not be used as a sensitive probe in cellular applications. Although retinal fluorescence was exploited in imaging of a single HEK293 cell,[9] it is far from being a universal tool in confocal microscopy; 2. In order to achieve a concentration independent system, two proteins of different pKas and absorptions were coupled to each other so that the ratio of the absorptions at two different wavelengths would lead to a ratiometric (and thus concentration independent) readout.
The plan required the ability to alter the absorption of the chromophore based on interactions with titratable amino acid side chains in the binding pocket. Thus, the chromophore had to be sensitive to changes in electrostatic potential. Our earlier experience with the cyanine dye family demonstrated that the extreme nature of its push-pull resonance, a feature that is often observed in fluorescent molecules, suppresses its ability for significant change in absorption, no matter the electrostatic nature of close by residing amino acids.[8j] It was hypothesized that the ideal chromophore should possess a moderate push-pull system, such that it would retain its ability to respond to changes in polarity of its immediate environment.[8j] Ultimately, we explored the julolidine retinal analog (JRA) to serve as an entry to this area. The spectroscopic characterization of JRA and its imine/iminium forms are illustrated in Figure 1. The nitrogen atom provides the desired conjugation, yet, the intervening aryl ring attenuates the resonance, yielding a fluorophore that is more responsive to polarity changes. With the choice of fluorophore in hand, the study was divided into three separate phases; first through appropriate mutations, it is required to raise the pKa of the iminium such that the fluorophore remains protonated across a large range of pHs, and thus fluorescent during measurement; second, placing of a titratable amino acid in a judicious position is required to achieve the desired wavelength alteration as a result of changes in the protonation state; third, the ability of the protein/fluorophore complex to function as a ratiometric pH sensor needs to be demonstrated.
Figure 1.
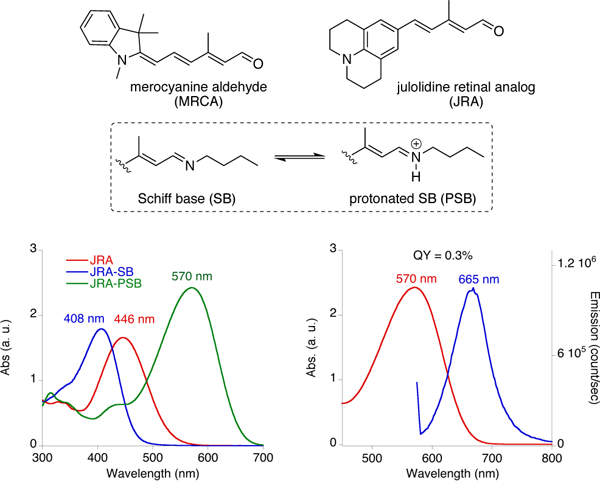
Chemical structures of merocyanine aldehyde (MRCA) and julolidine retinal analog (JRA), along with UV-vis spectra of JRA, its SB and PSB with n-butylamine and the fluorescence emission spectrum of JRA-PSB when excited at 570 nm.
Altering the pKa of the PSB
hCRBPII mutants bind JRA through the formation of an iminium (protonated Schiff base, PBS) between the chromophore’s aldehyde functionality and the protein’s active site lysine residue (Q108K). To achieve the required bathochromic shift, the imine must be protonated as a PSB, otherwise, the absorption of the complex is below 450 nm. Thus, the first order of business was to obtain a protein/chromophore complex with sufficiently high pKa to ensure the PSB would remain intact during the course of the pH titration.
In the course of our previous investigations into controlling the pKa of the PSB for all-trans-retinal bound to Cellular Retinoic Acid Binding Protein II (CRABPII), a protein closely related to hCRBPII, two important factors were identified.[8c] First, the hydrophobicity of the binding cavity is unlikely to support the required protonated state, leading to observed low pKa values; thus a stabilizing interaction for the cation is needed. Second, based on our spectroscopic data, and supported through crystallographic analysis, the iminium geometry (cis or trans) strongly influences the pKa of the PSB, since it dictates the placement of the nitrogen atom in either a hydrophobic or hydrophilic region.
To begin our efforts, a hCRBPII mutant with a sufficiently high retinal-PSB pKa was chosen as the parent for protein redesign. The Q108K:K40L:T51V:R58F tetra-mutant (M1) complexed with retinal has a pKa of 8.7. The same mutant bound JRA, resulting in a similar pKa (8.8, M1, entry 1, Figures S1–S12 in the SI illustrate the pH titration and pKa for all mutants M1–M13 discussed in the text). Fortuitously, the M1/JRA complex yielded a well-diffracted crystal for structural analysis to guide further alterations. The JRA polyene chain is located in the hCRBPII cavity, similarly oriented as observed for all-trans-retinal bound to the same mutant (Figure 2a). Both chromophores adopt a cis-iminium geometry, stabilized by a water-mediated interaction with Q4. Because JRA is significantly shorter than all-trans-retinal, the hydrophobic R58F residue has moved further inside the cavity, shielding the chromophore from the aqueous media. The bulky julolidine ring fits well into the protein cavity after replacing a water molecule, Wat2, found in the corresponding all-trans-retinal bound crystal structure (Figure 2b).
