

Abstract
Background:
Clonal hematopoiesis of indeterminate potential (CHIP)-associated mutations increase the risk of atherosclerotic heart disease. Comorbidities significantly impact the prognosis of patients with myelodysplastic syndromes (MDS). The objective of this study was to determine the association and impact of CHIP-mutations with comorbidities in patients with MDS.
Methods:
We conducted a retrospective analysis of 566 consecutive patients with MDS at MD Anderson Cancer Center from August 2013 to December 2016. The Adult Comorbidity Evaluation-27 (ACE-27) scale assessed the severity of comorbid conditions. We used next-generation sequencing to detect the presence of CHIP mutations in bone marrow aspirates. Spearman’s correlation and logistic regression were used to determine the association between mutations and comorbidities.
Results:
Mutations in TET2, ASXL1, DNMT3A, JAK2, and TP53 were noted in 20%, 18%, 9%, 2%, and 21% of patients, respectively. Patients with DNMT3A and JAK2 mutations had higher likelihoods of prior history of myocardial infarction (MI) (odds ratio (OR) = 2.62; p = 0.03) and veno-occlusive disease (OR = 6.48; p = 0.02), respectively. TP53 was associated with prior history of malignancy. Patients with TET2 had no association with any comorbidity. A prognostic model including IPSS-R, ACE-27 score, and TP53 mutation (I-RAT) predicted median overall survival.
Conclusions:
In patients with MDS, the presence of CHIP-associated mutations is associated with comorbidities. DNMT3A and JAK2 mutations were associated with higher likelihoods of prior MI and thrombotic events. There was no association between comorbidity and TET2 mutation. Incorporating IPSS-R with ACE-27 and TP53 mutations improved outcome prediction in patients with MDS.
Keywords: myelodysplastic syndromes, clonal hematopoiesis of indeterminate potential, mutations, comorbidities
Precis for use in the Table of Contents:
The presence of clonal hematopoiesis of indeterminate potential (CHIP)-associated mutations was associated with comorbidities, including higher likelihood of prior myocardial infarction and thrombotic events, as well as worse overall prognosis in patients with myelodysplastic syndromes (MDS). Incorporating IPSS-R with ACE-27 scores and TP53 mutations improved outcome prediction.
Introduction:
Myelodysplastic syndromes (MDS) comprise a clinically and molecularly heterogeneous group of clonal hematopoietic stem cell disorders characterized by ineffective hematopoiesis and peripheral blood cytopenias.1,2 MDS is predominantly a disease of the elderly and approximately 70% of patients are over the age of 70.2 Multiple mutations in bone marrow (BM) cells have been described in MDS, with at least one mutation present in approximately 90% of patients.3,4
Mutations known to occur in MDS have been reported in peripheral blood of individuals with no apparent underlying MDS or other hematological disorder.5–8 This phenomenon is known as clonal hematopoiesis of indeterminate potential (CHIP).9 CHIP is associated with increased risk of developing myeloid disorders and cardiovascular disease (CVD).6–8,10,11 As in MDS, CHIP is more frequent with advancing age. The most commonly mutated genes associated with CHIP are DNMT3A, ASXL1, TET2, TP53, JAK2, and SF3B1.5–7,12,13 In MDS, mutations in TET2, ASXL1, and DNMT3A have been noted in > 10% of cases.3,4
Of importance, patients with MDS and comorbidities have worse outcomes independent of age as quantified by the international prognostic scoring system (IPSS) and the revised IPSS (IPSS-R).14,15 This suggests potential cross-talk between MDS and other systemic disorders such as CVD. Comorbidities and molecular abnormalities have been noted to be independent prognostic factors in patients with MDS.14,16 However, none of the current risk classifications include comorbidities and the impact of molecular mutations to predict survival in patients with MDS. Based on these concepts, we analyzed the relationship between CHIP-associated mutations and comorbidities in patients with MDS and investigated the impact of incorporating clinical factors and mutations into the IPSS-R classification.
Patients and Methods:
We conducted a retrospective cohort study that included consecutive patients with previously untreated MDS that presented to MD Anderson Cancer Center (MDACC) between August 2013 and December 2016. A total of 566 consecutive patients with MDS were reviewed. Information regarding their comorbidities was extracted using the ACE-27, a validated 27-item comorbidity index for patients with cancer that has been previously studied in MDS.14,15,17 Derived from the Kaplan-Feinstein Comorbidity Index, the ACE-27 categorizes specific diseases into 3 levels of comorbidity: grade 1 (mild), grade 2 (moderate), and grade 3 (severe), according to individual organ(s) affected. An overall comorbidity score (none, 0; mild, 1; moderate, 2; and severe, 3) is assigned based on the highest ranked/graded single ailment. In cases in which two or more moderate ailments occur in different organ systems or disease groupings, the overall comorbidity score is designated as severe.
