

Summary
The tumor microenvironment (TME) polarizes tumor-infiltrating macrophages toward tumor support. Macrophage-abundant tumors are highly malignant and are the cause of poor prognosis and therapeutic resistance. In this study, we show that the prolyl hydroxylase (PHD) inhibitor FG-4592 (FG) inhibits tumor growth of macrophage-abundant tumors and prolongs mouse survival. FG not only normalizes tumor vessels and improves tumor oxygenation but also directly affects macrophages and activates phagocytosis through the PHD-hypoxia-inducible factor (HIF) axis. Remarkably, FG can promote phagocytic ability of the Ly6Clo subset of tumor-infiltrating macrophages, leading to tumor growth inhibition. Moreover, Ly6Cneg macrophages contributed to blood vessel normalization. Using a malignant tumor mouse model, we characterized macrophage function and subsets. Altogether, our findings suggest that the PHD inhibitor can promote the anti-tumor potential of macrophages to improve cancer therapy.
Subject Areas: Microenvironment, Immune Response, Cancer
Graphical Abstract
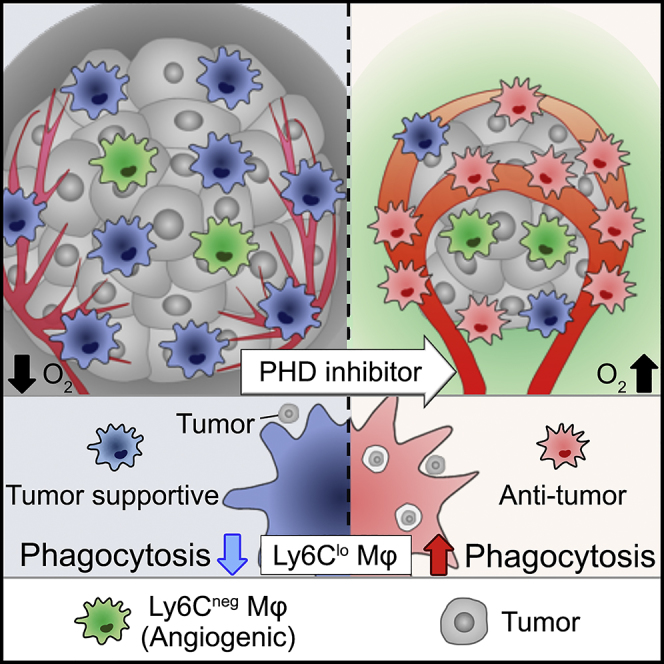
Highlights
-
•
PHD inhibitor treatment inhibits tumor growth and prolongs survival time of mice
-
•
Regulating the PHD-HIF pathway can alter the tumor-infiltrating macrophage phenotype
-
•
PHD inhibitor activates the tumor phagocytic ability of Ly6Clo macrophages
Microenvironment; Immune Response; Cancer
Introduction
Tumor vessel structure differs from normal vessel structure in terms of short lumen diameter, irregular sprouting, and poor tight junction formation. This leads to leaky tumor vessels with low blood flow (Jain, 2014). These tumor microenvironments (TMEs) lead to hypoxia and high interstitial pressure, wherein immune cells exhibit impaired cytotoxicity and pathogen recognition systems. Therefore, repair of blood vessel function and normalization of the microenvironment in tumors are expected to enhance anti-tumor immunity (Quail and Joyce, 2013, Tian et al., 2017, Park et al., 2016).
Macrophages (Mφs) are the most abundant immune cells in tumors (Movahedi et al., 2010). Moreover, tumor Mφs, referred to as tumor-associated macrophages (TAMs), have been canonically classified into the inflammatory (M1) or anti-inflammatory (M2) phenotype. M1 Mφs are characterized as having high phagocytic ability and inhibitory activity toward tumor growth (Guerriero et al., 2017). In contrast, M2 Mφs, which are strongly polarized by the TME, promote tumor progression and metastasis (Henze and Mazzone, 2016, Mosser and Edwards, 2008). In addition, M2 Mφs exclude cytotoxic lymphocytes from tumors such as lung carcinomas, melanomas, and colorectal carcinomas, which are classified as TAM-abundant tumors exhibiting a low number of T cells. These phenomena are associated with tumor malignancy and result in therapeutic resistance (Binnewies et al., 2018). Therefore, there is a need for new therapeutic strategies against these tumors.
Hypoxia inducible factors (HIFs) are master regulators of the cellular response to hypoxia (Imtiyaz and Simon, 2010). HIF stability is post-transcriptionally regulated by oxygen availability through prolyl hydroxylase (PHD). If the oxygen concentration is reduced, PHDs become inactive, resulting in HIF accumulation (Semenza, 2012). HIF-1 is widely expressed and is detected in virtually all innate and adaptive immune populations including Mφs. For Mφs, the function of HIF-1 is to increase aggregation, invasion, and motility and drive the expression of proinflammatory cytokines (Nizet and Johnson, 2009, Palazon et al., 2014). Conversely, for cancer cells, HIFs control the expression of crucial genes involved in proliferation and metastasis of cancer cells (Keith et al., 2011). Therefore, the effect of simultaneously raising HIF levels in both immune cells and cancer cells on tumors remains unclear. Moreover, some reports have argued that normalization of tumor vessels results in accelerated tumor progression (Du et al., 2008, Stockmann et al., 2008). Therefore, it is also unclear whether vessel normalization is beneficial for tumor progression. In this study, we examined the effect of the PHD inhibitor on tumor growth by administering FG-4592 (FG), which is currently under investigation in a phase 3 clinical trial for the treatment of anemia in chronic kidney disease (Besarab et al., 2016, Provenzano et al., 2016).
