

Abstract
The brain is particularly sensitive to changes in energy supply. Defects in glucose utilization and mitochondrial dysfunction are hallmarks of nearly all neurodegenerative diseases and are also associated with the cognitive decline that occurs as the brain ages. Chronic neuroinflammation driven by glial activation is commonly implicated as a contributing factor to neurodegeneration and cognitive impairment. Human immunodeficiency virus (HIV) disrupts normal brain homeostasis and leads to a spectrum of HIV-associated neurocognitive disorders (HAND). HIV activates stress responses in the brain and triggers a state of chronic neuroinflammation. Growing evidence suggests that inflammatory processes and bioenergetics are interconnected in the propagation of neuronal dysfunction. Clinical studies of HIV-infected individuals and basic research support the notion that HIV creates an environment in the CNS that interrupts normal metabolic processes at the cellular level and collectively alter whole brain metabolism. In this review, we highlight reports of abnormal brain metabolism from clinical studies and animal models. We also describe diverse CNS cell-specific changes in bioenergetics associated with HIV. Moreover, we propose that attention should be given to adjunctive therapies that combat sources of metabolic dysfunction as a mean to improve and prevent neurocognitive impairments.
Keywords: HIV, brain, aging, metabolism, bioenergetics, mitochondria
Graphical Abstract
1. Introduction
Human immunodeficiency virus (HIV) is widely recognized for its deleterious impact on the immune system. Fortunately, the introduction and implementation of combination antiretroviral therapy (CART) has transformed HIV infection into a manageable chronic disease instead of the death sentence it was when it emerged thirty-five years ago. Although CART has been extremely successful in reducing viral replication in the periphery to undetectable levels in many people with HIV (PWH), HIV reservoirs still persist. The central nervous system (CNS) is a target organ system of HIV and neuroinvasion occurs shortly after infection. A major consequence of persistent HIV infection in the CNS is the development of HIV-associated neurocognitive disorders (HAND), which is estimated to effect 30–60% of PWH (Heaton et al., 2011). HAND is a research diagnosis based on neuropsychological testing and a functional assessment of activities of daily living. At minimum, a person with HIV is diagnosed with HAND when they show abnormal test performance (< 1 standard deviation (SD) below best available population norms) on at least 2 cognitive domains. The most common domains impacted in the CART era include deficits in processing speed, executive function, and memory retrieval deficits [1, 2]. However, there is considerable heterogeneity in the patterns of cognitive impairment demonstrated [3], which can be impacted by numerous factors including viral suppression and adherence to antiretroviral medication [2]. Classification of HAND comprises a range of cognitive impairments increasing in severity. Asymptomatic neurocognitive impairment (ANI) is defined as mild cognitive difficulties involving two cognitive domains that fall below 1 SD of population norms without overt functional impairment in daily life activities. Similarly, mild neurocognitive disorders (MND) are marked by cognitive impairments in two cognitive domains that score below 1 SD of norms, yet mild to moderate interference in daily functioning is observed. HIV-associated dementia (HAD), the most severe form of HAND, requires a score below 2 SD of population norms with significant functional impairments [4, 5].
It is widely accepted that HIV enters the brain via infected macrophage/monocytes and lymphocytes that cross the blood brain barrier (BBB). Once inside the brain, infected monocytes/macrophages facilitate productive infection and release free virions into the brain parenchyma that infect neighboring microglia and to some degree, astrocytes. This infection leads to the release of neurotoxic viral proteins and to the activation of resident glia. There is substantial evidence that this inflammatory response is a major driver for the development and progression of HAND. Although neurons are not infected by HIV, the pathophysiology of HAND ultimately impacts neurons. Loss of synaptic complexity, neuronal damage and death are documented during HIV infection [6–8]. Much attention has been given to direct neurotoxicity associated with the release of viral proteins from infected cells and indirect neurotoxicity mediated by the inflammatory response mounted by both infected and uninfected non-neuronal cells. However, recently it has become increasingly important to examine how neurotoxic factors and inflammation are associated with alterations in cellular metabolism in the context of neurodegenerative disorders.
The brain is a complex organ requiring a vast amount of energy to sustain its basic functions. At baseline, the brain consumes over 20% of the oxygen and 25% of the glucose taken in by the body, although it makes up only 2% of the body’s weight [9–11]. This is a 10-fold greater energy requirement than any other organ. Glucose is the primary energy substrate of the brain. The majority of glucose is used to transduce energy through glycolysis and mitochondrial oxidative phosphorylation to generate the large amounts of ATP required to maintain membrane potentials, facilitate synaptic transmission and other neural cell activities. The brain undergoes a steady decline in energy metabolism during normal aging. Since the brain requires substantial amounts of energy, a shortage of metabolic supply can contribute to cognitive decline. There is a body of evidence demonstrating that a loss of energy homeostasis may be an early event that primes the CNS for functional impairments. Chronic HIV infection is associated with metabolic disturbances in the brain, even in PWH on effective CART regimens [12–15]. In this review, we aim to summarize the current understanding of how HIV infection contributes to energy disturbances involved in HAND pathology.
2. Disturbances in brain bioenergetics and its metabolites appear in clinical studies as well as in animal models of HIV infection
The energy requirements of the brain are very high and require tight and balanced regulation to maintain an uninterrupted supply of energy substrates. In addition to glucose, cells of the CNS can utilize other energy substrates such as lactate, pyruvate, glutamate, glutamine, fatty acids, and ketone bodies to support their energy demands [16]. Metabolic dysfunction and cerebral hypometabolism have been extensively reviewed in the context of several neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS) [17–20], thus, providing clear evidence for a connection between neurological disease and altered energy metabolism. Metabolic changes in the brain are a hallmark of HIV infection [12–15, 21, 22]. Proton magnetic resonance spectroscopy (1H MRS) provides a sensitive and noninvasive method for the detection of metabolic changes in the brain. Specific brain metabolites, such as N-acetylaspartate (NAA) and myo-inositol (MI) are commonly used as markers for neuronal health or glial activation, respectively. NAA is synthesized by neurons from acetyl-CoA and aspartate within the mitochondria. Fluctuations in NAA closely mirror ATP levels and likely reflect impaired mitochondrial energy production rather than neuronal cell loss [23]. Conversely, MI is primarily present in glial cells and functions as an osmolyte that maintains glial cell volumes. Activated glia have increased cell volumes and thus tend to have elevated MI, making it a suitable marker for glial activation [24]. These compounds are often reported in relation to brain creatine (Cr) levels to provide metabolic information specific to each cell population. Several groups have reported that regardless of effective HIV suppression with CART, PWH exhibit a marked increase in MI/Cr at both acute and chronic time points of HIV infection [14, 25]. Interestingly, increased MI/Cr often precedes signs of neurological impairment. Moreover, NAA is notably reduced in PWH who present with cognitive impairment [26]. These findings highlight that despite effective CART, glial inflammation during subclinical stages of HIV infection is followed by neuronal injury that contributes to cognitive impairments. High metabolic demands by activated astrocytes and microglia likely explain, in part, increased Cr levels. Subsequently, reduced NAA levels suggest neuronal mitochondrial injury likely as a result of ongoing neuroinflammation. Similarly, in the rapid progression macaque model of neuroAIDS and in humanized mice infected with HIV, 1H MRS revealed that NAA was decreased, whereas, brain Cr was elevated throughout the disease progression suggesting high energy turnover [12, 27]. One study employed 1H MRS and functional MRI (fMRI) to correlate metabolite concentration with blood oxygenation level-dependent (BOLD) signals[21]. A significant positive correlation was found between markers of glial activation (MI and Cr) and BOLD signal strength in the working memory network in HIV-positive individuals. These findings provide additional support that cognitive impairments are linked with glial energetic abnormalities.
In AD, a number of brain imaging studies using FDG-PET tracer have shown that brain glucose uptake is significantly diminished prior to altered cognitive function or pathological signs such as brain atrophy {Bateman, 2012, Clinical and biomarker changes in dominantly inherited Alzheimer’s disease;de Leon, 2007, Imaging and CSF studies in the preclinical diagnosis of Alzheimer’s disease;Mosconi, 2005, Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD;Chen, 2013, Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies}. Similarly, in PET studies of HIV patients virologically suppressed on CART, varying levels of reduced glucose uptake have been identified in various regions of the brain including the frontal cortex and within the anterior cingulate cortex [28, 29]. In line with imaging studies, cerebrospinal fluid (CSF) metabolomics of PWH expand further on the aberrant changes in brain bioenergetics associated with HIV infection [13, 22]. In PWH, signs of enhanced anaerobic metabolism are associated with improved cognitive status. Anaerobic glycolysis utilizes glucose for the synthesis of ATP and lactate. Lactate can be utilized by neurons as an energy source. Increased lactate production resulting from enhanced anaerobic glycolysis appears to contribute toward improved cognitive function in HIV-infected patients [13]. Conversely, enhanced mitochondrial respiration and aerobic glycolysis marked by accumulation of acetate and citrate and TCA cycle intermediates seem to contribute to declining cognitive status [13]. Cr is tightly linked to aerobic glycolysis. Therefore, increased Cr along with the accumulation of citrate and acetate provides added support that aerobic glycolysis rate is enhanced in cognitively impaired PWH[13]. Proteomic analyses of autopsied brain tissue of HAD patients show a large portion of the altered proteins are involved in the glycolytic and oxidative phosphorylation pathways [30]. Collectively, this supports the notion that metabolic abnormalities are associated with HAND and points to mitochondrial dysfunction as a major factor.