In addition to demographic data including age, gender, and race, information on IPSS risk groups, survival, and other disease characteristics was collected. All patients submitted BM aspirates at the time of presentation. Using DNA extracted from each aspirate, mutational analysis was conducted using an amplicon-based next-generation sequencing panel targeting the entire coding regions of 28 genes frequently mutated in myeloid neoplasms and CHIP, as previously described.18 Although CHIP has been defined as the presence of mutation with VAF of ≥ 2%, only mutations with a minimum of 5% allelic burden and 250x bi-directional coverage were considered based on the limit of detection/sensitivity of our assay. We only considered mutations that have been previously reported to be pathogenic either in COSMIC ID or other databases or in the literature. Variants of uncertain significance were not included.
For the purposes of this study, mutations in DNMT3A, TET2, ASXL1, JAK2, and TP53 genes were considered to be CHIP-associated.6,7,9,19
Spearman’s correlation coefficient was used to assess the association between CHIP-associated gene mutations as binary variables by the presence of mutations and continuous variables by variant allele frequency (VAF) and comorbidities. We also used logistical regression to calculate odds ratios (OR) to determine the association between the presence of and the VAF of each mutation and comorbidities. To incorporate the CHIP-associated mutations and the ACE-27 score into IPSS-R classification, the whole cohort was randomly divided into a training cohort and a validation cohort at a 3:1 ratio. Overall survival was defined as time in months from the date of MDS diagnosis to the date of last follow-up or death regardless of cause. Multiple imputation was performed for missing variables to reduce bias. A backwards multivariate Cox proportional hazards model was performed with internal validation by the bootstrapping method to identify prognostic factors. A risk classification that included the IPSS-R and ACE-27 scores, with the presence of CHIP mutations (I-RAT) was created with K-adaptive partitioning for optimal grouping and its cutoff. Akaike information criteria was calculated for the model comparison of IPSS-R to I-RAT. The I-RAT model was assessed with the validation cohort. Data extraction was cross-checked for accuracy by two of the investigators (K.N. and S.P.).This study was conducted following the ethical guidelines of MDACC.
Results:
Patient characteristics:
Five hundred and sixty-six patients with MDS were included in this study. Patient characteristics are listed in Table 1. Three hundred and seventy six patients (66%) were male and 464 (82%) were white. Median age at presentation was 69 years (range: 22 – 93). Median time from diagnosis to referral to MDACC was 0.8 months (range: 0 – 167.5). Median duration of follow-up was 7.5 months (range: 1 – 168). A total of 350 (62%) patients had an IPSS of low or intermediate 1; complex karyotype was noted in 128 (23%) patients. Notably, 139 (25%) patients had received chemotherapy and/or radiation therapy for prior malignancy and were determined to have therapy-related MDS. Two hundred and seventy three (48%) patients received front-line hypomethylating agent based therapy, 11 (2%) patients received chemotherapy, and 29 (5%) patients received other therapies (i.e., ruxolitinib, luspatercept, lenalidomide, anti-thymocyte globulin with cyclosporine, and prednisone); the remaining 253 (45%) patients were observed prior to frontline therapy. Eighty-one of the 253 patients that were observed had high risk disease. However presence of active infections and other comorbidities precluded them from active therapy. Some opted to be treated at home. Remainder of the patients on observation had low risk disease and continued to be observed off therapy till the date of last follow-up for this study.
Table 1.
Patient demographics, N = 566.
| Parameter | Total N = 566 | Training N = 435 | Validation N = 131 | |||
|---|---|---|---|---|---|---|
| Median age, yr24 | 69 (22–93) | 68 (21–92) | 70 (24–91) | |||
| Male, no. (%) | 376 (66) | 296 (68) | 80 (61) | |||
| Median WBC, x109/L24 | 3.8 (0.5–209.5) | 3 (0.5–10.2) | 11.6 (2.9–209.5) | |||
| Median hemoglobin, g/dl24 | 9.6 (5.1–16.5) | 9.5 (5.1–16.5) | 10.0 (6.1–15.5) | |||
| Median platelets, x109/L24 | 85 (5–783) | 84 (5–432) | 87 (6–783) | |||
