

Abstract
Prediction of human pharmacokinetics (PK) based on preclinical information for antibody–drug conjugates (ADCs) provide important insight into first‐in‐human (FIH) study design. This retrospective analysis was conducted to identify an appropriate scaling method to predict human PK for ADCs from animal PK data in the linear range. Different methods for projecting human clearance (CL) from animal PK data for 11 ADCs exhibiting linear PK over the tested dose ranges were examined: multiple species allometric scaling (CL vs. body weight), allometric scaling with correction factors, allometric scaling based on rule of exponent, and scaling from only cynomolgus monkey PK data. Two analytes of interest for ADCs, namely total antibody and conjugate (measured as conjugated drug or conjugated antibody), were assessed. Percentage prediction errors (PEs) and residual sum of squares (RSS) were compared across methods. Human CL was best estimated using cynomolgus monkey PK data alone and an allometric scaling exponent of 1.0 for CL. This was consistently observed for both conjugate and total antibody analytes. Other scaling methods either underestimated or overestimated human CL, or produced larger average absolute PEs and RSS. Human concentration‐time profiles were also reasonably predicted from the cynomolgus monkey data using species‐invariant time method with a fixed exponent of 1.0 for CL and 1.0 for volume of distribution. In conclusion, results from this retrospective analysis of 11 ADCs indicate that allometric scaling of CL with an exponent of 1.0 using cynomolgus monkey PK data alone can successfully project human PK profiles of an ADC within linear range.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Prediction of pharmacokinetics (PK) based on nonclinical data provides an important tool for selecting a first‐in‐human (FIH) dose and estimating the safety margin for new therapies. Multiple prediction methods have been developed and widely used for both small molecules and monoclonal antibodies; however, prediction of human PK for antibody–drug conjugates (ADCs) has not been systemically examined.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addressed the question of the appropriate scaling method to predict human PK from nonclinical data for ADCs by systemically examining current available methods.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Allometric scaling of CL using cynomolgus monkey alone with an exponent of 1.0 successfully predicts human clearance for both conjugate and total antibody analytes of an ADC with linear PK.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This publication enables better prediction of human PK for ADCs from animal PK data to inform FIH study design.
Antibody–drug conjugates (ADCs) are a novel class of therapeutic agents consisting of a monoclonal antibody (mAb) covalently bound with a cytotoxic drug through a chemical linker. ADCs are designed to preferentially deliver a potent cytotoxic drug to tumor cells via tumor‐specific or overexpressed cell surface antigens. After binding to the cell surface antigen, the ADC is internalized by tumor cells, where it undergoes lysosomal degradation, leading to the release of the cytotoxic drug, and, thus, cell death. Targeted delivery of cytotoxic drugs to tumors enables ADCs to potentially harness and improve their antitumor effect while minimizing their impact on normal tissues, thereby optimizing their benefit‐risk profile.
To date, four ADCs have received US Food and Drug Administration approval.1 The first of these, gemtuzumab ozogamicin (Mylotarg Wyeth Pharmaceuticals Inc, A subsidiary of Pfizer Inc, Philadelphia, PA 19101, a CD33‐directed ADC), was approved in 2001 for the treatment of acute myelogenous leukemia. It was withdrawn from the market in June 2010 as it was linked to a serious and potentially fatal liver condition known as veno‐occlusive disease. Gemtuzumab ozogamicin was resubmitted for approval with a fractionated dosing regimen and was subsequently approved by the US Food and Drug Administration in September 2017. Three other approved ADCs are brentuximab vedotin (Adcetris Seattle Genetics, Inc. Bothell, WA 98021, a CD30‐directed ADC) for the treatment of Hodgkin lymphoma and systemic anaplastic large cell lymphoma, trastuzumab emtansine (KadcylaGenentech, Inc. A Member of the Roche Group, 1 DNA Way South San Francisco, CA 94080‐4990, a human epidermal growth factor 2–direct ADC) for treating human epidermal growth factor 2–positive metastatic breast cancer, and inotuzumab ozogamicin (Besponsa Wyeth Pharmaceuticals Inc, A subsidiary of Pfizer Inc, Philadelphia, PA 19101, a CD22‐direct ADC) for the treatment of adults with relapsed or refractory B‐cell precursor acute lymphoblastic leukemia. Additionally, numerous ADCs are at preclinical and clinical development with different cytotoxic drugs, linkers and drug‐antibody ratios (DARs) being explored.2
Given the complex structure of an ADC with both large and small‐molecule components, a typical pharmacokinetic (PK) assessment of an ADC involves an analysis of multiple analytes in circulation, including ADC conjugate, total antibody (sum of conjugated and unconjugated antibodies), and unconjugated drug.3 There are two alternative ways to measure the ADC conjugate, namely conjugated antibody and conjugated drug. Conjugated antibody assay measures the concentration of antibody molecules with one or more cytotoxic drugs attached, whereas conjugated drug assay measures the total concentration of cytotoxic drug that is conjugated to the antibody. Selection of appropriate conjugate assay will depend on the ADC linker (cleavable vs. noncleavable), assay behavior, and recovery across different DAR species.3
ADCs have a unique elimination pathway, as compared with mAbs and small molecules. ADCs are typically cleared through two alternative pathways, namely proteolytic degradation and deconjugation.4 Similar to mAbs, ADC clearance (CL) through proteolytic degradation is driven primarily by catabolism mediated by target‐specific or nonspecific cellular uptake followed by lysosomal degradation, whereas deconjugation CL is usually mediated by enzymatic or chemical cleavage of the linker leading to the release of the cytotoxic drug from the ADC.
Prediction of human PKs to help estimate dose and dosing regimens is important during clinical development, especially prior to first‐in‐human studies, as drug efficacy and toxicity are usually linked to drug exposure. Often, it also supports an early assessment of efficacious doses and manufacturing feasibility. Allometric scaling is one of the commonly used approaches to predict human PK parameters for both small molecules5, 6 and monoclonal mAbs.7, 8 A fundamental assumption of allometric scaling is that anatomic, physiologic, and biochemical processes are similar across animal species and human and, thus, vary as a function of body weight. Numerous excellent studies and reviews have been published regarding human CL prediction using the simple allometric scaling method and many other approaches for small‐molecule drugs.9 Deng et al.7 showed that for mAbs, multiple species allometric scaling might not be the optimal method for projecting human PK. A better prediction of human CL was achieved for mAbs based on cynomolgus monkey PK data alone and an allometric scaling exponent of 0.85 for CL. Given ADCs are composed of both large and small molecule components, allometric scaling approaches established for small molecules and mAbs using data from animal species may not be directly applied to ADCs. We have identified 11 ADCs with PK data available from both animals (e.g., mice, rats, or cynomolgus monkeys) and humans (Table 1 ). The objective of the current study was to conduct a retrospective analysis using a data set of the 11 ADCs at the dose ranges demonstrating linear PKs to identify an appropriate scaling method to predict human PKs from nonclinical PK data for ADCs. Human concentration‐time profiles of an ADC were also projected using the species‐invariant time method and then compared with observed clinical data.
Table 1.