Results
FG Treatment Inhibits Tumor Growth in Lewis Lung Carcinoma and B16F10 Tumors
To examine the effects of the PHD inhibitor on tumors, we used a syngeneic murine tumor model of the Lewis lung carcinoma (LLC) cell line. FG-treated mice showed significantly inhibited tumor growth (Figures 1A–1C) that was dose-dependent (Figures S1A and S1B). To examine whether this tumor growth inhibition was also exhibited in other tumor models, B16F10 melanoma and MC38 colon tumor models were used. B16F10 tumor growth was significantly inhibited by FG treatment (Figures 1D–1F), but there were no significant differences in inhibition in the MC38 tumor model (Figures S1C and S1D). Furthermore, repeated FG treatment inhibited tumor growth and prolonged survival; however, the tumor regrew on day 22 after a single FG treatment (Figures 1G and 1H). To determine whether FG induced apoptosis or inhibited proliferation of tumor cells, tumor tissues were stained with anti-cleaved caspase 3 and anti-Ki-67 antibodies (Figures 1I–1L and S1E–S1H) and a cell proliferation assay was performed in vitro (Figure S1I); these experiments revealed no significant differences after FG treatment. Therefore, FG treatment-induced tumor growth inhibition was not due to apoptosis or inhibited proliferation in tumor cells. We also analyzed cytokine concentrations in blood plasma and found that the cytokine signatures remained largely unaffected by FG treatment (Figure S1J). Taken together, these results suggest that FG may not directly affect tumor cells and plasma cytokines.
Figure 1.

FG Treatment Inhibits Tumor Growth in LLC and B16F10 Tumors
(A) LLC tumor growth curves. Treatment with vehicle (Veh) or FG (3 mg; treated on day 10; n = 15).
(B) LLC tumor weight on day 16 (n = 15).
(C) Images of LLC tumors on day 16. Scale bar, 1 cm.
(D) B16F10 tumor growth curves. Treatment with Veh or FG (3 mg; treated on day 10); n = 8.
(E) B16F10 tumor weight on day 16 (n = 8).
(F) Images of B16F10 tumors on day 16. Scale bar, 1 cm.
(G) LLC tumor growth curves. Treatment with Veh (n = 11), FG (3 mg; treated once on day 10; n = 9), or FG twice (3 mg; treated on day 10 and 16; n = 11).
(H) Kaplan-Meier curves showing events-free survival rate of vehicle-, 3 mg FG once-, or 3 mg FG twice-treated mice (n = 10).
(I) Immunofluorescence (IF) images of cleaved caspase-3 (CC3; red)-stained sections of LLC tumors on day 12. Scale bar, 50 μm.
(J) Quantification of the CC3+ cell ratio of total cells on day 12 (n = 3).
(K) IF images of Ki-67 (red)-stained sections of LLC tumors on day 12. Scale bar, 50 μm.
(L) Quantification of the Ki-67+ cell ratio of total cells on day 12 (n = 3).
Data represent means ±SEM; two-way ANOVA (A, D, and G); Mann-Whitney test (B, E, J, and L); Log rank test (H). ns, not significant; *p < 0.05; **p < 0.01; ****p < 0.0001. See also Figure S1.
FG Treatment Improves TME
To investigate the indirect ways in which FG might inhibit tumor growth, we tested whether FG affected tumor vessels and the TME; TME normalization is known to have an anti-tumor effect and can reeducate immune cells (Quail and Joyce, 2013). Remarkably, we found that FG treatment drastically altered vessel structures, as tumor vessel density was significantly decreased, whereas the vessel luminal area was increased (Figures 2A and 2B). We then examined the reversibility of this alteration and found that tumor vessel structure of one-time FG-treated tumors reverted to that of vehicle control tumor vessels on day 19 (Figures S2A and S2B); however, tumors treated with FG repeatedly sustained normal vessel structure even by day 23 (Figures S2C and S2D). To assess vessel maturation, we examined tight junction formation and pericyte coverage rate with ZO-1 and NG2, respectively, whose levels were significantly increased after FG treatment (Figures 2C–2F). We also evaluated the effect of FG treatment on vessel structure in other tumor models and found that the B16F10 tumor model also exhibited changes to vessel structure following treatment (Figures S2E and S2F). Even the MC38 tumor model, in which no significant difference in tumor growth inhibition was observed after FG treatment, showed changes in vessel structure (Figures S2G and S2H). To assess whether the reconstituted tumor vessel had normal function, we evaluated tumor vessel function with high-molecular-weight dextran. Tumor tissue perfusion was recovered after FG treatment (Figure 2G, low-power field). Dextran leakage was observed in the vehicle controls as a hazy green area; in contrast, FG-treated tumor vessels showed recovered vessel function (Figure 2G, high-power field). Furthermore, hypoxic regions were significantly reduced in FG-treated mice (Figures 2H and 2I). Taken together, these results suggest that FG treatment induces TME improvement through tumor vessel normalization.
Figure 2.

FG Treatment Improves TME
(A) IF images of CD31-stained (red) sections of LLC tumors. Scale bar, 100 μm.
(B) Quantification of vessel density and vessel lumen area in LLC tumors (n = 6).
(C) IF images of ZO-1 (green) and CD31 (red)-stained sections of LLC tumors. Scale bar, 50 μm.
(D) Quantification of the ZO-1+ area ratio in the CD31+ area (n = 5).
(E) IF images of NG2 (green) and CD31 (red)-stained sections of LLC tumors. Scale bar, 50 μm.
(F) Quantification of the NG2+ area ratio in the CD31+ area (n = 5).
(G) Images of blood perfusion (low-power field; scale bar, 1 mm) and blood leakage (high-power field; scale bar, 100 μm) in LLC tumor tissues using FITC-conjugated dextran.
(H and I) IF images and quantification of the tumor hypoxic region by anti-pimonidazole staining in LLC tumors (anti-CD31 antibody, red; anti-pimonidazole antibody, green; n = 6). Arrowheads indicate pimonidazole-positive area. Scale bar, 100 μm.
Data represent means ±SEM; Mann-Whitney test (B, D, F, and I). **p < 0.01. See also Figure S2.