| Median % of BM blasts24 | 4 (0–19) | 4 (0–19) | 5 (0–18) | |||
| Cytogenetics, no. (%) | ||||||
| Diploid | 267 (30) | 178 (41) | 89 (68) | |||
| −5/5q- | 130 (23) | 35 (8) | 4 (3) | |||
| −7/7q- | 48 (9) | 43 (10) | 5 (4) | |||
| −5/5q- and −7/7q- | 43 (8) | 41 (9) | 2 25 | |||
| 20q- | 31 (6) | 27 (6) | 4 (3) | |||
| +8 | 44 (8) | 37 (9) | 7 (5) | |||
| Abn 11q | 18 (3) | 16 (4) | 2 25 | |||
| Others | 71 (13) | 53 (12) | 18 (14) | |||
| Complex | 128 (23) | 122 (28) | 6 (5) | |||
| MDS subcategory by WHO 2016 | ||||||
| MDS single lineage dysplasia | 40 (7) | 32 (7) | 8 (7) | |||
| MDS ringed sideroblasts | 59 (10) | 44 (10) | 15 (12) | |||
| MDS multi-lineage dysplasia | 128 (23) | 100 (23) | 28 (23) | |||
| MDS excess blasts | 198 (35) | 158 (36) | 40 (32) | |||
| Chronic myelomonocytic leukemia | 110 (19) | 85 (19) | 25 (20) | |||
| MDS unclassified | 25 (4) | 17 (4) | 8 (7) | |||
| MDS-del(5q) | 6 (1) | 6 (1) | 0 | |||
| IPSS risk group, no. | ||||||
| Low | 118 (21) | 79 (18) | 39 (30) | |||
| Intermediate-1 | 233 (41) | 179 (41) | 54 (41) | |||
| Intermediate-2 | 169 (30) | 136 (31) | 33 (25) | |||
| High | 46 (8) | 41 (9) | 5 (4) | |||
| IPSS revised risk group, no. (%) | ||||||
| Very low | 50 (9) | 41 (9) | 9 (7) | |||
| Low | 157 (27) | 107 (25) | 43 (33) | |||
| Intermediate | 127 (22) | 86 (20) | 41 (31) | |||
| High | 111 (20) | 87 (20) | 24 (18) | |||
| Very high | 128 (23) | 114 (26) | 14 (11) | |||
| ACE-27 | ||||||
| None | 101 (18) | 80 (18) | 21 (17) | |||
| Mild | 214 (38) | 175 (40) | 39 (32) | |||
| Moderate | 146 (26) | 107 (24) | 39 (32) | |||
| Severe | 105 (19) | 80 (18) | 25 (20) | |||
| CHIP-associated mutations, no. (%); median variant allele frequency (%) | ||||||
| No. (%) | Median | No. (%) | Median | No. (%) | Median | |
| DNMT3A | 50 (9) | 29.9 | 42 (10) | 27.5 | 8 (6) | 44.7 |
| TET2 | 112 (20) | 43.7 | 62 (14) | 38.6 | 50 (38) | 47.8 |
| ASXL1 | 104 (18) | 20.6 | 68 (16) | 17.9 | 36 (28) | 32.5 |
| JAK2 | 1125 | 35.9 | 825 | 27.1 | 325 | 39.7 |
| TP53 | 117 (21) | 36.3 | 110 (25) | 35.6 | 7 (5) | 48.7 |
Comorbidities in MDS by ACE-27:
Baseline ACE-27 comorbidity scores at presentation were as follows: none, 101 patients (17%); mild, 214 (38%); moderate, 146 (26%); and severe, 105 (19%). This was consistent with previously reported data.14 Overall 66% of the patients in this cohort were diagnosed with CVD and related diseases. Hypertension was the most common comorbidity, documented in 57% of all patients. History of prior malignancy was the second most common comorbidity, noted in 34% of the patients (prior solid tumor in 75%, myeloma in 13%, and lymphoma in 18% of the patients) (Figure 1A).
Figure 1. Comorbidities by systems and CHIP-associated mutations.

A: Baseline comorbidities, measured by ACE-27, were observed in the patient cohort. Abbreviations: ACE-27, Adult comorbidity evaluation-27; CVS, cardiovascular system; Resp, respiratory system; GI, gastrointestinal system; Endo, endocrine system; Neuro, neurological system; Psych, psychiatric system; Rheum, rheumatologic system; Malign, malignancy; Subs Abuse, substance abuse. B: Comorbidities and CHIP-associated mutations were observed at baseline and measured by ACE-27. Patients with indicated gene mutations (Yes/blue bars) were compared to those without (No/red bars). P-values were calculated by student t-test comparing incidence of each comorbidity among patients with and without the indicated mutations. Abbreviations: MI, myocardial infarction; Thromb, thrombosis; Resp, respiratory; Endo, endocrine.
Gene mutation characteristics:
Of the 566 total patients, 316 (56%) had a mutation in 1 or more CHIP-associated genes (i.e., TET2, ASXL1, DNMT3A, JAK2, and TP53); the mutation frequencies in TET2, ASXL1, DNMT3A, JAK2, and TP53 were 20%, 18%, 9%, 2%, and 21% respectively (Table 1B and Supplemental Figure 1). Among patients with DNMT3A mutations were 21 with canonical R882 mutations and 29 with non-canonical mutations outside of the 882 codon. Among MDS patients with evaluated CHIP-associated mutations, 9 (3%) had mutations in 3 CHIP-associated genes, 60 (19%) in 2, and 247 (78%) had a mutation in 1 CHIP-associated gene. One hundred and three (32.6%) of these patients had at least one additional non-CHIP-associated mutation among the tested genes. Two hundred and fifty (44%) MDS patients had no mutations on the evaluated CHIP-associated genes. The median VAF for each of these genes is shown in Table 1.