Figure 2.
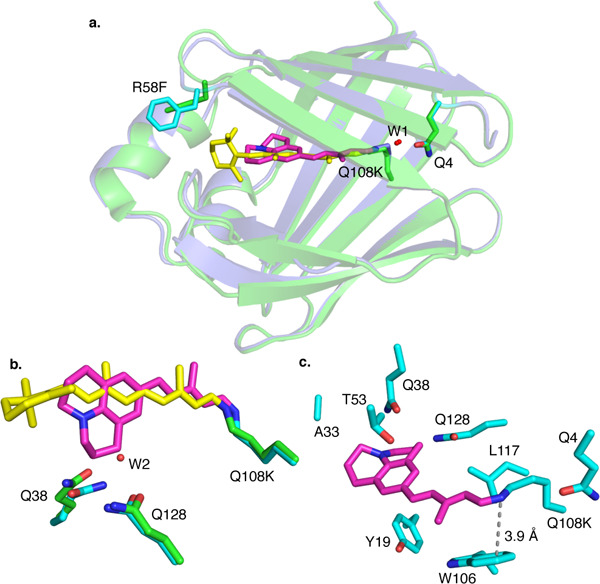
a. Crystal structures of hCRBPII-Q108K:K40L:T51V:R58F (M1) bound with all-trans-retinal (PDB ID 5F58) and JRA (PDB ID 6C7Z); b. Zoomed view of the two chromophores in the same two crystal structures; c. Residues mutated during the course of this study are highlighted.
The crystal structure of the tetra-mutant was used as a guide to achieve the desired increase in the pKa of the bound chromophore. As expected, an ionically interacting carboxylate in the PSB region can effectively stabilize the positive charge on the nitrogen atom, and result in an increased pKa. Leu117 was identified as a position ideally suited to achieve the latter upon its mutation to either Glu or Asp (see Figure 2c for relative positioning of nearby amino acids to the PSB). Gratifyingly, addition of the L117E mutation to the parent hCRBPII tetramutant led to a substantial increase in the observed pKa (9.8, M2, Table 1). As expected, the increased electrostatic interaction led to a blueshift in the absorption of the complex. Next, we aimed to influence the pKa of the iminium by removal of water-mediated interactions with Q4. As was previously observed, the Q4F mutation in many of our constructs leads to much higher protein expression levels. The M3-hexamutant exhibited a welcomed increase in pKa (10.4, Table 1, entry 3), presumably by altering the iminium geometry to have a more favorable interaction with L117E.
Table 1.
Mutants of hCRBPII; towards a ratiometric pH sensor.
| Mutant | hCRBPII Mutant | λmaxa (nm) |
λmaxb (nm) |
t1/2 (min) |
pKa | pHc | ERd | %QYe |
|---|---|---|---|---|---|---|---|---|
| M1 | Q108K:K40L:T51V:R58F | 644 | 644 | 38.6 | 8.8 | - | - | 6 |
| M2 | Q108K:K40L:T51V:R58F:L117E | 585 | 615 | 5.6 | 9.8 | 5–7 | 4.05 | 14 |
| M3 | Q108K:K40L:T51V:R58F:L117E:Q4F | 594 | 630 | 8.2 | 10.4 | 6–8 | 3.07 | 10 |
| M4 | Q108K:K40L:T51V:R58F:L117E:Q4F:W106F | 598 | 644 | n.d. | 9.6 | 4.5–7 | 1.89 | n.d. |
| M5 | Q108K:K40L:T51V:R58F:L117E:Q4F:Q38A | 584 | 624 | n.d. | 7.8 | 7–9 | n.d. | n.d. |
| M6 | Q108K:K40L:T51V:R58F:L117E:Q4F:Q38F | 635 | 643 | n.d. | 7.9 | n.r. | n.r. | n.d. |
| M7 | Q108K:K40L:T51V:R58F:L117E:Q4F:Q38A:Q128L | 645 | 645 | n.d. | 5.8 | n.r. | n.r. | n.d. |
| M8 | Q108K:K40L:T51V:R58F:L117E:Q4F:Q128E | 594 | 636 | 13.3 | 9.2 | 5–8 | 1.97 | n.d. |
| M9 | Q108K:K40L:T51V:R58F:L117E:Q4F:Q38E | 619 | 651 | 8.4 | 8.4 | 4–6 | 1.16 | n.d. |
| M10 | Q108K:K40L:T51V:T53C:R58W:T29L:A33W:Q4F:L117E | 594 | 630 | 3.0 | 10.6 | 6–8 | 2.12 | 10 |
| M11 | Q108K:K40L:T51V:T53C:R58W:T29L:A33W:Y19W:Q4F:L117E | 608 | 643 | 1.2 | 11.2 | 5–7 | 2.05 | 19 |
| M12 | Q108K:K40L:T51V:T53C:R58W:T29L:A33W:Q4F:L117D | 555 | 630 | 0.58 | 10.6 | 6–8 | 7.83 | 18 |
| M13 | Q108K:K40L:T51V:T53C:R58W:T29L:A33W:Y19W:Q4F:L117D | 575 | 643 | 0.33 | 10.7 | 5–7 | n.d. | 21 |
λmax of absorbance at basic pH.