3. Calcium Dysregulation and Mitochondrial Dysfunction
Calcium (Ca2+) is a fundamental signal messenger and is involved in almost all pathways of cellular physiology. Ca2+ signaling in the brain is regulated by a vast number of stimuli and serves a multitude of diverse functions including neurite outgrowth, synaptogenesis, synaptic transmission, plasticity, and survival[31]. In addition, Ca2+ plays an important role in the bioenergetics of neuronal function. In stimulated neurons, an increase in intracellular Ca2+ precedes any changes in O2 consumption rates [32]. Along with the mitochondria, the endoplasmic reticulum (ER) serves as one of the main Ca2+ storage organelles and is vital for Ca2+ homeostasis. The ER and mitochondria are physically associated and functionally linked to one another. Transfer of Ca2+ from the ER to mitochondria stimulates oxidative metabolism, thus, metabolically energizing the mitochondria. The mitochondrial-associated ER membrane plays an important role in the maintenance of cellular homeostasis by regulating Ca2+ transfer and energy metabolism [33]. Mitochondria have the capacity to store large amounts of Ca2+ in the matrix and serve as a Ca2+ buffering system. Mitochondrial Ca2+ accumulation is tightly regulated and it is intimately involved in the synthesis of ATP. Ca2+ activates several enzymes involved in the tricarboxylic acid (TCA) cycle and the electron transport chain (ETC) relies on Ca2+ to generate ATP [31]. Dysregulation of Ca2+homeostasis and signaling can disrupt energy homeostasis and has been implicated in neurological diseases, including HAND.
HIV-1 proteins, including Tat and gp120, induce Ca2+ dysregulation in both neurons and glial cells, as indicated by abnormal and disproportionate Ca2+ influx and increased intracellular Ca2+ release [34, 35]. This ultimately leads to elevated cytosolic free Ca2+ levels. Shifts in intracellular Ca2+ homeostasis considerably interrupts normal cellular function and induces dysregulation, injury and death of neurons and glial cells. The HIV protein, Tat has been shown to alter intracellular Ca2+ levels by triggering its release from the ER via the inositol-1,4,5-trisphosphate (IP3) receptor (IP3R) in rat hippocampal neurons [36]. Subsequently, the progressive elevation of free intracellular Ca2+ is taken up by the mitochondria. This results in mitochondrial Ca2+ overload and accumulation of reactive oxygen species (ROS) and oxidative stress. In this context, Tat-induced neuronal apoptosis was found to be mediated by dysregulation of Ca2+ signaling [36]. In fact, it was demonstrated in mouse striatal neuron cultures that Tat increases intracellular Ca2+ and induces instability in mitochondrial inner membrane potential leading to synaptic injury [34]. The HIV protein, gp120 has similar effects in glia cultures. For example, one report found that in a subset of astrocytes, gp120 induced substantial Ca2+ released from Ca2+ stores while also triggering Ca2+ influx across the plasma membrane [37]. Overall, this resulted in significantly elevated intracellular Ca2+ in these cells [37]. Taken together, it is possible that dysregulation of Ca2+ signaling by HIV and/or viral proteins may be upstream mediators of the disruptions in mitochondrial function and subsequently result in harmful hypermetabolic conditions.
Mitochondria are exceptionally dynamic organelles that undergo fusion, fission, and trafficking to meet the energetic demands of the cell. Mitochondria are best known for the generation of ATP via the TCA cycle and oxidative phosphorylation (OXPHOS) by means of the ETC located in the inner mitochondrial membrane. Neural tissue depends on mitochondria to produce the tremendous amount of ATP required for neuronal excitability, synaptic signaling, and alternations in neuronal function and structure. However, more than only the powerhouse of the cell, mitochondria also play fundamental roles in the regulation of Ca2+ homeostasis, signal transduction, stress response, and cell survival [38]. Mitochondria are crucial for the proper functioning of neural tissue and their dysfunction has been speculated to be the foundation for the development of numerous neurodegenerative diseases. Despite distinct phenotypes, PD, AD, Huntington Disease (HD), and Amyotrophic Lateral Sclerosis (ALS) share a common theme: impaired mitochondrial function. This includes defects in mitochondrial energy production, size, shape, distribution, movement, and turnover [39]. Similarly, there is in vivo and in vitro evidence indicating comparable mitochondrial abnormalities are associated with HIV infection [40, 41]. In PWH on CART, CSF metabolic profiles revealed augmented levels of succinate typically implicated in mitochondrial dysfunction, as well as markers of oxidative stress, and the accumulation of metabolic waste as contributors to HAND [22]. In HIV + brains, post-mortem gene expression analyses revealed that HIV-associated neurocognitive impairments are related to gene pathways involved in mitochondrial functioning being significantly down regulated [42]. This finding was also recapitulated in HIV-1 transgenic rats, which express seven out of nine HIV viral proteins [43]. Villenueve et al reported significant changes in synaptic mitochondria isolated from HIV-1 transgenic rats that included abnormalities in expression of ETC complex subunits [44]. In addition, increases in protein expression of TCA cycle and fatty acid metabolic processes were noted. This is supported by their findings that HIV-1 Tg rats had higher oxygen consumption rates than littermate controls [44]. Taken together, this indicates global brain mitochondrial functioning is perturbed during HIV infection. However, mitochondrial activity may vary across distinct cellular compartments and brain regions.
Distinct alterations in mitochondrial morphology are associated with CNS HIV infection [45]. Mitochondrial size was increased in the frontal cortex of HAND patients suggesting mitochondrial fusion is preferred over fission in these individuals. This was supported by decreases in mitochondrial fission protein, dynamin-1 like (DNM1L) and increases in mitochondrial fusion protein, mitofusin 1 (MFN1). Importantly, these changes were specifically identified in neuronal mitochondria[45]. In response to various stresses, mitochondrial hyperfusion protects cells and supports mitochondrial ATP synthesis[46]. Numerous studies point to a loss and/or gain of function in mitochondrial biology to contribute to HAND. It is commonly observed that mitochondrial functioning is often disrupted while the generation of ROS is increased during HIV neuropathogenesis. Studies clearly show an increase in oxidative and nitrosative stress early in HIV infection and throughout the progression of HAND [47–49]. These findings highlight the complexity of bioenergetics in the brain over the course of HIV infection. Even though brain tissue and CSF provide important insights into the overall state of the brain, upon closer examination, it is apparent that each cellular compartment and brain region have unique responses to HIV infection and this is demonstrated through different metabolic responses.
4. Cell-Specific Energy Changes during HIV infection
Bioenergetically, the brain is not a uniform organ. Neurons and microglia are highly aerobic and depend heavily on mitochondrial respiration. However, oligodendrocytes and astrocytes are predominantly glycolytic. These opposing energetic profiles create a reciprocal energy relationship within the CNS. Glycolysis in astrocytes and oligodendrocytes results in lactate production, which is in turn used by neurons to fuel the TCA cycle and oxidative phosphorylation. This complicates the study of the overall energy dynamics of the CNS. It is therefore important to consider the cell-specific metabolic changes related to HIV infection of the CNS. Despite viral suppression, infected cells may express and release HIV proteins, many of which are neurotoxic including gp120, Tat, Nef, and Vpr. There is a large body of evidence from primary neuronal cultures illustrating that HIV proteins cause direct injury to neurons without any contribution of non-neuronal cells (Table 1). Furthermore, HIV proteins also alter glial cell function.
Table 1.