ADCs and their properties used in human PK prediction
| ADCs | Target | mAb isotype | Drug | Linker | Average DAR (distribution) | Conjugation | Indication |
|---|---|---|---|---|---|---|---|
| DMUC5754A30 | MUC‐16 | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | Ovarian, pancreatic |
| DNIB0600A15 | Napi2b | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | Ovarian, lung |
| DEDN6526A31 | ETBR | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | Melanoma |
| DMOT4039A32 | MsLN | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | Ovarian, Pancreatic |
| Polatuzumab vedotin23, 33 | CD79b | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | NHL |
| Pinatuzumab vedotin33, 34 | CD22 | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | NHL |
| DSTP3086S35 | Steap1 | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | Prostate |
| ADC1 | NR | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | NR |
| ADC2 | NR | Humanized IgG1 | MMAE | VC | ~ 3.5 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | NR |
| T‐DM120 | HER2 | Humanized IgG1 | DM1 | MCC | ~3.5 (0–8) | Through lysines | HER2 + MBC |
| Brentuximab vedotin19, 36 | CD30 | Chimeric IgG1 | MMAE | VC | ~4.0 (0, 2, 4, 6, 8) | Through reduced interchain disulfide bonds | HL, ALCL |
ADC, antibody–drug conjugate; ALCL, anaplastic large‐cell lymphoma; DAR, drug‐antibody ratio; HER2, human epidermal growth factor 2; HL, classical Hodgkin lymphoma; IgG1, immunoglobulin‐G1; mAb, monoclonal antibody; MBC, metastatic breast cancer; MCC, 4‐[N‐maleimidomethyl] cyclohexane‐1‐carboxylate; MMAE, monomethyl auristatin E; NHL, non‐Hodgkin lymphomas; NR, not reported; PK, pharmacokinetic; VC, valine‑citrulline.
Methods
ADCs with PK data available in both animals (e.g., mice, rats, or cynomolgus monkeys) and humans were included in the analysis. Drug product materials used for animal and clinical studies were similar with respect to DAR, aggregation, and unconjugated drug levels. For ADCs that are cleared significantly via target‐mediated mechanisms, CL at doses that saturated the target‐mediated CL pathway was used for the analysis. Two analytes of interest for ADCs, namely total antibody (sum of conjugated and unconjugated antibodies) and conjugate (measured as either conjugated drug or conjugated antibody) were assessed. For the conjugate analyte, only the ADCs with the same conjugate measurement (either as conjugated antibody or as conjugated drug) between animals and humans were included in the analysis. Given most of ADCs have not measured conjugate CL in rodents with only cynomolgus monkey and human PK data available for the analysis, scaling method from cynomolgus monkey data only were examined for the conjugate analyte. Human PK prediction for unconjugated drugs was not assessed in the present study, due to low circulating levels of unconjugated drugs that often lead to incomplete PK profiles in animals or humans. All animal experiments were conducted in accordance with national and institutional guidelines for the care and use of laboratory animals.
Multiple species allometric scaling
CL of total antibody analyte in each animal species was plotted against the animal body weight (BW) on a log–log scale using the following allometric equation:
| (1) |
where a is the coefficient and x is the exponent of the allometric equation, as calculated from the intercept and slope of the linear regression line, respectively.
Allometric scaling with correction factors
Total antibody CL in each animal species was multiplied by the maximum life‐span potential (MLP; Eq. (2)) or brain weight (BrW, Eq. (3)) of the animal species, and then the product was plotted as a function of BW on a log–log scale according to the following equation:
| (2) |
| (3) |
where b and c are the coefficients, and y and z are the exponents. Standard BrWs used for correction were 0.36 g for mice, 1.8 g for rats, 63 g for cyno monkeys, and 1.4 kg for humans.10 The MLP for each animal species was calculated as a function of their respective BW and BrW, according to the following equation11:
| (4) |
Allometric scaling based on rule of exponents
Rule of exponents (ROEs) proposed by Mahmood12 for mAbs was applied to total antibody CL prediction for eight ADCs where CL data were available for at least two animal species. According to the ROEs, MLP as a correction factor was used when exponents of simple allometry by BW are <0.71, whereas BrW correction was used when exponents of simple allometry are >1.
Estimation of scaling exponent and projection of human CL based on cynomolgus monkey PK data only
Human CL of conjugate and total antibody of each ADC were predicted based on cynomolgus monkey data using the following allometric equation:
| (5) |
where w is the scaling exponent for CL. Based on the observed human CL and cynomolgus monkey CL and the typical BWs of cynomolgus monkeys and humans, w for conjugate or total antibody analyte for each ADC was calculated using Eq. (5). Additionally, human CL was calculated using Eq. (5) with a fixed exponent of 0.75, 0.85, or mean of w across ADCs and compared with the observed data. Exponent of 0.75 is a commonly used scaling factor to predict human CL across species.13 Exponent of 0.85 was selected based on the work done by Deng et al.7 on the mAb CL scaling.
Projection of human PK profiles using cynomolgus monkey data only
Using DNIB0600A, an anti‐Napi2b valine‐citrulline‐monomethyl auristatin E (vc‐MMAE) ADC (Table 1 ), as an example, the concentration‐time profiles of ADC and total antibody in cynomolgus monkeys following 0.3 and 1.0 mg/kg i.v. administration of DNIB0600A were transformed to human concentration‐time profiles using the species‐invariant time method14 described by the following equations:
| (6) |
| (7) |
DNIB0600A exhibited linear PK over the dose ranges tested in cynomolgus monkeys and humans.15 The allometric equations used scaling exponents estimated as above for CL and 1.0 for volume (V), respectively. The scaled bi‐exponential PK profiles were fit to a two compartmental model i.v. bolus input to estimate the PK parameters, which served as input for Monte‐Carlo simulations in humans. The simulations were performed for 1,000 subjects at the recommended phase II dose (2.4 mg/kg) of DNIB0600A using NONMEM 7 (version 7.3.0; ICON Development Solutions, Ellicott City, MD). Interindividual variability on CL and Vc was assumed to be 30%, based on observed interindividual variability of these parameters for human ADCs (data on file). The simulated PK profiles were overlaid and compared with the observed PK data.
Statistical analysis
Percentage prediction errors (PEs), which are ((CLhuman, predicted − CLhuman, observed)/CLhuman, observed) × 100% for overprediction and ((CLhuman, predicted − CLhuman, observed)/CLhuman,predicted) × 100% for underprediction, were used to assess the prediction performance.16 Twofold differences on CL will be translated into PE = 100% and −100% for overprediction and underprediction, respectively. Average |PE| (APE), is calculated using the equation below to assess the overall predictability for each scaling method.
| (8) |
where |PEi| is the absolute PE.
Additionally, residual sum of squares (RSS) were calculated using the following equation and compared with assess overall goodness of the prediction across scaling methods:
| (9) |
Dividing the available molecules into a test and validation data set was considered but not implemented because the small data set in current analysis (n = 11 ADCs) makes it impossible to conduct a robust analysis by further dividing the data set.
Results
Eleven ADCs with PK data available for both animals (e.g., mice, rats, or cynomolgus monkeys) and humans were included in this analysis (Table 1 ). Inotuzumab ozogamicin and gemtuzumab ozogamicin were not included in the analysis as these two molecules exhibited nonlinear and/or time‐dependent PK in humans over clinically tested doses.17, 18 Of the 11 ADCs, 10 ADCs are vc‐MMAE ADCs, which use a dipeptide (valine‐citrulline (vc)) linker conjugated to the cytotoxic drug, MMAE, via solvent accessible thiols present in mAb cysteines; whereas T‐DM1 uses DM1 as the cytotoxic drug conjugated to trastuzumab via the noncleavable 4‐[N‐maleimidomethyl] cyclohexane‐1‐carboxylate linker. Conjugation of MMAE or DM1 to mAbs through reduced interchain disulfide cysteine or lysine residues results in a heterogeneous mixture of conjugated antibodies, with DARs ranging from 0−8, and with an average DAR of ~ 3.5 or 4 for the 11 ADCs (Table 1 ). ADCs with site‐specific conjugation were not included in the present analysis due to lack of availability of PK data from animals and/or humans.
