

Abstract
A new thermal isomerization of polyynes is described. Benzyne intermediates substituted by a C(RR’)OR” substituent adjacent to one of the benzyne sp-hybridized carbons give rise to products in which the OR” moiety has migrated to the proximal benzyne carbon. This process likely proceeds via sequential formation of multiple reactive intermediates: an initial thermally generated benzyne, a strained benzoxetenonium ion, and an o-quinone methide. As some examples demonstrate, the overall transformation can be quite efficient. The mechanism of this novel reaction is further supported by experiments and DFT calculations.
Graphical abstract
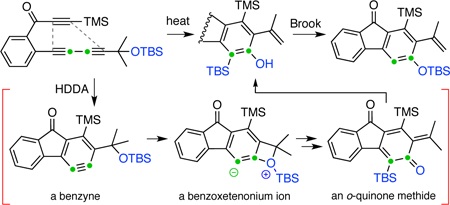
Quinone methides (QMs) are reactive intermediates of interest from the perspectives of preparative, mechanistic, and structural organic chemistry as well as their roles in chemical biology (e.g., biosynthetic pathways, prodrug cleavage, and electrophilic capture of biological nucleophiles). 1 QMs are often generated by eliminative processes of phenol derivatives or by photochemical reactions (including reversible generation in photochromic molecules). Besides these two methods, benzyne, itself a versatile reactive intermediate, can also generate o-QMs by reaction with the C=O π-bond in carbonyl-containing functional groups (Figure 1a). 2 For example, Yoshida and co-workers demonstrated a coupling reaction of two arynes with aldehydes to give 9-arylxanthene derivatives through QM intermediates. They, as well as Miyabe’s group, have also reported insertion reactions of arynes into formamide to afford coumarin derivatives following in situ trapping of the o-QM by esters, nitriles, and ketones.
Figure 1.
Generation of o-quinone methides via benzynes.
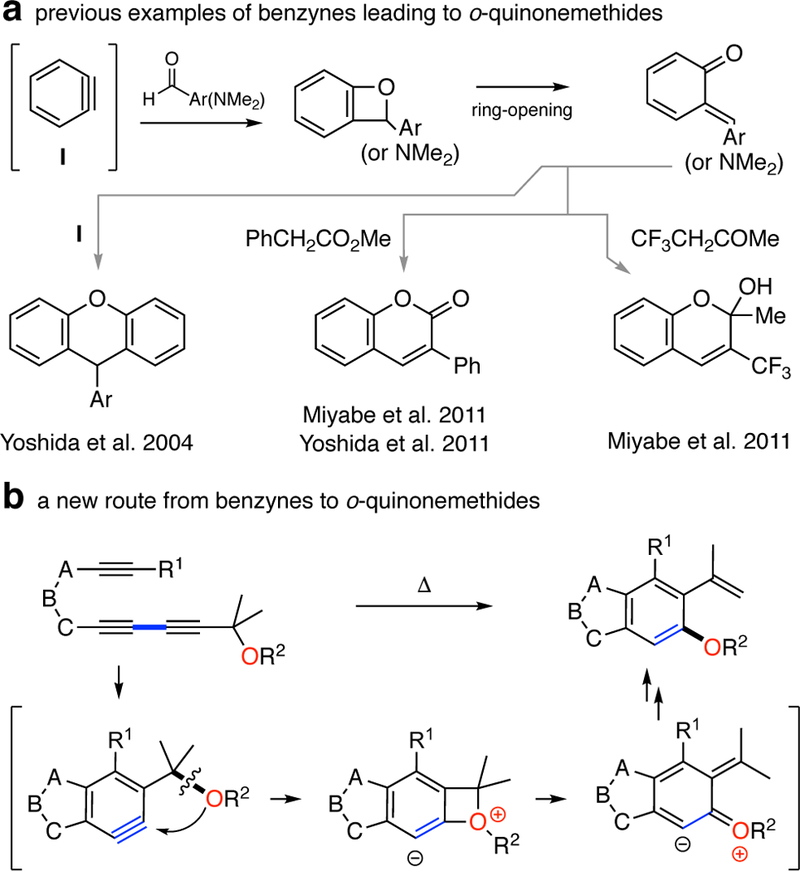
We now report a reaction (Figure 1b) that represents an alternative mechanistic paradigm for producing o-QMs in which a thermally generated benzyne derivative bearing a benzylic ether group can produce a four-membered benzoxetenonium 1,3-zwitterion (middle structure in the brackets in Figure 1b) by an intramolecular nucleophilic attack to the strained aryne bond. Electrocyclic ring-opening of the oxetene ring3 affords an o-substituted o-QM intermediate, and a subsequent hydrogen atom migration gives a formal C—O bond insertion product. In this process, the steric bulk of the R1 group is crucial, possibly facilitating passage through the strained four-membered ring transition state geometry that leads to the strained four-membered zwitterion.4 Indeed, when R1 is a hydrogen, the reaction proceeds through other pathways. 