λmax of absorbance at acidic pH.
pH range of different ratios of emission, excited at 594 nm and at 633 nm.
ER: emission ratio; the highest ratio of two collected emission spectra achieved with excitation wavelengths at 594 nm and at 633 nm.
Quantum yields (QY) were calculated using 565 nm excitation and was based on Oxazine 1 as a standard. n.r., non-ratiometric behavior during pH titration. n.d., not determined.
Having increased the pKa of the PSB to a sufficiently high level, we continued to investigate other factors that were believed to have a role in controlling the basicity of the iminium. The crystal structure in Figure 2c, and also structures from other mutants of the same protein, suggest that W106 participates in a π-cation stabilization of the PSB, thus aiding to maintain the higher pKa of the complex. Not surprisingly, mutation to the corresponding Phe led to a decrease in the pKa to 9.6 (M4, Table 1).
Another region of intrigue was near the julolidine ring, where the aforementioned water molecule, usually positioned between Q38 and Q128 in the all-trans-retinal bound structures, was replaced by the methylene of the six-member ring. Consequently, the bound JRA, and in particular, the nitrogen atom attached to the aromatic ring, is in close proximity of the latter two Gln residues. It was hypothesized that changes to the polarity in this region could alter the wavelength of absorption and also the pKa of the PSB by altering the ability for the nitrogen atom to participate in conjugation. Q38A and Q38F mutants M5 and M6 decreased the pKa to 7.8 and 7.9, respectively.[8b] Interestingly, changing both Gln residues to hydrophobic amino acids led to the lowest pKa observed in this series (5.8, M7).
Achieving wavelength regulation
An effective pH indicator requires the presence of a titratable proton. In our design, a titratable functional group in the form of a carboxylic acid, placed near the iminium was envisioned such that in its two different states it would interact to a different extent with the iminium. In principle, the carboxylate should lead to a hypsochromic shift in absorption of the hCRBPII/JRA complex, as it would reduce the conjugation of the cationic charge due to increased ionic interaction. Conversely, the neutral carboxylic acid would have attenuated interaction with the iminium, thus leading to a larger degree of cationic conjugation along the polyene, resulting in a bathochromic shift in the absorption spectrum.
The latter hypothesis was put to test with the mutants described above. The L117E mutation places the carboxylic acid in proximity of the iminium (~4.4 A from the nitrogen atom, based on the crystal structure in Figure 2c). A control experiment using the M1/JRA complex, not possessing the carboxylate residue, showed a steady PSB absorption at 644 nm over a large pH range (5.6 to 10.2, see Figure 3). On the other hand, as illustrated in Figure 3b, the L117E containing M2 pentamutant shows two distinct λmax values at pH 5.0 (615 nm) and at pH 8.0 (581 nm). This is in agreement with the mechanism that was previously proposed for the wavelength regulation of retinylidene: a more neutral electrostatic environment around the chromophore provides a more red-shifted retinal-PSB.[8b]
Figure 3.