As shown in Tables 2 and 3 , the CL of total antibody for 11 ADCs ranged from ~ 5–10 mL/day/kg in mice (n = 7), ~ 7–10 mL/day/kg in rats (n = 3), ~ 5–28 mL/day/kg in cynomolgus monkeys (n = 11), and ~ 5–21 mL/day/kg in humans (n = 11) within their linear range. For conjugate analyte, the clearance of conjugate ranged from ~ 10–30 mL/day/kg in cynomolgus monkeys (n = 8) and ~ 9–27 mL/day/kg in humans (n = 8) within their linear range (Table 4 ).
Table 2.
Predicted human CL for total antibody analyte of ADCs using scaling from multiple nonclinical species
| ADCa , b | Observed CL (mL/day/kg) | Multiple species allometric scalinge | Maximum life potential as correction factore | Brain weight as correction factore | Rule of exponentsg | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse | Rat | Cyno | Human | x e | CLpred | PEf (%) | y e | CLpred | PEf (%) | z e | CLpred | PEf (%) | CLpred | PEf (%) | |
| DNIB0600Ac | 9.00 | ND | 13.3 | 12.2 | 1.08 | 16.7 | 36.6 | 1.49 | 15.6 | 27.7 | 2.08 | 15.0 | 22.9 | 14.99 | 22.9 |
| DMOT4039Ac | 9.50 | ND | 27.6 | 20.0 | 1.21 | 51.3 | 156 | 1.62 | 47.9 | 140 | 2.21 | 46.1 | 131 | 46.11 | 131 |
| Polatuzumab vedotinc | 5.09 | ND | 6.00 | 14.5 | 1.03 | 6.6 | −119 | 1.44 | 6.19 | −134 | 2.03 | 5.95 | −144 | 5.95 | −144 |
| Pinatuzumab vedotinc | 6.10 | ND | 9.40 | 13.8 | 1.08 | 12.1 | −14.1 | 1.50 | 11.3 | −22.0 | 2.08 | 10.9 | −26.8 | 10.88 | −26.8 |
| DSTP3086S | 9.90 | 9.50 | 13.4 | 8.20 | 1.06 | 15.0 | 83.4 | 1.47 | 11.7 | 42.6 | 2.06 | 10.1 | 23.4 | 10.12 | 23.4 |
| ADC1c | 6.60 | ND | 10.5 | 10.8 | 1.09 | 13.8 | 27.3 | 1.50 | 12.9 | 19.1 | 2.09 | 12.4 | 14.6 | 12.38 | 14.6 |
| T‐DM1 | 8.00 | 6.5 | 4.60 | 4.90 | 0.89 | 3.36 | −45.8 | 1.30 | 2.61 | −87.7 | 1.89 | 2.26 | −117 | 3.36 | −45.8 |
| Brentuximab vedotind | ND | 9.00 | 14.6 | 10.6 | 1.18 | 25.4 | 139 | 1.82 | 45.9 | 333 | 2.53 | 64.5 | 509 | 64.5 | 509 |
| APE | 77.7 | 101 | 124 | 115 | |||||||||||
| RSS | 1,337 | 2,136 | 3,693 | 2,908 | |||||||||||
ADC, antibody–drug conjugate; APE, average absolute value of percentage prediction error (|PE|); BrW, brain weight; BW, body weight; CL, clearance; cyno, cynomolgus monkeys; MPL, maximum life‐span potential; ND, no data; PK, pharmacokinetic; ROE, rule of exponent; RSS, residual sum of square.
aAll ADCs except for brentuximab vedotin are humanized immunoglobulin‐G1 (IgG1) antibodies. Brentuximab vedotin is a chimeric IgG1 antibody. bUse reported body weight for mice (20 g), rats (250 g), cyno (3.5 kg), and humans (70 kg). cOnly mice and cynomolgus monkey PK data are available. Regression was done based on two species. dOnly rat and cyno PK data are available. Regression was done based on two species. eMultiple species allometric scaling: CL = a · BWx; allometric scaling with MLP as correction factor: MLP · CL = b · BWy; allometric scaling with BrW as correction factor: BrW · CL = c · BWz; where a, b, and c is the coefficient and x, y, and z is the exponent of the allometric equation. fPEs = ((CLhuman, predicted − CLhuman, observed)/CLhuman, observed) × 100% for overprediction and ((CLhuman, predicted − CLhuman, observed)/CLhuman, predicted) × 100% for underprediction. gROE proposed by Mahmood12 for monoclonal antibodies: according to ROE, MLP as a correction factor was used when exponents of simple allometry are <0.71, whereas BrW correction was used when exponents of simple allometry are > 1.
Table 3.
Predicted human clearance for total antibody analyte of ADCs using scaling from only cynomolgus monkey PK data with different fixed exponents of CL
| ADCa | Observed CL (mL/day/kg) | Scaling from cyno data using a fixed exponent of CL of 0.75 | Scaling from cyno data using a fixed exponent of CL of 0.85 | Scaling from cyno data using a fixed exponent of CL of 1.0 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Cyno | Human | w a | CLpred | PE (%) | CLpred | PE (%) | CLpred | PE (%) | |
| DMUC5754A | 16.0 | 15.6 | 0.99 | 7.6 | −105 | 10.2 | −52.9 | 16 | 2.6 |
| DNIB0600A | 13.3 | 12.2 | 0.97 | 6.3 | −93.7 | 8.5 | −43.5 | 13.3 | 9.0 |
| DEDN6526A | 11.2 | 21.3 | 1.22 | 5.3 | −302 | 7.2 | −196 | 11.2 | −90.2 |
| DMOT4039A | 27.6 | 20.0 | 0.89 | 13.1 | −52.7 | 17.6 | −13.6 | 27.6 | 38.0 |
| Polatuzumab vedotin | 6.00 | 14.5 | 1.29 | 2.80 | −418 | 3.80 | −282 | 6 | −142 |
| Pinatuzumab vedotin | 9.40 | 13.8 | 1.13 | 4.40 | −214 | 6.00 | −130 | 9.4 | −46.8 |
| DSTP3086S | 13.4 | 8.2 | 0.84 | 6.30 | −30.2 | 8.60 | 4.65 | 13.4 | 63.4 |
| ADC1 | 10.5 | 10.8 | 1.01 | 5.00 | −116 | 6.70 | −61.2 | 10.5 | −2.86 |
| ADC2 | 8.80 | 9.6 | 1.03 | 4.10 | −134 | 5.60 | −71.4 | 8.8 | −9.09 |
| T‐DM1 | 4.60 | 4.9 | 1.02 | 2.20 | −123 | 2.90 | −69.0 | 4.6 | −6.52 |
| Brentuximab vedotin | 14.6 | 10.6 | 0.89 | 6.90 | −53.6 | 9.30 | −14.0 | 14.6 | 37.7 |
| APE | 149 | 85.3 | 40.7 | ||||||
| RSS | 716 | 462 | 297 | ||||||
ADC, antibody–drug conjugate; APE, average absolute value of percentage prediction error (|PE|); CL, clearance; cyno, cynomolgus monkeys; PE, percentage prediction errors, which was calculated by ((CLhuman, predicted − CLhuman, observed)/CLhuman, observed) × 100% for overprediction and ((CLhuman, predicted − CLhuman, observed)/CLhuman, predicted) × 100% for underprediction; PK, pharmacokinetic; RSS, residual sum of square.
The exponent w for the total antibody were back calculated based on the observed mean CL in cyno and in humans, mean ± SD of w = 1.03 ± 0.14.
Table 4.