FG Treatment Induces Mφ Infiltration in Tumors and Inhibits Tumor Growth through Mφs
Next, we examined whether TME improvement by FG leads to changes in the immune cell response. We found that FG-treated tumors showed an increased CD45+ leukocyte cell population ratio (Figure 3A) with a significantly increased CD45+ CD11b+ F4/80+ Mφ cell population ratio (Figure 3B); in contrast, the T cell population did not show significant changes and had a low population of tumor-infiltrating T cells (Figure 3C). Furthermore, T cells displayed no significantly activated profiles as measured by CD69 and CD25 expression (Figure S3J). An increased number of Mφs was also observed in the tumor tissue by immunofluorescence staining (Figures 3D and 3E). These findings indicate that the LLC tumor was TAM abundant and contained few activated T cells. To determine whether FG treatment promoted proliferation of tumor-infiltrating Mφs, tumor tissues were stained with anti-F4/80 anti-Ki-67 antibodies. There were no significant differences in Mφ proliferation in tumor tissue after FG treatment (Figures S3C and S3D). Furthermore, we analyzed other immune cell contents in circulating blood and tumor tissue and found no significant differences between vehicle and FG treatment (Figures S3E and S3F).
Figure 3.

FG Treatment Induces Mφ Infiltration in Tumors and Inhibits Tumor Growth through Mφs
(A–C) Quantification of tumor-infiltrating (A) CD45+ (n = 12–13), (B) CD11b+F4/80+ (n = 8), and (C) CD4+ and CD8+ (n = 5) cell ratios in LLC tumors by flow cytometric analysis.
(D) IF imaging of F4/80 (red)-stained sections of LLC tumors. Scale bar, 50 μm.
(E) Quantification of F4/80+ cell number per mm2 of tumors (n = 5).
(F−I) Flow cytometric quantification of (F) Ly6Chi, Ly6Clo, and Ly6Cneg Mφ ratios in LLC tumors (n = 6), (G) the ratio of tumor-infiltrating CD11b+F4/80+ cells/CD45+ cells in B16F10 tumors (n = 4), (H) Ly6Chi, Ly6Clo, and Ly6Cneg Mφs ratios in B16F10 tumors (n = 4), and (I) CD4+ and CD8+ cell lymphocyte ratios in B16F10 tumors (n = 4).
(J) Tumor growth curves of the LLC tumor mouse model treated with liposome control or clodronate liposome and vehicle or 3 mg FG (relative to vehicle liposome controls; n = 4).
(K) IF images of F4/80 (red)-stained sections of LLC tumors. Scale bar, 50 μm.
(L) Quantification of F4/80+ cell number per mm2 of tumors (n = 3).
Data represent means ±SEM. Mann-Whitney test (A–C and E–I); two-way ANOVA (J); unpaired Student's t test (L). ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S3.
Further analysis was carried out on this tumor-infiltrating Mφ population, which would be polarized into M1 or M2 Mφs, by using CD80 as an M1 marker and CD206 as an M2 marker (Muller et al., 2017). However, these markers were expressed in both vehicle- and FG-treated tumor Mφs, and there were no differences between Mφs in either treatment group (Figure S3A). As recent studies demonstrated that TAMs simultaneously express both M1 and M2 markers (Azizi et al., 2018, Muller et al., 2017, Peterson et al., 2016), we focused on other cell surface markers, whose expression levels reflected the differentiated state of TAMs. Most TAM subpopulations arise from the Ly6C+ population of circulating monocytes. Ly6C is downregulated during the differentiation of monocytes into TAMs (Franklin et al., 2014); however, there have been few reports on how the functions differ when TAMs are subdivided by Ly6C. Therefore, we divided these tumor-infiltrating Mφs using Ly6C into three populations, Ly6Chi, Ly6Clo, and Ly6Cneg (Figure S3A). The Ly6Clo Mφ ratio in the total Mφ population increased by FG treatment (Figure 3F). Moreover, the morphology of Ly6Clo Mφs from FG-treated tumors was different from Ly6Clo Mφs from vehicle-treated tumors (Figure S3B). It is possible that these Mφs formed phagosomes.
We also analyzed tumor-infiltrating immune cells in the B16F10 and MC38 tumor models. The ratio of Ly6C Mφ fractions in the B16F10 tumor showed a similar tendency as the LLC mouse tumor-infiltrating Mφs (Figures 3G and 3H). However, the ratio of tumor-infiltrating Ly6C Mφ fractions in the MC38 tumor, in which no significant difference in tumor growth occurred after FG treatment, was different than that of the LLC and B16F10 tumors (Figures S3G and S3H). T cell infiltration to the tumor was lower in the LLC mouse model than in B16F10 and MC38 (Figures 3C, 3I, and S3I). In addition, T cells showed a slightly activated profile in the tumor tissues (Figure S3K) and circulating blood (Figure S3M) of B16F10 tumor-bearing mice after FG treatment compared with that of the LLC mouse model (Figures S3J and S3L). Therefore, we selected the LLC mouse model for further experiments to focus on tumor-infiltrating Mφs.
Among the three different tumor inoculation models, vascular normalization by FG was qualitatively similar, whereas tumor growth inhibition by FG was evident in the LLC model and B16F10 model but not in the MC38 model. Such differences may arise from the population of infiltrating Mφs. Therefore, we examined whether FG affected the ability of tumor-infiltrating Mφs to inhibit tumor growth. To test this, tumor-infiltrating Mφs were depleted by liposome clodronate before FG administration. Liposome control and FG-treated tumors showed inhibited tumor growth compared with that of vehicle-treated tumors. However, tumor growth was not significantly different between the vehicle- and FG-treated tumors in the clodronate treatment group (Figure 3J). Mφ depletion efficiency was evaluated by immunostaining; Mφs showed an approximate 80% depletion compared with that in liposome control tumor tissues and spleen (Figures 3K, 3L, and S3N). These results indicate that FG affects the ability of tumor-infiltrating Mφs to inhibit tumor growth.