5 The highly substituted benzyne intermediate is accessed conveniently from a triyne precursor via a cycloisomerization event, namely, the hexadehydro-Diels-Alder (HDDA) reaction,6 and therefore, the overall cascade is essentially a net thermal isomerization of the triyne.
The first time we encountered this unusual transformation was upon heating the ynone substrate 1a. The fluorenone derivative 2a was produced in excellent yield (Chart 1). We proposed a mechanism to account for this outcome, which is shown in Figure 2. Initially, triyne 1a undergoes a HDDA reaction to generate benzyne 3, which is trapped by the pendant benzylic silyl ether to give the benzoxetenonium species 4. Silyl migration to the anionic carbon in this zwitterion (a retro-Brook-like rearrangement) could occur at this stage, although the stereoelectronic features for that process appear to be much more favorable following ring-opening to the O-silylated o-QM 5. Moreover, that electrocyclic opening itself might be expected to be quite rapid because it is accompanied by delocalization of the cationic portion of the zwitterion (cf. the computed TS2 in Figure 3, which suggests that this process is barrierless). A [l,5]-hydrogen atom shift in the neutral QM7 6 yields the o-silylated phenol 7. We presumed that under the reaction conditions for this experiment (150 °C, 14 h), this species had rearranged (see later discussion) to the observed product 2a. Similar thermal Brook rearrangements of o-silylated phenols have been reported.8 Due to the size of each of the two substituents at OR2 the termini of triyne 1 (blue and red colors), the ease of these HDDA cycloisomerizations—necessarily the rate-limiting step in each case—required more elevated temperatures to form the benzyne compared to analogous ynone-triyne substrates.9 The transformation was shown to be general, as evidenced by the examples of substrates 1b-h leading to 2b-h. Propargylic substitution other than geminal dimethyl in 1 is tolerated (cf. 2d-e). The R2 group can be alkyl in addition to a silyl substituent (2f). When it is acetyl (cf. 1h), two products, 2h and 2h’, were observed. The latter could arise by either an intermolecular transfer or an intramolecular, Fries-like process.
Chart 1.