Predicted human clearance for ADC conjugate analyte using scaling from only cynomolgus monkey PK data with different fixed exponents of CL
| ADC | Observed CL (mL/day/kg) | Scaling from cyno data using a fixed exponent of CL of 0.75 | Scaling from cyno data using a fixed exponent of CL of 0.85 | Scaling from cyno data using a fixed exponent of CL of 1 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Cyno | Human | w a | CLpred | PE (%) | CLpred | PE (%) | CLpred | PE (%) | |
| DMUC5754Ab | 28.4 | 25.7 | 0.966 | 13.4 | −91.8 | 18.1 | −42.0 | 28.4 | 10.5 |
| DNIB0600Ab | 25.1 | 18.0 | 0.889 | 11.9 | −51.3 | 16.0 | −12.5 | 25.1 | 39.4 |
| DEDN6526Ab | 18.3 | 22.0 | 1.06 | 8.67 | −154 | 11.7 | −88.0 | 18.3 | −20.2 |
| DMOT4039Ab | 30.0 | 27.0 | 0.965 | 14.2 | −90.1 | 19.1 | −41.4 | 30.0 | 11.1 |
| DSTP3086Sb | 26.0 | 17.4 | 0.866 | 12.3 | −41.5 | 16.6 | −4.82 | 26.0 | 49.4 |
| ADC2b | 21.0 | 17.6 | 0.942 | 9.91 | −77.6 | 13.4 | −31.3 | 21.0 | 19.3 |
| T‐DM1c | 10.1 | 8.68 | 0.950 | 4.78 | −81.6 | 6.44 | −34.8 | 10.1 | 16.4 |
| Brentuximab vedotinc | 18.5 | 23.9 | 1.09 | 8.75 | −173 | 11.8 | −103 | 18.5 | −29.2 |
| APE | 95.1 | 44.7 | 24.4 | ||||||
| RSS | 860 | 399 | 197 | ||||||
ADC, antibody–drug conjugate; APE, average absolute value of percentage prediction error (|PE|); CL, clearance; cyno, cynomologus monkeys; PE, percentage prediction errors, which was calculated by ((CLhuman, predicted − CLhuman, observed)/CLhuman, observed) × 100% for overprediction and ((CLhuman, predicted − CLhuman, observed)/CLhuman, predicted) × 100% for underprediction; PK, pharmacokinetic; RSS, residual sum of square.
aThe exponent w for the conjugate were back calculated based on the observed mean CL in cyno and in humans, mean ± SD of w = 0.966 ± 0.077. bConjugate was measured as conjugated drug; cConjugate was measured as conjugated antibody.
Human clearance prediction for total antibody analytes
Multiple species allometric scaling and allometric scaling method with correction factors
For total antibody analyte, there are eight ADCs with observed CL from humans and ≥2 nonclinical species available for the multiple species allometric scaling method. T‐DM1 and DSTP3086S have nonclinical PK from three animal species, whereas the remaining six ADCs have nonclinical PK data from only two animal species. Compared with observed human CL, multiple species allometric scaling overestimated human CL for five ADCs and underestimated human CL for three ADCs with %|PE| values ranging from 14–156% (Figure 1 ; Table 2 ). Three of eight ADCs had %|PE| value >100%, which is out of the twofold range of observed human CL (Figure 1 ; Table 2 ). Incorporation of correction factors (e.g., MLP or BrW) improved the total antibody CL prediction and decreased the %|PE| values for four ADCs in comparison with multiple species allometric scaling; however, incorporation of MLP or BrW as correction factors made the prediction worse for the remaining four ADCs (Figure 1 ; Table 2 ).
Figure 1.
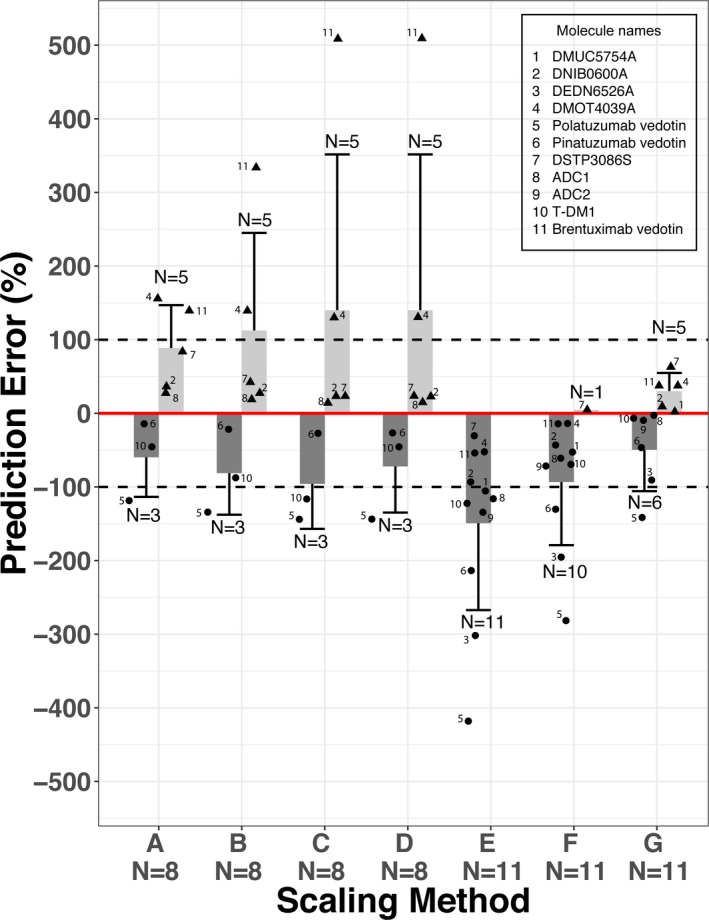
Accuracy of allometric scaling of human clearance (CL) of total antibody analytes for 11 antibody–drug conjugates (ADCs) from observed clearance using various scaling methods. (A) Multiple species allometric scaling, (B) allometric scaling with maximum life potential as correction factor, (C) allometric scaling with brain weight as correction factor, (D) allometric scaling based on rule of exponent, (E) scaling from cynomolgus monkey using a fixed exponent of clearance of 0.75, (F) scaling from cynomolgus monkey using a fixed exponent of clearance (CL) of 0.85, and (G) scaling from cynomolgus monkey using a fixed exponent of CL of 1.0. No rodent pharmacokinetic data are available for DMUC5754A, DEDN6526A, and ADC2, therefore, allometric scaling from cynomolgus monkey alone were performed for these three ADCs. Percentage prediction errors (PEs) is calculated as ((CLhuman, predicted − CLhuman, observed)/CLhuman, observed) × 100% for overprediction ( ) and ((CLhuman, predicted − CLhuman, observed)/CLhuman, predicted) × 100% for underprediction (
) and ((CLhuman, predicted − CLhuman, observed)/CLhuman, predicted) × 100% for underprediction ( ), respectively. The solid red line represents %PE = 0. The dashed lines represent %PE = 100% or −100% (i.e., twofold difference on CL). Light grey bar (
), respectively. The solid red line represents %PE = 0. The dashed lines represent %PE = 100% or −100% (i.e., twofold difference on CL). Light grey bar ( ) and whisker: mean + SD of PE for ADCs with positive PE (i.e., overprediction of human CL); dark grey bar (
) and whisker: mean + SD of PE for ADCs with positive PE (i.e., overprediction of human CL); dark grey bar ( ) and whisker: mean − SD of PE for ADCs with negative PE (i.e., underprediction of human CL); N above or below the whisker is number of ADCs with positive PE or negative PE; solid triangle (▲) represents individual PE for ADCs with positive PE (i.e., overprediction of human CL); solid dot (●) represents individual PE for ADCs with negative PE (i.e., underprediction of human CL).