Comorbidities by mutations:
We assessed comorbidities at presentation using ACE-27 and by the presence and VAF of CHIP-associated mutations (Supplemental Tables 1 and 2 and Supplemental Figure 2). Within the cardiovascular system (CVS) portion of the assessment, patients harboring the DNMT3A mutation were noted to have a higher frequency of prior history of myocardial infarction (MI) than those without the mutation (14% vs 6%; p = 0.03) (Figure 1B). However the presence and VAF level of DNMT3A mutation were only weakly associated with MI. Using the logistical regression method, in patients with DNMT3A mutation, the odds of having a history of MI was 2.62 (95% CI 1.095–6.246; p = 0.03). None of the other comorbidity systems were noted to have any significant association with DNMT3A mutation.
The presence and VAF of ASXL1 mutations were weakly and negatively associated with history of solid malignancy including melanoma. Additionally, the VAF of ASXL1 mutation was weakly associated with morbid obesity. JAK2, the least common of the CHIP-associated mutations was associated with a higher frequency of prior history of veno-occlusive disease (18% vs 4%; p = 0.01) with an OR of 6.48 (95% CI 1.375–30.477; p = 0.02). Despite being the second most frequently mutated gene observed in this cohort of patients, TET2 failed to show a significant association with any of the comorbidity systems. As expected, TP53 mutation was highly associated with a history of prior malignancy: solid tumor, 44% vs 21% (p < 0.001); myeloma, 9% vs 3% (p = 0.003); and lymphoma, 17% vs 3% (p < 0.001). Additionally, patients with disorders of the respiratory and endocrine systems (e.g., diabetes mellitus) were most likely harbor TP53 mutations: 16% vs 9% (p = 0.02) and 27% vs 16% (p = 0.01), respectively. The presence and VAF level of TP53 mutation was only weakly associated with these comorbidities.
The number of CHIP mutations was not associated with a specific comorbidity system except weakly for morbid obesity (R, 0.094; p = 0.025) (Supplemental Table 3). Though not statistically significant, we noted a trend towards higher ACE-27 scores and increased number of mutations (p = 0.074). A significant association was however observed between higher ACE-27 scores and harboring CHIP-associated mutation(s). Among 105 patients with severe ACE-27 scores, 71 (68%) carried CHIP-associated mutations (p = 0.016). When dividing for age (≤ 65 years versus > 65 years), a trend was noted between harboring DNMT3A mutation and prior history of MI in patients > 65 years of age (R, 0.099; p = 0.059), whereas there was no correlation noted in those ≤65 years (R, 0.113; p = 0.112). Similarly, a significant association was noted between harboring JAK2 and prior history of veno-occlusive disease in patients > 65 years of age (R, 0.270; p < 0.001). This association was not observed in those ≤ 65 years.
We also applied our previously reported prognostic model for overall survival (OS) in MDS to this cohort of MDS patients. The model integrates age, ACE-27 score, and the IPSS.14 Patients were categorized into 3 risk categories/scores: low, intermediate, and high. Based on this prognostic model, the median OS of patients with low risk scores was 43.0 months, 23.0 months with an intermediate risk score, and 9.0 months with a high risk score (p < 0.001). When applied to the current MDS cohort, patients with CHIP-associated mutations had a significantly higher risk score compared to those without CHIP-associated mutations (p = 0.002). Patients harboring CHIP mutations were categorized as follows: low, 55 (18%) patients; intermediate, 139 (44%) patients; and high, 121 (38%) patients. These scores were compared to those from patients with no CHIP mutations: low, 49 (20%); intermediate 139 (56%); and high 61 (24%). This translated into a significantly lower survival in carriers of CHIP mutation compared to the non-carriers (26.1 months vs 65.8 months; p = 0.005) (Figure 2A). However, when patients carrying TP53 mutations were excluded from the prognostic model, the impact of CHIP mutations on survival disappeared (Figure 2B).
Figure 2. Survival curves by CHIP-associated mutations.