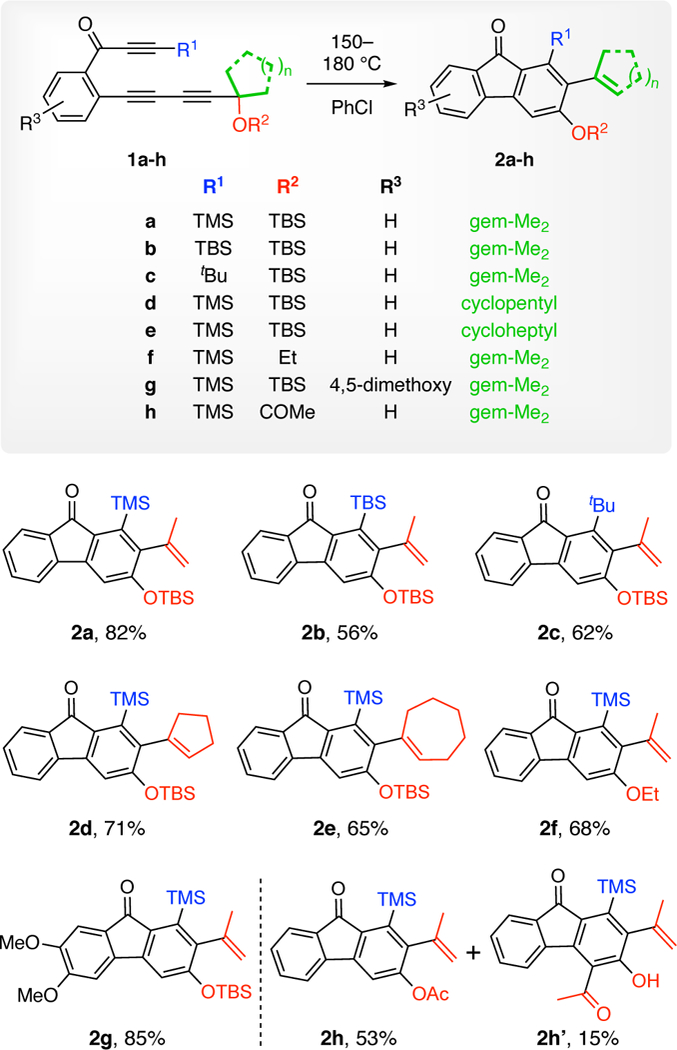
The major products (2a-h) isolated from reactions of triyne precursors 1a-h.a
a Yields are for isolated, purified (SiO2) material.
Figure 2.
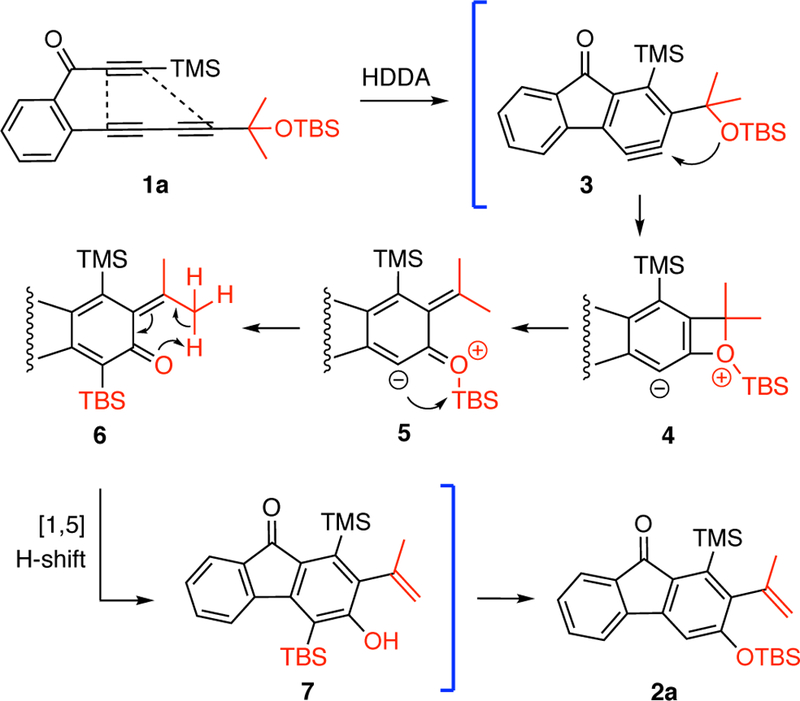
A proposed mechanism for the conversion of 1a to 2a.
Figure 3.