) and whisker: mean − SD of PE for ADCs with negative PE (i.e., underprediction of human CL); N above or below the whisker is number of ADCs with positive PE or negative PE; solid triangle (▲) represents individual PE for ADCs with positive PE (i.e., overprediction of human CL); solid dot (●) represents individual PE for ADCs with negative PE (i.e., underprediction of human CL).
Allometric scaling based on rule of exponents
ROE was originally proposed by Mahmood8 for mAbs with nonclinical PK data available from three animal species. Given six of eight total antibody analyte PK were available from only two species, ROE was applied to the ADCs with nonclinical PK data from two or three species as long as multiple species allometric scaling can be performed. BrW correction was applied to seven of eight ADCs with exponents of multiple species allometric scaling >1. Scaling based on ROE improved total antibody CL prediction for four ADCs while making the prediction worse for three ADCs in comparison of multiple species allometric scaling. Three of eight ADCs had %|PE| > 100% (Figure 1 ; Table 2 ).
Scaling of CL and V in humans based on cynomolgus monkey data only
There are 11 ADCs with observed total antibody data from cynomolgus monkeys and humans. The scaling exponent, w, which is derived from Eq. (5) using the observed cynomolgus monkeys and human CL values of total antibody, ranged from 0.84–1.29 for the 11 ADCs (Table 3 ), with a mean ± SD value of 1.03 ± 0.14. A scaling exponent of 1.0 was, therefore, used for projecting human CL for total antibody analytes using cynomolgus monkey data alone, in addition to scaling exponents of 0.75 and 0.85.
As shown in Figure 1 and Table 3 , scaling from cynomolgus monkey data only using a fixed exponent of 0.75 underestimated the human total antibody CL with negative PE for all 11 ADCs. Seven of 11 ADCs had %|PE| value >100%. Scaling from cynomolgus monkey data only using a fixed exponent of 0.85 improved the prediction and decreased the %|PE| values for all 11 ADCs (Figure 1 ; Table 3 ); however, this method still underestimated human CL with negative PE for 10 of 11 ADCs. Scaling using a fixed exponent of 1.0 further improved the prediction and removed the systemic bias of underestimation with human CL of five ADCs overestimated and six underestimated. All 11 ADCs, except polatuzumab vedotin (see Discussion section), have human CL prediction within twofold of observation (Figure 1 ; Table 3 ). Among the seven scaling methods, scaling from only cynomolgus monkey PK with a fixed exponent of 1.0 demonstrated the smallest APE and had the least RSS, the two alternative statistical measures of overall goodness of prediction (Tables 2 and 3 ).
Human CL prediction for conjugate analytes
For the conjugate analyte, only the ADCs with the same conjugate measurement (either as conjugated antibody or as conjugated drug) in animals and humans were included in the analysis. Conjugated antibody was measured for brentuximab vedotin19 and T‐DM1,20 whereas conjugated drug was measured for the remaining six vc‐MMAE ADCs. The scaling exponent, w, which is derived from Eq. (5) using the observed cynomolgus monkey and human CL values of ADC conjugate, ranged from 0.866–1.09 for the eight ADCs (Table 4 ), with a mean ± SD value of 0.966 ± 0.077. A scaling exponent of 1.0 was, therefore, used for projecting human CL for conjugate analytes using cynomolgus monkey data alone. Additionally, scaling exponents of 0.75 and 0.85 were also explored.
As shown in Table 4 , scaling from cynomolgus monkey data only using a fixed exponent of 0.75 or 0.85 consistently underestimated the conjugate CL in humans with negative PE for all eight ADCs (Figure 2 ; Table 4 ). Scaling using a fixed exponent of 1.0 improved the prediction and removed the systemic bias of underestimation. Consistent observation was made regardless of conjugate analyte measurement methods (either as conjugated antibody or as conjugated drug). The predicted human CL using a fixed exponent of 1.0 was within 50% of the observed data for all the eight ADCs (Figure 2 ; Table 4 ). Among the three scaling methods, scaling from only cynomolgus monkey PK with a fixed exponent of 1.0 demonstrated the smallest APE and had the least RSS, the two alternative statistical measures of overall goodness of prediction (Table 4 ).
Figure 2.
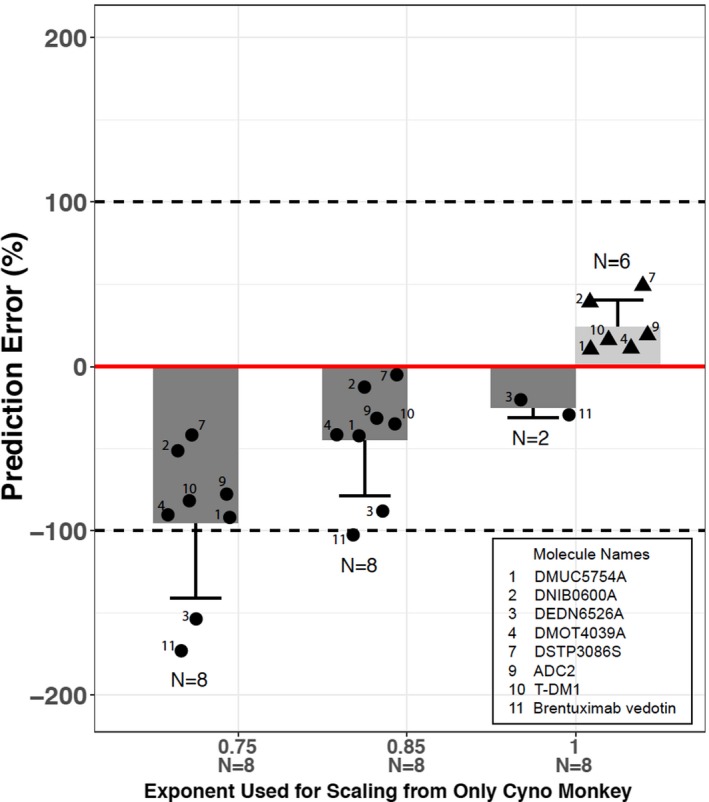
Accuracy of allometric scaling of human clearance (CL) of conjugate analytes for eight antibody–drug conjugates (ADCs) from observed clearance using various scaling exponents based on cynomolgus monkey data only. Percentage prediction errors (PEs) is calculated as ((CLhuman, predicted − CLhuman, observed)/CLhuman, observed) × 100% for overprediction ( ) and ((CLhuman, predicted − CLhuman, observed)/CLhuman, predicted) × 100% for underprediction (
) and ((CLhuman, predicted − CLhuman, observed)/CLhuman, predicted) × 100% for underprediction ( ), respectively. The solid red line represents %PE = 0. The dashed lines represent %PE = 100% or −100% (i.e., twofold difference on CL). Light grey bar (
), respectively. The solid red line represents %PE = 0. The dashed lines represent %PE = 100% or −100% (i.e., twofold difference on CL). Light grey bar ( ) and whisker: mean + SD of PE for ADCs with positive PE (i.e., overprediction of human CL); dark grey bar (
) and whisker: mean + SD of PE for ADCs with positive PE (i.e., overprediction of human CL); dark grey bar ( ) and whisker: mean − SD of PE for ADCs with negative PE (i.e., underprediction of human CL); N above or below the whisker is number of ADCs with positive PE or negative PE; solid triangle (▲) represents individual PE for ADCs with positive PE (i.e., overprediction of human CL); solid dot (●) represents individual PE for ADCs with negative PE (i.e., underprediction of human CL).