Summary of the effect of various HIV proteins on metabolic pathways in cells of the CNS (↑ increase, ↓ decrease, UNK – unknown).
| Tat | gp120 | Vpr | Nef | reference | |
|---|---|---|---|---|---|
| Mitochondrial Integrity | |||||
| Morphology | ↑ fragmentati on | ↑ fragmentation | ↑ fragmentation | ↑ fragmentation | [54, 59, 64, 83] |
| Membrane Potential | ↓ neurons ↑ astrocytes ↓ microglia |
↓ neurons and astrocytes | ↓ neurons | ↓ neurons | [59, 61–64, 81, 83, 84, 90, 153] |
| Oxidative Capacity | ↑ astrocytes ↓ microglia |
↓ neurons ↓ oligodendrocyt es ↑ microglia |
UNK | UNK | [54, 60, 83, 84, 90, 111, 152] |
| ATP Levels | ↓↑ neurons ↑ astrocytes ↓ microglia |
↓ neurons ↑ microglia |
↓ neurons | ↓ neurons | [44, 54, 60–65, 83, 84, 152] |
| ROS Generation | ↑ | ↑ | ↑ | ↑ | [63, 64, 80, 81] |
| Glycolysis | |||||
| Glycolytic Rate | ↓ microglia | ↑ microglia | UNK | UNK | [90, 152] |
| Lactate Production | ↓ astrocytes | UNK | UNK | UNK | [84] |
| Calcium Signaling | |||||
| Dysregulation of Ca2+ | ↑ Ca2+ influx ↑ Ca2+ levels |
↑ Ca2+ influx ↑ Ca2+ levels |
↑ Ca2+ influx ↑ Ca2+ levels |
UNK | [34, 35, 37, 82, 84, 110, 166] |
| ER stress | ↑ neurons ↑ astrocytes |
↑ astrocytes | ↑ neurons | UNK | [77–79] |
4.1. Neurons
Neuronal survival is dependent on mitochondrial integrity and functionality. Neurons rely on mitochondrial OXPHOS to meet their high energy needs. In fact, there are several reports that reveal neurons express very low levels of key glycolytic enzymes such as 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase-3 (Pfkfb3) thus, supporting the preference for OXPHOS in neurons [50, 51]. Stimulation of neurons with Ca2+ results in rapid ATP consumption followed by an immediate rise in mitochondrial oxidative respiration pointing to the importance of OXPHOS [52]. A lack of ATP can compromise neuronal function even without cell death and contribute to cognitive impairments.
Although neurons are not infected by HIV-1, it is evident that their functioning can be significantly impacted by HIV-1 proteins and proinflammatory factors released in response to chronic infection. It is well established that various HIV proteins cause disruptions in neuronal energy homeostasis [34, 53–58]. A recent report found that gp120 and Tat induce mitochondrial fragmentation and decrease mitochondrial membrane potential in human primary neurons [59]. Loss of mitochondrial membrane potential is associated with mitochondrial damage and diminished ATP synthesis. In line with this, rat primary cerebrocortical cells exposed to gp120 exhibited a significant reduction in oxidative respiration and loss of neuronal ATP [54, 60]. Mouse neurons treated with HIV Vpr also displayed mitochondrial depolarization and reduced ATP production [61, 62]. There are however conflicting reports of Tat increasing and decreasing ATP production in neuronal cultures [44, 63–65]. One group reported in rat cortical neurons that ATP levels increased with low and high doses of Tat after 24 hours and after 48 hours [63]. The authors explain this as likely a result of increased ATP production rather than diminished consumption of ATP [63]. This is in contrast to the findings of another group that found in rat striatal neurons, Tat reduced ATP under physiological oxygen conditions [64]. Nevertheless, in all instances Tat-associated changes in ATP production precedes cell death. Tat also inhibits ATP synthase suggesting another aspect of mitochondrial dysfunction [57, 66]. These findings provide strong evidence that HIV proteins can directly impact neuronal bioenergetics and contribute to neuronal dysfunction. However, to have a full understanding of the mechanisms in which HIV may be influencing neuronal energy homeostasis, it is also necessary to consider the importance of glia metabolic support and investigate ways in which HIV may also disrupt other CNS cell populations further depriving neurons of energy substrates.
4.2. Glia
More than merely accessory cells to neurons, glia are now recognized as essential to neuronal homeostasis, survival and brain function. Unfortunately, few studies have addressed the pathological implications of energetic disturbances in glial cells and the consequences this may have on neurons. The glial component of the brain consists of astrocytes, microglia and oligodendrocytes. Even though there are differences among brain regions, in the entire brain there is an approximately 1:1 ratio of glia to neurons [67]. Oligodendrocytes are reported to be the most abundant glial cell type, followed by astrocytes, and microglia [68]. The intimate metabolic coupling of neurons and glia is crucial to normal brain function. Defects in glial cell energy homeostasis are reported during HIV infection, and thus, may contribute to the pathogenesis of HAND.
4.2.1. Astrocytes
Astrocytes have emerged as a key contributor to energy uptake, delivery, production, utilization and storage in the CNS. Although only accounting for between 5–15% of the brain’s energy expenditure, reports suggest that astrocytes take up a disproportionally high amount of glucose in relation to their energy demands [10, 69]. In contrast to neurons, astrocytes are highly glycolytic in nature and readily take up glucose. Astrocytes have lower levels of oxidative metabolism when compared to neurons and instead favor the production of lactate over pyruvate for the TCA cycle. Astrocytes are the predominant suppliers of lactate in the CNS. Lactate derived from astrocytes is released into the brain milieu for neuronal uptake, thereby allowing neurons to generate high levels of ATP while circumventing the glycolytic pathway. This is the central point of the astrocyte-neuronal lactate shuttle (ANLS) model that describes the metabolic coupling of neurons and astrocytes such that activated neurons stimulate increased astrocytic glucose uptake, glycolysis, and lactate production in astrocytes that is shuttled back to neurons to be utilized for their metabolic needs. Considering the extensive metabolic coupling between neurons and astrocytes, any alterations in astrocytic energy pathways could have deleterious effects on neuronal function. One interesting report found age-dependent increases in oxidative metabolism and mitochondrial biogenesis in astrocytes [70]. This increase in mitochondrial aerobic metabolism was demonstrated to coincide with a functional switch in astrocytes from a neurotrophic phenotype to neurotoxic [70]. It may be surmised that metabolic changes in astrocytes, which occur as a function of age, may be prematurely induced the setting of HIV infection of the CNS. This agrees with reports of earlier onset of neurocognitive deficits in this population as well as evidence of comparable CSF metabolites profiles between PWH (median age 40) and older HIV negative control patients (median age 57), suggesting accelerated aging contributes to HAND [22, 71]. Enhanced mitochondrial respiration reduces the astrocytes’ ability to supply energy substrates to neurons and instead provide support their own metabolic needs [70]. This notion is supported by evidence that inflammatory reactions in astrocytes in response to infection or stress is metabolically expensive and may stimulate mitochondrial metabolism to meet their energy demands [72]. Considering the chronic neuroinflammatory state in PWH, it is important to address how astrocyte neurotrophic function may be affected.
Astrocytes come into direct contact with HIV-infected cells that enter the CNS by crossing the BBB. To some extent, astrocytes are permissive to HIV infection, but this likely does not develop into a productive infection. However, astrogliosis is a common pathological feature in HIV-1 infected brains [73, 74], triggered by HIV proteins as well as by proinflammatory products released by infected or stressed cells [75, 76]. There is also evidence of HIV-induced ER stress in astrocytes contributing to mitochondrial dysfunction [77]. ER stress and enhanced oxidative stress are detected in astrocytes exposed to HIV proteins, including Tat, gp120 and Vpr [78–81], suggesting that mitochondrial fitness is likely disturbed under conditions of ER dysfunction. In vitro, HIV-infected astrocytes demonstrate diminished mitochondrial integrity resulting in release of cytochrome C into the cytoplasm and altered intracellular Ca2+ signaling [82]. However, the impact of HIV directly on mitochondrial dynamics in astrocytes is largely understudied. Acute exposure of astrocytes to HIV-1 induced mitochondrial permeability transition pore opening and loss of mitochondrial membrane potential [83]. Similarly, gp120 alone increased mitochondrial membrane depolarization [81]. Recently, our group was the first to show that Tat protein induces a metabolic shift in astrocytes from glucose utilization to fatty acid oxidation resulting in enhanced Ca2+ uptake into the mitochondria facilitating enhanced mitochondrial respiration, increased ATP levels, and reduced lactate production [84]. This indicates that enhanced aerobic respiration in astrocytes may be contributing to HAND by misappropriating energy substrates essential for neurons. Neuronal metabolism, which is already constrained by factors associated with HIV, is further deprived of astrocytic metabolic support exacerbating neuronal energy deficits and impairments.
4.2.2. Microglia
Microglia are the only resident cell in the brain parenchyma that can support productive HIV infection and therefore are likely a major contributor to neurotoxicity observed during HIV infection. Microglia are often referred to as the sentinel cells of the CNS and represent 5–20% of the adult brain. Microglia have the capacity to migrate, proliferate and phagocytize. Under physiological conditions, microglia exist in their “resting” state characterized by ramified morphology, and use these highly branched processes to survey the environment for the presence of pathogens or cellular debris. Upon exposure to pathological triggers, microglia transition into an amoeboid phenotype and quickly mobilize to the site of injury to initiate an innate immune response.
HIV infects microglia in vivo and in vitro. In response to direct HIV infection or exposure to viral particles, microglia become activated and release various soluble molecules including nitric oxide, superoxide anions, quinolinic acid, viral proteins Tat, gp120, and Vpr, chemokines, and proinflammatory cytokines including TNF-α and IL-1β [85]. In PET studies using [11C]-PK11195, a marker of the translocator protein expressed by activated microglia, HIV-infected patients show increased detection of microglial activation in several cortical regions even when they are neurocognitively asymptomatic [86].