FG Treatment Inhibits Tumor Growth through Mφs via the PHD-HIF Axis
PHD inhibitors impair prolyl hydroxylation and proteasome degradation of HIFs, which results in upregulation of the HIF signaling pathway (Hoppe et al., 2016). Therefore, we examined whether the FG-induced inhibition of tumor growth resulted from inhibition of the PHD-HIF axis. To genetically mimic the FG drug reaction, Mφ-specific Von Hippel Lindau (VHL)-knockout mice (VHLfl/fl LysM-Cre) were employed; VHLfl/fl LysM-Cre exhibits HIF upregulation (Kaelin, 2008). An LLC tumor transplantation model of VHLfl/fl LysM-Cre mice exhibited similar results as the FG-treated LLC tumor transplant mouse model, namely, tumor growth inhibition (Figures 4A–4C). Furthermore, the Ly6Clo Mφ ratio in the total tumor-infiltrating Mφ population was increased in VHLfl/fl LysM-Cre mice (Figure 4D). Vessel density was also reduced, and the vessel lumen was found extended (Figures S4A and S4B). To examine the off-target effects of FG, an LLC tumor transplantation model of VHLfl/fl and VHLfl/fl LysM-Cre mice were treated with/without FG. In VHLfl/fl mice, FG treatment inhibited tumor growth compared with that of vehicle control (Figures S4C and S4D). However, no significant difference was observed in tumor growth between vehicle- and FG-treated tumors of VHLfl/fl LysM-Cre mice (Figures 4E, 4F, and S4E). Therefore, these results suggest that FG can inhibit tumor growth through Mφs via the PHD-HIF axis. To evaluate the effects of the PHD inhibitor on Mφs, we performed qPCR analysis of bone marrow-derived Mφs (BMDMs) and found that FG-treated BMDMs showed upregulated HIF downstream gene expression (Figure 4G). Previous studies have demonstrated that HIF-1 participates in the M1 polarization of Mφs in an infectious mice model (Andrejeva and Rathmell, 2017, Semba et al., 2016). To assess this in a tumor model, Mφ-specific HIF-1-knockout mice (HIF1fl/fl LysM-Cre) were employed. In HIF1fl/fl mice, FG treatment inhibited tumor growth compared with that of vehicle treatment (Figures S4F and S4G). However, tumor growth was not significantly different between the vehicle- and FG-treated tumors of HIF1fl/fl LysM-Cre mice (Figures 4H, 4I, and S4H). Taken together, these results indicate that the PHD inhibitor affects the HIF signaling pathway, especially the HIF-1 signaling pathway, in Mφs.
Figure 4.

FG Treatment Inhibits Tumor Growth through Mφs via the PHD-HIF Axis
(A) Tumor growth curves of the LLC tumor model of Vhlfl/flLysM-Cre−/− (VHLfl/fl; n = 9) or Vhlfl/flLysM-Cre+/- (VHLfl/flLysM-Cre; n = 7) mice.
(B) Tumor weight on day 16 (n = 7–9).
(C) Images of tumors on day 16. Scale bar, 1 cm.
(D) Flow cytometric analysis of Ly6Chi, Ly6Clo, and Ly6Cneg Mφ ratios in the LLC tumor model (n = 6).
(E) Tumor growth curves of the LLC tumor model of Vhlfl/flLysM-Cre+/- (VHLfl/flLysM-Cre) mice. Treatment with Veh or FG (3 mg; treated on day 10); n = 3–4.
(F) Tumor weight on day 16 (n = 3–4).
(G) qPCR analysis of bone marrow-derived macrophages (BMDMs) treated with/without FG.
(H) Tumor growth curves of the LLC tumor model of HIF-1fl/flLysM-Cre+/- (HIF1fl/flLysM-Cre) mice. Treatment with Veh or FG (3 mg; treated on day 10); n = 4.
(I) Tumor weight on day 16 (n = 4).
Data represent means ± SEM; two-way ANOVA (A, E, and H); Mann-Whitney test (B, D, F, and I); paired t test (G). ns, not significant; *p < 0.05; **p < 0.01. See also Figure S4.
FG Directly Activates Phagocytosis in Mφs
Mφs play an important role in the innate immune system by directly inhibiting tumor growth through phagocytosis (Fiumara et al., 1997, Guerriero et al., 2017, Ubil et al., 2018). Therefore, we hypothesized that FG treatment induces Mφs to activate phagocytosis and thereby inhibit tumor growth. To examine Mφ-engulfed tumor cells in the FG-treated tumor tissue, we used the GFP-labeled LLC tumor model and harvested tumors 48 h after FG administration; these FG-treated tumors did not show normalized tumor vessels (Figures S2I and S2J) and there was a significant increase in tumor-phagocytic Mφs (GFP+ Mφs; Figures 5A and 5B). Furthermore, we did not observe significant differences in cleaved caspase-3 and Ki-67 expression between vehicle and FG-treated tumors (Figures 1I–1L). Taking these results into consideration, we theorized that tumor growth was not inhibited because of apoptosis but possibly because of phagocytosis due to an increased number of phagocytic Mφs within the tumor.
Figure 5.

FG Directly Activates Phagocytosis in Mφs
(A) IF images of tumor-phagocytic Mφs. Tumor sections were stained with anti-GFP antibody (green) and anti-F4/80 antibody (red). Arrowheads indicate GFP+F4/80+ cells. Scale bar, 20 μm.
(B) Quantification of phagocytic (GFP+F4/80+) cell number in tumors (n = 5).
(C) Schematic diagram of the in vitro bead phagocytosis assay.
(D) Images of the in vitro BMDM bead phagocytosis assay. BMDMs were stained with CellVue Claret reagent (red); FITC-beads are shown in green. Arrowheads indicate phagocytic Mφs. Scale bar, 20 μm.
(E) Quantification of the phagocytic BMDM ratio. Three independent experiments were performed with two replicates.