) and whisker: mean − SD of PE for ADCs with negative PE (i.e., underprediction of human CL); N above or below the whisker is number of ADCs with positive PE or negative PE; solid triangle (▲) represents individual PE for ADCs with positive PE (i.e., overprediction of human CL); solid dot (●) represents individual PE for ADCs with negative PE (i.e., underprediction of human CL).
The volume of distribution at steady state (Vss) of total antibody and conjugate analytes used in the study were observed to be ~ 89 and ~ 80 mL/kg, respectively, in both cynomolgus monkeys and humans, and seemed to be proportional to the BW (data not shown). Therefore, an exponent of 1.0 was used for scaling the Vss of total antibody and conjugate analyte using cynomolgus monkey data alone.
Projection of PK profiles in humans based on cynomolgus monkey data only
Using DNIB0600A as an example, the predicted human concentration‐time data for ADC conjugate and total antibody were scaled from cynomolgus monkeys using species‐invariant time method with exponent of 1.0 for CL and 1.0 for V (see Eqs. (6) and (7)). The predicted human concentration time profiles for DNIB0600A total antibody obtained by Monte‐Carlo simulation based on the projected human PK parameters, were consistent with observed phase I data (Figure 3 a; Table S1 ). Although a trend of underprediction was observed for DNIB0600A conjugate analyte, the predicted human concentration‐time profiles of DNIB0600A conjugate were largely consistent with observed data with majority of the observed concentrations falling within 95% prediction interval (Figure 3 b; Table S2 ).
Figure 3.
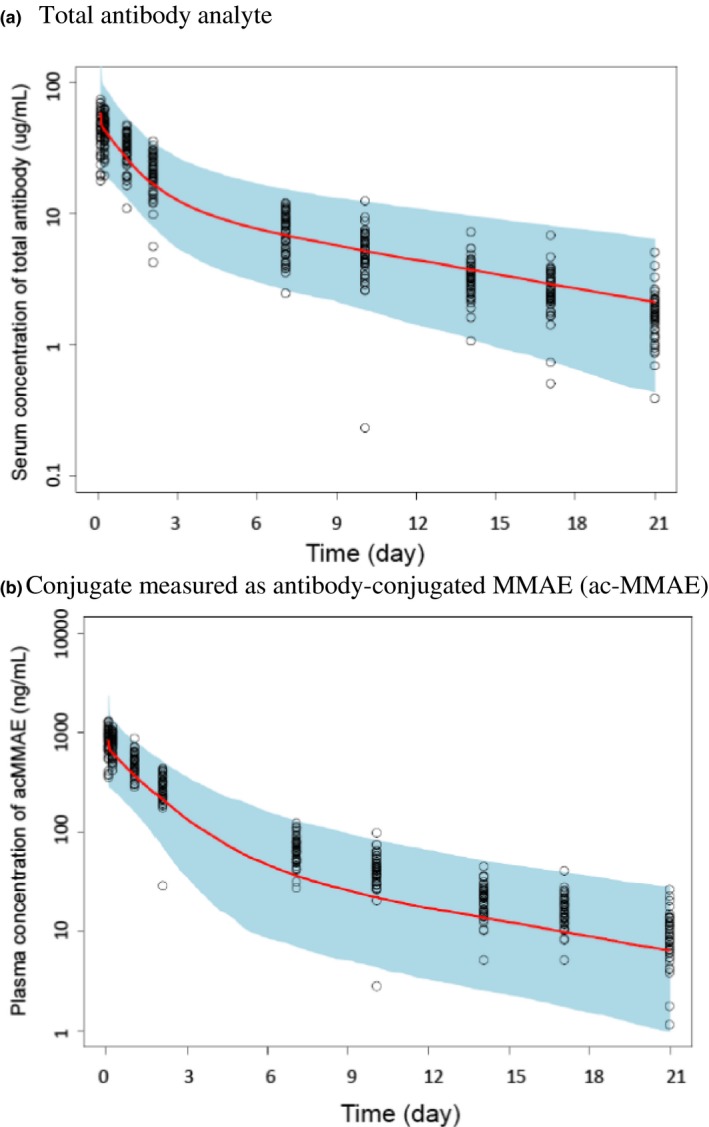
Observed (open circle) and predicted concentration‐time profiles of DNIB0600A at 2.4 mg/kg [median ( ), 2.5th to 97.5th percentile (blue shaded area)] in humans. (a) Total antibody analyte, (b) conjugate measured as antibody‐conjugated MMAE (ac‐MMAE). Concentration‐time profiles of total antibody or ac‐MMAE were scaled from cynomolgus monkey using species‐invariant time method approach with exponent of 1.0 and 1.0 for clearance and volume, respectively.
), 2.5th to 97.5th percentile (blue shaded area)] in humans. (a) Total antibody analyte, (b) conjugate measured as antibody‐conjugated MMAE (ac‐MMAE). Concentration‐time profiles of total antibody or ac‐MMAE were scaled from cynomolgus monkey using species‐invariant time method approach with exponent of 1.0 and 1.0 for clearance and volume, respectively.
Discussion
To date, there have been very few examples published on ADC interspecies scaling. Haddish‐Berhane et al.21 proposed to use the allometric exponent of 1 to predict human CL of an ADC from monkey data based on monkey and human PK comparison of three ADCs (i.e., T‐DM1, brentuximab vedotin, and inotuzumab ozogomycin). However, the detailed assessment to support the proposed scaling factor of 1 was not presented. Additionally, studies with two ADCs using ImmunoGen's SPDB as the linker and DM4 as the cytotoxic drug showed that 1 as an allometric exponent on CL worked well for one ADC SAR566658, whereas 0.75 worked better for another ADC SAR3419.22 Here, we systemically examined the human PK prediction for 11 ADCs using multiple scaling methods. Allometric scaling using three species for two ADCs and two species for six ADCs (Table 2 ) showed that projected total antibody CL values of most ADCs were generally inconsistent with observed values. The use of correction factors, such as BrW and MLP, as well as the application of ROE, did not improve the estimations with APE and RSS still relatively high (Figure 1 , Table 2 ). Allometric scaling from a single species (cynomolgus monkey) using a fixed exponent of 0.75 or 0.85 consistently underestimated the human CL for both analytes: conjugate analyte for eight ADCs and total antibody analyte for 11 ADCs (Figures 1 and 2 ). Notably, scaling from cynomolgus monkey only with a fixed scaling exponent of 1.0 on CL and V and the species‐invariant time method clearly demonstrated that this method can be reliably used to project human CL and concentration time profiles for both analytes prior to initiating first‐in‐human trials. In fact, as shown in Tables 2 , 3 , 4 , this method resulted in the lowest APE and RSS for both conjugate and total antibody analytes compared with other commonly used scaling methods.
It was noted that CL for total antibody analyte for polatuzumab vedotin were consistently underpredicted with %|PE| above 100% regardless of the methods used. This may be partly because total antibody CL of polatuzumab vedotin is not fully saturated at 2.4 mg/kg in some of the patients, especially for patients with high baseline B cell counts. Polatuzumab vedotin is a vc‐MMAE ADC against human CD79b receptor, a surface antigen that is expressed on B cells and B cell‐derived malignancies, including non‐Hodgkin lymphomas.23 Given polatuzumab vedotin does not cross react with cynomolgus monkey CD79b,24 PK of polatuzumab vedotin is linear in cynomolgus monkey with the total antibody CL reflecting nonspecific proteolytic degradation. In a phase I study in patients with relapse/refractory non‐Hodgkin lymphoma, most of the patients have low baseline B cell counts due to prior therapy, as a result, the PK of polatuzumab vedotin is approximately linear over the dose tested in clinics; however, a trend of faster CL was observed in individual patients with higher B cell counts and tumor burden,25 indicating target‐mediated CL may contribute to the CL of polatuzumab vedotin besides nonspecific protein degradation. Consequently, we conducted a sensitivity analysis to remove the patients with detectable baseline B cell counts. As expected, the observed human CL for total antibody analyte of polatuzumab vedotin was decreased and the accuracy of the human CL prediction was improved with %|PE| of 63% using scaling from cynomolgus monkeys only.