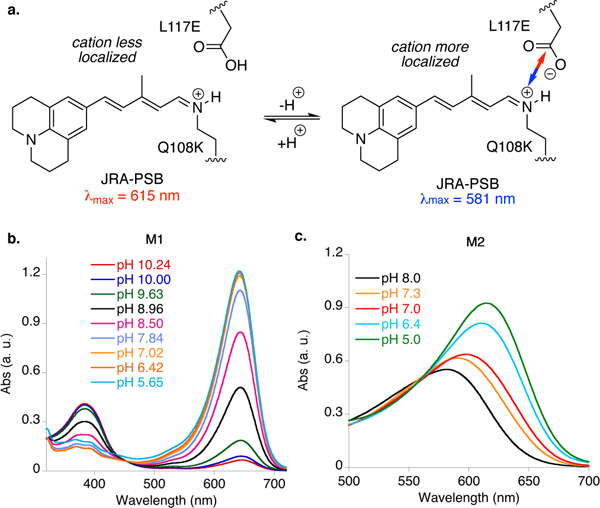
a. The putative mechanism for the response to protonation of the nearby amino acid L117E indicates that a weaker ionic interaction leads to a bathochromic shift. The difference in absorption can be exploited to discern the protonation level, and thus the pH, when correlated to a standard curve. b. The λmax of M1, lacking the nearby carboxylic acid to the PSB, does not change in response to changes in the pH. c. On the other hand, M2, having the L117E mutation, shows a substantial shift in absorption upon titration of the M2/JRA complex.
Having a hCRBPII/JRA complex that responds to pH with more than 30 nm change in absorption, paved the way for a ratiometric pH sensor. Excitation of the complex at both regions would lead to differing total emission. The strength of the emission signal (the total photon count) due to excitation at each wavelength would depend on the population of each species, i.e., neutral L117E vs. its anionic form. Thus, the ratio of emissions measured at each excitation would provide a concentration independent, ratiometric analysis for pH. We next focused on obtaining the most ideal reporter, based on achieving the largest possible resolution in absorption of the two states (neutral and anionic forms of the titratable group), while maintaining fast binding kinetics and high pKa of the PSB.
Optimization of the ratiometric probe
To optimize the wavelength separation between the two states of the pH reporter, we borrowed principles from our previous work with all-trans-retinal bound to hCRBPII.[8b] Briefly stated, we disclosed factors that enable wavelength regulation leading to more than 200 nm shift in the absorption of the retinylidene chromophore. We considered placing two charged residues simultaneously at the ends of the chromophore. The proper combination of multiple charged residues along the JRA-PSB might allow for a more substantial change in the absorption between the two forms, yielding better spectral resolution. Alternatively, this can also provide pH probes of various working ranges based on the pKa value of the side chain. In this context, a working pH range is defined as ± 1 unit from the pKa of the titratable group.
M8 and M9, listed in Table 1, incorporate additional glutamates in place of Gln residues at positions 128 and 38, respectively. As mentioned above, these residues are positioned close to the end of the chromophore. Having two Glu residues on opposite ends of the protein-bound chromophore enhanced the wavelength shift during titration, although this result was not universally observed for M9 and many of the mutants with two distinct titratable groups (data not shown). Curiously, hydrophobic changes were less predictable, as seen with the effects of the Q38A (M5, 40 nm shift) and the Q38F (M6, 8 nm shift).
The most interesting result was obtained while performing parallel studies to increase the rate of complex formation. Not only a faster maturing system has operational advantages, enabling the researcher to carry out experiments without delay, but also more consequentially, a fast reacting mutant outcompetes off-target imine formations that lead to potential background issues. Mutational hotspots discovered previously to enhance the rate of iminium formation for binding of all-trans-retinal and MCRA were tested with JRA.[8d,8j] The most effective mutations were, A33W, T29L, T53C, and R58W. Gratifyingly, the combination of these mutations with L117E as the sole carboxylate in the vicinity of the chromophore, not only increased rate of binding, but also maintained the wavelength resolution at acidic and basic pHs (see M10 and M11, Table 1). The most interesting result was obtained for the protein containing the L117D mutation (M12) rather than L117E. A 75 nm shift of the λmax was observed during the titration from pH 5 to pH 8 (Figure 4b), in contrast to 36 nm for the same construct having the L117E mutation (compare M12 with M10). Further mutation of Y19W, also key to increased rates of PSB formation, led to the M13 decamutant. Along with fast kinetics for PSB formation (t½= 20 sec), this mutant possesses well resolved absorption maxima for both neutral and anionic states of L117D, (68 nm shift observed between pHs 5 to 7), while maintaining the necessary high iminium pKa (10.7).
Figure 4.