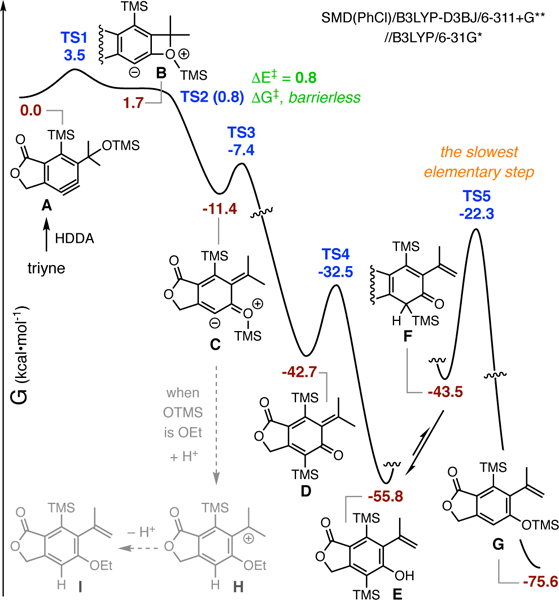
Computed energy profile (See Figure S1 in Supporting Information for more details).
DFT calculations [SMD(PhCl)/B3LYP-D3BJ/6– 311+G**//B3LYP/6–31G*, Figure 3] were used to further explore the mechanistic thinking laid out in Figure 2. A slightly simplified and symmetrized [e.g., TMS instead of TBS and an ester linker (cf. 8a, later)] set of structures was used. The HDDA-produced benzyne A is seen to cyclize to the benzoxetenonium ion B with a very low barrier (TS1, 3.5 kcaL·mol−1), although this strained species is 1.7 kcaL·mol−1 higher in free energy than the benzyne. This proceeds by way of an extremely facile, indeed, barrierless, electrocyclic ring-opening to afford the more stable zwitterion C. Subsequent retro-Brook reaction within the zwitterion C results in formation of the o-QM D via another low-barrier process (TS3, ΔG≠ = 4.0 kcal·mol−1). A 1,5-hydrogen atom shift in o-QM 004 gives the phenol E via TS4 (ΔG≠ = 10.2 kcal·mol−1). The keto tautomer F is seen to be less stable than the phenol by only 12.3 kca·mol−1, although we do not know the specifics of the mediator that is promoting this symmetry-forbidden 1,3-hydrogen atom migration. By contrast, processes like the 1,3-silyl migration of F to the silyl ether product G are known to be symmetry allowed and concerted (cf. TS5, ΔG≠ = 21.2 kcal·mol−1).10 Finally, note that when the R group attached to the oxonium ion is not a silyl substituent (e.g., Et in 2f), Et migration is not observed. Instead, a protonation/deprotonation sequence via the benzylic carbenium ion H is presumed to lead to products like I (i.e., 2f).
To address the question of whether the transformation of 1 to 2 is tolerant of triyne precursors containing other types of linkers, we examined the HDDA substrates 8a-d (Chart 2). Once again we observed that the benzyne formation step in these substrates is slower than expected based on analogous triynes that we have previously studied having the same linker but a less bulky substituent (CH2R) on the terminal diyne.9 The trapping nucleophile is not limited to oxygen. The tertiary amino group in substrate 8b migrated to give 9b in an analogous fashion to the reaction of the ethoxylated analog (cf. 1f to 2f), now by accessing a four-membered benzazetene species. In the case of 8c, the silylated phenol product 9c was formed cleanly (cf. 2h’). We do not have a good explanation for the reluctance of this compound to undergo the Brook rearrangement (it remained intact even following recording of the melting point (= 235–237 °C).
Chart 2.