Given the greater sequence homology observed between nonhuman primates and humans as compared with rodents, the binding epitope, binding affinity to antigen, binding affinity to neonatal Fc receptor (FcRn), tissue cross‐reactivity profiles, and disposition and elimination pathways of mAbs are similar in nonhuman primates and humans.26 Therefore, it is not entirely unexpected that scaling of CL for ADCs from cynomolgus monkey alone provides the better human PK predictions than scaling using multiple animal species, just like what has been reported for mAbs.7, 8 It is worth noting that scaling exponent for ADCs from cynomolgus monkey PK data alone is different from that of mAbs (1.0 vs. 0.85). The apparent differences in scaling exponent between ADCs and mAbs are not fully understood but could be partly due to the differences in elimination pathways between the two. It was noted that CL of ADC total antibody analytes was usually faster and more variable across ADCs than CL of mAbs (~ 5–21 vs. ~ 3–6 mL/day/kg in humans for ADCs vs. mAbs7), indicating that the mAb component of the ADCs may undergo additional elimination pathway on top of target‐mediated and nonspecific proteolytic degradation that a typical mAb was eliminated through. In fact, Lyon et al.27 showed that ADCs with hydrophobic drug linkers (e.g., vc‐MMAE) undergo selective uptake by nonparenchymal cells (i.e., sinusoidal endothelium and Kupffer cells) of the liver, thus contributing to accelerated clearance of ADC total antibody analyte as compared with mAbs. Additionally, ADC conjugate analytes also undergo deconjugation CL pathway, which is usually mediated by enzymatic or chemical cleavage of the linker leading to the release of the cytotoxic drug from the ADCs.4, 28 It is possible that scaling for these additional CL pathways (e.g., selective hepatic uptake due to hydrophobicity or deconjugation clearance) from cynomolgus monkeys to humans is different from that of typical mAbs, thus resulting in different scaling exponents between ADCs and mAbs.
It is worth noting the limitation of the current analysis that only includes 11 ADCs with 10 of them using the same linker and cytotoxic drug (i.e., vc‐MMAE). Additionally, the conjugation chemistry used for the 11 ADCs results in a heterogeneous mixture of conjugated antibodies with an average DAR of 3–4 (Table 1 ). Given DAR distribution, linker‐cytotoxic drug and conjugation chemistry may impact the elimination of an ADC,4, 28 it remained to be assessed whether the current empirical scaling method is applicable for ADCs with site‐specific conjugation, different linker‐cytotoxic drugs, and DAR distribution.2
There are different methods for predicting concentration‐time profiles in humans from preclinical to clinical. Concentration‐time profiles in humans could be predicted by a compartmental PK approach using human PK parameters scaling up from nonclinical data. Although allometric scaling methods are often used in the prediction of human PK parameters (i.e., CL and Vss values), there has been little done on the systemic evaluation of scaling of distribution CL and/or micro constant (e.g., k 12 and k 21), which are required for projecting multiexponential PK profiles (concentration–time curves). The species‐invariant time method provides an alternative approach for the projection of full human PK profiles with specific dose and dosing regimens, which is valuable in drug development as drug efficacy and toxicity are usually linked to drug exposure.
A platform model published recently by Kagedal et al.29 showed that the PK of ADC conjugate measured as antibody‐conjugated‐MMAE was largely comparable across different vc‐MMAE ADCs, especially when the target‐mediated CL is saturated. Given the PK similarity of vc‐MMAE ADCs and majority of ADCs evaluated being vc‐MMAE ADCs, we believe that DNIB0600A, an anti‐Napi2b vc‐MMAE ADC (Table 1 ), serves as good representation of vc‐MMAE ADCs to evaluate the robustness of the species‐invariant time method to project human PK profiles based on cynomolgus monkey data only.
In summary, this analysis, based on a data set of 11 ADCs, demonstrates that scaling using a binding species, such as cynomolgus monkeys, is pharmacologically appropriate for therapeutic ADCs that demonstrate linear CL. Allometric scaling of CL using cynomolgus monkey alone with an exponent of 1.0 provided a good estimate of human CL for both ADC conjugate and total antibody analytes. Concentration‐time profiles of conjugate and total antibody analytes for ADCs in humans were also projected reasonably well based on PK data in cynomolgus monkeys using the species‐invariant time method.
Funding
The reported nonclinical and clinical studies were funded by Genentech, Inc./F. Hoffmann‐La Roche.
Conflict of Interest
C.L., Cindy Z., R.D., D.Li, D.L., B.L., Crystal Z., Z.L., D.M., S.C., D.S., B.W., P.A., D.L., S.P., S.G., and A.K. are paid employees of Genentech, Inc., a stockholder for Roche Holding Ltd. Y.G. is a paid consultant of Genentech, Inc.
Author Contributions
C.L., C.Z. (Cindy Zhang), and A.K. wrote the manuscript. C.L., R.D., D.L., S.P., S.G., and A.K. designed the research. C.L., D.L., D.L., B.L., C.Z. (Crystal Zhang), D.M., S.C., D.S., B.W., P.A., and D.L. performed the research. C.L., C.Z. (Cindy Zhang), R.D., Y.G., Z.L., and A.K. analyzed the data.
Supporting information
Table S1. Median, 2.5th, and 97.5th percentile of observed and predicted concentrations of total antibody over time in humans following the first dose of DNIB0600A at 2.4 mg/kg.
Table S2. Median, 2.5th, and 97.5th percentile of observed and predicted concentrations of conjugate analyte (measured as antibody‐conjugated MMAE (ac‐MMAE)) over time in humans following the first dose of DNIB0600A at 2.4 mg/kg.
Acknowledgments
Support for third‐party editing assistance was provided by Anshin Biosolutions.
Contributor Information
Chunze Li, Email: li.chunze@gene.com.
Amrita V. Kamath, Email: kamath.amrita@gene.com.