(F) Images of the in vitro BMDM and LLC cell phagocytosis assay. BMDMs were stained with CellVue Claret reagent (red); LLC cells were stained with CFSE (green) and Hoechst (blue). Arrowheads indicate phagocytic Mφs. Scale bar, 20 μm.
(G) Quantification of the phagocytic BMDM ratio. Three independent experiments were performed.
(H) Schematic diagram of the ex vivo bead phagocytosis assay.
(I) Image of the ex vivo phagocytosis assay. Ly6Clo Mφs were stained with CellVue Claret reagent (red); FITC-beads shown in green. Arrowheads indicate phagocytic Mφs. Scale bar, 20 μm.
(J) Quantification of the phagocytic Ly6Clo Mφ ratio (n = 5).
Data represent means ± SEM. Statistical analysis was performed using the Mann–Whitney test. *p < 0.05; **p < 0.01. See also Figure S5.
To further verify the direct effects of FG on Mφs, a phagocytosis assay was performed using BMDMs in vitro (Figure 5C). The level of phagocytic Mφs increased in BMDMs treated with FG (Figures 5D and 5E). In addition, we cocultured BMDM and LLC in vitro and found that the phagocytic ability of tumor cells in BMDMs was activated by FG treatment (Figures 5F and 5G). Activated BMDMs continuously engulfed LLC cells (Figure S5A). Therefore, our results indicate that FG directly affects Mφs.
Next, we analyzed the duration of these FG direct effects on Mφs. We changed the culture medium 12 h after adding FG and then evaluated phagocytic activity 5 days later (Figure S5B). The phagocytic activity of Mφs observed following FG administration was absent by day 5 (Figures S5C and S5D), indicating that the direct effects of FG on Mφs were lost at least 5 days later. We also harvested GFP-labeled LLC tumors 7 days after FG treatment (on day 16) when the TME had improved and then examined the tumors for tumor-phagocytic Mφs in vivo. The increase in tumor-phagocytic Mφs was maintained in-FG treated tumor tissues (Figures S5E and S5F). Thus, these results imply that TME improvement prolongs the anti-tumor effects of Mφs.
To determine which Ly6C Mφ populations activate tumor phagocytosis after FG treatment, we analyzed GFP+ Mφs levels, which were subdivided by Ly6C (Figure S5G). The ratio of GFP+ Mφs in the Ly6C-positive population (Ly6Chi and Ly6Clo) were found increased on day 16 (Figures S5H). To further test this, three separate fractions of Mφs (Ly6Chi, Ly6Clo, and Ly6Cneg Mφs) were sorted from tumor tissues on day 16, and a phagocytosis assay was performed ex vivo (Figures 5H and S5I). Ly6Clo Mφs sorted from FG-treated tumors showed a significantly higher ratio of phagocytic Mφs than that of Ly6Clo Mφs sorted from vehicle-treated tumors (Figures 5I and 5J). The Ly6Chi Mφ populations had a high ratio of phagocytic Mφs in both groups and there were no significant differences between them, but there was a similar tendency toward increased phagocytosis as in the Ly6Clo Mφ population after FG treatment (Figures S5J and S5K). Meanwhile, the Ly6Cneg Mφ populations showed no difference in tumor phagocytosis between those sorted from vehicle- and FG-treated tumors (Figures S5L and S5M). These data suggest that FG activates the tumor-phagocytic ability of Ly6Clo Mφs.
Activation of Mφs via FG Treatment Inhibits Tumor Growth
To examine whether activation of Mφs by FG treatment inhibits tumor growth in a mouse model, Mφs were sorted from vehicle- or FG-treated tumors on day 16. Sorted Mφs were transplanted into day 10 tumor-bearing mice into the tumor locus (Figure 6A). Histological identification of transplanted macrophages indicated that the transplanted cells were widely distributed within the tumor (Figure S6A). Surprisingly, Ly6Clo Mφs from mice subjected to FG-treated tumor transplantation showed inhibited tumor growth (Figures 6B–6D). We also performed the same experiment for Ly6Cneg Mφs from vehicle- and FG-treated tumors. Both Ly6Cneg Mφ-transplanted tumors showed no significant difference in tumor growth (Figures S6B and S6C). Interestingly, these Mφ-transplanted tumors induced changes to the vessel structure; specifically, tumor vessel density was significantly decreased, whereas the vessel luminal area increased (Figures S6D–S6F). Furthermore, ZO-1 and NG2 expression was significantly increased in Ly6Cneg Mφ-transplanted tumors (Figures S6G–S6J). These findings suggest that the Ly6Cneg Mφ population contributes to tumor vessel normalization.
Figure 6.

Activation of Mφs via FG Treatment Inhibits Tumor Growth
(A) Schematic diagram of sorted Ly6Clo Mφs transplanted into the tumor locus.
(B) Tumor growth curves of the LLC tumor mouse models with PBS-injected or transplanted Ly6Clo Mφs from vehicle- or FG-treated mouse tumors (n = 6). **p < 0.01 vs. PBS and Veh Ly6Clo Mφ (two-way ANOVA).
(C) Tumor weight on day 16 (n = 6). *p = 0.06 (Mann-Whitney test).
(D) Images of the tumor on day 16. Scale bar, 1 cm.
(E) Schematic diagram of sorted Ly6Clo Mφ transplantation; cells were treated with/without FG before transplantation into the tumor locus.
(F) Tumor growth curves of LLC tumor mouse models with PBS-injected or transplanted FG treated with/without Ly6Clo Mφs on day 10 (n = 5). *p < 0.05 vs. PBS; ***p < 0.001 vs. Ly6Clo Mφ + Veh (two-way ANOVA).
(G) Tumor weight on day 16 (n = 5). *p < 0.05 versus Ly6Clo Mφ + Veh (unpaired Student's t test).
(H) Images of the LLC tumor on day 16. Scale bar, 1 cm.