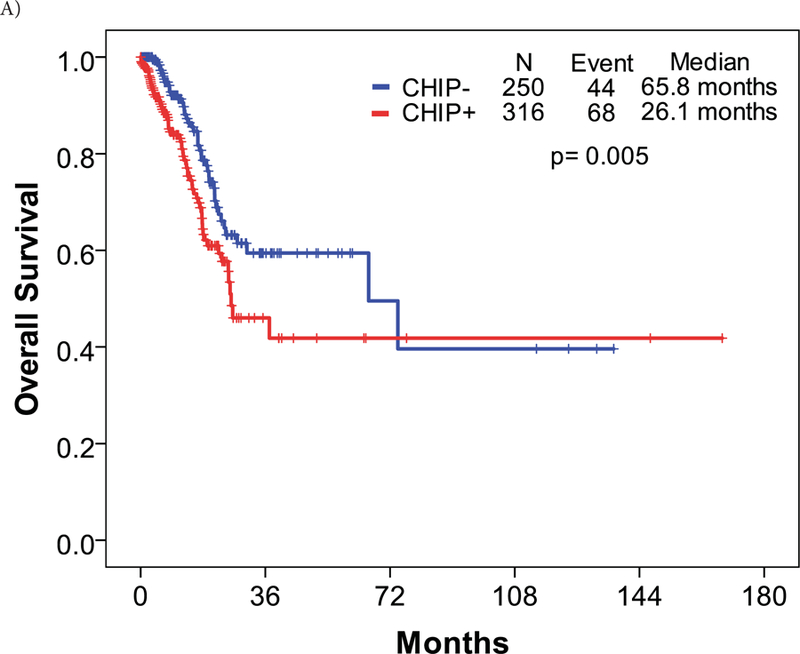
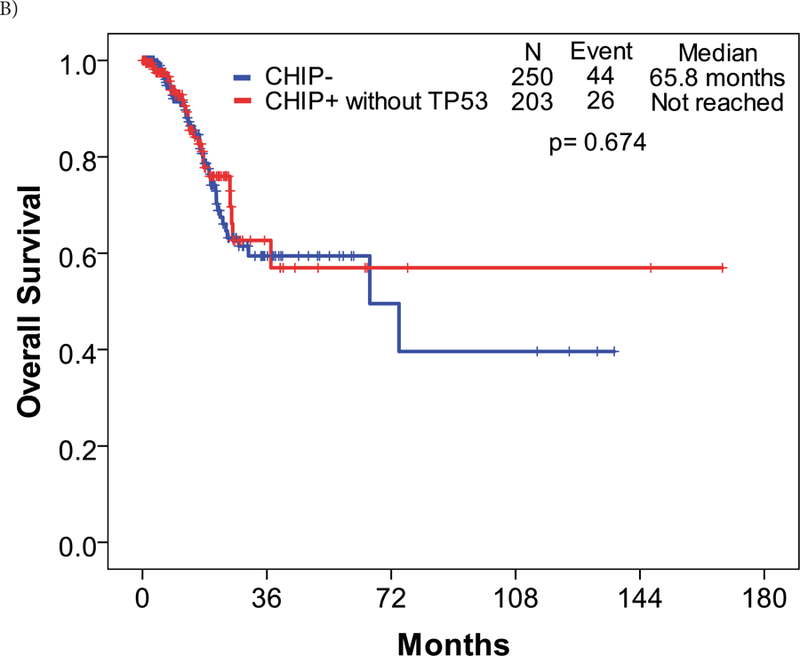
A: Patients harboring CHIP-associated mutations had slightly lower overall survival compared to those without the mutations. B: When TP53 mutations were removed from the overall survival calculations, the effect of CHIP-associated mutations on survival was non-significant.
Building a prognostic model of CHIP mutations and ACE-27 for survival:
Patient characteristics of the assigned training and validation cohorts after randomization are described in Table 1. A backward multivariate proportional hazards model identified the classification of cytogenetic, BM blast percentage, hemoglobin, and platelet count by IPSS-R, ACE-27 score, and the presence of TP53 mutation (I-RAT) as prognostic factors for OS (Table 2). The prognostic significance of TP53 mutation for OS was constant regardless of VAF by univariate Cox proportional hazard model (Supplemental Figure 4). Each prognostic factor was internally validated with the bootstrapping method (Supplemental Table 4). Moreover, when we excluded the presence of TP53 mutation from our multivariate proportional hazards model above, the IPSS-R and ACE-27 retained their prognostic impact on OS (Table 2).The risk scoring of I-RAT was developed following the ratio of each hazard ratio which was rounded to the nearest 0.5 (Table 3). K-adaptive partitioning identified 5 risk groups (Very Low, Low, Intermediate, High, and Very High) with the optimal cutoffs of total points at 2, 5.5, 8, and 9.5 (Supplemental Table 5). The I-RAT model showed lower Akaike information criteria of 778.818 compared to that of IPSS-R with 786.7214, which confirmed the superiority of I-RAT to the current IPSS-R. The C-statistic for the I-RAT model is 0.822 (standard error=0.02). Among the training cohort, survival curves between risk groups with IPSS-R ACE-27 TP53 were more well-separated than those of IPSS-R risk groups (Supplemental Figure 3). The I-RAT risk classification was validated with preserved rank HR along with higher risk categories (Supplemental Table 6). With the whole cohort, survival curves by the I-RAT classification showed better separation between cohorts with statistical significance than those of IPSS-R classification (Figure 3).
Table 2.