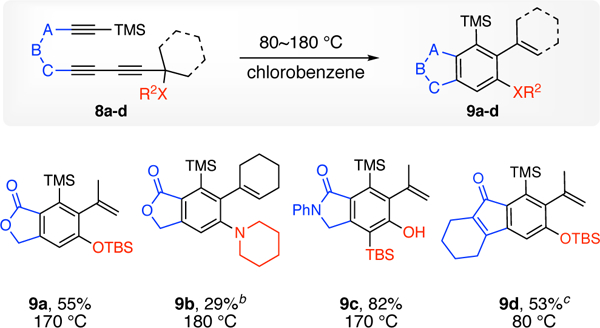
The major products (9a-d) isolated from triyne precursors 7a-da.
aYields are for isolated, purified (SiO2) material. bDCB was the reaction solvent; 9b was the major compound seen in the crude product mixture (1H NMR analysis). cChloroform was the reaction solvent.
1H NMR spectroscopy was used to monitor the progress of the reaction of 1a (Figure 4a). The consumption of 1a proceed with a half-life that is estimated to be 47 min at 150 °C in chlorobenzene. As a comparison, the cyclization half-life of another ynone substrate with the same linker structure is ca. 5 h at 80 °C.9 Intriguingly, at an early stage (30 min) of conversion, we observed quite clean the formation of phenol 7. As the reaction proceeded (60 and 110 min), the signal of silyl ether 2a started to appear and increase. Eventually (350 min), most of 7 was isomerized to the thermodynamically more stable constitutional isomer 2a. This NMR study clearly suggests that phenol 7 indeed is a metastable intermediate, the initially formed product arising from rearomatization of the o-QM 6, which then undergoes rearrangement to 2a.8
Figure 4.
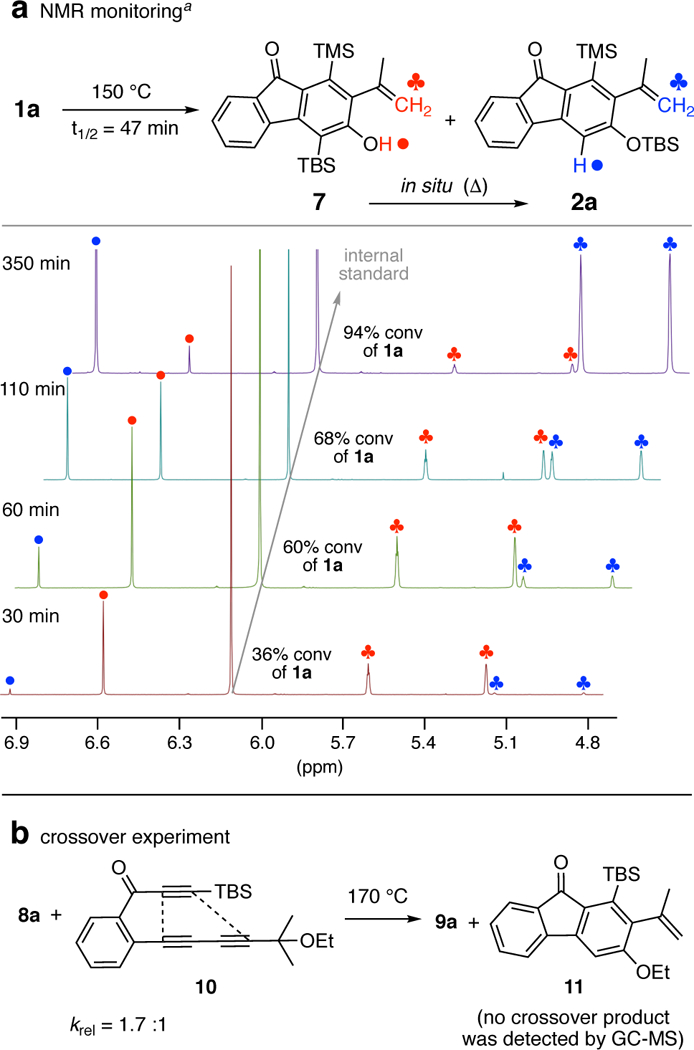
1H NMR monitoring and crossover experiment. a1,3,5-Trimethoxybenzene was used as an internal standard (IS) to monitor the conversion of 1a; the indicated %conversions were measured by integration of a unique aromatic resonance for 1a vs. the IS.