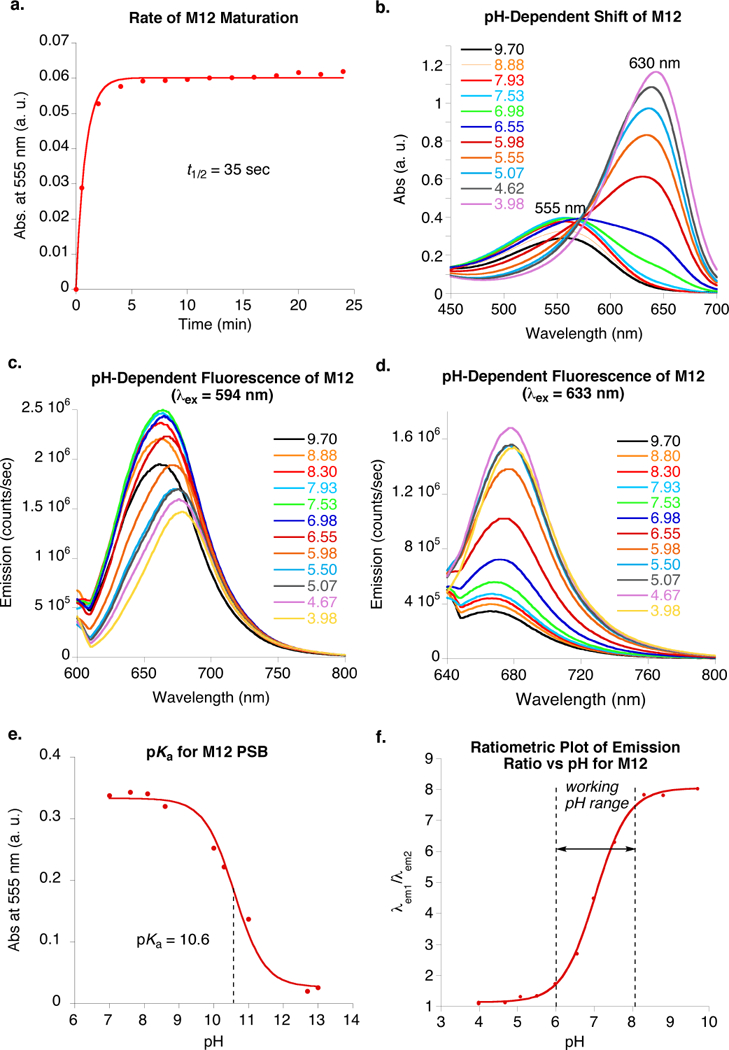
Spectroscopic properties of the M12/JRA complex (note: pH titration and pKa traces for all other mutants are provided in the SI, Figures S1–S12). a. PSB formation of M12 with JRA, with an apparent half-life of 35 sec is complete within 2 min at room temperature. b. The M12/JRA complex has a pH-dependent 75 nm wavelength shift in absorption. c. Emission intensity decreases in response to decreasing pH for excitation at 594 nm. d. Emission intensity increases in response to decreasing pH for excitation at 633 nm. e. pKa titration curve for M12. f. Standard curve of emission ratio (excitation at 594 nm and 633 nm)as a function of pH, leading to a ratiometric pH measurement.
Next, a set of our ratiometric pH sensors were tested at various pH values to generate the required standard curve for analysis. With an eye towards the future for use of these probes in vivo with laser scanning confocal microscopy, excitation wavelengths present in most common commercial instruments were utilized. Spectroscopic behavior of M12, characteristic of most of our mutants, is summarized in Figure 4. The rate of binding and maturation to the PSB is fast (Figure 4a). As described above, M12 exhibits a large wavelength shift as a function of pH (Figure 4b), while maintaining the PSB during the course of acid/base titration (pKa = 10.6, Figure 4e). As depicted in Figures 4c and 4d emission at each pH was recorded after excitation at λex1 = 594 nm (λem1 = 600–800 nm) and at λex2 = 633 nm (λem2 = 640–800 nm). The emission spectra were integrated and the ratio, λem1/λem2, was plotted as a function of the pH (Figure 4f). Importantly, the changes in the pH alter the absorption spectra as expected, but not the emission λmax. Standard curves thus obtained, exhibit the typical sigmoidal shape of a pKa titration curve. Important to note also is the working pH range, determined as highlighted in Figure 4f (dashed lines). While all the mutants showed a shift in absorption during titration, the working pH ranges were different. Mutants listed in Table 1 have a range from pH 4.5 to pH 9, thus covering the entire physiologically relevant segment.
Accuracy in measurements relies not only on a well resolved set of absorption maxima, but also on a large dynamic range of ratios. Thus, a large change in the emission ratio would ensure higher accuracy as compared to smaller changes, since the changes would be more reliably measured. The emission ratio (ER) listed in Table 1, highlights this for each mutant. As one would expect, M12 with the highest wavelength shift (75 nm) gave the highest ratio of the two fluorescent forms (ER = 7.83, M12, Table 1 and Figure 4f).
The quantum yields for all the constructs developed in this study have been listed in Table 1. Not unexpectedly, some of the bathochromically shifted mutants showed diminished quantum yield, although M13, considered one of the best candidates as a ratiometric probe, exhibits the highest quantum yield (21%). It is important to note that JRA (the aldehyde) is not fluorescent, and its PSB generated with n-butylamine is weakly fluorescent, having a 0.3% quantum yield. This ensures small background fluorescence due to free chromophore in solution. Furthermore, the bathochromic shift of the protein/chromophore complex, due to PSB formation, shifts the fluorescence observation window away from the region in which the free chromophore fluoresces.