Backward multivariate Cox proportional hazards model for overall survival: A) training and B) TP53 excluded cohorts.
| A) Training | P | HR | 95% Confidence interval of HR | |
|---|---|---|---|---|
| Lower | Upper | |||
| IPSS-R: prognostic factor | ||||
| Cytogenetic | 0.002 | 1.409 | 1.132 | 1.755 |
| Percentage of blasts in BM | <0.001 | 1.504 | 1.222 | 1.851 |
| Hemoglobin | 0.034 | 1.461 | 1.028 | 2.075 |
| Platelet count | 0.000 | 2.629 | 1.556 | 4.440 |
| TP53 mutation | 0.012 | 2.118 | 1.179 | 3.804 |
| ACE-27 | 0.005 | 1.376 | 1.101 | 1.720 |
| B) TP53 excluded | P | HR | 95% Confidence interval of HR | |
| Lower | Upper | |||
| Cytogenetic | 0.003 | 1.400 | 1.121 | 1.749 |
| Percentage of blasts in BM | <0.001 | 1.517 | 1.205 | 1.910 |
| Hemoglobin | 0.001 | 2.109 | 1.354 | 3.286 |
| Platelet count | 0.018 | 2.006 | 1.129 | 3.565 |
| ACE-27 | 0.011 | 1.403 | 1.081 | 1.820 |
Table 3.
Risk scoring of IPSS-R, ACE-27, and TP53 (I-RAT) models.
| 0 | 1 | 1.5 | 2 | 3 | 4 | |
|---|---|---|---|---|---|---|
| Cytogenetics | Very Good | Good | Intermediate | Poor | Very Poor | |
| BM blasts | <=2 | >2-<5% | 5–10% | >10% | ||
| Hemoglobin | >=10 | 8-<10 | <8 | |||
| Platelets | >=100 | 50–100 | <50 | |||
| TP53 mutation | No | Yes | ||||
| ACE-27 | None | Mild | Moderate | Severe |
Figure 3. Risk stratification and overall survival of the entire cohort.
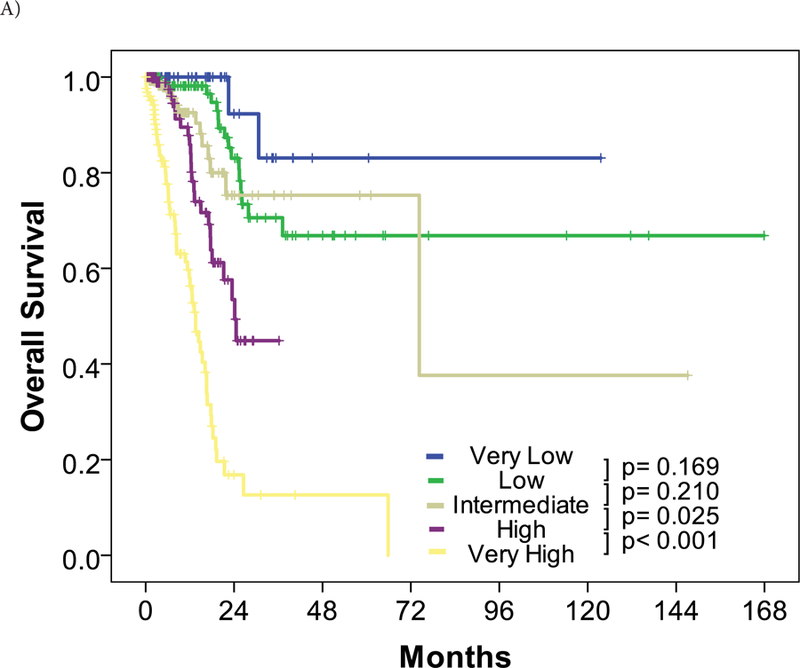
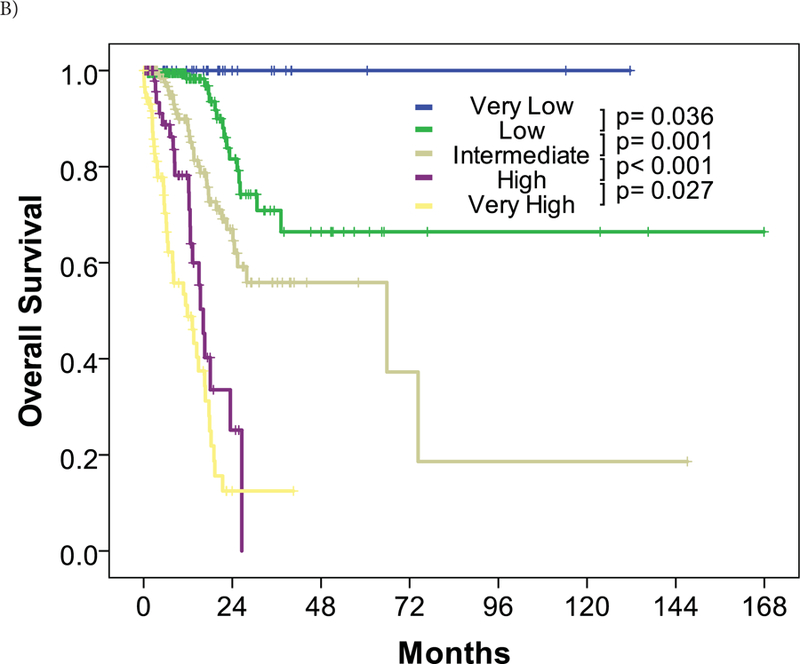
Overall survival was stratified by risk for the entire cohort by (A) the IPSS-R classification and (B) the I-RAT model.