The role of metabolic reprogramming in the regulation of innate immune responses has been under much investigation. Evidence shows that under non-activated conditions, microglia are likely to rely on oxidative metabolism [87]. However, upon stimulation, microglia switch from oxidative metabolism to glycolytic metabolism to support their shift in activation state [87, 88]. In murine BV-2 microglia cells, lipopolysaccharide (LPS) stimulation led to the upregulation of pro-inflammatory genes. Under these conditions, the cells increased glucose uptake and shifted from oxidative metabolism towards glycolytic metabolism [89]. These cells were unresponsive to various mitochondrial stressors, suggesting a loss of mitochondrial function [87]. Activated BV-2 cells exhibit decreased oxygen consumption rates and increased lactate release highlighting the shift from oxidative metabolism towards glycolytic metabolism [89]. It is proposed that during this shift, cells preferentially utilize glycolysis rather than oxidative phosphorylation in an effort to preserve and generate the metabolic resources needed to meet the demands associated with cellular proliferation and activation while still producing a sufficient supply of ATP. Microglial metabolism and mitochondrial dysfunction, in the context of HIV infection, is largely understudied. However, one group observed that Tat significantly reduced mitochondrial membrane potential in mouse primary microglia [90]. Additionally, reductions in cellular energetics was made evident by decreased basal and maximal respiration, decreased ATP production, and reduced extracellular acidification, a measure of glycolysis [90]. These results demonstrate that Tat mediates mitochondrial dysfunction in microglia. Furthermore, mitochondrial dysfunction can also trigger an inflammatory response in microglia [91]. Thus, Tat-mediated mitochondrial dysfunction may fuel chronic inflammation in response to HIV infection of microglia.
4.2.3. Oligodendrocytes
Oligodendrocytes (OLs) synthesize myelin, a protein- and lipid- rich membrane that promotes fast axonal conduction. However, the myelin sheath restricts assess of axons to extracellular metabolites [92, 93]. Therefore, in addition to aiding action potential propagation, oligodendrocytes provide energy substrates to the axons that they sheath [94, 95]. Reminiscent of the astrocyte-neuron lactate shuttle, there also exists an oligodendrocyte-axon metabolic coupling in which cytoplasmic “myelinic” channels and monocarboxylate transporters allow energy substrates, such as lactate and pyruvate, to be transferred from oligodendrocytes to neurons to generate ATP [96, 97]. Oligodendrocytes have high energy demands and are therefore highly sensitive to energy deprivation [98]. This can ultimately lead to insufficient metabolic support to neurons and neuronal dysfunction. Disruption of oligodendrocyte-axon coupling may contribute to disease pathogenesis. In the case of MS, a demyelinating disorder of the CNS, metabolic dysfunction is commonly associated with disease pathogenesis [99]. Several groups have shown that disruption of oligodendrocyte lactate supply leads to axonal dysfunction and neuronal degeneration [94]. Oligodendrocytes display similar rates of glycolysis as astrocytes [96, 100]. However, during development, oligodendrocytes use oxidative phosphorylation to meet the high energy demands needed to synthesize myelin. As oligodendrocytes mature, a metabolic shift occurs from oxidative metabolism to glycolysis to generate ATP. Metabolic stress leads oligodendrocytes to use their energy substrates for survival rather than myelin maintenance [101]. Clinical observations of myelin loss and white matter damage are frequently reported in PWH. Many studies show preferential damage to the white matter in the HIV-1 infected brain [102–105]. This points to oligodendrocyte injury as another neuropathological consequence of HIV infection. Although there are some reports in vitro of HIV infection in oligodendrocytes [106], there is no evidence to support these findings in vivo [107, 108]. Therefore, similar to neurons, HIV-induced oligodendrocyte injury likely occurs from both exposure to HIV proteins and secondary inflammatory responses [109–112]. Both gp120 and Tat cause increased intracellular Ca2+ in oligodendrocyte cultures to levels which ultimately lead to cellular injury [110]. gp120 hampers the ability of oligodendrocytes to reduce the tetrazolium salt MTT, which is used as an indicator of mitochondrial activity [111]. This suggests that oligodendrocytes may exhibit functional impairments in mitochondrial activity however, this is a research area that requires much more exploration.
5. Additional factors which may influence metabolism in PWH
5.1. Combination Antiretroviral Therapy (CART)
Although the use of CART has reduced the incidence of HAD, the prevalence of the milder forms of HAND remain high [113, 114]. Antiretrovirals (ARV) compounds target key steps in the viral replication cycle including viral entry inhibitors, nucleoside reverse transcriptase inhibitors (NRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI), protease inhibitors (PI), fusion inhibitors (FI), chemokine receptor CCR5 blockers, and integrase strand transfer inhibitors (INSTI). A combination of these compounds is used to assist in preventing the emergence of drug-resistant HIV mutant strains. There is a growing body of evidence that components of CART can be neurotoxic upon long-term use. Studies following patients after discontinuation of CART revealed an unexpected improvement in cognition [115]. Additionally, another study found that those on a CART regime with higher CNS penetrance-effectiveness (CPE) resulted in more effective suppression of viral replication in the CSF, yet had poorer performance on neurological testing [116]. This implies that CART may have neurotoxic effects and contribute to cognitive impairments. An MRS study revealed that PWH taking NRTIs had lower levels of NAA in the frontal white matter compared to HIV negative individuals and PWH on a different CART regime [117]. Longer periods of NRTIs treatment were also associated with diminished NAA in these individuals [117]. In effort to gain further understanding of potential CART neurotoxicity, two animal models were utilized: 1) SIV-infected pigtail macaques either receiving no CART or receiving early CART and 2) adult rats administered various combinations of ARV drugs [118]. Results from these studies indicate CART alone causes synaptic damage[118]. In vitro, studies further demonstrate ARV toxicity. In rat mixed neuronal-glial cerebrocortical cultures, exposure to individual or combinations of antiretrovirals compounds reduced synaptophysin and MAP2 expression suggesting damage to dendrites and presynaptic terminals [60]. Neuronal damage caused by ARV manifests as beading, simplification of the dendritic processes, and neuronal shrinkage [119]. Signs of altered energy profiles and mitochondrial toxicity by ARV are also reported. Accumulation of ROS in response to various ARV drugs occurs in neurons, astrocytes and oligodendrocyte precursor cells [118, 120, 121]. Mitochondrial membrane potential and morphology are also altered by various ARV [83]. Furthermore, neuronal ATP was significantly diminished after exposure to ARV compounds [60]. Exposure to various ARV drugs disrupts presynaptic striatal nerve terminals mitochondrial function by reducing maximal mitochondrial respiration along with a drop in ATP production [122]. Efavirenz (EFV) is an NNRTI is which widely prescribed as an anti-HIV agent however, many patients experience CNS-related side effects including impaired concentration and cognitive deficits [123]. Several groups have demonstrated that EFV causes mitochondrial alterations in neuronal cell lines and primary neuron cultures, including decreased ATP production, and mitochondrial fragmentation and depolarization [124, 125]. Considering this evidence, one group assessed the effects of EFV on neuronal and astrocyte bioenergetics [123]. They reported reduced mitochondrial membrane potential, decreased mitochondrial respiration, and increased ROS generation in both neuronal and astrocyte cultures [123]. Interestingly, EFV diminished neuronal ATP while increasing astrocyte ATP levels. This was a result of EFV-induced activation of AMPK leading to the upregulation of glycolysis in astrocytes [123]. Another study exposed human astrocytes to a combination of ARV compounds and found increased glucose utilization, enhanced glycolysis, and increased mitochondrial metabolism suggesting that astrocytes undergo an enhanced energy state during ARV treatment [121]. Unsurprisingly, in addition to the mitochondrial dysfunction associated with ARV, ER stress was also induced by these compounds [126]. The neurotoxicity of ARV drugs varies depending on the drug class and the individual drug. Therefore, as efforts to increase the efficacy of CNS penetrance of ARV gains more attention, it is important to further explore the impact these compounds have on brain metabolism and subsequently cognition.