Conclusions
In conclusion, we describe the engineering of a protein/chromophore complex as a fluorescently active pH sensor. The described pH probe operates ratiometrically. The design takes advantage of changes in the absorption profile of a bound julolidine based retinal analog, as a function of titrating a carboxylic acid side chain in proximity to the bound iminium. The stronger ionic interaction between the carboxylate and the iminium hypsochromically shifts the absorption in excess of 70 nm, as compared to the neutral carboxylic acid state. Ratio of emissions obtained through excitation of the protein complex at two wavelengths corresponding to the absorption maxima, leads to a concentration independent readout of pH. Disappointingly, JRA was not cell permeable, and thus is not suitable for cellular applications. Nonetheless, this study demonstrates a proof-of-principle strategy for developing a fluorophore-based ratiometric pH sensor. Future endeavors will focus on altering the structure of the fluorophore such that it is cell permeable and thus capable of in vivo applications.
Experimental Section
General:
UV/Vis spectra were recorded using a Cary 300 Bio WinUV, Varian Spectrometer. Fluorescence spectra were recorded with a Fluorolog®−3 spectrofuorometer (HORIBA, Ltd.). All DNA oligos were synthesized by Integrated DNA Technologies on a 25 nmole scale. pET17b plasmids containing the genes for M1-M13 hCRBPII mutants expressed in BL21 (DE3)pLysS competent cells and proteins were purified as described before.[8b] The absorption coefficients (ε) for all mutants were measured by the method previously reported by Gill and von Hippel.[10] The ε for the proteins in this study were as follows: M1(ε) 27,200 M−1cm−1; M2(ε) 27,400 M−1cm−1; M3(ε) 28,000 M−1cm−1; M4(ε) 27,800 M−1cm−1; M5(ε)25,400 M−1cm−1; M6(ε) 27,800 M−1cm−1; M7(ε) 28,300 M−1cm−1; M8(ε) M−1cm−1; M9(ε) 28,100 M−1cm−1; M10(ε) 37,000 M−1cm−1; M11(ε) 45,000 M−1cm−1; M12(ε) 35,900 M−1cm−1; M13(ε) 41,100 M−1cm−1.
Site-directed mutation:
The hCRBPII-pET17b plasmid was used for mutagenesis following QuickChange site-directed mutagenesis kit protocol (Agilent Technologies). PCR conditions for amplification of mutants are specified below using a Bio-Rad iCycler thermal cycler: Total reaction volume: 50 μL, 70 ng of DNA plasmid template, 20 pmol forward primer, 20 pmol reverse primer, 5 μL pfu buffer (10x), 1 μL pfu turbo polymerase, 1 μL dNTP, balance of volume was DNase free water. The PCR cycler was programmed as follows: 94 °C initial denaturing, followed by 20 rounds of [20 sec at 94 °C denaturing, 45 sec annealing at 5° below Tm, 210 sec at 72 °C elongation. Final elongation at 72 °C for 10 min concluded the PCR reaction. Primers used for the mutants are as follows: Q4F [Forward: 5′-G ACG AGG GAC TTC AAT GGA ACC-3′; Reverse: 5′-GGT TCC ATT GAA GTC CCT CGT C-3′]; Y19W [Forward: 5′-CTTTGAGGGCTGGATGAAGGC-3′; Reverse: 5′-GCCTTCATCCAGCCCTCAAAG-3′ −3′; T29L [Forward: 5′-GAT TTT GCC CTG CGC AAG ATT GC-3′; Reverse: 5′-GC AAT CTT GCG CAG GGC AAA ATC-3′]; A33W [Forward: 5′-CGC AAG ATT TGG GTA CGT CTC AC-3′; Reverse: 5′-GT GAG ACG TAC CCA AAT CTT GCG-3′], K40L:Q38A [Forward: 5′-CGTCTCACTGCGACGCT GGTTATT G-3′; Reverse: 5′-CAATAACCAGCGTCGCAGTGAGACG-3’]; K40L:Q38E [Forward: 5′-CGTCTCACTGAGACGCTGGTTATTG-3′; Reverse: 5′-CAATAACCAG CGTCTCAGTGAGACG-3’]; T53C [Forward: 5′-CAAGACAAAATGCACTAGCACATTCCG-3′; Reverse: 5′-CGGAATGTGCTAGTGCATTTTGTCTT G-3’]; R58W [Forward: 5’-CTAGCACATTCTGGAACTATGATGTG-3’; Reverse: 5’-CACATCATAGTT CCAGAATGTGCTAG-3’]; E72W [Forward: 5’-GTA GAG TTT GAC TGG TAC ACA AAG AGC-3’; Reverse: 5’-GCT CTT TGT GTA CCA GTC AAA CTC TAC-3’]; Q108K:W106F [Forward: 5’-GAACCGCGGCTTCAAGAAGTGG-3’; Reverse:5’-CCACTTCTTGAAGCCGCGGTTC-3’]; L117E [Forward:5’-CAAGCTGTACGACGAGCTGACC-3’; Reverse: 5’-GGTCAGCTCGTCGTACAGCTTG-3’]; L117D [Forward:5’-CAAGCTGTACGAGGAGCTGACC-3’; Reverse: 5’-GGTCAGCTCCTCGTACAGCTTG-3’]; Q128E [Forward:5’-CAGGTGTGCCGTGAGGTGTTCAAAAAG-3’; Reverse: 5’-CTTTTTGAACACCTCACGGCACACCTG-3’]; Q128L [Forward:5’-CAGGTGTGCCGTCTGGTGTTCAAAAAG-3’; Reverse: 5’-CTTTTTGAA CACCAGACGGCACACCTG-3’]. 