Data represent means ± SEM. See also Figure S6.
Next, we examined whether Ly6Clo Mφs, which were isolated from vehicle tumors and exposed in vitro to FG, could inhibit tumor growth in the tumor mouse model. Ly6Clo Mφs sorted from vehicle-treated tumors were cultured with/without FG for 12 h and transplanted into tumor-bearing mice in the tumor locus (Figure 6E). FG-treated Ly6Clo Mφ-transplanted tumors showed significantly inhibited tumor growth (Figures 6F–6H). Thus, our results demonstrate that FG directly affects Ly6Clo Mφs and induces their activation, thereby inhibiting tumor growth.
Discussion
In this study, we demonstrated that FG treatment inhibits tumor growth by activating tumor-infiltrating Mφs and normalizing tumor vessels via the PHD-HIF axis. We also characterized the tumor-infiltrating Mφ population into three types, Ly6Clo, Ly6Cneg, and Ly6Chi.
The Ly6Clo Mφ population showed the most drastic change in phenotype after FG treatment. Tumor growth inhibition by FG was observed in the LLC and B16F10 tumor models, but the MC38 tumor model, which contained fewer Ly6Clo Mφs, did not show tumor growth inhibition following FG treatment. A recent study showed that Ly6Clo Mφs along the tumor margins can prevent cytotoxic lymphocyte infiltration into the tumor core (Beatty et al., 2015). The lower the activity of cytotoxic lymphocytes in the tumor, the higher the malignancy of the tumor; consequently, the rate of tumor growth is thought to be faster (Fridman et al., 2012). In fact, LLC and B16F10 tumors had a higher rate of tumor growth than did MC38 in this study. Taking these factors into consideration, the Ly6Clo Mφ population may contribute to tumor growth. More importantly, our results showed that FG treatment can alter the Ly6Clo Mφ phenotype to a phagocytic phenotype, which would show activity against the tumor.
We further found that Ly6Cneg Mφs may be associated with tumor vessel normalization. When Ly6Cneg Mφs were transplanted into tumors, tumor-infiltrating Mφs isolated from both vehicle- and FG-treated tumors altered tumor vessel structure. In addition, vessel formation was strongly induced in Ly6Cneg Mφs isolated from FG-treated tumors. These findings suggest that FG treatment also affects Ly6Cneg Mφ, leading to tumor vessel normalization.
Ly6Chi Mφs isolated from vehicle-treated tumors exhibited higher phagocytic ability than that of the Ly6Clo and Ly6Cneg Mφ populations in vitro. Furthermore, our results showed that FG may also activate the phagocytic ability of Ly6Chi Mφs. Unfortunately, owing to their fragile expanded cell structure after engulfing tumor cells, it was too difficult to purify and isolate live Ly6Chi Mφs from tumors in the transplantation experiment.
Recent studies demonstrated that TAMs simultaneously express both M1 and M2 markers (Azizi et al., 2018, Muller et al., 2017, Peterson et al., 2016). In this study we used CD80 as an M1 cell surface marker and CD206 as an M2 cell surface marker. Consistent with recent reports, these markers were expressed in both vehicle- and FG-treated tumor-infiltrating Mφs, and there were no differences. We analyzed other cell surface markers, such as MHCII, but we were unable to observe any changes in expression after FG treatment. Furthermore, we examined gene expression levels in BMDMs treated with/without FG. FG-treated BMDMs showed upregulation of HIF downstream gene expression (VEGF, GLUT1, etc.) as well as M1 marker genes (iNOS, MHCII, IL-1β, IL-6, and TNF-α) and M2 marker genes (Arginase-1 and IL-10). Thus, it was too difficult to distinguish and determine Mφ polarization using existing markers. The mixed phenotype of Mφs, which consists of the inflammatory (M1) and anti-inflammatory (M2) phenotypes, may depend on the balance of each molecule.
A previous study demonstrated that heterozygous deficiency of Phd2 in endothelial cells does not affect vessel lumen size but normalizes the endothelial barrier and pericyte coverage (Leite de Oliveira et al., 2012). Most of our results are in agreement with previous reports, but one thing was different; the vessel lumen area was found increased in this study. We previously showed that the PHD inhibitor elongates tumor vessel diameter (Koyama et al., 2017) and suggested that the tumor vessels were reconstituted after PHD inhibitor treatment. This may have resulted from PHD inhibitor treatment, which affects not only endothelial cells but also Mφs and other cells.
Several studies have previously reported that tumor vessel normalization either did not changed the tumor size (Cantelmo et al., 2016, Mazzone et al., 2009) or increased tumor size along with change in apoptosis and/or proliferation (Folkman, 1971, Krzywinska et al., 2017). On the other hand, other studies have reported that vessel normalization alters the TME and activates anti-tumor immunity; therefore, vascular normalization is emerging as a strategy for enhancing cancer therapy (Quail and Joyce, 2013, Henze and Mazzone, 2016, Park et al., 2016). In the present study, the expression levels of the apoptosis marker, cleaved caspase-3, and the proliferation marker, Ki-67, did not change in the FG-treated tumors. FG treatment not only led to normalization of tumor vessels and improved the TME but also directly activated phagocytosis in Mφs. In addition, several studies have reported that Mφs play an important role in the innate immune system by directly inhibiting tumor growth through phagocytosis without inducing apoptosis (Fiumara et al., 1997, Guerriero et al., 2017, Ubil et al., 2018). Therefore, we argue that the tumor growth inhibition in FG-treated tumors is contributed by phagocytosis due to an increased number of phagocytic Mφs within the tumor.
HIF stabilization in CD8+ T cells results in increased expression and release of important cytolytic molecules, such as granzyme B and perforin (Doedens et al., 2010, Palazon et al., 2014, Palazon et al., 2017). This suggests that T cells function in tumor growth inhibition. Consistent with this, we observed that T cells were slightly activated in B16F10 tumor tissues and circulating blood of B16F10 tumor-bearing mice after FG treatment. Therefore, in the B16F10 tumor model, FG may affect not only tumor-infiltrating Mφs but also T cells. However, unexpectedly, T cell activation was not observed in the LLC tumor model and Mφs were the primary factors after FG treatment. Further studies are needed to elucidate the role of the PHD-inhibitor on T cells.