5.2. Drugs of Abuse
Substance abuse is a major comorbidity associated with HIV infection and there is evidence that drug use may have an additive or synergetic effect on the progression and severity of HAND [127]. Similar to HIV, drugs of abuse target the CNS and alter both neuronal and glial function. Clinical studies suggest that PWH who abuse drugs have more rapidly progressing disease, with higher viral loads, and increased cognitive impairment [128–132]sa. In the SIV-macaque model, exposure to opioids and methamphetamine (meth) increased brain and CSF viral loads [133, 134]. Cocaine, methamphetamines, and opioids have been demonstrated to increase HIV replication in rodent models and in vitro [135–138] thus, supporting a role for drugs of abuse to potentially increase neuroinflammation and neurodegeneration and possibly disrupt brain metabolism. Independently, drugs of abuse have been demonstrated to alter brain metabolism during active use and during periods of abstinence [139–145]. Drug use alone is associated with significant microgliosis [146]. These changes are proposed to contribute to drug seeking, cravings, and relapse behaviors and therefore brain metabolism may play a major role in the neurobiology of addiction. Changes in brain metabolism are also observed in meth users [147]. Furthermore, cocaine reduces regional brain glucose uptake and glucose metabolism in cocaine abusers and in mouse models of chronic cocaine use [148, 149]. Moreover, in the hippocampus of long-term cocaine users downregulation of mitochondrial oxidative phosphorylation genes was noted [150]. This suggests that energy pathways are diminished in drug users and may be further exacerbated in PWH who abuse drugs and are already in a hypometabolic state. In mice, it was found that cocaine induced a metabolic switch from glycolysis to fatty acid oxidation and ketone metabolism leading to enhanced activation of the TCA cycle, oxidative respiration, and a drastic increase in consumption of ATP and acetyl-CoA in the nucleus accumbens [151]. In CHME-5, an immortalized microglial cell line, cocaine with HIV infection or exposure to HIV gp120 protein had an additive effect on cell energetics by increasing ATP concentrations, glycolysis, and oxidative phosphorylation [152]. This is likely to accommodate the enhanced energy requirements as the microglia transition to an activated phenotype. In rat hippocampal neurons, cocaine enhanced the Tat-induced decrease in mitochondrial membrane potential and exacerbated ROS production [153]. Conversely, exposure to Tat and cocaine enhanced mitochondrial metabolism in astrocytes increasing ATP levels [84]. In the case of meth, significant loss of ATP levels in rat cerebrocortical neuronal cells has been reported and this is aggravated in the presence of gp120 [60]. Astrocytes respond to meth by altered mitochondrial morphology through increased fusion which resulted in increased oxidative capacity and ATP levels [83]. Striatal neuronal cultures treated with morphine and Tat interact to enhance mitochondrial dysfunction and increase intracellular Ca2+ [34]. It is apparent that drugs of abuse and HIV interact to disturb bioenergetics within the brain.
5.3. Aging
As previously highlighted, the success of CART has prolonged the lives of PWH to near normal life expectancy [154], so that PWH are now becoming an aging population. Living longer with HIV puts this population at risk for developing HAND, and at greater risk for pathological changes associated with aging. Normal aging is associated with decreased normal brain functioning [155] resulting in impairments in attention and memory relative to younger adults [156] although, this is not indicative of overt neurological disease. However, in PWH reports of deviations from the normal trajectory of brain aging has introduced the idea that HIV may lead to accelerated aging [22, 157–159]. One study comparing a large cohort of HIV positive males to HIV negative males reported the median age of first neurocognitive impairment in HIV positive men was 48 years old compared to 57 years old in the uninfected men [71]. This suggests that in PWH even on an effective CART regime, these disorders present at a younger age than are observed in HIV negative control groups [71]. In a longitudinal study assessing the effects of aging and HIV on memory, investigators found that HIV and age interact to produce greater declines in verbal memory over time [160]. Furthermore, PWH have a higher burden of cognitive impairments with advancing age than uninfected individuals without HIV [161]. In a cross-sectional analysis of CSF metabolomics, many of the metabolites altered in the PWH (median age 40) overlapped with older HIV negative control patients (median age 57) [22]. These metabolites include markers of glia activation (myo-inositol), mitochondrial dysfunction (succinate), oxidative stress (urate and hypoxanthine), and metabolic waste products (ketone bodies) [22]. Moreover, rapid brain aging appears to be associated with altered metabolism in PWH as they age [22].
6. Concluding Remarks
Taken together, HIV infection has a multifactorial impact on brain energetics at the cellular level leading to disruptions in whole brain metabolic homeostasis. HIV infection of the CNS not only triggers a cycle of chronic inflammation which enhances the energy demands of glial cells but also hampers the ability of glial cells to support neurons, while simultaneously dampening neuronal energy pathways needed to maintain the necessary energy homeostasis to facilitate normal CNS functioning. Intriguingly, HIV induces cell-specific alterations that exert major global effects on the brain. HIV causes chronic inflammation in microglia which release virions, viral proteins, and cytokines that can act on astrocytes and oligodendrocytes to augment their energy pathways. Oligodendrocytes and astrocytes react by shifting away from neurotrophic metabolic support of neurons and instead fail to sustain neuronal needs. Neurotoxicity associated with HIV infection directly challenges neuronal metabolism under conditions where neurons are already being deprived of metabolic substrates. Overall, it is likely that altered brain energy dynamics result in neurocognitive impairments associated with HIV infection. Viral eradication, the ultimate cure for HIV infection, remains elusive. Therefore, efforts to restore the energy balance in the brain through approaches that increase energy substrate bioavailability including, but not limited to implementation of a ketogenic diet [65, 162, 163], intranasal insulin treatment [164], or exercise [165] coupled with use of anti-inflammatory drugs may provide therapeutic benefits to PWH.
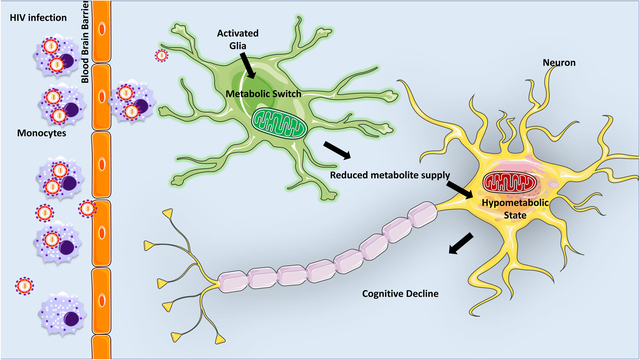
Figure 1.

Proposed model of overall metabolic status of CNS during HIV infection. HIV enters the CNS likely in HIV-infected peripheral monocytes. Infected microglia undergo a proinflammatory response. Subsequently, HIV particles, viral proteins, and proinflammatory mediators released by microglia can activate other microglia and neighboring astrocytes thus creating a state of chronic inflammation. Maintenance of HIV-induced inflammation is metabolically expensive and thus, induces a metabolic switch in glia cells. As astrocytes and oligodendrocytes work to fulfill their own enhanced energy requirements, these cells are less able to provide metabolic support to neurons. Compromised glia support in combination with direct neurotoxicity of HIV challenges neuronal metabolism, which can lead to neuronal energy deficits, hinder neuronal fitness and present as neurocognitive impairments. (Abbreviations: OXPHOS- oxidative phosphorylation, ATP-adenosine triphosphate, MMP- mitochondrial membrane potential, LDH-lactate dehydrogenase, FAO-fatty acid oxidation, TCA-tricarboxylic acid, NAA- N-acetylaspartate, ROS-reactive oxygen species).
Highlights.
Abnormal brain metabolic profiles of people with HIV coincide with cognitive deficits.
HIV-associated stressors (viral particles, viral proteins and cytokines) directly disrupt normal glial metabolism that compromises their ability to support neuronal metabolism
Additional factors such as combination antiviral therapy, aging and substance abuse can exacerbate HIV-associated metabolic disturbances and accelerate disease progression.
Acknowledgements:
This work was supported by the National Institutes of Health R01MH107340, P01DA037830, P30MH092177.