Discussion:
We conducted a retrospective cohort study to evaluate the association between CHIP-associated mutations and comorbidities in patients with MDS. To our knowledge, this is the first study evaluating a possible association between these mutations and comorbidities affecting patient prognosis in MDS. As previously described, we used the ACE-27 since it has been validated to study comorbidities in cancer patients, including those with MDS.14,15,17 Using the ACE-27, we found that disorders of the CVS, followed by a history of prior malignancy, were the two most common comorbid ailments. This was consistent with our previous report.14 Contrary to what has been previously reported by other groups3,4, our cohort had a higher frequency of patients with TP53 mutations. This was mainly due to the higher proportion of therapy related patients being referred to our cancer center from not only outside the institution but also within the institution.
While patients with MDS may ultimately succumb to complications arising from cytopenias, such as infections or bleeding, or from progression of disease to acute leukemia, a significant number die from concurrent comorbidities.11 The association of CHIP with CVDs is well described in the literature.10,11 Carriers of CHIP mutations are noted to be at double the risk of developing coronary artery disease compared to the non-carriers. Moreover, causality has been established between TET2 mutation and development of aortic atherosclerotic lesions in murine models.10,20 This is thought to be from increased expression of chemokines and cytokines by TET2 mutant macrophages of hematopoietic origin, leading to increased inflammation and accelerated atherosclerosis.10,20–22 Recent data indicated an increased risk of cardiovascular mortality with increased longevity in MDS, and data from our center reported that 44% of patients with lower-risk MDS patients died from cardiovascular causes.11,23 In the current study we found this to be true as well, particularly in MDS patients with DNMT3A mutations. Consistent with previous reports, disorders of the CVS were the most commonly encountered comorbidities our MDS cohort.14,15 The DNMT3A mutation was significantly associated with a higher likelihood of prior history of MI (14% vs 6%; p = 0.03) with an OR of 2.62. This association was seen independent of age, especially in patients ≤ 65 years. Of interest, TET2 mutations were not associated with CVD in this cohort. Similarly, ASXL1 mutations did not associate with a prior history of cardiovascular events. Upon evaluating the VAF of DNMT3A, we found only a weak association between clone size and increased frequency of prior history of CVD. This is contrary to what has been described by Jaiswal, et al in patients with CHIP mutations.10 One possible factor contributing to this difference is the small number of patients with prior cardiovascular events in our cohort: 38 of 566 patients.
More recently Cook, et al demonstrated a higher incidence of gastrointestinal reflux disease associated with DNMT3A mutations, and chronic pulmonary disease associated with TET2 mutations.21 In our cohort we found patients with TP53 mutations had a higher likelihood of prior history of respiratory disorders (16% vs 9%; p = 0.02) and diabetes mellitus (27% vs 16%; p = 0.01) rather than TET2. It’s possible that this association may have been confounded by prior history of malignancy and type of therapy used to treat these patients. Of the 19 patients that had disease of the respiratory system and were also TP53 mutated, 10 patients were therapy-related. Similarly, of the 32 patients with disease of the endocrine system and with TP53 mutation, 15 were noted to have therapy-related MDS. As expected, a high proportion of the patients with TP53 mutations also had a history of prior malignancy, most frequently a prior solid malignancy followed by lymphoma and then myeloma. These findings highlight the impact of comorbidities, particularly those of the CVS in CHIP and MDS, suggesting a common underlying pathophysiology. Brunner, et al recently showed in patients with MDS surviving > 5 years after diagnosis, the risk of dying from CVD was comparable to dying from the underlying MDS (26.9% vs 29.5%).11 This advocates for early intervention and optimization of the modifiable cardiovascular risk factors, such as hypertension, obesity, and hyperlipidemia in patients with CHIP and lower-risk MDS.
Comorbidities measured by ACE-27 and the presence of TP53 mutation are independent prognostic factors for survival. The prognostic impact on survival by comorbidities and the presence of TP53 mutations reported here is consistent with prior publications.14,16 In the current study, CHIP-associated mutations lost their prognostic impact on survival when TP53 mutations were excluded. ACE-27 and the IPSS-R, as expected, retained their impact on OS after excluding TP53 mutations from the multivariate proportional hazards model. The incorporation of comorbidities and TP53 to the current standard IPSS-R classification resulted in a better prognostic model (I-RAT) that was validated with a separate dataset after randomization of the entire cohort.