Finally, to rule out the possibility of a bimolecular silyl or alkyl group transfer, we carried out a crossover experiment using triynes 8a and 10, whose cyclization half-lives are comparable. No evidence of either of the two possible crossover products was observed after full consumption of both 8a and 10 (Figure 4b). This supports the mechanism in which an intramolecular retro-Brook rearrangement of 5 produces o-QM 6 (Figure 2).
In conclusion, we have shown that certain polyyne substrates can efficiently undergo a tandem thermal isomerization via an o-QM intermediate. This represents a new pathway of formation of this class of intermediate. The scope of the reaction was briefly demonstrated (Charts 1-2), and a plausible mechanistic pathway was proposed (Figure 2). Furthermore, DFT calculations and mechanistic studies provided additional support of the proposed mechanism (Figure 3–4). This represents another example of the HDDA reaction serving as a platform to support fundamentally new types of reactivity.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the Institute of General Medical Sciences of the U.S. Department of Health and Human Services (R35 GM127097) and the National Science Foundation (CHE-1665389). Most of the NMR data were obtained with an instrument acquired through the NIH Shared Instrumentation Grant program (S10OD011952).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures for all new reactions; spectroscopic characterization data for all new compounds; results of DFT computations (3D structures, geometries, and energies of all minima and maxima); copies of 1H and 13C NMR spectra (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Van De Water RW; Pettus TR R o-Quinone Methides: Intermediates Underdeveloped and Underutilized in Organic Synthesis. Tetrahedron 2002, 58, 5367–5405. [Google Scholar]; (b) Ferreira SB; Silva FDCD; Pinto AC; Gonzaga DTG; Ferreira VF Syntheses of Chromenes and Chromanes via o-Quinone Methide Intermediates. J. Heterocycl. Chem. 2009, 46, 1080–1097. [Google Scholar]; (c) Singh MS; Nagaraju A; Anand N; Chowdhury S ortho-Quinone Methide (o-QM): A Highly Reactive, Ephemeral and Versatile Intermediate in Organic Synthesis. RSC Adv. 2014, 4, 55924–55959. [Google Scholar]
- (2).(a) Yoshida H; Watanabe M; Fukushima H; Ohshita J; Kunai AA 2:1 Coupling Reaction of Arynes with Aldehydes via o-Quinone Methides: Straightforward Synthesis of 9-Arylxanthenes. Org. Lett. 2004, 6, 4049–40051. [DOI] [PubMed] [Google Scholar]; (b) Yoshida H; Ito Y; Ohshita J Three-component Coupling Using Arynes and DMF: Straightforward Access to Coumarins via ortho-Quinone Methides. Chem. Commun. 2011, 47, 8512–8514. [DOI] [PubMed] [Google Scholar]; (c) Yoshioka E; Kohtani S; Miyabe H A Multicomponent Coupling Reaction Induced by Insertion of Arynes into the C=O Bond of Formamide. Angew. Chem., Int. Ed. 2011, 50, 6638–6642. [DOI] [PubMed] [Google Scholar]
- (3).(a) Adam W; Hadjiarapoglou L; Peters K; Sauter M Dimethyldioxirane Epoxidation of Benzofurans: Reversible Thermal and Photochemical Valence Isomerization between Benzofuran Epoxides, Quinone Methides, and Benzoxetenes. J. Am. Chem. Soc. 1993, 115, 8603–8608. [Google Scholar]; (b) Tomioka H; Matsushita T Benzoxetene. Direct Observation and Theoretical Studies. Chem. Lett. 1997, 399–400. [Google Scholar]