References
- 1.Maki PM, et al. , Cognitive function in women with HIV: findings from the Women’s Interagency HIV Study. Neurology, 2015. 84(3): p. 231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubin LH, et al. , Cognitive trajectories over 4 years among HIV-infected women with optimal viral suppression. Neurology, 2017. 89(15): p. 1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouillette MJ, et al. , Identifying Neurocognitive Decline at 36 Months among HIV-Positive Participants in the CHARTER Cohort Using Group-Based Trajectory Analysis. PLoS One, 2016. 11(5): p. e0155766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antinori A, et al. , Updated research nosology for HIV-associated neurocognitive disorders. Neurology, 2007. 69(18): p. 1789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saylor D, et al. , HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat Rev Neurol, 2016. 12(4): p. 234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Everall IP, et al. , Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol, 1999. 9(2): p. 209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gray F, et al. , Prominent cortical atrophy with neuronal loss as correlate of human immunodeficiency virus encephalopathy. Acta Neuropathol, 1991. 82(3): p. 229–33. [DOI] [PubMed] [Google Scholar]
- 8.Sa MJ, et al. , Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol, 2004. 107(2): p. 97–110. [DOI] [PubMed] [Google Scholar]
- 9.Belanger M, Allaman I, and Magistretti PJ, Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab, 2011. 14(6): p. 724–38. [DOI] [PubMed] [Google Scholar]
- 10.Attwell D and Laughlin SB, An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab, 2001. 21(10): p. 1133–45. [DOI] [PubMed] [Google Scholar]
- 11.Mink JW, Blumenschine RJ, and Adams DB, Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol, 1981. 241(3): p. R203–12. [DOI] [PubMed] [Google Scholar]
- 12.Ratai EM, et al. , Brain creatine elevation and N-Acetylaspartate reduction indicates neuronal dysfunction in the setting of enhanced glial energy metabolism in a macaque model of neuroAIDS. Magn Reson Med, 2011. 66(3): p. 625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dickens AM, et al. , Cerebrospinal fluid metabolomics implicate bioenergetic adaptation as a neural mechanism regulating shifts in cognitive states of HIV-infected patients. AIDS, 2015. 29(5): p. 559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young AC, et al. , Cerebral metabolite changes prior to and after antiretroviral therapy in primary HIV infection. Neurology, 2014. 83(18): p. 1592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rottenberg DA, et al. , Abnormal cerebral glucose metabolism in HIV-1 seropositive subjects with and without dementia. J Nucl Med, 1996. 37(7): p. 1133–41. [PubMed] [Google Scholar]
- 16.Zielke HR, Zielke CL, and Baab PJ, Direct measurement of oxidative metabolism in the living brain by microdialysis: a review. J Neurochem, 2009. 109 Suppl 1: p. 24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z and Zhong C, Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol, 2013. 108: p. 21–43. [DOI] [PubMed] [Google Scholar]
- 18.Abolhassani N, et al. , Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech Ageing Dev, 2017. 161(Pt A): p. 95–104. [DOI] [PubMed] [Google Scholar]
- 19.Peng S, Eidelberg D, and Ma Y, Brain network markers of abnormal cerebral glucose metabolism and blood flow in Parkinson’s disease. Neurosci Bull, 2014. 30(5): p. 823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathur D, et al. , Perturbed glucose metabolism: insights into multiple sclerosis pathogenesis. Front Neurol, 2014. 5: p. 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ernst T, Chang L, and Arnold S, Increased glial metabolites predict increased working memory network activation in HIV brain injury. Neuroimage, 2003. 19(4): p. 1686–93. [DOI] [PubMed] [Google Scholar]
- 22.Cassol E, et al. , Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. AIDS, 2014. 28(11): p. 1579–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bates TE, et al. , Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport, 1996. 7(8): p. 1397–400. [PubMed] [Google Scholar]
- 24.Chang L, et al. , Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J Neuroimmune Pharmacol, 2013. 8(3): p. 576–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harezlak J, et al. , Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS, 2011. 25(5): p. 625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul RH, et al. , Relative sensitivity of magnetic resonance spectroscopy and quantitative magnetic resonance imaging to cognitive function among nondemented individuals infected with HIV. J Int Neuropsychol Soc, 2008. 14(5): p. 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boska MD, et al. , Associations between brain microstructures, metabolites, and cognitive deficits during chronic HIV-1 infection of humanized mice. Mol Neurodegener, 2014. 9: p. 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen AB, et al. , Cerebral FDG-PET scanning abnormalities in optimally treated HIV patients. J Neuroinflammation, 2010. 7: p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Towgood KJ, et al. , Regional cerebral blood flow and FDG uptake in asymptomatic HIV-1 men. Hum Brain Mapp, 2013. 34(10): p. 2484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou L, et al. , First evidence of overlaps between HIV-Associated Dementia (HAD) and non-viral neurodegenerative diseases: proteomic analysis of the frontal cortex from HIV+ patients with and without dementia. Mol Neurodegener, 2010. 5: p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zündorf G and Reiser G, Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal, 2011. 14(7): p. 1275–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gleichmann M, et al. , Simultaneous single neuron recording of O2 consumption, [Ca2+]i and mitochondrial membrane potential in glutamate toxicity. J Neurochem, 2009. 109(2): p. 644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi T, et al. , MAM: more than just a housekeeper. Trends Cell Biol, 2009. 19(2): p. 81–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fitting S, et al. , Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na⁺ influx, mitochondrial instability, and Ca²⁺ overload. J Neurosci, 2014. 34(38): p. 12850–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitting S, et al. , Opiate addiction therapies and HIV-1 Tat: interactive effects on glial [Ca²⁺]i, oxyradical and neuroinflammatory chemokine production and correlative neurotoxicity. Curr HIV Res, 2014. 12(6): p. 424–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kruman II, Nath A, and Mattson MP, HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol, 1998. 154(2): p. 276–88. [DOI] [PubMed] [Google Scholar]
- 37.Ciardo A and Meldolesi J, Effects of the HIV-1 envelope glycoprotein gp120 in cerebellar cultures. [Ca2+]i increases in a glial cell subpopulation. Eur J Neurosci, 1993. 5(12): p. 1711–8. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP, Gleichmann M, and Cheng A, Mitochondria in neuroplasticity and neurological disorders. Neuron, 2008. 60(5): p. 748–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schon EA and Przedborski S, Mitochondria: the next (neurode)generation. Neuron, 2011. 70(6): p. 1033–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett GJ, Doyle T, and Salvemini D, Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol, 2014. 10(6): p. 326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Opii WO, et al. , Oxidative stress and toxicity induced by the nucleoside reverse transcriptase inhibitor (NRTI)−-2’,3’-dideoxycytidine (ddC): relevance to HIV-dementia. Exp Neurol, 2007. 204(1): p. 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levine AJ, et al. , Systems analysis of human brain gene expression: mechanisms for HIV-associated neurocognitive impairment and common pathways with Alzheimer’s disease. BMC Med Genomics, 2013. 6: p. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Repunte-Canonigo V, et al. , Gene expression changes consistent with neuroAIDS and impaired working memory in HIV-1 transgenic rats. Mol Neurodegener, 2014. 9: p. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villeneuve LM, et al. , HIV-1 transgenic rats display mitochondrial abnormalities consistent with abnormal energy generation and distribution. J Neurovirol, 2016. 22(5): p. 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fields JA, et al. , HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol Dis, 2016. 86: p. 154–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomes LC, Di Benedetto G, and Scorrano L, During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol, 2011. 13(5): p. 589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uzasci L, Nath A, and Cotter R, Oxidative stress and the HIV-infected brain proteome. J Neuroimmune Pharmacol, 2013. 8(5): p. 1167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakamura H, Masutani H, and Yodoi J, Redox imbalance and its control in HIV infection. Antioxid Redox Signal, 2002. 4(3): p. 455–64. [DOI] [PubMed] [Google Scholar]
- 49.Turchan J, et al. , Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology, 2003. 60(2): p. 307–14. [DOI] [PubMed] [Google Scholar]
- 50.Herrero-Mendez A, et al. , The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol, 2009. 11(6): p. 747–52. [DOI] [PubMed] [Google Scholar]
- 51.Almeida A, Moncada S, and Bolaños JP, Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol, 2004. 6(1): p. 45–51. [DOI] [PubMed] [Google Scholar]
- 52.Hayakawa Y, et al. , Rapid Ca2+-dependent increase in oxygen consumption by mitochondria in single mammalian central neurons. Cell Calcium, 2005. 37(4): p. 359–70. [DOI] [PubMed] [Google Scholar]
- 53.Stevens PR, et al. , Creatine protects against mitochondrial dysfunction associated with HIV-1 Tat-induced neuronal injury. Curr HIV Res, 2014. 12(6): p. 378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avdoshina V, et al. , The HIV Protein gp120 Alters Mitochondrial Dynamics in Neurons. Neurotox Res, 2016. 29(4): p. 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah A and Kumar A, HIV-1 gp120-Mediated Mitochondrial Dysfunction and HIV-Associated Neurological Disorders. Neurotox Res, 2016. 30(2): p. 135–7. [DOI] [PubMed] [Google Scholar]