The I-RAT model stratified the cohort into 5 risk groups compared to the IPSS-R classification. Approximately 40% of patients were classified into lower or higher risk categories. Notably, no patient in the very low risk category by the I-RAT died regardless of cause, with the median follow-up of 10.5 months. The incorporation of clinical factors and molecular abnormalities contributed to better prognostic classification in patients with MDS.
The work presented here has several limitations. Our results may have two potential confounders: we did not account for transfusion burden and our study population was a heterogeneous group, including those who have received prior therapy. Because of the nature of the retrospective study and the diversity of the cohort, we were unable to account for transfusions and other treatments that occurred outside of our study site. Additionally, observations regarding the precise time when mutations may have emerged prior to or after the emergence of associated cardiovascular risk factors in each patient were not possible, because they occurred prior to presentation at our clinic and baseline observations. Also, ACE-27 was solely measured at baseline and the follow-up of 7.5 months was relatively short. One result of this study was improvement upon current prognostic models to better evaluate patient prognosis at the time of presentation. A longer follow-up might show additional relationships between MDS and comorbidities. Finally, we focused on mutations in the five most commonly observed CHIP-associated genes: ASXL1, DMNT3A, JAK2, TET2, and TP53. Mutations in the other CHIP-associated genes may have biological importance with regards to comorbidities, however including additional CHIP-associated genes would reduce the statistical significance of the observed mutations.
In summary, this study shows that CHIP-associated mutations are associated with a higher frequency of prior history of cardiovascular events and poor prognosis in patients with MDS, particularly DNMT3A mutations associated with MI. Contrary to other reported studies, we did not find the same association with TET2 mutations. Moreover, we noted JAK2 to be significantly associated with prior history of veno-occlusive disease. These findings emphasize the importance of screening for life-style and other risk factors in order to accurately evaluate the risk of cardiovascular events in patients with CHIP and MDS. The incorporation of comorbidities by ACE-27 and the presence of TP53 mutations substantially improve upon our current prognostic classification of patients with MDS. This model needs to be prospectively validated in other cohorts.
Supplementary Material
Acknowledgments
Funding: This work was supported in part by the University of Texas MD Anderson Cancer Center Support Grant CA016672, the University of Texas MD Anderson MDS/AML Moon Shot, and Leukemia Texas.
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med 2009;361(19):1872–1885. [DOI] [PubMed] [Google Scholar]
- 2.Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood 2008;112(1):45–52. [DOI] [PubMed] [Google Scholar]
- 3.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014;28(2):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013;122(22):3616–3627; quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371(26):2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20(12):1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malcovati L, Galli A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017;129(25):3371–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 2017;377(2):111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunner AM, Blonquist TM, Hobbs GS, et al. Risk and timing of cardiovascular death among patients with myelodysplastic syndromes. Blood Adv 2017;1(23):2032–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buscarlet M, Provost S, Zada YF, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017;130(6):753–762. [DOI] [PubMed] [Google Scholar]
- 13.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012;44(11):1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naqvi K, Garcia-Manero G, Sardesai S, et al. Association of comorbidities with overall survival in myelodysplastic syndrome: development of a prognostic model. J Clin Oncol 2011;29(16):2240–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daver N, Naqvi K, Jabbour E, et al. Impact of comorbidities by ACE-27 in the revised-IPSS for patients with myelodysplastic syndromes. Am J Hematol 2014;89(5):509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364(26):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piccirillo JF, Costas I., Claybour P, et al. The measurement of comorbidity by cancer registries. J registry management 2003;30:8–14. [Google Scholar]
- 18.Kanagal-Shamanna R, Luthra R, Yin CC, et al. Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget 2016;7(12):14251–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol 2011;29(5):504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355(6327):842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook EK IT, Young S, Rosen G, Jamali M, et al. Feeding the Fire: The comorbid and inflammatory backdrop of clonal hematopoiesis of indeterminate potential (CHIP) by mutation subtype. 59th Annual American Society of Hematology Meeting Dec 2017 Abstract no 426 2017. [Google Scholar]
- 22.Tall AR, Levine RL. Cardiovascular disease: Commonality with cancer. Nature 2017;543(7643):45–47. [DOI] [PubMed] [Google Scholar]
- 23.Dayyani F, Conley AP, Strom SS, et al. Cause of death in patients with lower-risk myelodysplastic syndrome. Cancer 2010;116(9):2174–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018;32(2):520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.H Z, Y W, L N, N N, K2 C Monovar: single-nucleotide variant detection in single cells. Nature Methods 2016;13(6):505–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.