- (4).Gupta S; Lin Y; Xia Y; Wink D; Lee D Alder-Ene Reactions Driven by High Steric Strain and Bond Angle Distortion to Form Benzocyclobutenes. Chem. Sci. 2019, DOI: 10.1039/c8sc04277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Xiao X; Woods BP; Xiu W; Hoye TR Benzocyclobutadienes: An Unusual Mode of Access Reveals Unusual Modes of Reactivity. Angew. Chem., Int. Ed. 2018, 57, 9901–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Miyawaki K; Suzuki R; Kawano T; Ueda I Cycloaromatization of a Non-Conjugated Polyenyne System. Tetrahedron Lett. 1997, 38, 3943–3946. [Google Scholar]; (b) Bradley AZ; Johnson RP Thermolysis of 1,3,8-Nonatriyne: Evidence for Intramolecular [2 + 4] Cycloaromatization to a Benzyne Intermediate. J. Am. Chem. Soc. 1997, 119, 9917–9918. [Google Scholar]; (c) Tsui JA; Sterenberg BT A Metal Templated 4 + 2 Cycloaddition Reaction of an Alkyne and a Diyne To Form a 1,2-Aryne. Organometallics 2009, 28, 4906–4908. [Google Scholar]; (d) Yun SY; Wang K-P; Lee N-K; Mamidipalli P; Lee D Alkane C-H Insertion by Aryne Intermediates with a Silver Catalyst. J. Am. Chem. Soc. 2013, 135, 4668–4671. [DOI] [PubMed] [Google Scholar]; (e) Hoye TR; Baire B; Niu D; Willoughby PH; Woods BP The hexadehydro-Diels-Alder reaction. Nature, 2012, 490, 208–212. For a review: [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Diamond OJ; Marder TB Methodology and applications of the hexadehydro-Diels-Alder (HDDA) reaction. Org. Chem. Front. 2017, 4, 891–910. [Google Scholar]
- (7).Bai W-J; David JG; Feng Z-G; Weaver MG; Wu K-L; Pettus TRR The Domestication of ortho-Quinone Methides. Acc. Chem. Res. 2014, 47, 3655–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Copper GD Preparation and Thermal Rearrangement of Poly(trimethylsilyl)phenols. J. Org. Chem. 1961, 26, 925–929. [Google Scholar]; (b) Eastham SA; Ingham SP; Hallett MR; Herbert J; Quayle P; Raftery J A Formal Synthesis of Aflatoxin B2: a Dötz Benzannulation Approach. Tetrahedron Lett. 2006, 47, 2299–2304. [Google Scholar]; (c) Austin WF; Zhang Y; Danheiser RL A Benzannulation Strategy for the Synthesis of Phenols and Heteroaromatic Compounds Based on the Reaction of (Trialkylsilyl)vinylketenes with Lithium Ynolates. Tetrahedron 2008, 64, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schön W; Messinger J; Solodenko W; Kirschning A Synthetic Approaches towards 4-Functionalized Estrone Derivatives. Synthesis 2012, 44, 3822–3828. [Google Scholar]
- (9).Woods BP; Baire B; Hoye T R Rates of Hexadehydro-Diels-Alder (HDDA) Cyclizations: Impact of the Linker Structure. Org. Lett. 2014, 16, 4578–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Brook AG; MacRae DM; Limburg WW Stereochemistry of the Formation and Thermal Rearrangement of ß-Ketosilanes. J. Am. Chem. Soc. 1967, 89, 5493–5495. [Google Scholar]; (b) Slutsky J; Kwart H The Kinetics, Stereochemistry, and Mechanisms of the Silaallylic and Silapropynylic Rearrangements. J. Am. Chem. Soc. 1972, 95, 8678–8685. [Google Scholar]; (c) Yamabe T; Nakamura K; Shiota Y; Yoshizawa K; Kawauchi S; Ishikawa M Novel Aspects of the [1,3] Sigmatropic Silyl Shift in Allylsilane. J. Am. Chem. Soc. 1997, 119, 807–815. [Google Scholar]; (d) Takahashi M; Kira M J. Am. Chem. Soc. 1999, 121, 8597–8603. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.