20 units of Dpn I restriction enzyme (New England BioLabs®) was added to the PCR product for DNA template digestion and the reaction mixture was incubated at 37 °C for 1 h. The solution was then transformed to XL-1 blue E. coli competent cells and the plasmid was isolated, cultured, and then purified using a Promega Wizard Plus SV miniprep DNA purification kit (A 1330) following the suggested protocols.
UV-Vis and Kinetic measurements of hCRBPII/JRA PSB formation:
hCRBPII/JRA-PSB formation was followed by UV-vis in PBS buffer (pH=7.3) at 23 °C. pH was verified each time before recording the spectrum. The experiment was performed with a final protein concentration of 20 μM, and 0.5 equivalents of JRA. The spectra were recorded immediately after mixing the protein and chromophore. Absorption peaks of SB, PSB and free chromophore were observed over time. In order to measure the kinetics of binding, two absorption measurements were performed per sec. The concentration of hCRBPII/JRA was plotted as a function of time and fit to a second order rate equation as previously described.[8j,11]
PSB pKa determination:
The PSB peak was monitored with UV-vis at different pH with acidification/basification.The protein in PBS buffer (pH = 7.3) was incubated with 0.5 equivalents of JRA until PSB formation was completed. The concentration of JRA was verified based on the absorbance value and extinction coefficient in ethanol (ε=30,000 M−1cm−1). The solution was acidified with citric acid buffer to a pH ~4 and then titrated with NaOH. UV-vis spectra were collected at each pH point. The titration was continued until the PSB was converted to SB. Absorbance values at the λmax were plotted as a function of pH. The scatter plot obtained as such was fitted to a sigmoidal curve for pKa determination, using the following equation:
Where ΔAo is the total absorbance change of the PSB peak between the most acidic and most basic pH, and ΔA is the difference of the absorption between the value measured at each pH vs the absorption at the most basic pH.
Fluorescence pH-titration:
The proteins with pH-dependent UV-vis shifts were evaluated for ratiometric emission using a Fluorolog-3 fluorimeter. A solution of protein-JRA complex in PBS buffer (with absorbance no more than 0.2) was titrated from ~12 to ~4 using 1M citric acid buffer (pH = 3.5). At each pH point spectra were collected at two excitation wavelengths (594 nm and 633 nm). The emission for excitation at 594 nm was integrated from 600–800 nm and the emission for excitation at 633 nm was integrated from 640–800 nm. The final peaks were integrated and divided. The ratio of the integrated emissions was plotted as a function of pH and fitted to the following equation:
Where FR is a ratio of integrated emission spectra excited at 594 nm and at 633 nm.
Quantum Yield Measurement:
The quantum yields were determined by comparing the proportional gradients of the unknown sample and fluorescent standards Oxazine-170 (purchased from Acros Organics) or Oxazine-1 (purchased from Exciton) using the following equation:[12]
Where “grad” is equal to the slope of a plot relating the integrated emission (y axis) to the absorbance (x axis) using a minimum of three different concentrations per sample; η is the refractive index of the solvent used for the fluorescence readings.