References
- 1. Hedrich, W.D. , Fandy, T.E. , Ashour, H.M. , Wang, H. & Hassan, H.E. Antibody‐drug conjugates: pharmacokinetic/pharmacodynamic modeling, preclinical characterization, clinical studies, and lessons learned. Clin. Pharmacokinet. 57, 687–703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Polakis, P. Antibody drug conjugates for cancer therapy. Pharmacol. Rev. 68, 3–19 (2016). [DOI] [PubMed] [Google Scholar]
- 3. Kaur, S. , Xu, K. , Saad, O.M. , Dere, R.C. & Carrasco‐Triguero, M. Bioanalytical assay strategies for the development of antibody‐drug conjugate biotherapeutics. Bioanalysis 5, 201–226 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Kamath, A.V. & Iyer, S. Challenges and advances in the assessment of the disposition of antibody‐drug conjugates. Biopharm. Drug Dispos. 37, 66–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiou, W.L. , Robbie, G. , Chung, S.M. , Wu, T.C. & Ma, C. Correlation of plasma clearance of 54 extensively metabolized drugs between humans and rats: mean allometric coefficient of 0.66. Pharm. Res. 15, 1474–1479 (1998). [DOI] [PubMed] [Google Scholar]
- 6. Feng, M.R. , Lou, X. , Brown, R.R. & Hutchaleelaha, A. Allometric pharmacokinetic scaling: towards the prediction of human oral pharmacokinetics. Pharm. Res. 17, 410–418 (2000). [DOI] [PubMed] [Google Scholar]
- 7. Deng, R. et al Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs 3, 61–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ling, J. et al Interspecies scaling of therapeutic monoclonal antibodies: initial look. J. Clin. Pharmacol. 249, 1382–1402 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Zou, P. et al Applications of human pharmacokinetic prediction in first‐in‐human dose estimation. AAPS J. 14, 262–281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davies, B. & Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 10, 1093–1095 (1993). [DOI] [PubMed] [Google Scholar]
- 11. Cho, C.Y. et al Pharmacokinetic scaling of bisphenol A by species invariant time methods. Xenobiotica 32, 925–934 (2002). [DOI] [PubMed] [Google Scholar]
- 12. Mahmood, I. Pharmacokinetic allometric scaling of antibodies: application to the first‐in‐human dose estimation. J. Pharm. Sci. 98, 3850–3861 (2009). [DOI] [PubMed] [Google Scholar]
- 13. West, G.B. & Brown, J.H. The origin of allometric scaling laws in biology from genomes to ecosystems: towards a quantitative unifying theory of biological structure and organization. J. Exp. Biol. 208, 1575–1592 (2005). [DOI] [PubMed] [Google Scholar]
- 14. Dedrick, R.L. Animal scale‐up. J. Pharmacokinet. Biopharm. 1, 435–461 (1973). [DOI] [PubMed] [Google Scholar]
- 15. Xu, J. et al Pharmacokinetics of an anti‐body‐drug conjugate (ADC) – DNIB0600A in a phase I study in patients with platinum‐resistant ovarian cancer (OC)/non‐small cell lung cancer (NSCLC). ASCO Annual Meeting (2013) May 31, 2013, Chicago, IL (2013).
- 16. Tang, H. & Mayersohn, M. A global examination of allometric scaling for predicting human drug clearance and the prediction of large vertical allometry. J. Pharm. Sci. 95, 1783–1799 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Advani, A. et al Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B‐cell non‐Hodgkin's lymphoma: results of a phase I study. J. Clin. Oncol. 28, 2085–2093 (2010). [DOI] [PubMed] [Google Scholar]
- 18. Dowell, J.A. , Korth‐Bradley, J. , Liu, H. , King, S.P. & Berger, M.S. Pharmacokinetics of gemtuzumab ozogamicin, an antibody‐targeted chemotherapy agent for the treatment of patients with acute myeloid leukemia in first relapse. J. Clin. Pharmacol. 41, 1206–1214 (2001). [DOI] [PubMed] [Google Scholar]
- 19. Center for Drug Evaluation and Research . Clinical Pharmacology and Biopharmaceutics Review (Adcetris, Silver Spring, MD, 2011) <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125388Orig1s000ClinPharmR.pdf>. Accessed August 19, 2018. [Google Scholar]
- 20. Girish, S. et al Clinical pharmacology of trastuzumab emtansine (T‐DM1): an antibody‐drug conjugate in development for the treatment of HER2‐positive cancer. Cancer Chemother. Pharmacol. 69, 1229–1240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haddish‐Berhane, N. et al On translation of antibody drug conjugates efficacy from mouse experimental tumors to the clinic: a PK/PD approach. J. Pharmacokinet. Pharmacodyn. 40, 557–571 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Bouillon‐Pichault, M. et al Translational model‐based strategy to guide the choice of clinical doses for antibody‐drug conjugates. J. Clin. Pharmacol. 57, 865–875 (2017). [DOI] [PubMed] [Google Scholar]
- 23. Palanca‐Wessels, M.C. et al Safety and activity of the anti‐CD79B antibody‐drug conjugate polatuzumab vedotin in relapsed or refractory B‐cell non‐Hodgkin lymphoma and chronic lymphocytic leukaemia: a phase 1 study. Lancet Oncol. 16, 704–715 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Zheng, B. et al In vivo effects of targeting CD79b with antibodies and antibody‐drug conjugates. Mol. Cancer Ther. 8, 2937–2946 (2009). [DOI] [PubMed] [Google Scholar]
- 25. Lu, D. et al Population Pharmacokinetics and Exposure‐Response Assessment of Anti‐CD79B Antibody Drug Conjugate in Patients: Interim Analysis Results – Poster PII 095. American College of Clinical Pharmacology Annual Meeting (2015). 10.1002/cpt.52 [DOI]
- 26. Deng, R. , Jin, F. , Prabhu, S. & Iyer, S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin. Drug Metab. Toxicol. 8, 141–160 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Lyon, R.P. et al Reducing hydrophobicity of homogeneous antibody‐drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 33, 733–735 (2015). [DOI] [PubMed] [Google Scholar]
- 28. Lin, K. & Tibbitts, J. Pharmacokinetic considerations for antibody drug conjugates. Pharm. Res. 29, 2354–2366 (2012). [DOI] [PubMed] [Google Scholar]
- 29. Kagedal, M. et al Platform model describing pharmacokinetic properties of vc‐MMAE antibody‐drug conjugates. J. Pharmacokinet. Pharmacodyn. 44, 537–548 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Liu, J.F. et al Phase I study of safety and pharmacokinetics of the anti‐MUC16 antibody‐drug conjugate DMUC5754A in patients with platinum‐resistant ovarian cancer or unresectable pancreatic cancer. Ann. Oncol. 27, 2124–2130 (2016). [DOI] [PubMed] [Google Scholar]
- 31. Li, C. et al Clinical pharmacokinetics of DEDN6526A, an anti‐endothelin B receptor (ETBR) antibody‐ drug conjugate (ADC) in patients (pts) with metastatic or unresectable melanoma: results from a first‐in‐human phase I study. American Association of Pharmaceutical Scientists National Biotechnology Conference June 8, 2015, San Francisco, CA (2015).
- 32. Weekes, C.D. et al Phase I study of DMOT4039A, an antibody‐drug conjugate targeting mesothelin, in patients with unresectable pancreatic or platinum‐resistant ovarian cancer. Mol. Cancer Ther. 15, 439–447 (2016). [DOI] [PubMed] [Google Scholar]
- 33. Fuh, F.K. et al Anti‐CD22 and anti‐CD79b antibody‐drug conjugates preferentially target proliferating B cells. Br. J. Pharmacol. 174, 628–640 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Advani, R.H. et al Phase I study of the anti‐CD22 antibody‐drug conjugate pinatuzumab vedotin with/without rituximab in patients with relapsed/refractory B‐cell non‐Hodgkin lymphoma. Clin. Cancer Res. 23, 1167–1176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang, B. et al Pharmacokinetics (PK) of DSTP3086S, anti‐Steap1 antibody drug conjugate (ADC) in patients with Metastatic Castration‐Resistant Prostate Cancer: Results from a first‐in‐human Phase I study. American College of Clinical Pharmacology Annual Meeting (2017). September 17, 2017, San Diego, CA
- 36. Center for Drug Evaluation and Research . Pharmacology Review. (Adcetris, Silver Spring, MD, 2011) <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125388Orig1s000PharmR.pdf>. Accessed August 19, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Median, 2.5th, and 97.5th percentile of observed and predicted concentrations of total antibody over time in humans following the first dose of DNIB0600A at 2.4 mg/kg.
Table S2. Median, 2.5th, and 97.5th percentile of observed and predicted concentrations of conjugate analyte (measured as antibody‐conjugated MMAE (ac‐MMAE)) over time in humans following the first dose of DNIB0600A at 2.4 mg/kg.