We have previously shown that the PHD-HIF axis induces angiogenesis and promotes tissue wound healing (Koyama et al., 2017, Takaku et al., 2012). In the present study, we used Vhl LysM Cre mice to confirm the main working mechanism of FG. VHL was associated with prolyl hydroxylated HIFs and promoted HIF degradation (Kaelin, 2008, Takeda et al., 2010). VHLfl/fl LysM-Cre mice showed a similar phenotype as the FG-treated tumor, where tumor growth was inhibited, the Ly6Clo Mφ ratio in the total tumor-infiltrating Mφs increased, and tumor vessels were normalized. Thus, the PHD inhibitor may affect not only endothelial cells but also tumor-infiltrating Mφs via the PHD-HIF axis. Moreover, HIF-1α drives a metabolic shift toward glycolysis and is associated with M1 polarization in an infectious mice model (Semba et al., 2016). In this study, we showed that tumor growth was not significantly different between vehicle- and FG-treated tumors in HIF-1fl/flLysMCre mice, suggesting that HIF-1 may polarize Mφs to an M1-like phenotype even in tumor-infiltrating Mφs. Therefore, tumor-infiltrating Mφs may change their phenotypes to that of anti-tumor Mφs via the PHD-HIF axis after treatment with the PHD inhibitor.
In this study, we showed that FG inhibits the growth of tumors that are TAM abundant and contain few activated T cells in a subcutaneous tumor model. This tumor immune microenvironment is associated with tumor malignancy and results in therapeutic resistance (Binnewies et al., 2018). Therefore, we chose this subcutaneous tumor model to identify the therapeutic strategies against these tumors. However, our model does not completely reproduce the local TME. Tissue-resident Mφs respond to diverse environmental signals and have distinct global expression profiles (Lavin et al., 2014). Therefore, orthotopic or genetically engineered mouse models are important for reproducing the local TME and evaluating the effect of FG treatment on tissue-resident Mφs. However, we hope that our study may lead to the development of the therapeutic strategy against TAM-abundant tumors that contain few activated T cells.
In conclusion, we demonstrated that FG inhibits tumor growth by activating tumor-infiltrating Mφs and normalizing tumor vessels and the TME via the PHD-HIF axis. In particular, FG treatment altered the Ly6Clo Mφs phenotype, which is associated with tumor malignancy, to an anti-tumor phenotype. Therefore, the PHD inhibitor can potentially be utilized for promoting the anti-tumor potential of Mφs to improve cancer therapy.
Limitations of the Study
In this study, we showed that FG treatment inhibited tumor growth by activating the phagocytic ability of tumor-infiltrating Mφs via the PHD-HIF axis. However, we were unable to analyze the gene expression of activated tumor-infiltrating Mφs in vivo. This is because activated tumor-infiltrating Mφs had already engulfed LLC cells, and thus any internal controls were unsuccessful. Therefore, we alternatively examined changes in gene expression using FG-treated BMDMs. We are working on analyzing gene expression changes of activated tumor-infiltrating Mφs after FG treatment for future studies. In addition, we chose a subcutaneous tumor injection model because we wanted to assess the effect of FG treatment on tumors that are TAM abundant and contain few activated T cells. Orthotopic or genetically engineered mouse models would be needed to precisely reproduce the local TME and evaluate the effects of FG on tissue-resident Mφs.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Y. Nishida for technical assistance. Flow cytometric experiments and analysis were performed at the Research Support Platform of Osaka City University Graduate School of Medicine. This work was supported by a Grant-in-Aid for Research Activity Start-up (grant number JP17H07024 to S.N.), Grant-in-Aid for Young Scientists (grant number JP17K15459 to S.M.), and Grant-in-Aid for Scientific Research (grant number JP17K08602 and JP17K08958 to S.T. and K.M., respectively) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author Contributions
S.N. and S.M. performed the majority of the experiments and data analysis. M.S. performed the gene editing experiment. T.Y. and S.K. performed the mice breeding and histological experiments. N.T. performed part of the VHLf/f LysM-Cre mice experiments. M.T. performed part of the MC38 transplant experiments. S.N., S.M., M.S., K.M., and S.T. designed experiments. S.N., S.M., M.S., T.Y., S.K., N.T., M.T., Y.M., J.U., T.N., K.M., and S.T. interpreted the data and provided necessary materials. S.N., S.M., and S.T. drafted the manuscript. S.M. and S.T. conceived the concept of the study and supervised the research. All authors reviewed the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.08.033.