Supplementary Material
Acknowledgements
We are grateful to the NIH (GM101353) for generous funding.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Putney LK, Barber DL, J. Biol. Chem. 2003, 278, 44645–44649; [DOI] [PubMed] [Google Scholar]; b) Wang HY, Singh D, Fliegel L, J. Biol. Chem. 1997, 272, 26545–26549. [DOI] [PubMed] [Google Scholar]
- [2].a) Aerts RJ, Durston AJ, Moolenaar WH, Cell 1985, 43, 653–657; [DOI] [PubMed] [Google Scholar]; b) Boonsirichai K, Sedbrook JC, Chen RJ, Gilroy S, Masson PH, Plant Cell 2003, 15, 2612–2625; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Busa WB, Crowe JH, Science 1983, 221, 366–368; [DOI] [PubMed] [Google Scholar]; d) Grainger JL, Winkler MM, Shen SS, Steinhardt RA, Dev. Biol. 1979, 68, 396–406; [DOI] [PubMed] [Google Scholar]; e) Lagadic-Gossmann D, Huc L, Lecureur V, Cell Death Differ. 2004, 11, 953–961; [DOI] [PubMed] [Google Scholar]; f) Lee HC, Johnson C, Epel D, Dev. Biol. 1983, 95, 31–45; [DOI] [PubMed] [Google Scholar]; g) Monshausen GB, Bibikova TN, Messerli MA, Shi C, Gilroy S, Proc. Natl. Acad. Sci. USA 2007, 104, 20996–21001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bizzarri R, Serresi M, Luin S, Beltram F, Anal. Bioanal. Chem. 2009, 393, 1107–1122. [DOI] [PubMed] [Google Scholar]
- [4].Lossau H, Kummer A, Heinecke R, PollingerDammer F, Kompa C, Bieser G, Jonsson T, Silva CM, Yang MM, Youvan DC, MichelBeyerle ME, Chem. Phys. 1996, 213, 1–16. [Google Scholar]
- [5].Bencina M, Sensors 2013, 13, 16736–16758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Craggs TD, Chem. Soc. Rev. 2009, 38, 2865–2875. [DOI] [PubMed] [Google Scholar]
- [7].Zimmer M, Chem. Soc. Rev. 2009, 38, 2823–2832. [DOI] [PubMed] [Google Scholar]
- [8].a) Wang WJ, Geiger JH, Borhan B, Bioessays 2014, 36, 65–74; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang WJ, Nossoni Z, Berbasova T, Watson CT, Yapici I, Lee KSS, Vasileiou C, Geiger JH, Borhan B, Science 2012, 338, 1340–1343; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Berbasova T, Nosrati M, Vasileiou C, Wang WJ, Sing K, Lee S, Yapici I, Geiger JH, Borhan B, J. Am. Chem. Soc. 2013, 135, 16111–16119; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Berbasova T, Santos EM, Nosrati M, Vasileiou C, Geiger JH, Borhan B, ChemBioChem 2016, 17, 407–414; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Crist RM, Vasileiou C, Rabago-Smith M, Geiger JH, Borhan B, J. Am. Chem. Soc. 2006, 128, 4522–4523; [DOI] [PubMed] [Google Scholar]; f) Lee KSS, Berbasova T, Vasileiou C, Jia XF, Wang WJ, Choi Y, Nossoni F, Geiger JH, Borhan B, Chempluschem 2012, 77, 273–276; [Google Scholar]; g) Nosrati M, Berbasova T, Vasileiou C, Borhan B, Geiger JH, J. Am. Chem. Soc. 2016, 138, 8802–8808; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Vasileiou C, Vaezeslami S, Crist RM, Rabago-Smith M, Geiger JH, Borhan B, J. Am. Chem. Soc. 2007, 129, 6140–6148; [DOI] [PubMed] [Google Scholar]; i) Vasileiou C, Wang WJ, Jia XF, Lee KSS, Watson CT, Geiger JH, Borhan B, Proteins-Structure Function and Bioinformatics 2009, 77, 812–822; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Yapici I, Lee KSS, Berbasova T, Nosrati M, Jia XF, Vasileiou C, Wang WJ, Santos EM, Geiger JH, Borhan B, J. Am. Chem. Soc. 2015, 137, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Kralj JM, Hochbaum DR, Douglass AD, Cohen AE, Science 2011, 333, 345–348; [DOI] [PubMed] [Google Scholar]; b) Maclaurin D, Venkatachalam V, Lee H, Cohen AE, Proc. Natl. Acad. Sci. USA 2013, 110, 5939–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gill SC, von Hippel PH, Anal. Biochem. 1989, 182, 319–326. [DOI] [PubMed] [Google Scholar]
- [11].Hori Y, Norinobu T, Sato M, Arita K, Shirakawa M, Kikuchi K, J. Am. Chem. Soc. 2013, 135, 12360–12365. [DOI] [PubMed] [Google Scholar]
- [12].Rurack K, Spieles M, Anal. Chem. 2011, 83, 1232–1242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.