- 56.De Simone FI, et al. , HIV-1 Tat and Cocaine Impair Survival of Cultured Primary Neuronal Cells via a Mitochondrial Pathway. J Neuroimmune Pharmacol, 2016. 11(2): p. 358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Norman JP, et al. , HIV-1 trans activator of transcription protein elicits mitochondrial hyperpolarization and respiratory deficit, with dysregulation of complex IV and nicotinamide adenine dinucleotide homeostasis in cortical neurons. J Immunol, 2007. 178(2): p. 869–76. [DOI] [PubMed] [Google Scholar]
- 58.Rozzi SJ, et al. , Human Immunodeficiency Virus Promotes Mitochondrial Toxicity. Neurotox Res, 2017. 32(4): p. 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teodorof-Diedrich C and Spector SA, Human Immunodeficiency Virus Type-1 gp120 and Tat Induce Mitochondrial Fragmentation and Incomplete Mitophagy in Human Neurons. J Virol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanchez AB, et al. , Antiretrovirals, Methamphetamine, and HIV-1 Envelope Protein gp120 Compromise Neuronal Energy Homeostasis in Association with Various Degrees of Synaptic and Neuritic Damage. Antimicrob Agents Chemother, 2016. 60(1): p. 168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitayama H, et al. , Human immunodeficiency virus type 1 Vpr inhibits axonal outgrowth through induction of mitochondrial dysfunction. J Virol, 2008. 82(5): p. 2528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, et al. , HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology, 2017. 117: p. 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perry SW, et al. , HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol, 2005. 174(7): p. 4333–44. [DOI] [PubMed] [Google Scholar]
- 64.Tiede LM, et al. , Oxygen matters: tissue culture oxygen levels affect mitochondrial function and structure as well as responses to HIV viroproteins. Cell Death Dis, 2011. 2: p. e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hui L, et al. , Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J Neurochem, 2012. 122(2): p. 382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lecoeur H, et al. , HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis, 2012. 3: p. e282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rose J, et al. , Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology, 2017. 391: p. 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.von Bartheld CS, Bahney J, and Herculano-Houzel S, The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J Comp Neurol, 2016. 524(18): p. 3865–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barros LF, et al. , Preferential transport and metabolism of glucose in Bergmann glia over Purkinje cells: a multiphoton study of cerebellar slices. Glia, 2009. 57(9): p. 962–70. [DOI] [PubMed] [Google Scholar]
- 70.Jiang T and Cadenas E, Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell, 2014. 13(6): p. 1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mateen FJ, et al. , Neurologic disorders incidence in HIV+ vs HIV- men: Multicenter AIDS Cohort Study, 1996–2011. Neurology, 2012. 79(18): p. 1873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson AR, Milner JJ, and Makowski L, The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev, 2012. 249(1): p. 218–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vitkovic L and da Cunha A, Role for astrocytosis in HIV-1-associated dementia. Curr Top Microbiol Immunol, 1995. 202: p. 105–16. [DOI] [PubMed] [Google Scholar]
- 74.Desplats P, et al. , Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology, 2013. 80(15): p. 1415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou BY, et al. , Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci, 2004. 27(3): p. 296–305. [DOI] [PubMed] [Google Scholar]
- 76.Deshpande M, et al. , Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox Res, 2005. 7(3): p. 183–92. [DOI] [PubMed] [Google Scholar]
- 77.Nooka S and Ghorpade A, HIV-1-associated inflammation and antiretroviral therapy regulate astrocyte endoplasmic reticulum stress responses. Cell Death Discov, 2017. 3: p. 17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fan Y and He JJ, HIV-1 Tat Induces Unfolded Protein Response and Endoplasmic Reticulum Stress in Astrocytes and Causes Neurotoxicity through Glial Fibrillary Acidic Protein (GFAP) Activation and Aggregation. J Biol Chem, 2016. 291(43): p. 22819–22829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shah A, et al. , HIV-1 gp120 induces type-1 programmed cell death through ER stress employing IRE1α, JNK and AP-1 pathway. Sci Rep, 2016. 6: p. 18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shah A, et al. , HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis, 2013. 4: p. e850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang Y, et al. , Cocaine potentiates astrocyte toxicity mediated by human immunodeficiency virus (HIV-1) protein gp120. PLoS One, 2010. 5(10): p. e13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eugenin EA and Berman JW, Cytochrome C dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. J Neurochem, 2013. 127(5): p. 644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borgmann K and Ghorpade A, Methamphetamine Augments Concurrent Astrocyte Mitochondrial Stress, Oxidative Burden, and Antioxidant Capacity: Tipping the Balance in HIV-Associated Neurodegeneration. Neurotox Res, 2018. 33(2): p. 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Natarajaseenivasan K, et al. , Astrocytic metabolic switch is a novel etiology for Cocaine and HIV-1 Tat-mediated neurotoxicity. Cell Death Dis, 2018. 9(4): p. 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.González-Scarano F and Martín-García J, The neuropathogenesis of AIDS. Nat Rev Immunol, 2005. 5(1): p. 69–81. [DOI] [PubMed] [Google Scholar]
- 86.Garvey LJ, et al. , Increased microglia activation in neurologically asymptomatic HIV-infected patients receiving effective ART. AIDS, 2014. 28(1): p. 67–72. [DOI] [PubMed] [Google Scholar]
- 87.Orihuela R, McPherson CA, and Harry GJ, Microglial M1/M2 polarization and metabolic states. Br J Pharmacol, 2016. 173(4): p. 649–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gimeno-Bayón J, et al. , Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res, 2014. 92(6): p. 723–31. [DOI] [PubMed] [Google Scholar]
- 89.Voloboueva LA, et al. , Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett, 2013. 587(6): p. 756–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thangaraj A, et al. , HIV-1 TAT-mediated microglial activation: role of mitochondrial dysfunction and defective mitophagy. Autophagy, 2018. 14(9): p. 1596–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.West AP, Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology, 2017. 391: p. 54–63. [DOI] [PubMed] [Google Scholar]
- 92.Nave KA, Myelination and support of axonal integrity by glia. Nature, 2010. 468(7321): p. 244–52. [DOI] [PubMed] [Google Scholar]
- 93.Nave KA, Myelination and the trophic support of long axons. Nat Rev Neurosci, 2010. 11(4): p. 275–83. [DOI] [PubMed] [Google Scholar]
- 94.Lee Y, et al. , Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature, 2012. 487(7408): p. 443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meyer N, et al. , Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep, 2018. 22(9): p. 2383–2394. [DOI] [PubMed] [Google Scholar]
- 96.Funfschilling U, et al. , Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature, 2012. 485(7399): p. 517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Philips T and Rothstein JD, Oligodendroglia: metabolic supporters of neurons. J Clin Invest, 2017. 127(9): p. 3271–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dewar D, Underhill SM, and Goldberg MP, Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab, 2003. 23(3): p. 263–74. [DOI] [PubMed] [Google Scholar]
- 99.Adiele RC and Adiele CA, Metabolic defects in multiple sclerosis. Mitochondrion, 2017. [DOI] [PubMed] [Google Scholar]
- 100.Amaral AI, et al. , Characterization of glucose-related metabolic pathways in differentiated rat oligodendrocyte lineage cells. Glia, 2016. 64(1): p. 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rone MB, et al. , Oligodendrogliopathy in Multiple Sclerosis: Low Glycolytic Metabolic Rate Promotes Oligodendrocyte Survival. J Neurosci, 2016. 36(17): p. 4698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen Y, et al. , White matter abnormalities revealed by diffusion tensor imaging in non-demented and demented HIV+ patients. Neuroimage, 2009. 47(4): p. 1154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Corrêa DG, et al. , Diffusion tensor MR imaging of white matter integrity in HIV-positive patients with planning deficit. Neuroradiology, 2015. 57(5): p. 475–82. [DOI] [PubMed] [Google Scholar]
- 104.Gongvatana A, et al. , White matter tract injury and cognitive impairment in human immunodeficiency virus-infected individuals. J Neurovirol, 2009. 15(2): p. 187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wohlschlaeger J, et al. , White matter changes in HIV-1 infected brains: a combined gross anatomical and ultrastructural morphometric investigation of the corpus callosum. Clin Neurol Neurosurg, 2009. 111(5): p. 422–9. [DOI] [PubMed] [Google Scholar]
- 106.Albright AV, et al. , HIV-1 infection of cultured human adult oligodendrocytes. Virology, 1996. 217(1): p. 211–9. [DOI] [PubMed] [Google Scholar]
- 107.Takahashi K, et al. , Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann Neurol, 1996. 39(6): p. 705–11. [DOI] [PubMed] [Google Scholar]
- 108.Sharpless N, et al. , The restricted nature of HIV-1 tropism for cultured neural cells. Virology, 1992. 191(2): p. 813–25. [DOI] [PubMed] [Google Scholar]
- 109.Hauser KF, et al. , HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia, 2009. 57(2): p. 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zou S, et al. , Oligodendrocytes Are Targets of HIV-1 Tat: NMDA and AMPA Receptor-Mediated Effects on Survival and Development. J Neurosci, 2015. 35(32): p. 11384–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bernardo A, Agresti C, and Levi G, HIV-gp120 affects the functional activity of oligodendrocytes and their susceptibility to complement. J Neurosci Res, 1997. 50(6): p. 946–57. [DOI] [PubMed] [Google Scholar]
- 112.Kimura-Kuroda J, Nagashima K, and Yasui K, Inhibition of myelin formation by HIV-1 gp120 in rat cerebral cortex culture. Arch Virol, 1994. 137(1–2): p. 81–99. [DOI] [PubMed] [Google Scholar]