Supplemental Information
References
- Andrejeva G., Rathmell J.C. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 2017;26:49–70. doi: 10.1016/j.cmet.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty G.L., Winograd R., Evans R.A., Long K.B., Luque S.L., Lee J.W., Clendenin C., Gladney W.L., Knoblock D.M., Guirnalda P.D. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology. 2015;149:201–210. doi: 10.1053/j.gastro.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besarab A., Chernyavskaya E., Motylev I., Shutov E., Kumbar L.M., Gurevich K., Chan D.T., Leong R., Poole L., Zhong M. Roxadustat (FG-4592): Correction of anemia in incident dialysis patients. J. Am. Soc. Nephrol. 2016;27:1225–1233. doi: 10.1681/ASN.2015030241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantelmo A.R., Conradi L.C., Brajic A., Goveia J., Kalucka J., Pircher A., Chaturvedi P., Hol J., Thienpont B., Teuwen L.A. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell. 2016;30:968–985. doi: 10.1016/j.ccell.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens A.L., Stockmann C., Rubinstein M.P., Liao D., Zhang N., DeNardo D.G., Coussens L.M., Karin M., Goldrath A.W., Johnson R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R., Lu K.V., Petritsch C., Liu P., Ganss R., Passegue E., Song H., Vandenberg S., Johnson R.S., Werb Z. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumara A., Belfiore A., Russo G., Salomone E., Santonocito G.M., Ippolito O., Vigneri R., Gangemi P. In situ evidence of neoplastic cell phagocytosis by macrophages in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 1997;82:1615–1620. doi: 10.1210/jcem.82.5.3909. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Franklin R.A., Liao W., Sarkar A., Kim M.V., Bivona M.R., Liu K., Pamer E.G., Li M.O. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman W.H., Pages F., Sautes-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- Guerriero J.L., Sotayo A., Ponichtera H.E., Castrillon J.A., Pourzia A.L., Schad S., Johnson S.F., Carrasco R.D., Lazo S., Bronson R.T. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543:428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze A.T., Mazzone M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Invest. 2016;126:3672–3679. doi: 10.1172/JCI84427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe G., Yoon S., Gopalan B., Savage A.R., Brown R., Case K., Vasanji A., Chan E.R., Silver R.B., Sears J.E. Comparative systems pharmacology of HIF stabilization in the prevention of retinopathy of prematurity. Proc. Natl. Acad. Sci. U S A. 2016;113:E2516–E2525. doi: 10.1073/pnas.1523005113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiyaz H.Z., Simon M.C. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 2010;345:105–120. doi: 10.1007/82_2010_74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R.K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin W.G., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Keith B., Johnson R.S., Simon M.C. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer. 2011;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S., Matsunaga S., Imanishi M., Maekawa Y., Kitano H., Takeuchi H., Tomita S. Tumour blood vessel normalisation by prolyl hydroxylase inhibitor repaired sensitivity to chemotherapy in a tumour mouse model. Sci. Rep. 2017;7:45621. doi: 10.1038/srep45621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinska E., Kantari-Mimoun C., Kerdiles Y., Sobecki M., Isagawa T., Gotthardt D., Castells M., Haubold J., Millien C., Viel T. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017;8:1597. doi: 10.1038/s41467-017-01599-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y., Winter D., Blecher-Gonen R., David E., Keren-Shaul H., Merad M., Jung S., Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite de Oliveira R., Deschoemaeker S., Henze A.T., Debackere K., Finisguerra V., Takeda Y., Roncal C., Dettori D., Tack E., Jonsson Y. Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell. 2012;22:263–277. doi: 10.1016/j.ccr.2012.06.028. [DOI] [PubMed] [Google Scholar]
- Mazzone M., Dettori D., de Oliveira R.L., Loges S., Schmidt T., Jonckx B., Tian Y.M., Lanahan A.A., Pollard P., de Almodovar C.R. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahedi K., Laoui D., Gysemans C., Baeten M., Stange G., Van den Bossche J., Mack M., Pipeleers D., In’t Veld P., De Baetselier P. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- Muller S., Kohanbash G., Liu S.J., Alvarado B., Carrera D., Bhaduri A., Watchmaker P.B., Yagnik G., Di Lullo E., Malatesta M. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18:234. doi: 10.1186/s13059-017-1362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizet V., Johnson R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009;9:609–617. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A., Goldrath A.W., Nizet V., Johnson R.S. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41:518–528. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A., Tyrakis P.A., Macias D., Velica P., Rundqvist H., Fitzpatrick S., Vojnovic N., Phan A.T., Loman N., Hedenfalk I. An HIF-1alpha/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. 2017;32:669–683.e5. doi: 10.1016/j.ccell.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.S., Kim I.K., Han S., Park I., Kim C., Bae J., Oh S.J., Lee S., Kim J.H., Woo D.C. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell. 2016;30:953–967. doi: 10.1016/j.ccell.2016.10.018. [DOI] [PubMed] [Google Scholar]
- Peterson T.E., Kirkpatrick N.D., Huang Y., Farrar C.T., Marijt K.A., Kloepper J., Datta M., Amoozgar Z., Seano G., Jung K. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. U S A. 2016;113:4470–4475. doi: 10.1073/pnas.1525349113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano R., Besarab A., Sun C.H., Diamond S.A., Durham J.H., Cangiano J.L., Aiello J.R., Novak J.E., Lee T., Leong R. Oral hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) for the treatment of anemia in patients with CKD. Clin. J. Am. Soc. Nephrol. 2016;11:982–991. doi: 10.2215/CJN.06890615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba H., Takeda N., Isagawa T., Sugiura Y., Honda K., Wake M., Miyazawa H., Yamaguchi Y., Miura M., Jenkins D.M. HIF-1alpha-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016;7:11635. doi: 10.1038/ncomms11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G.L. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann C., Doedens A., Weidemann A., Zhang N., Takeda N., Greenberg J.I., Cheresh D.A., Johnson R.S. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–818. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaku M., Tomita S., Kurobe H., Kihira Y., Morimoto A., Higashida M., Ikeda Y., Ushiyama A., Hashimoto I., Nakanishi H. Systemic preconditioning by a prolyl hydroxylase inhibitor promotes prevention of skin flap necrosis via HIF-1-induced bone marrow-derived cells. PLoS One. 2012;7:e42964. doi: 10.1371/journal.pone.0042964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N., O'Dea E.L., Doedens A., Kim J.W., Weidemann A., Stockmann C., Asagiri M., Simon M.C., Hoffmann A., Johnson R.S. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L., Goldstein A., Wang H., Ching Lo H., Sun Kim I., Welte T., Sheng K., Dobrolecki L.E., Zhang X., Putluri N. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017;544:250–254. doi: 10.1038/nature21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubil E., Caskey L., Holtzhausen A., Hunter D., Story C., Earp H.S. Tumor-secreted Pros1 inhibits macrophage M1 polarization to reduce antitumor immune response. J. Clin. Invest. 2018;128:2356–2369. doi: 10.1172/JCI97354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.