- 113.Heaton RK, et al. , HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology, 2010. 75(23): p. 2087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heaton RK, et al. , HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol, 2011. 17(1): p. 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robertson KR, et al. , Neurocognitive effects of treatment interruption in stable HIV-positive patients in an observational cohort. Neurology, 2010. 74(16): p. 1260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marra CM, et al. , Impact of combination antiretroviral therapy on cerebrospinal fluidz HIV RNA and neurocognitive performance. AIDS, 2009. 23(11): p. 1359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schweinsburg BC, et al. , Brain mitochondrial injury in human immunodeficiency virus-seropositive (HIV+) individuals taking nucleoside reverse transcriptase inhibitors. J Neurovirol, 2005. 11(4): p. 356–64. [DOI] [PubMed] [Google Scholar]
- 118.Akay C, et al. , Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J Neurovirol, 2014. 20(1): p. 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Robertson K, Liner J, and Meeker RB, Antiretroviral neurotoxicity. J Neurovirol, 2012. 18(5): p. 388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jensen BK, et al. , Altered Oligodendrocyte Maturation and Myelin Maintenance: The Role of Antiretrovirals in HIV-Associated Neurocognitive Disorders. J Neuropathol Exp Neurol, 2015. 74(11): p. 1093–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cohen J, et al. , Astrocyte Senescence and Metabolic Changes in Response to HIV Antiretroviral Therapy Drugs. Front Aging Neurosci, 2017. 9: p. 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stauch KL, et al. , Central nervous system-penetrating antiretrovirals impair energetic reserve in striatal nerve terminals. J Neurovirol, 2017. 23(6): p. 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Funes HA, et al. , Neuronal bioenergetics and acute mitochondrial dysfunction: a clue to understanding the central nervous system side effects of efavirenz. J Infect Dis, 2014. 210(9): p. 1385–95. [DOI] [PubMed] [Google Scholar]
- 124.Purnell PR and Fox HS, Efavirenz induces neuronal autophagy and mitochondrial alterations. J Pharmacol Exp Ther, 2014. 351(2): p. 250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ciavatta VT, et al. , In vitro and Ex vivo Neurotoxic Effects of Efavirenz are Greater than Those of Other Common Antiretrovirals. Neurochem Res, 2017. 42(11): p. 3220–3232. [DOI] [PubMed] [Google Scholar]
- 126.Bertrand L and Toborek M, Dysregulation of Endoplasmic Reticulum Stress and Autophagic Responses by the Antiretroviral Drug Efavirenz. Mol Pharmacol, 2015. 88(2): p. 304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gill AJ and Kolson DL, Chronic inflammation and the role for cofactors (hepatitis C, drug abuse, antiretroviral drug toxicity, aging) in HAND persistence. Curr HIV/AIDS Rep, 2014. 11(3): p. 325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bell JE, et al. , HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain, 1998. 121 (Pt 11): p. 2043–52. [DOI] [PubMed] [Google Scholar]
- 129.Nath A, et al. , Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol, 2001. 7(1): p. 66–71. [DOI] [PubMed] [Google Scholar]
- 130.Langford D, et al. , Patterns of selective neuronal damage in methamphetamine-user AIDS patients. J Acquir Immune Defic Syndr, 2003. 34(5): p. 467–74. [DOI] [PubMed] [Google Scholar]
- 131.Lucas GM, et al. , Illicit drug use and HIV-1 disease progression: a longitudinal study in the era of highly active antiretroviral therapy. Am J Epidemiol, 2006. 163(5): p. 412–20. [DOI] [PubMed] [Google Scholar]
- 132.Rippeth JD, et al. , Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J Int Neuropsychol Soc, 2004. 10(1): p. 1–14. [DOI] [PubMed] [Google Scholar]
- 133.Marcondes MC, et al. , Methamphetamine increases brain viral load and activates natural killer cells in simian immunodeficiency virus-infected monkeys. Am J Pathol, 2010. 177(1): p. 355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Czub S, et al. , Modulation of simian immunodeficiency virus neuropathology by dopaminergic drugs. Acta Neuropathol, 2004. 107(3): p. 216–26. [DOI] [PubMed] [Google Scholar]
- 135.Kim SG, et al. , Cocaine-mediated impact on HIV infection in humanized BLT mice. Sci Rep, 2015. 5: p. 10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dhillon NK, et al. , Cocaine-mediated enhancement of virus replication in macrophages: implications for human immunodeficiency virus-associated dementia. J Neurovirol, 2007. 13(6): p. 483–95. [DOI] [PubMed] [Google Scholar]
- 137.Liang H, et al. , Methamphetamine enhances HIV infection of macrophages. Am J Pathol, 2008. 172(6): p. 1617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schweitzer C, et al. , Morphine stimulates HIV replication in primary cultures of human Kupffer cells. Res Virol, 1991. 142(2–3): p. 189–95. [DOI] [PubMed] [Google Scholar]
- 139.Lehrmann E, et al. , Transcriptional profiling in the human prefrontal cortex: evidence for two activational states associated with cocaine abuse. Pharmacogenomics J, 2003. 3(1): p. 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li Y, et al. , ¹H NMR-based metabonomics in brain nucleus accumbens and striatum following repeated cocaine treatment in rats. Neuroscience, 2012. 218: p. 196–205. [DOI] [PubMed] [Google Scholar]
- 141.Deng Y, et al. , (1) H-nuclear magnetic resonance-based metabonomic analysis of brain in rhesus monkeys with morphine treatment and withdrawal intervention. J Neurosci Res, 2012. 90(11): p. 2154–62. [DOI] [PubMed] [Google Scholar]
- 142.Hermann D, et al. , Dorsolateral prefrontal cortex N-acetylaspartate/total creatine (NAA/tCr) loss in male recreational cannabis users. Biol Psychiatry, 2007. 61(11): p. 1281–9. [DOI] [PubMed] [Google Scholar]
- 143.Fonseca R, et al. , Methamphetamine Induces Anhedonic-Like Behavior and Impairs Frontal Cortical Energetics in Mice. CNS Neurosci Ther, 2017. 23(2): p. 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim JE, et al. , Metabolic alterations in the anterior cingulate cortex and related cognitive deficits in late adolescent methamphetamine users. Addict Biol, 2018. 23(1): p. 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Crocker CE, et al. , Enduring changes in brain metabolites and executive functioning in abstinent cocaine users. Drug Alcohol Depend, 2017. 178: p. 435–442. [DOI] [PubMed] [Google Scholar]
- 146.Tomlinson GS, et al. , Upregulation of microglia in drug users with and without pre-symptomatic HIV infection. Neuropathol Appl Neurobiol, 1999. 25(5): p. 369–79. [DOI] [PubMed] [Google Scholar]
- 147.Volkow ND, et al. , Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am J Psychiatry, 2001. 158(3): p. 383–9. [DOI] [PubMed] [Google Scholar]
- 148.Thanos PK, et al. , The effects of cocaine on regional brain glucose metabolism is attenuated in dopamine transporter knockout mice. Synapse, 2008. 62(5): p. 319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Volkow ND, et al. , Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse, 1993. 14(2): p. 169–77. [DOI] [PubMed] [Google Scholar]
- 150.Zhou Z, et al. , Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc Natl Acad Sci U S A, 2011. 108(16): p. 6626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shao X, et al. , HMG-CoA synthase 2 drives brain metabolic reprogramming in cocaine exposure. Neuropharmacology, 2017. [DOI] [PubMed] [Google Scholar]
- 152.Samikkannu T, Atluri VS, and Nair MP, HIV and Cocaine Impact Glial Metabolism: Energy Sensor AMP-activated protein kinase Role in Mitochondrial Biogenesis and Epigenetic Remodeling. Sci Rep, 2016. 6: p. 31784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Aksenov MY, et al. , Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 dopamine receptor. Neurotoxicology, 2006. 27(2): p. 217–28. [DOI] [PubMed] [Google Scholar]
- 154.van Sighem AI, et al. , Life expectancy of recently diagnosed asymptomatic HIV-infected patients approaches that of uninfected individuals. AIDS, 2010. 24(10): p. 1527–35. [DOI] [PubMed] [Google Scholar]
- 155.Dumas JA, What is Normal Cognitive Aging? Evidence from Task-Based Functional Neuroimaging. Curr Behav Neurosci Rep, 2015. 2(4): p. 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Verhaeghen P and Cerella J, Aging, executive control, and attention: a review of meta-analyses. Neurosci Biobehav Rev, 2002. 26(7): p. 849–57. [DOI] [PubMed] [Google Scholar]
- 157.Cohen RA, Seider TR, and Navia B, HIV effects on age-associated neurocognitive dysfunction: premature cognitive aging or neurodegenerative disease? Alzheimers Res Ther, 2015. 7(1): p. 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Levine AJ, et al. , Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J Neurovirol, 2016. 22(3): p. 366–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pfefferbaum A, et al. , Accelerated aging of selective brain structures in human immunodeficiency virus infection: a controlled, longitudinal magnetic resonance imaging study. Neurobiol Aging, 2014. 35(7): p. 1755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Seider TR, et al. , Verbal memory declines more rapidly with age in HIV infected versus uninfected adults. J Clin Exp Neuropsychol, 2014. 36(4): p. 356–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sheppard DP, et al. , Elevated rates of mild cognitive impairment in HIV disease. J Neurovirol, 2015. 21(5): p. 576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cunnane SC, et al. , Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann N Y Acad Sci, 2016. 1367(1): p. 12–20. [DOI] [PubMed] [Google Scholar]
- 163.Stafstrom CE and Rho JM, The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol, 2012. 3: p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mamik MK, et al. , Insulin Treatment Prevents Neuroinflammation and Neuronal Injury with Restored Neurobehavioral Function in Models of HIV/AIDS Neurodegeneration. J Neurosci, 2016. 36(41): p. 10683–10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dufour CA, et al. , Physical exercise is associated with less neurocognitive impairment among HIV-infected adults. J Neurovirol, 2013. 19(5): p. 410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kruman II, Nath A, and Mattson MP, HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol, 1998. 154(2): p. 276–88. [DOI] [PubMed] [Google Scholar]